

Abstract
Brainstem A2/C2 catecholamine (CA) neurons in the solitary tract nucleus (NTS) are thought to play an important role in the control of food intake and other homeostatic functions. We have previously demonstrated that these neurons, which send extensive projections to brain regions involved in the regulation of appetite, are strongly and directly activated by solitary tract (ST) visceral afferents. Ghrelin, a potent orexigenic peptide released from the stomach, is proposed to act in part through modulating NTS CA neurons but the underlying cellular mechanisms are unknown. Here we identified CA neurons using transgenic mice that express enhanced green florescent protein driven by the tyrosine hydroxylase promoter (TH-EGFP). We then determined how ghrelin modulates TH-EGFP neurons using patch clamp techniques in a horizontal brain slice preparation. Ghrelin inhibited the frequency of spontaneous glutamate inputs (sEPSCs) onto TH-EGFP neurons, including cholecystokinin-sensitive neurons, an effect blocked by the GHSR1 antagonist, D-Lys-3-GHRP-6. This resulted in a decrease in the basal firing rate of NTS TH-EGFP neurons, an effect blocked by the glutamate antagonist NBQX. Ghrelin also dose-dependently inhibited the amplitude of ST afferent evoked EPSCs (ST-EPSCs) in TH-EGFP NTS neurons, decreasing the success rate for ST-evoked action potentials. In addition, ghrelin decreased the frequency of mini-EPSCs suggesting its actions are pre-synaptic to reduce glutamate release. Lastly, ghrelin’s inhibition of the ST-EPSCs was significantly increased by an 18 hour fast. These results demonstrate a potential mechanism by which ghrelin inhibits NTS TH neurons through a pathway whose responsiveness is increased during fasting.
Keywords: NTS, ghrelin, catecholamine, fasted, visceral, CCK
Introduction
The solitary tract nucleus (NTS) is the primary site through which gastrointestinal afferent information enters the brain (Browning and Travagli, 2010; Moran et al., 2001; Berthoud, 2008; Grill and Hayes, 2009). TheA2/C2 catecholamine (CA) neurons lie within the NTS and are thought to be important for the control of food intake as well as other homeostatic functions. Ingestion of a meal and gastric distention activate gene transcription in these neurons, as do the anorexigens cholecystokinin, PYY, serotonin and leptin (Rinaman et al., 1995; Monnikes et al., 1997; Willing and Berthoud, 1997; Rinaman et al., 1998; Blevins et al., 2008; Williams et al., 2008; Lam et al., 2009). Furthermore, NTS-CA neurons project to multiple brain regions important for appetite control, including the hypothalamus, nucleus accumbens and dorsal motor nucleus of the vagus (DMNV), making them ideally situated to pass on the afferent information about satiety to downstream targets (Sawchenko and Swanson, 1981; Wang et al., 1992; Rogers et al., 2003; Reyes and Van Bockstaele, 2006). Injection of saporin toxin conjugated to dopamine-beta-hydroxylase into the NTS ablates NTS-CA neurons (as well as presumably any catecholamine neurons projecting to the NTS (Ritter et al., 2001; Madden and Sved, 2003)), attenuating CCK and LiCl-induced satiety (Rinaman, 2003; Rinaman and Dzmura, 2007). However, the mechanisms by which appetite-controlling hormones regulate these neurons remain largely unknown.
Ghrelin is a potent orexigenic hormone produced by the upper GI tract (Kojima et al., 1999) whose circulating levels increase before a meal and decrease with feeding, and that is widely proposed as an important indicator of energy insufficiency (Castaneda et al., 2010; Kojima and Kangawa, 2010; Zigman and Elmquist, 2003; Cummings, 2006). Ghrelin stimulates food intake in part through regulation of hypothalamic neurons (Castaneda et al., 2010; Fry and Ferguson, 2010; Kojima and Kangawa, 2010; Zigman and Elmquist, 2003; Cummings, 2006). However, mRNA and protein for ghrelin’s receptor, growth hormone secretagogue receptor (GHSR), are also found in the NTS (Lin et al., 2004; Zigman et al., 2006) and injections of ghrelin directly into the dorsal vagal complex stimulates feeding (Faulconbridge et al., 2003). GHSRs are also expressed on vagal afferents and some reports suggest that vagal afferents are required for ghrelin’s effects on food intake (Date et al., 2002; Date et al., 2006), although this remains controversial (Arnold et al., 2006). The effects of ghrelin on food intake are lost in human patients with surgical procedures involving vagotomy (le Roux et al., 2005). Brainstem CA neurons have been reported to be critical for ghrelin-induced food intake (Date et al., 2006), but this is also controversial (Emanuel and Ritter, 2010) and how ghrelin regulates NTS-CA neurons is not clear.
Propagation of afferent information to downstream targets by NTS neurons depends on the translation of solitary tract (ST) afferent inputs into spike activity. We have previously shown that 90% of TH-EGFP neurons are directly activated by ST afferents (Appleyard et al., 2007). Here we determine how ghrelin modulates afferent activation of TH-EGFP neurons and whether its effects are altered by fasting.
Materials and Methods
NTS slices
Hindbrains of both male and female TH-EGFP mice (6–20 weeks old) were prepared as previously described (Appleyard et al., 2007). All animal procedures were conducted with the approval of the Animal Care and Use Committees at WSU and in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Guide). The hindbrain was removed and placed for one minute in cold (0–4° C) artificial cerebral spinal fluid composed of (mM): 125 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 10 dextrose, 2 CaCl2, and bubbled with 95%O2/5%CO2. The osmoloarity was adjusted to 301–305 mOsm using dextrose, except for the studies comparing fed and fasted mice where slices from both ad lib fed and fasted mice were incubated in solutions where the dextrose concentration was kept at 5mM and the osmolarity adjusted to 301–305mOsm using sucrose. The medulla was trimmed to a 2 cm block (rostral-caudal) centered on the obex. A wedge of tissue was removed from the ventral surface to align the ST with the cutting plane when mounted in a vibrating microtome (Leica VT-1000S). Slices (250 μm thick) cut with a sapphire knife (Delaware Diamond Knives) contained the ST in the same plane as the NTS. Slices were submerged in a perfusion chamber and all recordings performed at 31–35 ° C and pH 7.4. Neurons were visualized using an upright microscope (Olympus BX51). Recording electrodes were filled with a solution (mM): 10 NaCl, 130 K gluconate, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 HEPES, 2 NaATP, 0.2 NaGTP; pH 7.3; 297–301 mOsm. Neurons were recorded from NTS within 200 μm rostral or caudal from obex and medial to the ST - medial NTS (mNTS). Patch electrodes, 3–5 MΩ, were guided to neurons using differential interference contrast (DIC) optics illuminated with infrared light (Olympus B51). Voltage clamp recordings were made with an Axopatch 700B (Molecular devices), Digidata 1440A digitizer (Molecular devices) and pClamp 10 software (Molecular devices). Only neurons with holding currents not exceeding 50 pA at VH= −60 mV for the 15 minute control period (input resistance >150 MΩ) were studied further. Series resistance was monitored throughout the recordings and neurons were not included in further analysis if it exceeded 20 MΩ, or drifted >25%. Series resistance did not differ between control (ACSF) and ghrelin treatment. Synaptic currents were evoked with an ultrafine concentric bipolar stimulating electrode (50 μm ID, F. Haer) placed on the ST 1–3 mm from the recording electrode. Electrical stimuli were delivered from an isolated programmable stimulator (Isoflex stimulator, AMPI with Master-8, AMPI) triggered to deliver a burst of stimuli (5 – 50 Hz). All drugs were obtained from Tocris Cookson (Ballwin, MO) or Sigma (St Louis, MO). All membrane potentials reported are corrected for junction potentials.
TH immunofluorescent histochemistry
Mice (6–20 weeks) were anesthetized (2% tribromoethanol) and then perfused transcardially with 4% paraformaldehyde. After cryoprotection of the brainstem tissue with 30% sucrose, brains were frozen and sequential sections were prepared using a cryostat. All sections were maintained in the order they were cut to allow accurate assessment of the co-localization of TH staining and EGFP florescence in sub nuclei of the NTS. Sections were processed for immunofluorescence using standard techniques (Appleyard et al., 2005). Rabbit polyclonal anti-TH (Millipore, Billerica, MA) was used at a final dilution of 1:1000 (v/v). After rinsing, sections were incubated in donkey anti-rabbit immunoglobulin-G Cy3 (1:200) (Jackson Immuno-research Laboratories, West Grove, PA). Sections were mounted in rostral-caudal order and high resolution confocal images were acquired using an Olympus IX81 DSU spinning disk confocal microscope. Co-localization was then counted throughout the extent of the NTS and DMNV, including the co-localization in specific NTS sub nuclei.
Post-hoc staining for TH immunofluorescent and neurobiotin
Recordings were made from TH-EGFP neurons as described above but 1% neurobiotin (Invitrogen, Carlsbad, CA) was also included in the internal solution. At the end of the recording, current was injected into the neuron and then the recording pipette slowly removed. Brain slices were placed in 4% paraformaldehyde in PBS for at least 24 hours. Slices were then processed for immunoflorescence as described above. However, in addition to the anti-TH antibody a streptavidin conjugated to Alexa 647 (Invitrogen, Carlsbad, CA, 1:200) was used to stain for the presence of the neurobiotin. High resolution confocal images were again acquired using an Olympus IX81 DSU spinning disk confocal microscope.
Statistics
All data are presented as averages ± SEM. Ghrelin was determined to have had a significant effect on the ST-EPSC on any given cell using an unpaired Student’s t-test. Statistical comparisons of drug effects between groups (e.g. Fed vs Fasted) were made using unpaired Student’s t-test, one way ANOVA with Fisher’s PLSD post hoc analysis and Fisher’s Exact-test where appropriate (see individual results; Statview 4.57, Abacus Concepts and InStat3, GraphPad). The Kolmogorov-Smirnov test (KS-test) was used to determine the significance of the drug effect for individual neurons when analyzing the sEPSC and mEPSC data (mini analysis, Synaptosoft). P <0.05 indicated significant differences.
Results
Co-localization of EGFP with TH in TH-EGFP mice
We used transgenic mice expressing enhanced green fluorescent protein (EGFP) under the control of the tyrosine hydroxylase (TH) promoter to identify NTS CA neurons. In agreement with what we had previously reported (Appleyard et al., 2007) we found that overall TH co-localized with EGFP in 84% of NTS neurons from the TH-EGFP mice. However, we did find that the co-localization varied from region to region, for example more caudal NTS EGFP neurons were extensively co-localized with TH (90–100%), while the more rostral NTS neurons were less co-localized (50–70%). Co-localization was high (>84%) in the regions in which our recordings are made, within 200 μm rostral or caudal from obex and medial to the ST. We did observe regions of very low co-localization, particularly in the dorsal motor nucleus of the vagus (DMNV, 10N), which contained cells that expressed EGFP, but only a low percentage (24%) of which were immunopositive for TH. However, we do not record from these neurons in our studies. All TH-EGFP neurons could be easily visualized and identified for recordings. As an additional confirmation that the neurons we were recording from expressed TH we also performed post-hoc staining by including neurobiotin in our pipette. We found that neurobiotin/strepavidin was co-localized with both EGFP and TH immunostaining in 5/6 (83.3%) neurons examined.
Ghrelin inhibits spontaneous glutamate inputs onto TH-EGFP neurons
Application of 100nM ghrelin increased the inter-event interval of spontaneous glutamate inputs (sEPSCs) in 16out of 26TH-EGFP neurons tested (P<0.05; KS test, Figure 1B and C). The basal rate of sEPSCs is highly variable between TH-EGFP neurons with the frequency ranging from 0.5 to 12.6 Hz, as has been reported previously (Appleyard et al., 2007). However, across responsive neurons, ghrelin decreased the sEPSC frequency from 5.2±1.1 Hzin control (ACSF) to 3.1 ± 0.7 Hz (ghrelin) (n=16), with an average inhibition of 37 ± 3% that was partially reversed following a 10 minute wash to 24 ± 5% (Figure 1D, n=15). In contrast, ghrelin did not consistently change the amplitude of the sEPSCs (Figure 1E; Control: −54.3 ± 3.4 pA vs. Ghrelin: −52.0 ± 3.6 pA). The average frequency of the non-responsive neurons was 1.9 ± 0.4 Hz (Control) and 2.0 ± 0.5 Hz in ghrelin (n=10), significantly lower than the ghrelin responsive neurons (p<0.05, Student’s test).
Figure 1.

Ghrelin inhibits spontaneous EPSCs (sEPSCs) frequency in TH-EGFP neurons; an effect blocked by the GHSR antagonist D-Lys-3-GHRP-6. A. Visualization of the NTS horizontal brain-slice preparation from a TH-EGFP mouse using DIC (left) and florescence (right). Scale bar = 1 mM. B Representative traces of sEPSCs showing the frequency was inhibited by a 10 minute bath application of 100 nM ghrelin. VM = −60 mV. C. Graph showing ghrelin’s inhibition of the frequency of sEPSCs over time. D. Graph showing the average sEPSC frequency compared to control (ACSF) in the presence of ghrelin, D-Lys-3-GHRP-6 or both. E. A bar graph showing no change in the average sEPSC amplitude for each condition. * p< 0.01 vs. control (ACSF).
The GHSR receptor mediates ghrelin’s inhibition of the frequency of sEPSCs
Pre-incubation with the GHSR1 antagonist D-Lys-3-GHRP-6 (30uM) completely attenuated the effects of 100 nM ghrelin in 8 out of 9 TH-EGFP neurons (Figure 1D; P>0.05, KS test). Across neurons, this constituted a significant block of ghrelin’s effects (P<0.05, Fisher exact test, n=1/9 and n=16/26), with the average sEPSC frequency being 4.7 ± 1.0 Hz in control, 3.8 ± 0.8 Hz in D-Lys-3-GHRP-6 and 3.9 ± 1.0 Hz in D-Lys-3-GHRP-6 + ghrelin. There was no significant effect of D-Lys-3-GHRP-6 or D-Lys-3-GHRP-6 + ghrelin on the sEPSC amplitude across neurons (Figure 1E, n=8, p>0.05, Student’s t-test; Control = −41.6 ± 4.4 pA, D-Lys-3-GHRP-6 = −39.3 ± 4.5 pA, D-Lys-3-GHRP-6 + ghrelin = −38.3 ± 3.9 pA). D-Lys-3-GHRP-6 alone had no significant effect on the inter-event interval in 6/8 TH-EGFP neurons (KS test p>0.05), but significantly increased the inter-event interval in 2 neurons (KS test p<0.05). Although the underlying mechanism is not clear, this drug has been reported to have other actions in addition to its antagonism of the GHSR1 (Schioth et al., 1997; Depoortere et al., 2006; Erriquez et al., 2009).
Ghrelin inhibits ST-EPSCs in TH-EGFP neurons in a concentration-dependent manner
While there are several sources of glutamate inputs in the NTS, a major source is the solitary tract (ST) afferent fibers, including those coming from the GI tract. Cutting the brainstem slices horizontally preserves a lengthy segment of the ST in the same plane as the cell bodies of NTS (Figure 1A). This allows placement of the stimulating electrode on the visible ST at a sufficient distance from the recording area to minimize focal activation of local interneurons and interconnecting fibers. Brief shocks (100 μsec duration) passed through the stimulating electrode evoked excitatory postsynaptic currents (ST-EPSCs). As we have described previously ST-EPSCs in TH-EGFP neurons had nearly invariant latencies, few failures, frequency-dependent amplitude depression and were attenuated by the non-NMDA glutamate antagonists NBQX or CNQX (Appleyard et al., 2007). Bath application of 100 nM ghrelin significantly inhibited the amplitude of the ST-EPSC in 10/11 neurons tested (Figure 2A and B), from −237 ± 24pA in ACSF to −170 ± 21 pA in ghrelin, an effect partially reversed by wash to −197.4 ± 22pA (Figure 2B). The range of inhibition was from 10% to 63%, with the average being 30 ± 5% (Figure 2C, n=10). In contrast, continued perfusion in ACSF did not significantly change the ST-EPSC amplitude; control = −240 ± 37 pA vs 10 minute continued perfusion in ACSF = −241 ± 37 pA (Figure 2C, 0nM dose, n=9). Ghrelin did not significantly change either the input resistance (Responders: Control = 569±91MΩ,100 nM ghrelin = 610 ± 105 MΩ, n=10) or holding current (Responders: Control = −11.9 ± 1.1 pA, 100 nM ghrelin = −11.0 ± 1.1 pA, n=10) in TH-EGFP neurons consistent with ghrelin’s actions being predominately pre-synaptic.
Figure 2.

Ghrelin inhibits the amplitude of ST stimulated EPSCs in TH-EGFP neurons. A. Representative trace of two ST-stimulated EPSCs. ST activation evoked monosynaptic EPSCs in TH-EGFP neurons. VM = −60 mV. Ghrelin significantly inhibited the amplitude of the ST stimulated EPSCs. This effect was partially reversed following a 10 minute wash. B. A graph showing the effect of ghrelin on ST-EPCS amplitude over time in a representative neuron. C. Dose response relationship showing the average change in ST-EPSC amplitude compared to a 10 minute control exposure to ACSF (0nM) with increasing doses of ghrelin. The results are not cumulative, but reflect the result following a single 10 minute exposure of the TH-EGFP neuron to only one dose of ghrelin. n=7–11 for each dose. * p< 0.05 ** p<0.01 vs. control (ACSF), One Way ANOVA.
The effects of ghrelin were concentration dependent with 10 nM ghrelin producing a 20.4 ± 2% inhibition in 5/7 TH-EGFP neurons (ST-EPSC amplitude control = −244 ± 32 pA, 10nM ghrelin = −196 ± 27 pA, n=5) and 30 nM ghrelin producing a 25.5 ± 2% inhibition in 6/9 neurons (ST-EPSC amplitude control = −174 ± 16 pA, 30nM ghrelin = −130 ± 13 pA, n=6); compared to 100 nM ghrelin producing the 30 ± 5% inhibition in 10/11 neurons described above (Figure 2C; ACSF vs. 100 nM ghrelin p<0.001; ACSF vs. 30nM ghrelin p<0.001; ACSF vs. 10 nM ghrelin p<0.005, One way ANOVA). The EC50 for ghrelin’s inhibition of ST-EPSCs in TH-EGFP neurons was approximately 7nM.
Presynaptic actions of ghrelin on NTS-CA neurons
Ghrelin significantly increased the paired-pulse ratio (EPSC2 amplitude/EPSC1 amplitude when two shocks were applied 20 mS apart) in ghrelin responsive neurons. On average ghrelin increased the size of the PPR across neurons from 0.65 ± 0.05 in control (ACSF) to 0.82 ± 0.09 in ghrelin (p<0.05, Student’s t-test, n=10). A change in the paired pulse ratio is normally associated with a pre-synaptic mechanism of action, especially as post-synaptic AMPA receptor desensitization has little effect on the peak amplitude of ST-EPSCs in NTS neurons (Chen et al., 1999). To study this more closely we examined mini-EPSCS (mEPSCs) in the presence of 2uM TTX to block action potentials (Figure 3). The voltage was held at −60mV, the approximate reversal potential for chloride to isolate glutamatergic mEPSCs. Ghrelin significantly increased the inter-event interval in 7/8 NTS TH-EGFP neurons (Figure 3A and B, p<0.05 KS test), with an average frequency of 2.0 ± 0.3 Hz in TTX only to 1.4 ± 0.1 Hz in TTX + ghrelin, (n=7) and an average inhibition of 40 ± 5% (Figure 3C). In contrast, ghrelin had no consistent effect on mEPSC amplitude (Figure 4D; TTX only = −37.3 ± 3.6 pA; TTX + ghrelin = −36.9 ± 3.4 pA). Taken together with the lack of effect of ghrelin on either input resistance or holding current these data are consistent with ghrelin’s actions being predominately pre-synaptic.
Figure 3.
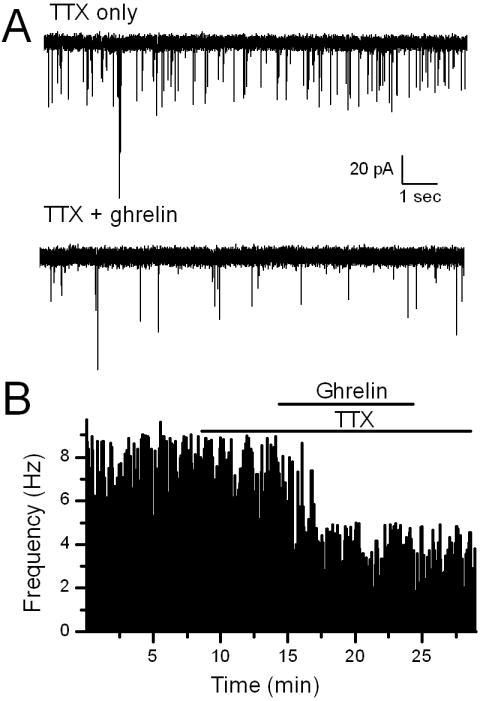
Ghrelin significantly decreased the frequency of miniature EPSCs in TH-EGFP neurons. A. Representative traces of mEPSCs from control (ACSF) conditions and in the presence of 100nM ghrelin. B. A graph showing the frequency of mEPSCs over time. Ghrelin (100 nM) significantly decreased the rate of mEPSCs in this example (p<0.05, KS test). vs. control (ACSF).
Figure 4.

Ghrelin decreases the firing rate of TH-EGFP neurons as well as ST-stimulated APs. A. Representative trace from a current clamp experiment showing the firing rate of a TH-EGFP neuron. Bath application of ghrelin (100nM) significantly reduced the basal firing rate of 5/6 TH-EGFP neurons. Ghrelin also hyperpolarized 3/6 neurons Average effect = 6.8±1.3 mV), including the example shown here. The effect was partially reversed following a wash. (n=5). Action potentials were truncated at +10mV. Arrow denotes where evoked APs were tested. B. Representative traces in current clamp following a train of 5 stimulations given to the ST at 0.2 Hz, each stimulation is indicated by a grey circle. Under control conditions the majority of trains resulted in an AP (I-O ratio = 0.87 ± 0.13 (n=11)). In the presence of ghrelin, stimulation of the ST failed to evoke an AP most of the time (I-O ratio was reduced in 7/11 neurons to 0.38 ± 0.14). This effect was partially reversed following wash (I-O ratio = 0.66 ± 0.12). C. Average I-O ratio or success rate of the ST stimulation resulting in an AP under control conditions (ACSF), 100 nM ghrelin and following a 10 minute wash (n=7). * p< 0.01 vs. control (ACSF), # p<0.01 vs. control and ghrelin. D. Average firing rate of TH-EGFP neurons in control (ACSF), 20 uM NBQX and NBQX + 100 nM ghrelin. * p< 0.01 vs. control (ACSF) (n=7). Ghrelin did not hyperpolarize any neurons in the presence of NBQX.
Ghrelin inhibits the basal firing rate of TH-EGFP neurons
As reported previously we found that the majority of TH-EGFP neurons fire at rest (Appleyard et al., 2007). Bath application of 100nM ghrelin significantly reduced the basal firing rate in 5/6TH-EGFP neurons from 2.6 ± 1.0 Hz to 0.9 ± 0.7Hz, with the average inhibition being 82.5 ± 10% (Figure 4A, p<0.01, Student’s t-test, n=5). This effect was partially reversed following wash to 56.3 ± 14%. To determine whether ghrelin inhibited the firing rate of TH-EGFP neurons through its effect to reduce excitatory glutamate inputs onto these neurons we examined whether the effects of ghrelin were attenuated by the glutamate antagonist NBQX. 20 μM NBQX alone inhibited the firing rate of 6/7TH-EGFP neurons from 4.2 ± 1.0 Hz to 3.2 ± 0.9 Hz (p<0.01, Student’s t-test, n=7), suggesting that spontaneous excitatory glutamate inputs do indeed alter the firing rate of these neurons, at least in our slice preparation. NBQX completely blocked the effects of ghrelin in 6/7neurons (Figure 4D, p>0.05, Student’s t-test). Across neurons NBQX blocked the effect of ghrelin on the firing rate in TH-EGFP neurons (p<0.05, Fishers exact test), with an average firing rate across neurons of 3.2 ± 0.9 Hz in NBQX alone and 3.1 ± 0.9 Hz when NBQX and ghrelin were co-applied. This data supports the hypothesis that ghrelin’s effects are primarily due to an inhibition of glutamate inputs onto these neurons. We also found that when ghrelin was applied alone it hyperpolarized 3/6 neurons an average of 6.8 ± 1.3 mV (including the example shown in Figure 4A), while no effect was seen in the other 3 neurons (Average change 0.4 ± 1.9 mV). However, ghrelin did not significantly change the membrane potential of any NTS TH-EGFP neurons tested when applied in the presence of NBQX (Control = −59.7 ± 1.3 mV, NBQX = −59.9 ± 1.0 mV, Ghrelin + NBQX = −58.5 ± 1.5 mV; n=7, p>0.05, Student’s t-test). As ghrelin did significantly reduce the firing rate of one TH-EGFP neuron in the presence of NBQX from 5.0 Hz to 2.8 Hz we cannot rule out an additional effect of ghrelin in a small population of TH-EGFP neurons.
Ghrelin inhibits ST-evoked APs in TH-EGFP neurons
To test whether ghrelin also inhibits the ability of ST afferents to generate an action potential in TH-EGFP neurons we measured the success rate or the input output ratio (I-O) of ST stimulation to action potential firing in TH-EGFP neurons. Under control conditions (ACSF) the I-O ratio was 0.87 ± 0.13 (n=11). Ghrelin significantly reduced the I-O ratio to 0.38 ± 0.14 in 7/11 TH-EGFP neurons (Figure 4B and C, p<0.01, Student’s t-test). This effect was partially reversed following wash (I-O ratio = 0.66 ± 0.12; p>0.05 from control, Student’s t-test, n=7).
Ghrelin inhibits ST-EPSCs in the minority of non-florescent NTS neurons from the TH-EGFP mice or POMC-EGFP NTS neurons
100nM ghrelin inhibited ST-EPSC amplitude in 3/9 non-green florescent neurons in the NTS of TH-EGFP mice. The average ST-EPSC amplitude in the responders was −382 ± 113 pA in control and −305 ± 94 pA in ghrelin, with an average inhibition of 20 ± 3% (n=3). The average ST-EPSC amplitude in non-responders was −246 ± 26 pA in control compared to −251 ± 28 pA in ghrelin (101±1% of control, n=6). Ghrelin had no significant effect on input resistance in EGFP negative neurons, even in the 3 responsive neurons (Control: 520 ± 19 mΩ, ghrelin: 496 ± 5 mΩ, n=3).
We next examined the effect of ghrelin on another population of NTS neurons the pro-opiomelanocortin (POMC) neurons, which were indentified using a POMC-EGFP transgenic mouse (Cowley et al., 2001). We have previously demonstrated that 80% of POMC-EGFP neurons receive direct strong inputs from ST-afferents and that CCK increases glutamate inputs onto these neurons (Appleyard et al., 2005). However, we found that 100nM ghrelin only had a significant effect in 2/9 POMC-EGFP neurons with the average ST-EPSC amplitude being −130 pA in control and −106 pA in ghrelin, an average inhibition of 19% (n=2). In non-responders the average amplitude was −236 ± 49 pA in control and −225 ± 49 pA in ghrelin (n=7). Again, ghrelin had no significant effect on input resistance (Control = 589 ± 60 mΩ, ghrelin = 571 ± 56 mΩ, n=9). These results demonstrate that ghrelin preferentially inhibits ST-EPSCs in TH-EGFP neurons over either non-TH or POMC-EGFP NTS neurons (TH vs. non TH p<0.05; TH vs. POMC p<0.01; Fisher’s exact test).
Ghrelin affects both CCK-sensitive and insensitive NTS TH-EGFP neurons
CCK has been shown to increase c-fos activation of NTS TH immunopositive neurons (Monnikes et al., 1997; Willing and Berthoud, 1997; Rinaman et al., 1998) and we and others have shown that CCK increases glutamate inputs onto TH neurons (Baptista et al., 2005; Appleyard et al., 2007). As ghrelin significantly decreased the frequency of sEPSCs, the opposite effect to CCK, we determined whether ghrelin inhibited spontaneous glutamate inputs onto TH-EGFP neurons that were also sensitive to CCK. Ghrelin significantly inhibited sEPSCs onto 4/6 CCK-sensitive TH-EGFP neurons an average of 32 ± 9% (Figure 5: CCK control: 1.3 ± 0.6 Hz, CCK: 6.6 ± 2.2 Hz; ghrelin control: 2.0 ± 0.9 Hz, ghrelin: 1.1 ± 0.5 Hz, p<0.05 for both CCK and ghrelin, KS test, n=4). Ghrelin also inhibited the sEPSC frequency in 3/5 CCK-insensitive TH-EGFP neurons by an average of 44 ± 1% (CCK Control 2.1 ± 0.8 Hz, CCK 2.1 ± 0.9 Hz; ghrelin control 3.8 ± 2.6 Hz, ghrelin 2.2 ± 1.5 hz, p<0.05 for ghrelin but not CCK, KS test, n=3).
Figure 5.

Ghrelin inhibits sEPSC frequency in CCK-sensitive and insensitive TH-EGFP neurons. A. Representative traces from control conditions (ACSF), following a bath application of ghrelin (100nM) and a subsequent application of CCK (100nM) in the same neuron. B. Graph showing the change in sEPSC frequency over time. This representative neuron responded first to ghrelin and then to CCK. Ghrelin had a significant effect in 4/6 CCK-sensitive and 3/5 CCK-insensitive neurons tested.
Fasting increases the size of ghrelin’s effects in NTS TH-EGFP neurons
Animals and people tend to feel hungrier and eat more in a fasted state. To examine whether the effects of ghrelin on the incoming afferents are increased following a fast, mice were either food-deprived for 18 hours (fasted) or maintained with ad lib access to food (fed), with both groups having ad lib access to water. Glucose concentrations are quite high in our normal ACSF at 10–15mM (10 mM glucose with additional glucose added to adjust the osmolarity of our solutions), which is fairly standard for brain slice recordings. However, given that both plasma and CSF glucose levels are generally lower and are further reduced during fasting (For review see Routh, 2002) we lowered the concentration of glucose we use in our ACSF solution to 5 mM for these experiments for slices from both fasted and fed animals in order to be closer to the physiological range(with sucrose substituted to maintain osmolarity). 5 mM glucose is still on the high side of physiological levels for fasting, even for plasma concentrations (Routh, 2002); however, it was the lowest concentration of glucose that maintained the health of the slices for the duration of the experiments. 5 of these experiments in each condition (10 total) were performed with a blind design, where the experimenter did not know whether the slices were from a fed or fasted animal. We found no significant difference between the results when the investigator was blind to the treatment group compared to when they knew the treatment group and so the results were combined. Ghrelin significantly inhibited the ST-EPSC amplitude in 8/12 neurons from −222 ± 33 pA to −171 ± 33 pA in slices taken from fed animals and maintained in ACSF with 5 mM glucose, an average inhibition of 26 ± 5%. In contrast, in neurons recorded from slices taken from fasted animals maintained in 5 mM glucose, ghrelin significantly inhibited the ST-EPSC amplitude in 10/14 neurons from −181 ± 12 pA to −80 ± 10 pA, an average inhibition of 54 ± 7%. Thus the average inhibition by ghrelin was significantly increased in slices prepared from fasted mice compared to slices prepared and maintained under exactly the same conditions from ad lib fed mice (Figure 6; p<0.01, Student’s t-test). We did not observe any significant differences between slices from fed animals maintained in 5 mM glucose (with sucrose added to keep 301–305 mOsm) or fed animals maintained in our normal conditions of approximately 10–15mM glucose in either ST-EPSC amplitude (−237.8 ± 24 pA vs. −222 ± 33 pA) or the size of ghrelin’s inhibition (30 ± 5% vs. 26 ± 5%) (p>0.05, Student’s t-test), suggesting that altering the glucose concentration our neurons were exposed to for the duration of our experiments did not alter ghrelin’s effect. We only saw a significant difference between slices from fed vs. fasted animals (both in 5mM glucose, p<0.01, Student’s t-test). Ghrelin also increased the PPR 1.25 ± 0.04 fold in fed animals vs. 1.71 ± 0.18 fold in fasted animals in slices maintained in 5 mM glucose (p<0.05, Student’s t test). In contrast, the input resistance of the cells was not significantly different between any of the groups (Fasted, 5 mM glucose: 511 ± 72 mΩ, Fed, 5mM glucose: 675 ± 88 mΩ vs. Fed, normal: 569 ± 91 mΩ).
Figure 6.
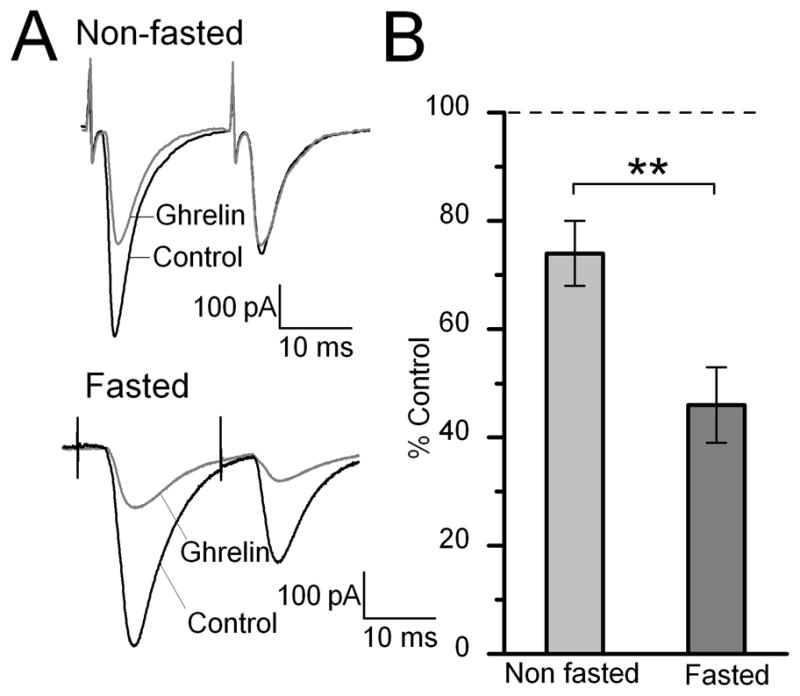
Fasting increased the size of ghrelin’s inhibition of ST-EPSCs in TH-EGFP neurons. A. Representative traces of two ST-stimulated EPSCs from non-fasted and fasted (18 hours) mice. All mice had ad lib access to water. All recordings were made under the same conditions in 5 mM glucose, with sucrose used to adjust the osmolarity. B. Graph showing the average affect of ghrelin on the ST-EPSC amplitude in fasted and non-fasted mice. ** p< 0.01 for non-fasted (n=12) vs. fasted (n=14). In contrast, we did not observe any significant difference between slices from fed animals maintained in 5 mM glucose or fed animals maintained in our normal conditions of approximately 15mM glucose (P>0.05, Student’s t-test).
Discussion
Catecholamine neurons in the NTS have been proposed by many groups to be important for the regulation of food intake (Monnikes et al., 1997; Willing and Berthoud, 1997; Rinaman et al., 1998). Recently it was suggested that they are also essential for the orexigenic effects of the hormone ghrelin (Date et al., 2006), yet little is known about the cellular mechanisms involved. Here we report four new findings that provide key information about how ghrelin regulates these neurons. First, ghrelin inhibits spontaneous glutamate inputs onto TH-EGFP neurons through activation of GHSR1. Second, ghrelin dose-dependently inhibits ST afferent activation of TH-EGFP neurons via a pre-synaptic mechanism to decrease glutamate release. Third, ghrelin inhibition of glutamate release reduces both sensory afferent evoked action potentials as well as the basal firing rate of these neurons. Lastly, ghrelin’s ability to inhibit the amplitude of ST-EPSCs in TH-EGFP neurons is increased following an 18 hour fast.
Ghrelin inhibits glutamate release from afferent terminals
ST afferent transmission is mediated primarily through glutamate acting at postsynaptic non-NMDA receptors (Andresen and Yang, 1990; Jin et al., 2003). Stimulation of the ST depolarizes the presynaptic terminal, increases calcium and triggers rapid release of glutamate (Chen et al., 1999; Kline et al., 2009). The probability of release from ST terminals is very high (Bailey et al., 2006), resulting in a high I-O ratio, or success rate to generate an AP, in NTS neurons (Appleyard et al., 2007; Bailey et al., 2007); but is associated with considerable use-dependent synaptic depression (Chen et al., 1999; Doyle and Andresen, 2001; Appleyard et al., 2007). Here we report that ghrelin acts with nM affinity to inhibit the amplitude of the ST evoked EPSC in TH-EGFP neurons, an action that translates into a reduced I-O ratio. We found ghrelin increased the PPR and decreased the frequency of mEPSCs (in the presence of TTX, with no change in input resistance or holding current) suggesting that the effects of ghrelin are pre-synaptic. This finding is consistent with data suggesting the GHSR is not expressed post-synaptically in TH immunoreactive NTS neurons (Zigman et al., 2006), but is found in vagal afferents (Date et al., 2002; Page et al., 2007).
In contrast to ghrelin, other Gq coupled receptors, such as CCK1 and oxytocin, increase transmitter release from ST afferent terminals (Baptista et al., 2005; Appleyard et al., 2007; Peters et al., 2008). Interestingly, ghrelin antagonizes the effects of CCK on the vagus nerve and CCK1 and GHSR receptors are co-localized in some afferent neurons (Date et al., 2005). Furthermore, our results demonstrate that ghrelin and CCK can inversely regulate glutamate release onto the same TH-EGFP neuron. The cellular mechanisms underlying the opposing effects of two Gq coupled receptors, potentially on the same terminal, remains to be determined.
In addition to its effects on ST evoked APs, ghrelin also reduces the spontaneous firing rate of TH-EGFP neurons. Spontaneous firing is dependent on a complex integration of the effects of different inputs (Silver, 2010), which for TH-EGFP neurons are likely to include ST inputs, other afferent inputs from the CNS and intra NTS inputs; in addition to the intrinsic properties of the TH-EGFP neuron (Appleyard et al., 2007) and direct effects of transmitters. We determined that NBQX, which blocks sEPSCs, reduces the spontaneous firing rate of TH-EGFP neurons and completely attenuates ghrelin’s effect. Therefore, altering spontaneous glutamate release appears to be a potential mechanism by which transmitters or hormones could dynamically adjust the firing threshold of TH-EGFP neurons, as has been suggested for other neurons (Lee et al., 2010; Sutton et al., 2006). Furthermore, this provides a mechanism by which ghrelin can decrease TH-EGFP neuronal activity even in the absence of afferent stimulation.
Physiological implications for ghrelin inhibiting afferent activation of NTS TH neurons
Ghrelin has been shown to reduce the mechanosensitvity of upper GI afferents in the mouse (Page et al., 2007) and decrease the firing rate of vagal afferents in the rat (Date et al., 2002), but the downstream targets of these afferents were unknown. Here we report that ghrelin inhibits glutamate release from ST terminals onto TH-EGFP neurons. In contrast, we found that ghrelin inhibited afferent activation of only a minority of non-TH NTS neurons, including identified POMC-EGFP neurons; although it is possible that ghrelin indirectly regulates inputs onto these non-catecholamine neurons in vivo. A proportion of vagal afferents have been reported to express the GHSR at the level of the nodose ganglia in the mouse (Page et al., 2007); our data suggest that these receptors are also expressed in the afferent terminals of these neurons.
Many groups have shown that treatments that cause satiety by increasing visceral afferent discharge, such as gastric distention and CCK, activate gene transcription in NTS TH neurons in vivo (Rinaman et al., 1995; Monnikes et al., 1997; Willing and Berthoud, 1997; Rinaman et al., 1998; Williams et al., 2008). Therefore, the ability of ghrelin to reduce afferent activation of TH-EGFP neurons could be one mechanism by which ghrelin stimulates food intake. Injections of ghrelin directly into the dorsal vagal complex (DVC) have been shown to increase food intake (Faulconbridge et al., 2003) and 4th ventricle injection of ghrelin induces relaxation of the proximal stomach (Kobashi et al., 2009) suggesting that ghrelin modulates appetite behaviors by its actions at the level of the DVC, presumably in parallel with its other sites of action such as the hypothalamus and ventral tegmental area (Castaneda et al., 2010; Fry and Ferguson, 2010; Cummings, 2006; Abizaid, 2009). However, the ST contains a broad array of sensory afferents and NTS TH neurons may participate in neuronal circuits widely influencing homeostatic functions that could be regulated by ghrelin (Lacroix and Rivest, 1997; Chan and Sawchenko, 1998; Van Bockstaele et al., 2001; Laorden et al., 2002; Krout et al., 2005; Hollis et al., 2004).
Signals that activate c-fos in TH NTS neurons also stimulate CA release at their projection sites (Buller and Day, 1996; Onaka, 2000; Ueta et al., 2000). Our results predict as ghrelin inhibits action potential firing in TH-EGFP neurons it will reduce the release catecholamines. This is consistent with the finding that ghrelin does not activate c-fos expression in NTS CA neurons (Emanuel and Ritter, 2010; Faulconbridge et al., 2008), but it is not consistent with the hypothesis that ghrelin stimulates catecholamine release in the arcuate nucleus (ARC) through activation of NTS-CA neurons (Date et al., 2006). However, it is possible that ghrelin increases levels of catecholamines in the hypothalamus through activation of a different population of brainstem catecholamine neurons (Emanuel and Ritter, 2010, Date et al., 2006).
NTS TH neurons make extensive projections to many nuclei, including the paraventricular (PVH) and arcuate (ARC) nuclei of the hypothalamus, nucleus accumbens and DMNV (Sawchenko and Swanson, 1981; Cunningham and Sawchenko, 1988; Sawchenko and Pfeiffer, 1988; Wang et al., 1992; Rogers et al., 2003; Reyes and Van Bockstaele, 2006; Balcita-Pedicino and Rinaman, 2007). Which projections are important for the effects of ghrelin remains to be determined, although brainstem CA projections to the PVH do not appear to be required for ghrelin’s effects on food intake (Emanuel and Ritter, 2010). Interestingly, we found a large variation in the size of ghrelin’s effects on TH-EGFP neurons and it is possible that neurons that project to some nuclei are more sensitive to ghrelin than others.
Fasting increases ghrelin’s effects on afferent inputs onto NTS-CA neurons
Ghrelin’s inhibitory effect on afferent drive onto NTS TH-EGFP neurons was increased following an 18 hour fast. This finding is consistent with other studies demonstrating that the synapse between sensory afferents and NTS neurons is highly plastic to a variety of both chronic and acute stimuli (Browning and Travagli, 2010; Kline, 2008). The mechanism(s) underlying this plasticity remains to be established, but could involve either changes in receptor number, intrinsic receptor efficacy or downstream signaling events. Vagal afferents appear to be very adaptable as the levels of several neurotransmitters and/or their receptors are altered during changes in energy state, such as fasting (For review see (Dockray, 2009)). The levels of the GHSR receptor mRNA transcripts are regulated by re-feeding and exposure to CCK (Sato et al., 2007) suggesting that the number of GHSR1 found in the pre-synaptic terminal could be altered following a fast. However, there is little change in GHSR1 protein levels in the nodose during fasting (Burdyga et al., 2006). Interestingly, fasting also increases ghrelin’s effects in the hypothalamus, at least at the level of c-fos activation (Tung et al., 2005; Scott et al., 2007), suggesting that adaptive changes occur in multiple brain regions to increase the appetite-stimulatory effects of ghrelin during a fast.
In summary, our data shows that ghrelin inhibits visceral afferent inputs onto NTS TH-EGFP neurons through a pre-synaptic mechanism at the afferent terminal to decrease glutamate release and decrease action potential generation. Furthermore, the size of ghrelin’s effect is increased following a fast. These results demonstrate a potential mechanism by which ghrelin could inhibit NTS-CA neurons via a pathway whose responsiveness can be adjusted to the energy state of the animal.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (DK083452). We would also like to thank Dr. Sue Ritter and Dr. James Peters for their helpful comments on this manuscript and Dr. Gary Wayman for his assistance with the confocal images.
References
- Abizaid Ghrelin and Dopamine: new insights on the peripheral regulation of appetite. J Neuroendocrinology. 2009;21:787–793. doi: 10.1111/j.1365-2826.2009.01896.x. [DOI] [PubMed] [Google Scholar]
- Andresen MC, Yang MY. Non-NMDA receptors mediate sensory afferent synaptic transmission in medial nucleus tractus solitarius. Am J Physiol. 1990;259:H1307–1311. doi: 10.1152/ajpheart.1990.259.4.H1307. [DOI] [PubMed] [Google Scholar]
- Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. J Neurosci. 2007;27:13292–13302. doi: 10.1523/JNEUROSCI.3502-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci. 2005;25:3578–3585. doi: 10.1523/JNEUROSCI.4177-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M, Mura A, Langhans W, Geary N. Gut vagal afferents are not necessary for the eating-stimulatory effect of intraperitoneally injected ghrelin in the rat. J Neurosci. 2006;26:11052–11060. doi: 10.1523/JNEUROSCI.2606-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Jin YH, Doyle MW, Smith SM, Andresen MC. Vasopressin inhibits glutamate release via two distinct modes in the brainstem. J Neurosci. 2006;26:6131–6142. doi: 10.1523/JNEUROSCI.5176-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TW, Hermes SM, Whittier KL, Aicher SA, Andresen MC. A-type potassium channels differentially tune afferent pathways from rat solitary tract nucleus to caudal ventrolateral medulla or paraventricular hypothalamus. J Physiol. 2007;582:613–628. doi: 10.1113/jphysiol.2007.132365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcita-Pedicino JJ, Rinaman L. Noradrenergic axon terminals contact gastric preautonomic neurons in the paraventricular nucleus of the hypothalamus in rats. J Comp Neurol. 2007;501:608–618. doi: 10.1002/cne.21267. [DOI] [PubMed] [Google Scholar]
- Baptista V, Zheng ZL, Coleman FH, Rogers RC, Travagli RA. Cholecystokinin octapeptide increases spontaneous glutamatergic synaptic transmission to neurons of the nucleus tractus solitarius centralis. J Neurophysiol. 2005;94:2763–2771. doi: 10.1152/jn.00351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR. Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol Motil. 2008;20(Suppl 1):64–72. doi: 10.1111/j.1365-2982.2008.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins JE, Chelikani PK, Haver AC, Reidelberger RD. PYY(3-36) induces Fos in the arcuate nucleus and in both catecholaminergic and non-catecholaminergic neurons in the nucleus tractus solitarius of rats. Peptides. 2008;29:112–119. doi: 10.1016/j.peptides.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning KN, Travagli RA. Plasticity of vagal brainstem circuits in the control of gastric function. Neurogastroenterol Motil. 2010 doi: 10.1111/j.1365-2982.2010.01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller KM, Day TA. Involvement of medullary catecholamine cells in neuroendocrine responses to systemic cholecystokinin. J Neuroendocrinol. 1996;8:819–824. doi: 10.1046/j.1365-2826.1996.05252.x. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Ghrelin receptors in rat and human nodose ganglia: putative role in regulating CB-1 and MCH receptor abundance. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1289–1297. doi: 10.1152/ajpgi.00543.2005. [DOI] [PubMed] [Google Scholar]
- Castaneda TR, Tong J, Datta R, Culler M, Tschop MH. Ghrelin in the regulation of body weight and metabolism. Front Neuroendocrinol. 2010;31:44–60. doi: 10.1016/j.yfrne.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Chan RK, Sawchenko PE. Organization and transmitter specificity of medullary neurons activated by sustained hypertension: implications for understanding baroreceptor reflex circuitry. J Neurosci. 1998;18:371–387. doi: 10.1523/JNEUROSCI.18-01-00371.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Horowitz JM, Bonham AC. A presynaptic mechanism contributes to depression of autonomic signal transmission in NTS. Am J Physiol. 1999;277:H1350–1360. doi: 10.1152/ajpheart.1999.277.4.H1350. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Cummings DE. Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol Behav. 2006;89:71–84. doi: 10.1016/j.physbeh.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Cunningham ET, Jr, Sawchenko PE. Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. J Comp Neurol. 1988;274:60–76. doi: 10.1002/cne.902740107. [DOI] [PubMed] [Google Scholar]
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123:1120–1128. doi: 10.1053/gast.2002.35954. [DOI] [PubMed] [Google Scholar]
- Date Y, Toshinai K, Koda S, Miyazato M, Shimbara T, Tsuruta T, Niijima A, Kangawa K, Nakazato M. Peripheral interaction of ghrelin with cholecystokinin on feeding regulation. Endocrinology. 2005;146:3518–3525. doi: 10.1210/en.2004-1240. [DOI] [PubMed] [Google Scholar]
- Date Y, Shimbara T, Koda S, Toshinai K, Ida T, Murakami N, Miyazato M, Kokame K, Ishizuka Y, Ishida Y, Kageyama H, Shioda S, Kangawa K, Nakazato M. Peripheral ghrelin transmits orexigenic signals through the noradrenergic pathway from the hindbrain to the hypothalamus. Cell Metab. 2006;4:323–331. doi: 10.1016/j.cmet.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Depoortere I, Thijs T, Peeters T. The contractile effect of the ghrelin receptor antagonist, D-Lys3-GHRP-6, in rat fundic strips is mediated through 5-HT receptors. Eur J Pharmacol. 2006;537:160–165. doi: 10.1016/j.ejphar.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Dockray GJ. The versatility of the vagus. Physiol Behav. 2009;97:531–536. doi: 10.1016/j.physbeh.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol. 2001;85:2213–2223. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Emanuel AJ, Ritter S. Hindbrain catecholamine neurons modulate the growth hormone but not the feeding response to ghrelin. Endocrinology. 2010;151:3237–3246. doi: 10.1210/en.2010-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erriquez J, Bernascone S, Ciarletta M, Filigheddu N, Graziani A, Distasi C. Calcium signals activated by ghrelin and D-Lys(3)-GHRP-6 ghrelin antagonist in developing dorsal root ganglion glial cells. Cell Calcium. 2009;46:197–208. doi: 10.1016/j.ceca.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Faulconbridge LF, Cummings DE, Kaplan JM, Grill HJ. Hyperphagic effects of brainstem ghrelin administration. Diabetes. 2003;52:2260–2265. doi: 10.2337/diabetes.52.9.2260. [DOI] [PubMed] [Google Scholar]
- Faulconbridge LF, Grill HJ, Kaplan JM, Daniels D. Caudal brainstem delivery of ghrelin induces fos expression in the nucleus of the solitary tract, but not in the arcuate or paraventricular nuclei of the hypothalamus. Brain Res. 2008;1218:151–157. doi: 10.1016/j.brainres.2008.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M, Ferguson AV. Ghrelin: central nervous system sites of action in regulation of energy balance. Int J Pept. 2010:2010. doi: 10.1155/2010/616757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill HJ, Hayes MR. The nucleus tractus solitarius: a portal for visceral afferent signal processing, energy status assessment and integration of their combined effects on food intake. Int J Obes (Lond) 2009;33(Suppl 1):S11–15. doi: 10.1038/ijo.2009.10. [DOI] [PubMed] [Google Scholar]
- Hollis JH, Lightman SL, Lowry CA. Integration of systemic and visceral sensory information by medullary catecholaminergic systems during peripheral inflammation. Ann N Y Acad Sci. 2004;1018:71–75. doi: 10.1196/annals.1296.008. [DOI] [PubMed] [Google Scholar]
- Jin YH, Bailey TW, Doyle MW, Li BY, Chang KS, Schild JH, Mendelowitz D, Andresen MC. Ketamine differentially blocks sensory afferent synaptic transmission in medial nucleus tractus solitarius (mNTS) Anesthesiology. 2003;98:121–132. doi: 10.1097/00000542-200301000-00021. [DOI] [PubMed] [Google Scholar]
- Kline DD. Plasticity in glutamatergic NTS neurotransmission. Respir Physiol Neurobiol. 2008;164:105–111. doi: 10.1016/j.resp.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Hendricks G, Hermann G, Rogers RC, Kunze DL. Dopamine inhibits N-type channels in visceral afferents to reduce synaptic transmitter release under normoxic and chronic intermittent hypoxic conditions. J Neurophysiol. 2009;101:2270–2278. doi: 10.1152/jn.91304.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobashi M, Yanagihara M, Fujita M, Mitoh Y, Matsuo R. Fourth ventricular administration of ghrelin induces relaxation of the proximal stomach in the rat. Am J Physiol Regul Integr Comp Physiol. 2009;296:R217–223. doi: 10.1152/ajpregu.00878.2007. [DOI] [PubMed] [Google Scholar]
- Kojima M, Kangawa K. Ghrelin: more than endogenous growth hormone secretagogue. Ann N Y Acad Sci. 2010;1200:140–148. doi: 10.1111/j.1749-6632.2010.05516.x. [DOI] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Krout KE, Mettenleiter TC, Karpitskiy V, Nguyen XV, Loewy AD. CNS neurons with links to both mood-related cortex and sympathetic nervous system. Brain Res. 2005;1050:199–202. doi: 10.1016/j.brainres.2005.04.090. [DOI] [PubMed] [Google Scholar]
- Lacroix S, Rivest S. Functional circuitry in the brain of immune-challenged rats: partial involvement of prostaglandins. J Comp Neurol. 1997;387:307–324. doi: 10.1002/(sici)1096-9861(19971020)387:2<307::aid-cne11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Lam DD, Zhou L, Vegge A, Xiu PY, Christensen BT, Osundiji MA, Yueh CY, Evans ML, Heisler LK. Distribution and neurochemical characterization of neurons within the nucleus of the solitary tract responsive to serotonin agonist-induced hypophagia. Behav Brain Res. 2009;196:139–143. doi: 10.1016/j.bbr.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laorden ML, Castells MT, Milanes MV. Effects of morphine and morphine withdrawal on brainstem neurons innervating hypothalamic nuclei that control the pituitary-adrenocortical axis in rats. Br J Pharmacol. 2002;136:67–75. doi: 10.1038/sj.bjp.0704684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Roux CW, Neary NM, Halsey TJ, Small CJ, Martinez-Isla AM, Ghatei MA, Theodorou NA, Bloom SR. Ghrelin does not stimulate food intake in patients with surgical procedures involving vagotomy. J Clin Endocrinol Metab. 2005;90:4521–4524. doi: 10.1210/jc.2004-2537. [DOI] [PubMed] [Google Scholar]
- Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 66:859–870. doi: 10.1016/j.neuron.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Matsumura K, Fukuhara M, Kagiyama S, Fujii K, Iida M. Ghrelin acts at the nucleus of the solitary tract to decrease arterial pressure in rats. Hypertension. 2004;43:977–982. doi: 10.1161/01.HYP.0000122803.91559.55. [DOI] [PubMed] [Google Scholar]
- Madden CJ, Sved AF. Cardiovascular regulation after destruction of the C1 cell group of the rostral ventrolateral medulla in rats. Am J Physiol Heart Circ Physiol. 2003;285:H2734–2748. doi: 10.1152/ajpheart.00155.2003. [DOI] [PubMed] [Google Scholar]
- Monnikes H, Lauer G, Arnold R. Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat. Brain Res. 1997;770:277–288. doi: 10.1016/s0006-8993(97)00865-2. [DOI] [PubMed] [Google Scholar]
- Moran TH, Ladenheim EE, Schwartz GJ. Within-meal gut feedback signaling. Int J Obes Relat Metab Disord. 2001;25(Suppl 5):S39–41. doi: 10.1038/sj.ijo.0801910. [DOI] [PubMed] [Google Scholar]
- Onaka T. Catecholaminergic mechanisms underlying neurohypophysial hormone responses to unconditioned or conditioned aversive stimuli in rats. Exp Physiol. 2000;85(Spec No):101S–110S. doi: 10.1111/j.1469-445x.2000.tb00013.x. [DOI] [PubMed] [Google Scholar]
- Page AJ, Slattery JA, Milte C, Laker R, O’Donnell T, Dorian C, Brierley SM, Blackshaw LA. Ghrelin selectively reduces mechanosensitivity of upper gastrointestinal vagal afferents. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1376–1384. doi: 10.1152/ajpgi.00536.2006. [DOI] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Kellett DO, Jordan D, Llewellyn-Smith IJ, Andresen MC. Oxytocin enhances cranial visceral afferent synaptic transmission to the solitary tract nucleus. J Neurosci. 2008;28:11731–11740. doi: 10.1523/JNEUROSCI.3419-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Van Bockstaele EJ. Divergent projections of catecholaminergic neurons in the nucleus of the solitary tract to limbic forebrain and medullary autonomic brain regions. Brain Res. 2006;1117:69–79. doi: 10.1016/j.brainres.2006.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. J Neurosci. 2003;23:10084–10092. doi: 10.1523/JNEUROSCI.23-31-10084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L, Dzmura V. Experimental dissociation of neural circuits underlying conditioned avoidance and hypophagic responses to lithium chloride. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1495–1503. doi: 10.1152/ajpregu.00393.2007. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Baker EA, Hoffman GE, Stricker EM, Verbalis JG. Medullary c-Fos activation in rats after ingestion of a satiating meal. Am J Physiol. 1998;275:R262–268. doi: 10.1152/ajpregu.1998.275.1.R262. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Hoffman GE, Dohanics J, Le WW, Stricker EM, Verbalis JG. Cholecystokinin activates catecholaminergic neurons in the caudal medulla that innervate the paraventricular nucleus of the hypothalamus in rats. J Comp Neurol. 1995;360:246–256. doi: 10.1002/cne.903600204. [DOI] [PubMed] [Google Scholar]
- Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol. 2001;432:197–216. doi: 10.1002/cne.1097. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Travagli RA, Hermann GE. Noradrenergic neurons in the rat solitary nucleus participate in the esophageal-gastric relaxation reflex. Am J Physiol Regul Integr Comp Physiol. 2003;285:R479–489. doi: 10.1152/ajpregu.00155.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh VH. Glucose-sensing neurons: Are they physiologically relevant? Physiology & Behavior. 2002;76:403–413. doi: 10.1016/s0031-9384(02)00761-8. [DOI] [PubMed] [Google Scholar]
- Sato M, Nakahara K, Miyazato M, Kangawa K, Murakami N. Regulation of GH secretagogue receptor gene expression in the rat nodose ganglion. J Endocrinol. 2007;194:41–46. doi: 10.1677/JOE-06-0078. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW. Central noradrenergic pathways for the integration of hypothalamic neuroendocrine and autonomic responses. Science. 1981;214:685–687. doi: 10.1126/science.7292008. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Pfeiffer SW. Ultrastructural localization of neuropeptide Y and galanin immunoreactivity in the paraventricular nucleus of the hypothalamus in the rat. Brain Res. 1988;474:231–245. doi: 10.1016/0006-8993(88)90438-6. [DOI] [PubMed] [Google Scholar]
- Schioth HB, Muceniece R, Wikberg JE. Characterization of the binding of MSH-B, HB-228, GHRP-6 and 153N-6 to the human melanocortin receptor subtypes. Neuropeptides. 1997;31:565–571. doi: 10.1016/s0143-4179(97)90002-0. [DOI] [PubMed] [Google Scholar]
- Scott V, McDade DM, Luckman SM. Rapid changes in the sensitivity of arcuate nucleus neurons to central ghrelin in relation to feeding status. Physiol Behav. 2007;90:180–185. doi: 10.1016/j.physbeh.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Silver RA. Neuronal arithmetic. Nat Rev Neurosci. 2010;11:474–489. doi: 10.1038/nrn2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tung YC, Hewson AK, Carter RN, Dickson SL. Central responsiveness to a ghrelin mimetic (GHRP-6) is rapidly altered by acute changes in nutritional status in rats. J Neuroendocrinol. 2005;17(6):387–93. doi: 10.1111/j.1365-2826.2005.01316.x. [DOI] [PubMed] [Google Scholar]
- Ueta Y, Kannan H, Higuchi T, Negoro H, Yamaguchi K, Yamashita H. Activation of gastric afferents increases noradrenaline release in the paraventricular nucleus and plasma oxytocin level. J Auton Nerv Syst. 2000;78:69–76. doi: 10.1016/s0165-1838(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Menko AS, Drolet G. Neuroadaptive responses in brainstem noradrenergic nuclei following chronic morphine exposure. Mol Neurobiol. 2001;23:155–171. doi: 10.1385/mn:23:2-3:155. [DOI] [PubMed] [Google Scholar]
- Wang ZJ, Rao ZR, Shi JW. Tyrosine hydroxylase-, neurotensin-, or cholecystokinin-containing neurons in the nucleus tractus solitarii send projection fibers to the nucleus accumbens in the rat. Brain Res. 1992;578:347–350. doi: 10.1016/0006-8993(92)90269-f. [DOI] [PubMed] [Google Scholar]
- Williams DL, Schwartz MW, Bastian LS, Blevins JE, Baskin DG. Immunocytochemistry and Laser Capture Microdissection for Real-time Quantitative PCR Identify Hindbrain Neurons Activated by Interaction Between Leptin and Cholecystokinin. J Histochem Cytochem. 2008;56:285–293. doi: 10.1369/jhc.7A7331.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing AE, Berthoud HR. Gastric distension-induced c-fos expression in catecholaminergic neurons of rat dorsal vagal complex. Am J Physiol. 1997;272:R59–67. doi: 10.1152/ajpregu.1997.272.1.R59. [DOI] [PubMed] [Google Scholar]
- Zigman JM, Elmquist JK. Minireview: From anorexia to obesity--the yin and yang of body weight control. Endocrinology. 2003;144:3749–3756. doi: 10.1210/en.2003-0241. [DOI] [PubMed] [Google Scholar]
- Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]