

Abstract
The first synthesis of a series of ketones naturally occurring in E. pallida is described. The natural distribution of these ketones among different Echinacea species is also reported.
Keywords: Echinacea, synthesis, diaetylenic ketones
Introduction
Echinacea angustifolia, Echinacea pallida and Echinacea purpurea are the main medicinal Echinacea species and have long been used to treat infections, aid in wound healing, and enhance the immune system [1]. In 2005, Echinacea products ranked among the top botanical supplements sold in the United States. In recent years, treatment of rhinoviruses has been the focus of several studies, a number of which have failed to show the efficacy of Echinacea [2]. Commercial Echinacea products often are mixtures of the three main medicinal species and there is no regulation of the concentrations of the chemical constituents. Among the chemical constituents of Echinacea species, the alkamides, the ketones, caffeic acid derivatives such as chicoric acid and the polyphenols are considered important for biological activity [3].
The ketones from E. pallida exhibit a range of biological activities. An amphotericin-B-resistant strain (D10) of C. albicans and Tricophyton mentagrophytes were susceptible to E. pallida var pallida root extract in the presence of UV light [4]. Bauer reported the use of a mixture of ketones and alkamides was useful as an immunostimulant [5]. Binns has reported that the ketones from E. pallida are potent antifungal agents [6]. Related polyacetylenic ketones have been reported by Bohlmann in Centaurea ferox roots [7].
Despite the potential importance of the ketones from E. pallida, few reports of synthesis of authentic standards or analogs have been reported. Recently, Shi Shun and Tykwinski have reviewed natural polyacetylenes [8]. Crombie and co-workers have reported elegant syntheses of natural amides using organometallic coupling reactions [9–11]. Wailes has also reported the synthesis of natural dienamides [12]. Kraus and Bae recently reported the first synthesis of ketone 1 [13]. We report herein the direct preparation of ketones 2, 3 and 4, shown in Scheme 1.
Scheme 1.
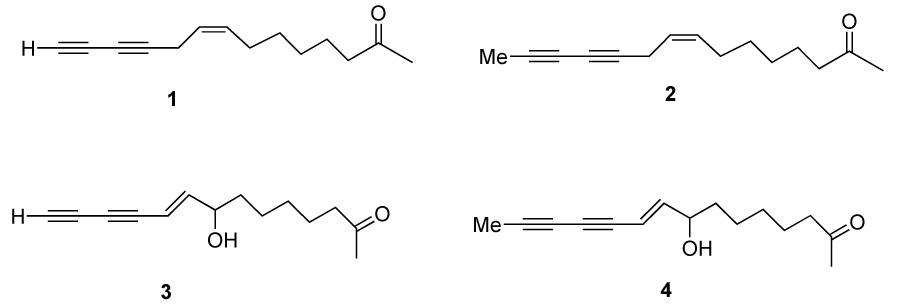
Results and Discussion
Syntheses
The synthesis route to ketones 3 and 4, found in E. pallida, is illustrated in Scheme 2. The known aldehyde 5 is treated with the anion derived from bis-trimethylsilyl diacetylene to generate an alcohol that can readily and selectively be converted into the olefinic acetylene 6 [14].
Scheme 2.
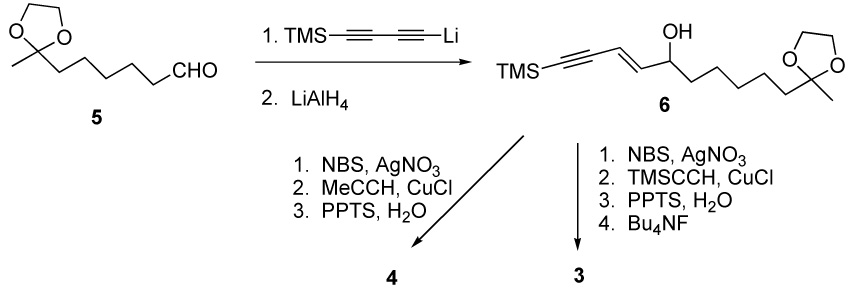
The selective reduction is the result of an intramolecular hydroalumination reaction which is directed by the hydroxyl group [15]. Compound 6 can be brominated by the conditions of Yun and Danishefsky [16], and then coupled [17] with either propyne or trimethylsilylacetylene. The ketal protecting group of the resulting diacetylene can be removed using mild aqueous acid (PPTS, H2O) to afford 4 in 27 % yield from 6. The silyl protecting group can be removed with tetrabutylammonium fluoride at ambient temperature to provide 3 in 30% overall yield from 6.
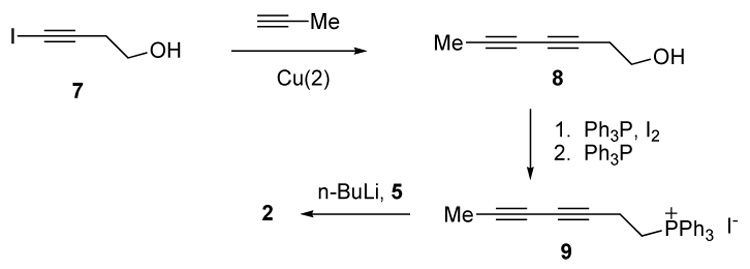
Although we reported the first synthesis of 1, the number of steps and low overall yield limit the amounts of this ketone that can be made available for biological testing. Therefore, a new synthetic route to ketones such as 1 and 2 was explored. We could shorten the synthesis significantly if the phosphonium salt 9 shown in Scheme 3 reacted efficiently with 5, an intermediate of our syntheses of compounds 3 and 4. Since a copper-catalyzed acetylene coupling reaction was likely to be successful with 4-iodo-3-butyn-1-ol (7) we introduced the acetylene coupling step at an early stage [18]. Thus, 4-iodo-3-butyn-1-ol was coupled with propyne in 81% yield. Diynol 8 was then converted into phosphonium salt 9 in two steps. Compound 9 underwent a cis-selective Wittig reaction with aldehyde 5 to generate an ene diyne in 54 % yield. This reaction gave the cis-alkene exclusively. Removal of the ketal protection group (PPTS, water) provided ketone 2 in 15% overall yield from 7.
Scheme 3.
Natural distribution of ketones 3 and 4
The distribution of these two ketones in accessions of six species of Echinacea (E. angustifolia, E. purpurea, E. pallida, E. sanguinea, E. simulata and E. tennesseensis) was determined by HPLC. Ketones 3 and 4 elute at 13.65 and 16.28 minutes, respectively (Figure 1). These two ketones demonstrate very similar UV spectra, both with λmax at 210 nm, which agrees with previously reported results [19].
Figure 1.
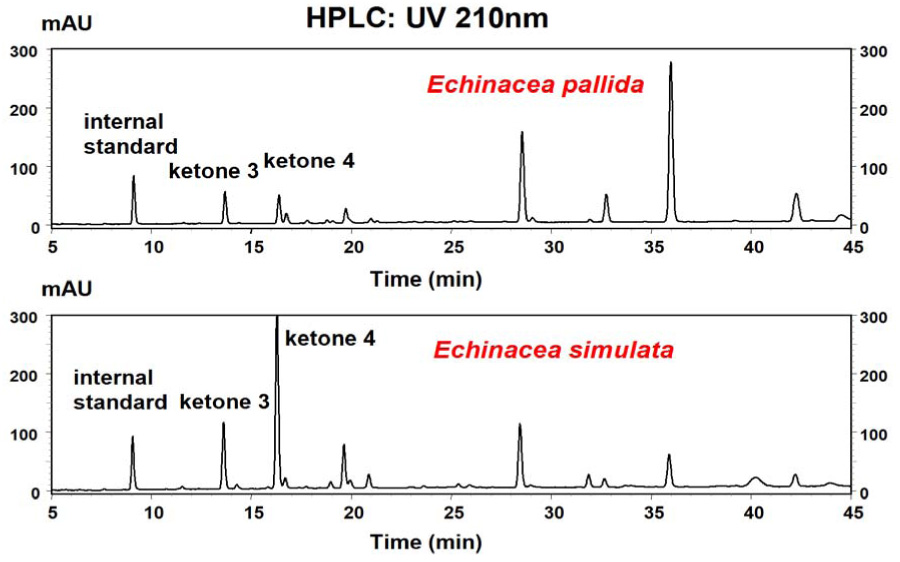
HPLC chromatograms obtained from a 95% ethanolic extracts of two-year-old roots from Echinacea pallida and Echinacea simulata, indicating ketones 3 and 4.
The mean levels of ketones 3 and 4 in two-year-old roots from six Echinacea species are presented in Figure 2. Our results show that of the six Echinacea species examined, ketones 3 and 4 present in at least two species: E. pallida and E. simulata.
Figure 2.
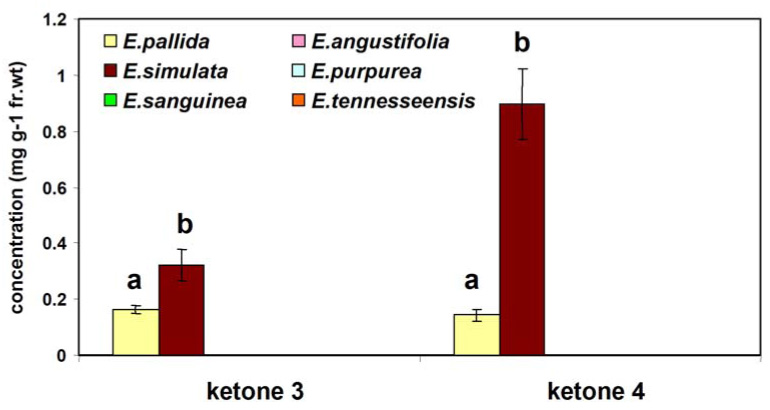
Concentrations of ketones 3 and 4 in two-year-old roots from six species of Echinacea. Error bars indicate standard deviations of means of triplicate experiments. For each ketone, different letters indicate a significant difference (p < 0.01).
Of these two species, significantly (p < 0.01) higher concentrations of both ketone 3 (0.32 ± 0.05 mg g−1 fr. wt) and ketone 4 (0.89 ± 0.12 mg g−1 fr. wt) were found in E. simulata, a species not typically used for commercial preparations. This information will be very useful for the standardization of Echinacea products.
Conclusions
The route described above for ketone 2 represents a significant improvement over our previous synthetic strategy. Our synthetic route to hydroxy ketones 3 and 4 is direct and quite flexible with regards to the preparation of analogous compounds. The observation that levels of ketones 3 and 4 are higher in E. simulata, a species of Echinacea not used commercially, demonstrates the value of studying the natural distribution of plant constituents using authentic standards.
Experimental
General
Unless stated otherwise, all reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) using 0.25 mm precoated silica gel F254 plates (Sigma-Aldrich). Column or flash chromatography were performed with the indicated solvents using silica gel (230–400 mesh) purchased from Dynamic Adsorbents, LLC. All melting points were obtained on a Laboratory Devices capillary melting point apparatus and are uncorrected. ¹H- and 13C-NMR spectra were recorded on a Bruker VXR-300 (300 MHz) or a Bruker VXR-400 (400 MHz) spectrometer. Chemical shifts are reported relative to internal chloroform (¹H, 7.26 ppm; 13C, 77.23 ppm). High resolution mass spectra were performed at the Iowa State University Mass Spectrometry Laboratory.
Plant material and extraction
Two-year-old fresh roots of 6 species of Echinacea, E. angustifolia (Accession PI 631285), E. purpurea (Accession PI 631307), E. pallida (Accession PI 631279), E. sanguinea (Accession PI 633672), E. simulata (Accession PI 631251) and E. tennesseensis (Accession PI 631350), provided by Dr. Mark P. Widrlechner at the USDA-ARS North Central Regional Plant Introduction Station, were studied to evaluate the natural distribution of ketones 3 and 4 in Echinacea species. The plant extraction method is the same as that in our previously published work [16]. Similarly, 7-hydroxy-(E)-N-isobutylundeca-2-ene-8,10-diynamide (C15H21O2) was added as an internal standard prior to extraction for quantification purposes. All experiments were performed in triplicate on independently extracted plant samples from three individual plants.
HPLC analysis
HPLC procedures were as previously described [20]. Ketones 3 and 4 were quantified at UV 210 nm based on the internal standard because they have the same UV absorption at 210 nm. The internal quantification method used here can account for variations in extraction efficiencies in different extracts. The limit of HPLC detection for each of the ketones is approximately 0.04 µg mL−1.
1-Trimethylsilyl-11-oxodocec-3-en-1-yn-5-ol ethylene ketal (6)
MeLi-LiBr complex (5 mL, 1.5 M soln in ether) was added to a solution of 1,4-bis(trimethylsilyl)-1,3-butadiyne (1.46 g, 7.5 mmol) in THF (10 mL) at 0 °C under Ar. The mixture was warmed to room temperature and stirred for 4 h. The mixture was then cooled down to −78 °C and aldehyde 5 (0.700 g, 3.76 mmol) in THF (4 mL) was added via cannula. The mixture was stirred for 1 h while warmed to rt. The reaction was quenched with sat NH4Cl (aq) and the aqueous layer was extracted with ethyl ether. The combined organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give the diynol (0.890 g, 77 % yield); ¹H-NMR (400 MHz, CDCl3) δ: 4.45–4.38 (m, 1H), 3.97–3.89 (m, 4H), 1.89–1.80 (brs, 1H), 1.74–1.60 (m, 4H), 1.48–1.32 (m, 6H), 1.31 (s, 3H), 0.19 (s, 9H).
To a solution of above diynol (0.600 g, 1.94 mmol) in diethyl ether (20 mL) was added LAH (0.088 g, 2.33 mmol) at 0 °C and the mixture was stirred for 2 h while warming to rt. The mixture was poured into ice cold water (5 mL) then extracted with diethyl ether. The organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give compound 6 (0.590 g, 98 % yield). ¹H-NMR (300 MHz, CDCl3) δ: 6.19 (dd, J= 15.9, 6.0 Hz, 1H), 5.72 (d, J= 15.9 Hz, 1H), 4.28–4.10 (m, 1H), 3.96–3.87 (m, 4H), 1.68–1.47 (m, 6H), 1.42–1.32 (m, 5H), 1.30 (s, 3H), 0.19 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ 147.0, 110.3, 110.0, 72.4, 66.1, 64.8, 39.3, 36.9, 29.8, 25.4, 24.2, 23.9, 15.5, 0.1; HRMS m/e (EI) for C17H30O3Si (M)+ calcd 310.1964, measured 310.1921.
8-Hydroxytetradeca-(9E)-ene-11,13-diyn-2-one (3)
To a solution of compound 6 (0.125 g, 0.40 mmol) in acetone (10 mL) was added N-bromosuccinimide (0.086 g, 0.48 mmol) and AgNO3 (0.004 g, 0.02 mmol) at rt. After stirring for 1 h at this temperature, the mixture was cooled to 0 °C and cold water (5 mL) was added. The aqueous layer was extracted with Et2O, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (hexanes-EtOAc) to give the corresponding bromoacetylene (0.055 g, 43% yield).
To a solution of trimethylsilylacetylene (0.052 mL, 0.37 mmol) and bromoacetylene (0.040 g, 0.126 mmol) in degassed piperidine (1 mL) was added CuCl (0.002 g, 0.013 mmol) at 0 °C. The mixture was stirred at rt for 0.5 h. The reaction was quenched with sat. NH4Cl(aq) (1 mL) and extracted with ethyl ether (3 × 10 mL). The organic phase was washed with brine (2 × 10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash chromatography to give the corresponding alcohol (0.035g, 83% yield).
To a solution of the above alcohol (0.030 g, 0.08 mmol) in water/acetone (1 mL/1 mL) was added PPTS (0.002 g, 0.008 mmol) at rt. The mixture was heated at 40 °C for 3 h and extracted with diethyl ether. The organic layer was washed with brine (2 × 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give the expected hydroxyketone (0.022 g, 95% yield).
To a solution of hydroxyketone (0.022 g, 0.076 mmol) in THF (5 mL) was added TBAF (0.114 mL, 1M soln in THF) at 0 °C. The mixture was stirred for 1h at rt and solvent was removed. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate, 2:1) to give compound 3 (0.015 g, 89 % yield). ¹H-NMR (400 MHz, CDCl3) δ: 6.34 (dd, J= 16.5, 5.6 Hz, 1H), 5.73 (d, J= 16Hz, 1H), 4.22–4.17 (m, 1H), 2.42 (t, J= 7.6 Hz, 2H), 2.41 (s, 1H), 1.60 (brs, 1H), 1.62–1.50 (m, 6H), 1.40–1.30 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 209.4, 150.6, 108.0, 74.3, 73.9, 72.1, 71.3, 68.3, 43.8, 36.8, 30.2, 29.2, 25.2, 23.8; HRMS m/e (EI) for C14H18O2 (M)+ calcd. 218.1307, measured 218.1229.
8-Hydroxy-pentadeca-(9E)-ene-11,13-diyn-2-one (4)
To a solution of compound 6 (0.125 g, 0.40 mmol) in acetone (10 mL) was added N-bromosuccinimide (0.086 g, 0.48 mmol) and AgNO3 (0.004 g, 0.02 mmol) at rt. After stirring for 1 hr at rt, the mixture was cooled to 0 °C and cold water (5 mL) was added. The aqueous layer was extracted with Et2O, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (hexane-EtOAc) to give the bromoacetylene (0.055 g, 43% yield).
Degassed piperidine (2 mL), bromoacetylene (0.060 g, 0.189 mmol) and CuCl (0.003 g, 0.019 mmol) were mixed in a sealed tube. The mixture was cooled to −78 °C and excess propyne gas (condensed to liquid, 2 mL) was added by blowing along the wall of the tube. The mixture was slowly warmed to rt. After stirring for 2h at rt, the mixture was cooled to −78 °C and the sealed tube was opened then slowly warmed to room temperature while excess propyne was evaporated. Sat. NH4Cl (aq) was added to the mixture then extracted with ethyl ether. The organic layer was washed with 10% HCl (aq), brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was dissolved in 1:1 water/acetone (2 mL) and treated with PPTS (0.003 g, 0.012 mmol) at rt. The mixture was heated at 40 °C for 3 h and extracted with diethyl ether. The organic layer was washed with brine (2 × 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate, 2:1) to give compound 4 (0.027 g, 63 % yield). ¹H-NMR (300 MHz, CDCl3) δ: 6.26 (dd, J= 15.9, 6.0 Hz, 1H), 5.71 (d, J= 15.9 Hz, 1H), 4.20–4.12 (m, 1H), 2.42 (t, J= 7.5 Hz, 2H), 2.13 (s, 3H), 1.98 (s, 3H), 1.62–1.49 (m, 4H), 1.39–1.24 (m, 4H); 13C-NMR (75 MHz, CDCl3) δ 209.4, 148.7, 109.1, 81.8, 80.4, 76.8, 72.2, 64.5, 43.8, 36.8, 30.1, 29.2, 25.2, 23.8, 4.8; HRMS m/e (EI) for C15H20O2 (M)+ calcd 232.1463, measured 232.1497.
3,5-Heptadiyn-1-ol (8)
Degassed piperidine (5.5 mL), 4-iodo-3-butynol (1.22 g, 6.2 mmol) and CuCl (0.061 g, 0.62 mmol) were mixedIn a tube. The mixture was cooled to −78 °C and excess propyne gas was added by blowing along the wall of the tube. The propyne was condensed to liquid (2 mL) in tube and the tube was sealed. The mixture was slowly warmed to rt. After stirring for 2 h at rt, the mixture was cooled to −78 °C and the sealed tube was opened. The mixture was warmed to rt slowly to evaporate excess propyne. NH4Cl (aq) (20 mL) was added to the mixture then extracted with Et2O (3 × 20 mL). Organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate= 2:1) to give compound 8 (538 mg, 81 % yield). ¹H-NMR (300 MHz, CDCl3) δ: 3.74–3.71 (m, 2H), 2.51 (t, J = 6.8 Hz, 2H), 1.91 (s, 3H).
Triphenyl(hepta-3,5-diynyl)phosphonium Iodide (9)
To a solution of imidazole (374 mg, 5.5 mmol) and Ph3P (1.44 g, 5.5 mmol) in Et2O–MeCN (12 mL/4 mL) was slowly added iodine (1.40 g, 5.5 mmol) at 0 °C. The resulting slurry was warmed to room temperature and then stirred for 20 min. The slurry was cooled to 0 °C and the alcohol 8 (538 mg, 4.9 mmol) was added in Et2O (10 mL) at 0 °C. The solution was slowly warmed to room temperature and then stirred for 1 h. The reaction was quenched by adding hexane (30 mL). The organic layer was washed with aq NaHCO3, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes–EtOAc, 2:1) to give the iodide (1.04 g, 98 % yield). ¹H-NMR (300 MHz, CDCl3) δ: 3.13 (t, J = 7.2 Hz, 2H), 2.77 (t, J = 7.2 Hz, 2H), 1.83 (s, 3H). To a solution of PPh3 (0.793 g, 3.03 mmol) in acetonitrile was added the above iodide (0.60 g, 2.75 mmol) and the mixture was boiled for 24 h, then cooled to rt and the solvent was removed to give 9 as a yellowish oil. (1.02 g, 78% yield). ¹H-NMR (400 MHz, CDCl3) δ: 7.76–7.53 (m, 15H), 3.86–3.80 (m, 2H), 2.79 (dt, J = 20.8, 6.4 Hz, 2H), 1.87 (s, 3H).
Pentadec-8Z-ene-11,13-diyn-2-one (2)
To a solution of compound 9 (612 mg, 1.19 mmol) in THF (10 mL) was added NaHMDS (1.19 mL, 1M in THF) at −78 °C. The mixture was stirred for 20 min at −78 °C, then aldehyde 5 (201 mg, 1.08 mmol) in THF (3 mL) was added by cannula. The mixture was slowly warmed to rt then stirred for 12 h. The reaction was quenched with NH4Cl (5 mL) and extracted with Et2O (20 mL). The organic layer was washed with water, brine, dried (MgSO4) and concentrated. The residue was purified via column chromatography (hexanes/ethyl acetate= 2:1) to give an enediyne (173 mg, 54 % yield). ¹H-NMR (400 MHz, CDCl3) δ: 5.46 (dt, J = 10.4, 7.2 Hz, 1H), 5.36 (dt, J = 10.4, 7.2 Hz, 1H), 3.96–3.88 (m, 4H), 2.98 (d, J = 7.2 Hz, 2H), 2.01 (td, J = 7.2, 6.0 Hz, 2H), 1.89 (s, 3H), 1.63–1.59 (m, 2H), 1.41–1.29 (m, 6H), 1.30 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 132.7, 122.9, 110.4, 75.0, 73.6, 65.3, 64.8, 64.7, 39.4, 29.7, 29.5, 27.3, 24.2, 24.0, 17.8, 4.4.
To a solution of the above compound (56 mg, 0.22 mmol) in 1:1 acetone/water (2 mL) was added PPTS (4.6 mg, 0.022 mmol) at rt. The mixture was heated at 40 °C for 3 h and extracted with ether. The organic layer was washed with brine (2 × 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography (hexanes/ethyl acetate, 2:1) to give compound 2 (43 mg, 93% yield). ¹H-NMR (400 MHz, CDCl3) δ: 5.48–5.34 (m, 2H), 2.96 (d, J = 7.2 Hz, 2H), 2.41 (t, J = 7.2 Hz, 2H), 2.12 (s, 3H), 2.01 (td, J = 7.2, 6.0 Hz, 2H), 1.88 (s, 3H), 1.60–1.51 (m, 2H), 1.42–1.22 (m, 6H); 13C-NMR (100 MHz, CDCl3) δ: 209.4, 132.5, 123.1, 74.9, 73.6, 65.3, 64.8, 43.9, 30.1, 29.2, 28.9, 27.2, 23.9, 17.8, 4.4; HRMS (EI): m/z calcd. for C15H20O : 216.1514; found: 216.1510.
Statistical analysis
Statistical analyses were performed using SAS software version 8.02 (SAS Institute Inc., Cary, NC). One-way analysis of variance followed by the Tukey test was used to compare means. Significance of difference was defined at p < 0.01.
Acknowledgments
We thank the National Institute of Health (Grant P01 ES12020) and the Office of Dietary Supplements for financial support through the Center for Research on Botanical Dietary Supplements at Iowa State University. We are especially grateful to Dr. Mark P. Widrlechner, USDA-ARS North Central Regional Plant Introduction Station, and Fredy R. Romero, Dept of Horticulture, ISU, for providing Echinacea plants.
Footnotes
Sample availability: Milligram quantities of 3 and 4 are available from the authors.
References
- 1.Wichtl M, editor. Herbal drugs and phytopharmaceuticals. A handbook for practice on a scientific basis. 3rd edn. Stuttgart: Medpharm Scientific Publishers; 2004. [Google Scholar]
- 2.Turner RB, Bauer R, Woelkart K, Hulsey TC, Gangemi JD. N. Engl. J. Med. 2005;353:341–348. doi: 10.1056/NEJMoa044441. [DOI] [PubMed] [Google Scholar]
- 3.Barnes J, Anderson LA, Gibbons S, Phillipson JD. J. Pharm. Pharmacol. 2005;57:929–954. doi: 10.1211/0022357056127. [DOI] [PubMed] [Google Scholar]
- 4.Merali S, Binns S, Paulin-Levasseur M, Ficker C, Smith M, Baum B, Brovelli E, Arnason JT. Pharm. Biol. 2003;41:412–420. [Google Scholar]
- 5.Bauer R, Wagner H. (LOMAPHARM, Rudolf Lohmann G.m.b.H. K.-G. Pharmazeutische Fabrik, Fed. Rep. Ger.) Ger. Offen. DE 3744571. 1989 [Google Scholar]
- 6.Binns SE, Purgina B, Bergeron C, Smith ML, Ball L, Baum BR, Arnason JT. Planta Med. 2000;66:241–244. doi: 10.1055/s-2000-8573. [DOI] [PubMed] [Google Scholar]
- 7.Bohlmann F, Rode KM, Zdero C. Chem. Ber. 1966;99:3544–3551. [Google Scholar]
- 8.Shi Shun ALK, Tykwinski RR. Angew. Chem. Int. Ed. 2006;45:1034–1057. doi: 10.1002/anie.200502071. [DOI] [PubMed] [Google Scholar]
- 9.Crombie L, Manzoor-I-Khuda M. J. Chem. Soc. 1957:2767–2777. [Google Scholar]
- 10.Crombie L, Fisher D. Tetrahedron Lett. 1985;26:2481–2484. [Google Scholar]
- 11.Crombie L, Horsham MA, Blade RJ. Tetrahedron Lett. 1987;28:4879–4882. [Google Scholar]
- 12.Wailes PC. Austral. J. Chem. 1959;12:173–189. [Google Scholar]
- 13.Kraus GA, Bae J, Schuster J. Synthesis. 2005:3502–3504. doi: 10.1055/s-2005-918418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellassoued M, Majidi A. Tetrahedron Lett. 1991;32:7253–7254. [Google Scholar]
- 15.Borden WT. J. Am. Chem. Soc. 1970;92:4898–4901. [Google Scholar]
- 16.Yun H, Danishefsky SJ. J. Org. Chem. 2003;68:4519–4522. doi: 10.1021/jo0341665. [DOI] [PubMed] [Google Scholar]
- 17.Brunel Y, Rousseau G. Tetrahedron Lett. 1995;36:2619–2622. [Google Scholar]
- 18.Dieter RK, Chen N, Watson RT. Tetrahedron. 2005;61:3221–3230. [Google Scholar]
- 19.Bauer R, Remiger P. Planta Med. 1989;55:367–371. doi: 10.1055/s-2006-962030. [DOI] [PubMed] [Google Scholar]
- 20.Wu L, Bae J, Kraus GA, Wurtele ES. Phytochemistry. 2004;65:2477–2484. doi: 10.1016/j.phytochem.2004.06.027. [DOI] [PubMed] [Google Scholar]