

Abstract
Many cortical synapses exhibit spike timing-dependent plasticity (STDP) in which the precise timing of presynaptic and postsynaptic spikes induces synaptic strengthening [long-term potentiation (LTP)] or weakening [long-term depression (LTD)]. Standard models posit a single, postsynaptic, NMDA receptor-based coincidence detector for LTP and LTD components of STDP. We show instead that STDP at layer 4 to layer 2/3 synapses in somatosensory (S1) cortex involves separate calcium sources and coincidence detection mechanisms for LTP and LTD. LTP showed classical NMDA receptor dependence. LTD was independent of postsynaptic NMDA receptors and instead required group I metabotropic glutamate receptors and calcium from voltage-sensitive channels and IP3 receptor-gated stores. Downstream of postsynaptic calcium, LTD required retrograde endocannabinoid signaling, leading to presynaptic LTD expression, and also required activation of apparently presynaptic NMDA receptors. These LTP and LTD mechanisms detected firing coincidence on ~25 and ~125 ms time scales, respectively, and combined to implement the overall STDP rule. These findings indicate that STDP is not a unitary process and suggest that endocannabinoid-dependent LTD may be relevant to cortical map plasticity.
Keywords: LTP, LTD, synaptic plasticity, endocannabinoid, barrel, metabotropic glutamate receptor
Introduction
Many neocortical synapses exhibit long-term potentiation (LTP) and long-term depression (LTD) in response to precise, millisecond-scale timing of presynaptic and postsynaptic action potentials, termed spike timing-dependent plasticity (STDP) (Markram et al., 1997; Egger et al., 1999; Feldman, 2000; Holmgren and Zilberter, 2001; Sjostrom et al., 2001; Froemke and Dan, 2002). In STDP, LTP is typically induced when presynaptic spikes lead postsynaptic spikes by 0 to ~20 ms, and LTD is induced when the order is reversed, for delays up to ~100 ms (Abbott and Nelson, 2000; Dan and Poo, 2004). STDP provides an inherently Hebbian, computationally powerful means of LTP and LTD induction in vivo and is strongly implicated in training-and deprivation-induced receptive field plasticity in sensory cortex (Dan and Poo, 2004; Feldman and Brecht, 2005).
The cellular signaling mechanisms underlying STDP induction are debated (Karmarkar et al., 2002; Shouval et al., 2002; Johnston et al., 2003; Dan and Poo, 2004). In standard models, postsynaptic NMDA receptors (NMDARs) are proposed to be the sole coincidence detector and primary calcium source for STDP, with pre–post firing order generating strong NMDAR-mediated calcium signals to drive LTP and post–pre firing order generating weaker signals to drive LTD (Shouval et al., 2002; Dan and Poo, 2004; Froemke et al., 2005). Such firing order and interval dependence of NMDAR currents can arise by several plausible mechanisms (Shouval et al., 2002; Johnston et al., 2003; Kampa et al., 2004; Froemke et al., 2005). However, some theoretical and experimental studies suggest that other coincidence detector(s) may be involved, specifically to mediate the LTD component of STDP (Karmarkar and Buonomano, 2002; Karmarkar et al., 2002; Sjostrom et al., 2003). Candidate coincidence detectors for LTD include inositol 1,4,5-trisphosphate (IP3) receptors, which contribute to cerebellar LTD (Wang et al., 2000), and presynaptic endocannabinoid (eCB) and NMDA receptors, which are implicated in spike timing-dependent LTD in visual cortex (Sjostrom et al., 2003). Whether most STDP rules involve a single, postsynaptic NMDAR-based coincidence detector or multiple coincidence detector pathways is not known.
We studied the cellular mechanisms for STDP at excitatory layer 4 synapses on layer 2/3 pyramidal cells (L4-L2/3 synapses) in developing rat somatosensory (S1) cortex. STDP at this synapse is strongly implicated in experience-dependent whisker map plasticity, and spike timing-dependent LTD in particular is proposed to underlie a common component of map plasticity, the downregulation of cortical responses to deprived sensory inputs (Allen et al., 2003; Celikel et al., 2004; Bender et al., 2006). Thus, STDP mechanisms are likely to be relevant for cortical plasticity and development.
Results showed that STDP at L4-L2/3 synapses involves two separate coincidence detection mechanisms. The LTP component was dependent on postsynaptic NMDARs and was driven by pre–post firing order at short intervals (5 to ~30 ms). The LTD component instead involved group I metabotropic glutamate receptors (mGluRs), calcium release from IP3R-gated stores, retrograde eCB signaling, and activation of non-postsynaptic, potentially presynaptic NMDARs and was driven by a much larger range (~125 ms) of timing intervals. Thus, the LTP and LTD components of STDP use different calcium sources and coincidence detection mechanisms.
Materials and Methods
All procedures were approved by the University of California, San Diego Institutional Animal Care and Use Committee. Slices (400 μm) containing the posteromedial barrel subfield (PMBSF) were prepared from Long–Evans rats [postnatal day 16 (P16) to P23], such that they contained one barrel from each whisker row, A–E (Finnerty et al., 1999). Rats were anesthetized with isoflurane and decapitated. The brain was rapidly removed in ice-cold Ringer’s solution (in mM: 119 NaCl, 26 NaHCO3, 11D-(+)-glucose, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 2.5 CaCl2). Slices were cut on a vibrating microtome (VT1000S; Leica, Wetzlar, Germany), pre-incubated in Ringer’s solution at 30°C for 30 min, and then incubated at room temperature (22–24°C) until use (1–7 h). All recordings were made at room temperature.
The PMBSF was identified by the presence of three to five large (250–450 μm) barrels in layer IV, visible under transillumination (Bender et al., 2003). A concentric bipolar stimulating electrode (FHC, Bowdoinham, ME) was placed at the base of an L4 barrel. Whole-cell recordings were made from L2/3 pyramidal cells in the same barrel column. A glass pipette (8–10 μm tip diameter) containing 5 mM bicuculline methiodide (BMI; Sigma, St. Louis, MO) in Ringer’s solution was placed in L2/3 within 100 μm of the recording electrode to block GABAA receptors (Castro-Alamancos et al., 1995; Feldman, 2000). Neurons with pyramidal shaped somata were selected for recording using infrared, differential interference contrast optics. All cells tested exhibited regular spiking responses to positive current injection, characteristic of pyramidal cells.
Whole-cell recording
Whole-cell recordings were made with 3–5 MΩ pipettes using an Axopatch 200B or AxoClamp 2B amplifier (Molecular Devices, Union City, CA). Recordings were filtered at 2 kHz and digitized at 5 kHz using a 12 bit data acquisition board (National Instruments, Austin, TX) and custom data acquisition and analysis routines running in Igor (Wavemetrics, Lake Oswego, OR). For current-clamp experiments, the internal solution contained the following (in mM): 116 potassium gluconate, 20 HEPES, 6 KCl, 2 NaCl, 0.5 EGTA, adjusted to pH 7.20 with KOH (mOsm to 290). Membrane potential was −84.1 ± 3.9 mV (SD; in a subset of 38 cells) after junction potential correction (−12 mV) and cells hyperpolarized by an average of ~4 mV during 45 min of recording. Cells were excluded if they depolarized by >10 mV. Input resistance was calculated from the response to a hyperpolarizing current step during each sweep. The mean input resistance was 126 ± 34 MΩ (SD; range, 75–200 MΩ), and the mean series resistance was 19 ± 5 MΩ (SD; range, 10–30 MΩ). Stimulus intensity was set to evoke small EPSPs (amplitude, 2.4 ± 0.7 mV, SD; n = 38). Only the initial slope (first 2–4 ms) of the EPSP was analyzed. Multicomponent EPSPs with well-isolated initial components were sometimes included (only the initial slope of the first component was analyzed). For voltage-clamp experiments, the internal solution contained the following (in mM): 108 D-gluconic acid, 108 cesium OH, 20 HEPES, 5 TEACl, 2.8 NaCl, 0.4 EGTA, adjusted to pH 7.25 with CsOH (290 mOsm).
Timing-dependent LTP and timing-dependent LTD induction protocols
EPSPs were measured at a constant rate of 0.1–0.167 Hz for a 6–12 min baseline period. To induce timing-dependent LTP (t-LTP) or timing-dependent LTD (t-LTD), single presynaptic and postsynaptic action potentials were paired at 0.2–0.25 Hz (100 repetitions). For most consistent t-LTP induction, the postsynaptic cell was depolarized (5–15 mV) during pairing (Lisman and Spruston, 2005). Postsynaptic spikes were evoked by somatic current injection (mean, 1.5 ± 0.2 nA for 5 ms). Pairing delay was defined as the delay between the peak of the postsynaptic spike and the onset of the EPSP.
For experiments in which the postsynaptic cell was hyperpolarized during STDP induction, 0.4–0.6 nA of hyperpolarizing current was injected for 50 ms, 2–3 ms after the beginning of the current pulse driving the somatic action potential.
EPSP slope was calculated from 50 consecutive sweeps immediately before the start of pairing (baseline) and compared with the 50 sweeps beginning 20 min after the end of pairing (test). LTP or LTD magnitude was defined as EPSP slope during test/EPSP slope during baseline.
Paired-pulse experiments
In current clamp, short bursts of EPSPs (five pulses at 25 Hz) were elicited every 60 s during baseline and postpairing periods. Pairing was performed with single pulses at 0.2–0.25 Hz (100–300 repetitions). Paired-pulse ratio (PPR) was quantified from the amplitudes of the first two EPSPs of the train. In experiments with lowered Ca2+, [Mg2+] was increased to maintain constant divalent ion concentration. For cyclothiazide (CTZ) experiments, two EPSPs at 25 Hz were elicited every 25 s while voltage clamping the postsynaptic cell at −70 mV.
Quantification of internal (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d]cyclohepten-5,10-imine maleate blockade of NMDA currents
NMDA: AMPA current ratios were compared between internal (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (iMK-801)-containing cells and interleaved control cells using two methods. First, NMDA and AMPA current amplitudes were measured at +40 mV (60 ms after current onset) and at −80 mV (10–15 ms after current onset), respectively, and the ratio of these amplitudes was calculated. Second, in a separate set of cells, NMDA and AMPA currents were both measured at a potential of −60 mV in low (0.4 mM) Mg2+ Ringer’s solution before and after D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5) (50 μM). NMDA currents were isolated by subtraction of currents recorded in D-AP5 from total synaptic current. AMPA currents were operationally defined as the current remaining in D-AP5 (which may include some kainate current). The ratio of NMDA and AMPA current integrals was calculated.
Drugs
D-AP5, anandamide (in Tocrisolve), (S)-α-methyl-4-carboxy-phenylglycine (MCPG), 2-methyl-6-(phenylethynyl)-pyridine hydrochloride (MPEP), and DL-threo-β-benzyloxyaspartic acid (TBOA) (all from Tocris Cookson, Ellisville, MO) and NiCl2 (Sigma) were bath applied. MK-801, BAPTA, and heparin (Sigma) were dissolved directly into the internal solution. Stock solutions of N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1 H-pyrazde-3-carboxamide (AM251), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), thapsigargin, nimodipine, cyclothiazide (Tocris Cookson), and ryanodine (Sigma) were made in DMSO and then dissolved in Ringer’s solution (final concentration of DMSO, 0.003%), except for the experiments in Figure 7, in which AM251 was dissolved in ethanol (EtOH) (final concentration of EtOH, 0.004%). Cells were incubated (1–3 h) in thapsigargin and ryanodine and continually bathed while recording. (5Z,8Z,11Z,14Z)-N-(4-Hydroxy-2-methylphenyl)-5,8,11,14-eicosatestraenamide (VDM-11) and 1,6-bis(cyclohexyloximinocarbonylamino) hexane (RHC80267) were dissolved in DMSO and then added to internal (final concentration of DMSO, 0.05%). When drugs were applied internally via the patch pipette, interleaved controls contained the appropriate vehicle.
Figure 7.

Spike-timing windows of pharmacologically isolated t-LTP and t-LTD. A, STDP measured in the presence of AM251 (open circles, individual cells; open triangles, means and SEM) and iMK-801 (closed circles, individual cells; closed triangles, means and SEM) to isolate t-LTP and t-LTD signaling pathways, respectively. B, STDP timing window under control conditions. Curve, Mean STDP at this synapse from previously published data (Feldman, 2000; Celikel et al., 2004); closed circles, new control cells that were interleaved with data in A; plus signs, other control cells from the present study (not interleaved with data in A); closed triangles, control means and SEM.
Statistics
Comparisons were made by unpaired two-tailed Student’s t test, unless otherwise noted. Data are presented as mean ± SE. The critical level of significance was p < 0.05.
Results
Spike timing-dependent LTP and LTD require postsynaptic calcium but are differentially sensitive to D-AP5
Presumed L4-L2/3 excitatory synapses were studied using just-suprathreshold extracellular stimulation in L4 and whole-cell recording from L2/3 pyramidal cells in rat S1 slices (P16–P23), as described previously (Feldman, 2000). To isolate excitatory synapses, local GABAA receptors were blocked by focal bicuculline methiodide (Castro-Alamancos et al., 1995). STDP was induced in current clamp using 100 pairings of single presynaptic and postsynaptic spikes at 0.2–0.25 Hz. Consistent with previous results (Feldman, 2000; Celikel et al., 2004), pre–post spike delays of 5 ms (pre-leading-post) and −25 ms (post-leading-pre) elicited robust t-LTP (EPSP slope after pairing relative to baseline, 1.27 ± 0.05; n = 20) (Fig. 1A) and t-LTD, respectively (0.72 ± 0.03; n = 40) (Fig. 1A). Presynaptic firing (0.2–0.25 Hz) without postsynaptic spikes caused no plasticity (0.94 ± 0.13; n = 8).
Figure 1.

t-LTP and t-LTD at L4-L2/3 synapses are sensitive to D-AP5 and BAPTA. A, Top left, Representative EPSPs before and after t-LTD induction. Top right, Representative EPSPs before and after t-LTP induction. Middle, Mean time course of t-LTP and t-LTD. Open circles, t-LTD; closed circles, t-LTP. Bottom, Mean Vm and input resistance to show stability of recordings. B, t-LTD and t-LTP are blocked by postsynaptic BAPTA (5 mM). C, Time course of blockade of NMDA currents by D-AP5 (50 μM). The arrow marks 5 min from the beginning of D-AP5 application. D, Mean effect of 5 min of D-AP5 application before pairing (open squares). Closed squares are interleaved controls. E, Five minute D-AP5 application blocks t-LTP but not t-LTD. The summary of the effect of 5 min of D-AP5 application before pairing on t-LTD and t-LTP is shown. Error bars are SEM. *p < 0.05.
t-LTD was blocked by BAPTA (5 mM) in the postsynaptic pipette (1.02 ± 0.06, n = 8; interleaved controls, 0.76 ± 0.04, n = 11; p < 0.01) (Fig. 1B) as was t-LTP (0.95 ± 0.05, n = 5; interleaved controls, 1.23 ± 0.06, n = 7; p < 0.01) (Fig. 1B). Thus, both t-LTP and t-LTD require postsynaptic calcium, like other forms of LTP and LTD (Artola and Singer, 1993; Malenka and Bear, 2004).
To test whether NMDA receptors were the relevant calcium source for t-LTP and t-LTD, as proposed in standard STDP models (Koester and Sakmann, 1998; Lisman, 2001; Malinow and Malenka, 2002; Froemke et al., 2005), we first applied the NMDA receptor antagonist D-AP5 (50 μM). D-AP5 blocked 95% of synaptically evoked NMDA receptor currents (measured as total charge of pharmacologically isolated currents) within 5 min of wash-in (Fig. 1C). Brief D-AP5 application (beginning 5 min before the start of pairing) completely blocked t-LTP (0.97 ± 0.07, n = 4; interleaved controls, 1.49 ± 0.10, n = 4; p < 0.01) (Fig. 1E) but, surprisingly, failed to affect t-LTD (0.77 ± 0.05, n = 8; interleaved controls, 0.75 ± 0.03, n = 9; p = 0.73) (Fig. 1D,E). D-AP5 did not by itself alter baseline EPSPs (1.02 ± 0.06; n = 5). These data suggest that t-LTP, but not t-LTD, requires NMDA receptor activation during pairing, and thus NMDA receptors may not be the relevant coincidence detectors for t-LTD.
Dependence on postsynaptic NMDA receptors
We performed two experiments to explicitly test whether t-LTP and t-LTD were differentially dependent on postsynaptic NMDA receptors during pairing. First, we reduced postsynaptic NMDA receptor currents with postsynaptic hyperpolarization, which robustly blocks classical, NMDA receptor-dependent LTP and LTD (Malinow and Miller, 1986; Mulkey and Malenka, 1992; Bolshakov and Siegelbaum, 1994). In voltage-clamp experiments, holding cells at −90 mV abolished 98% of pharmacologically isolated, synaptically evoked NMDA conductance (relative to maximal conductance at +40 mV), indicating that NMDA receptors at L4-L2/3 synapses show standard voltage dependence (Fig. 2A). To test the effect of hyperpolarization on t-LTD, a modified post-leading-pre pairing protocol was used in which negative current was injected after the postsynaptic spike, so that the neuron was robustly hyperpolarized (to less than −90 mV at the soma) by the time of the presynaptic spike. A −50 ms post–pre spike delay was used to allow adequate time for hyperpolarization. Hyperpolarization acutely increased EPSP slope by 39 ± 11%, indicating a substantial increase in driving force and effective hyperpolarization of the synapse, and decreased EPSP width, consistent with block of NMDA currents (Fig. 2C, inset). However, hyperpolarization did not affect the magnitude of t-LTD (0.75 ± 0 0.06, n = 15; interleaved controls, 0.73 ± 0.05, n = 5; p = 0.85) (Fig. 2D,E).
Figure 2.

Hyperpolarization does not block t-LTD. A, Voltage dependence of synaptically evoked NMDA receptor conductance at L4-L2/3 synapses (normalized to maximal conductance for each of 3 cells). Inset, Representative NMDA receptor currents (measured in 10 μm DNQX). Holding potentials are indicated. B, Standard protocol for t-LTD induction (50 ms post-leading-pre pairing). Pre, Time of extracellular presynaptic stimulation. C, Hyperpolarization (Hyperpol.) protocol for t-LTD induction. After initiating the postsynaptic spike, current was injected to strongly hyperpolarize the postsynaptic cell before arrival of the EPSP. Inset, Representative EPSPs recorded showing increased driving force during the hyperpolarization protocol compared with Vrest. D, Effect of hyperpolarization on t-LTD induction (50–60 ms post-leading-pre pairing). E, Summary of t-LTD magnitude. Hyp, Hyperpolarization.
In the second experiment, we blocked postsynaptic NMDARs by applying the irreversible channel blocker MK-801 internally via the patch pipette (Berretta and Jones, 1996; Humeau et al., 2003; Samson and Pare, 2005). In voltage clamp, iMK-801 (1 mM) reduced NMDA:AMPA current amplitude ratios (measured at +40 and −80 mV, respectively; see Materials and Methods) by 93% relative to interleaved cells with normal internal (iMK-801:0.06 ± 0.01, n = 6; interleaved controls, 0.90 ± 0.15, n = 11) (Fig. 3A). This reduction was comparable with that observed with bath-applied D-AP5 (Fig. 1C). iMK-801 did not act by spilling into the extracellular medium, because neighboring neurons recorded simultaneously <10 μm away had normal NMDA: AMPA current ratios (0.97 ± 0.29; n = 5) (Fig. 3A). iMK-801 also blocked NMDA currents at −60 mV, as measured by the ratio of pharmacologically isolated NMDA and AMPA currents in low (0.4 mM) Mg2+ Ringer’s solution (ratio of NMDA:AMPA current integrals for cells with iMK-801, 0.026 ± 0.138, n = 9; interleaved controls, 2.428 ± 0.465, n = 8; this represents a 99% decrease in NMDA:AMPA charge ratio) (Fig. 3B). Despite powerfully blocking NMDA currents, iMK-801 (starting 8–14 min before pairing) did not block t-LTD (0.58 ± 0.12, n = 6; interleaved controls, 0.76 ± 0.06, n = 7; p = 0.20) (Fig. 3C,D). However, iMK-801 did block t-LTP (0.77 ± 0.07, n = 4; interleaved controls, 1.23 ± 0.08, n = 10; p < 0.005) (Fig. 3E). Together, these experiments indicate that t-LTP requires activation of postsynaptic NMDA receptors during pairing, but t-LTD does not. Thus, another coincidence detector in addition to the NMDA receptor must detect post–pre firing intervals for t-LTD.
Figure 3.

Blockade of postsynaptic NMDA currents by internal MK-801 does not block t-LTD. A, iMK-801 (1 mM) substantially blocks NMDA currents measured at +40 mV. Top, Representative EPSCs measured in control cells and in the presence of 50 μM D-AP5 and 1 mM internal MK-801. Holding potentials are indicated. Bottom, Quantification of NMDA (amplitude of current at +40 mV at dark bar in A1) to AMPA (amplitude of current at −80 mV at outlined bar in A) current ratios in control, D-AP5, iMK-801 conditions and cells recorded with normal internal within 10 μm of cells recorded with iMK-801 (neighbors). B, iMK-801 also blocks NMDA currents at −60 mV. Top, Representative EPSCs recorded at −60 mV in low (0.4 mM) Mg 2+Ringer’s solution in control (top) and MK-801 (bottom) internals in normal Ringer’s solution, 50 μM D-AP5 and 10 μM DNQX. Bottom, Quantification of the NMDA:AMPA current integral ratio (see Materials and Methods) for all cells tested. C, iMK-801 does not block t-LTD. The net effect of iMK-801 on t-LTD is plotted with interleaved controls. Open circles, iMK-801; closed circles, interleaved controls. D, Summary of the effect of iMK-801 on t-LTD. E, iMK-801 does block t-LTP. Error bars show mean ± SEM. *p < 0.05.
Sources of postsynaptic calcium for t-LTD
To identify the source of postsynaptic calcium for t-LTD induction, we considered calcium release from internal stores, which is implicated in many forms of LTD (Kemp and Bashir, 2001). Incubation in thapsigargin (10 μM), which depletes calcium stores, blocked t-LTD (0.93 ± 0.05, n = 7; control, 0.65 ± 0.05, n = 6; p < 0.05) (Fig. 4A). Heparin (400 U/ml; applied in the patch pipette), a blocker of IP3R-mediated calcium release from stores (Ghosh et al., 1988; Khodakhah and Armstrong, 1997), completely abolished t-LTD (1.03 ± 0.11, n = 5; interleaved controls, 0.66 ± 0.06, n = 7; p < 0.005) (Fig. 4A,C). In contrast, ryanodine (100 μM in the patch pipette), a blocker of ryanodine receptors and calcium-induced calcium release (CICR) from internal stores, did not prevent t-LTD (0.66 ± 0.10; n = 5) (Fig. 4A). Ryanodine was effective in these experiments, because bath application of ryanodine (100 μM) partially blocked t-LTD (0.88 ± 0.05, n = 10; DMSO vehicle controls, 0.65 ± 0.05, n = 6; p < 0.05), consistent with previous findings that CICR in presynaptic terminals contributes to LTD (Unni et al., 2004). Thus, calcium from postsynaptic IP3R-dependent stores, but not ryanodine receptor-dependent stores, is required for t-LTD. Heparin did not block t-LTP (1.26 ± 0.14, n = 4; interleaved controls, 1.21 ± 0.07, n = 4; p = 0.78), suggesting that calcium for t-LTP and t-LTD derive from separate, NMDA receptor- dependent and IP3R-dependent sources, respectively.
Figure 4.

Calcium sources for t-LTD. A, Summary of effect of blocking calcium release from internal stores with thapsigargin (10 μM), heparin (400 U/ml), and ryanodine (100 μM) in the postsynaptic pipette on t-LTD. B, Effect of general mGluR antagonists MCPG (0.5–1 mM), LY341495 (100 μM), and a specific mGluR5 antagonist (MPEP, 10 μM) on t-LTD. C, Effect of heparin on t-LTD. D, Example of post–pre pairing (−24 ms) with postsynaptic cell resting at −60 mV between spikes. Top, Each point represents individual EPSPs. The dashed line represents average slope during baseline. Bottom, Inset, Average EPSPs before (1) and after (2) protocol. E, Mean effect of nimodipine (1–20 μM), − 60 mV resting between spikes, and NiCl2 (50 μM) on t-LTD with appropriate controls. Error bars show mean ± SEM. Ctrl, Control. *p < 0.05; **p < 0.01.
The source of IP3 for t-LTD may be group I mGluRs, which activate phospholipase C (PLC) to produce IP3 (Berridge, 1998). mGluRs and IP3 receptors mediate several forms of LTD (Oliet et al., 1997; Normann et al., 2000; Wang et al., 2000; Kemp and Bashir, 2001). Group I mGluRs are postsynaptic in the neocortex, with mGluR5 being the dominant subtype in pyramidal cells (Blue et al., 1997; Lopez-Bendito et al., 2002). Two broad-spectrum mGluR antagonists, (S)-MCPG (0.5–1 mM) and LY341495 (100 μM), blocked t-LTD induction (MCPG, 0.99 ± 0.07, n =4; interleaved controls, 0.68 ± 0.04, n =13; p < 0.005; LY341495, 0.86 ± 0.07, n =5; DMSO vehicle controls, 0.65 ±0.05, n =6; p < 0.05) (Fig. 4B). LTD was also blocked by the specific mGluR5 antagonist MPEP (10 μM, 0.98 ± 0.07, n = 7; interleaved controls, 0.75 ± 0.05, n = 5; p < 0.05) (Fig. 4B). Thus, t-LTD requires group I mGluR and IP3R signaling.
mGluR and IP3R-dependent LTD is often also dependent on calcium from voltage-sensitive calcium channels (VSCCs) (Oliet et al., 1997; Otani and Connor, 1998; Wang et al., 2000). In pyramidal cells, backpropagating action potentials activate low-voltage gated (T-type) and high-voltage gated (R, P/Q, and L-type) dendritic VSCCs, with R- and T-type channels accounting for a substantial portion of the calcium in L2/3 pyramids (Schiller et al., 1998; Koester and Sakmann, 2000; Sabatini et al., 2001; Waters et al., 2003). To determine whether L-type VSCCs are required for LTD, we applied the antagonist nimodipine (1–20 μM) and found that t-LTD was partially blocked (0.83 ± 0.03, n = 12; DMSO vehicle controls, 0.65 ± 0.05, n = 6; p < 0.05) (Fig. 4E). Bath application of 50 μM NiCl2, which blocks T- and R-type channels (Magee and Johnston, 1995), prevented t-LTD (1.08 ± 0.06, n = 6; interleaved controls, 0.68 ± 0.09, n = 4; p < 0.005) (Fig. 4E) but also substantially decreased baseline transmission (0.72 ± 0.06; n = 3), indicating that t-LTD was either occluded or blocked. Inactivation of postsynaptic T-type channels during pairing by holding the cell at −60 mV between postsynaptic spikes (Huguenard, 1996; Kavalali et al., 1997; Oliet et al., 1997) completely blocked t-LTD (1.17 ± 0.14, n = 10; interleaved controls, 0.71 ± 0.05, n = 5; p < 0.05) (Fig. 4D,E). Membrane potential was approximately −80 mV during baseline and test periods in this experiment. Together, these experiments suggest that postsynaptic T-type, and to a lesser extent L-type, calcium channels are required for t-LTD. The contribution of other calcium channel subtypes is not ruled out.
t-LTD requires retrograde endocannabinoid signaling
LTD at many synapses requires signaling by eCBs (Auclair et al., 2000; Gerdeman et al., 2002; Marsicano et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003; Huang et al., 2003; Sjostrom et al., 2003; Safo and Regehr, 2005). eCBs are phospholipids that are synthesized and released in response to postsynaptic depolarization, Ca2+ elevation, and/or mGluR signaling. eCBs diffuse retrogradely to activate cannabinoid type 1 (CB1) receptors on presynaptic terminals (Piomelli et al., 1998; Wilson and Nicoll, 2001, 2002; Freund et al., 2003). L2/3 pyramidal cells release eCBs during postsynaptic spiking (Trettel and Levine, 2003; Trettel et al., 2004), and several classes of cortical excitatory and inhibitory terminals express functional CB1 receptors (Auclair et al., 2000; Trettel and Levine, 2003; Sjostrom et al., 2003).
To determine whether t-LTD was CB1 receptor dependent, we applied the selective CB1 receptor antagonist AM251 (1–3 μM). AM251 did not alter baseline transmission (1.04 ± 0.07; n = 4) but completely blocked t-LTD (0.96 ± 0.07, n = 6; DMSO, 0.003%, vehicle controls, 0.65 ± 0.05, n = 6; p < 0.01) (Fig. 5A,B). Inclusion of the anandamide membrane transport blocker VDM-11 (10–20 μM) in the recording pipette, which inhibits eCB secretion from the postsynaptic neuron (Ronesi et al., 2004), also blocked t-LTD (0.99 ± 0.04, n = 5; DMSO vehicle controls, 0.61 ± 0.10, n = 4; p < 0.01) (Fig. 5B). RHC80267 (50 μM in the recording pipette), which blocks postsynaptic synthesis of the endocannabinoid 2-arachidonylglycerol (2-AG), also blocked t-LTD (0.90 ± 0.02, n = 6; vehicle controls, 0.68 ± 0.05, n = 7; p < 0.01) (Fig. 5B). In contrast, AM251 had no effect on t-LTP (1.25 ± 0.07, n = 5; ethanol, 0.004%, vehicle control, 1.26 ± 0.07, n = 5) (Fig. 5C). Thus, t-LTD requires eCB (presumably 2-AG) release from the postsynaptic neuron and activation of CB1 receptors, but t-LTP does not.
Figure 5.

Cannabinoid dependence of t-LTD. A, AM251 (3 μM) completely blocks t-LTD. t-LTD in the presence of AM251 (open circles), DMSO alone (closed circles), and normal Ringer’s solution (open triangles). B, Mean effect of AM251, VDM-11 (10–20 μM), and RHC80267 (50 μM) on t-LTD with relevant vehicle controls. C, AM251 has no effect on t-LTP. t-LTP in the presence of AM251 (open squares) and ethanol vehicle alone (closed squares). Right, Mean effect of AM251 on t-LTP. D, Anandamide (AEA) wash-in with presynaptic stimulation induces long-lasting depression. Open circles show cells with anandamide (40 μM) and subsequent AM251 wash- nat0.1 and 30Hz. Closed circles are interleaved controls with presynaptic stimulation alone. E, Mean effect of anandamide wash-in at 30 and 0.1 Hz, with postsynaptic BAPTA (5 mM; 30 Hz), AM251 (3 μM; 0.1 Hz), and MPEP (10 μM; 0.1 Hz). *p < 0.05.
To determine whether CB1 receptor activation was sufficient to drive LTD, we applied anandamide, an endogenous CB1 agonist (40 μM in Tocrisolve) while stimulating presynaptic axons with either 30 Hz bursts [five pulses per burst, 25–60 s interburst interval, as required for anandamide effects in the study by Sjostrom et al. (2003)] or single pulses at 0.1 Hz. Postsynaptic cells rested at approximately −80 mV without spiking during these experiments, and presynaptic stimulation alone evoked stable synaptic responses (Fig. 5C, closed circles). Wash-in of anandamide caused significant synaptic depression for both stimulation frequencies (30 Hz, 0.72 ± 0.05, n = 7; 0.1 Hz, 0.72 ± 0.06, n = 6) (Fig. 5E), and this reduction persisted after anandamide wash-out and subsequent wash-in of AM251 (30 Hz, 0.50 ± 0.02, n = 2; 0.1 Hz, 0.77 ± 0.04, n = 6) (shown combined in Fig. 5D). Thus, anandamide-induced synaptic depression was long lasting. Anandamide-induced synaptic depression was mediated via CB1 receptors, because it was blocked when AM251 was applied synchronously with anandamide (1.08 ± 0.03, n = 3; p < 0.01) (Fig. 5E). Anandamide-induced synaptic depression appeared to represent a presynaptic process, because it persisted when BAPTA (5 mM) was included in the postsynaptic recording pipette (30 Hz, 0.69 ± 0.06; n = 3) (Fig. 5E) and when MPEP (10 μM) was included in the bath to block mGluR5 (0.1 Hz, 0.69 ± 0.13, n = 4; p = 0.85) (Fig. 5E). Moreover, anandamide-induced synaptic depression was accompanied by an increase in PPR (measured at 33 ms interstimulus interval; baseline, 0.69 ± 0.09; 30–40 min after anandamide, 0.91 ± 0.10; n = 18; paired t test, p < 0.05). PPR did not change significantly in control experiments in which anandamide was not applied (baseline, 0.82 ± 0.10; 30–40 min later, 0.86 ± 0.20, n = 5; paired t test, p = 0.50). The change in PPR suggests that anandamide acted at presynaptic CB1 receptors to reduce presynaptic release probability, as occurs at other synapses (Wilson and Nicoll, 2002; Zucker and Regehr, 2002; Sjostrom et al., 2003; Brown et al., 2004).
Together, these results show that t-LTD requires, and is mimicked by, eCB signaling, but that t-LTP is independent of eCBs. eCB-dependent LTD also occurs in visual and entorhinal cortex (Auclair et al., 2000; Sjostrom et al., 2003) and a variety of non-cortical synapses (Gerdeman et al., 2002; Marsicano et al., 2002; Robbeetal., 2002; Chevaleyre and Castillo, 2003; Huangetal., 2003).
t-LTD is accompanied by changes in paired-pulse ratio
Involvement of retrograde eCB signaling suggests that t-LTD may be expressed presynaptically by a decrease in release probability (Brown et al., 2004; Sjostrom et al., 2004). To test this hypothesis, we measured PPR, which is commonly inversely correlated with release probability (Zucker and Regehr, 2002), before and after t-LTD induction. PPR was measured for the first two EPSPs in a short burst (five pulses, 25 Hz, 60 s interburst interval). Under normal conditions, slight paired-pulse depression was observed (0.79 ± 0.09; n = 8) (Fig. 6A,B), consistent with the known behavior of unitary L4-L2/3 synaptic connections (Feldmeyer et al., 2002). Reducing external calcium to 1.25 mM increased PPR (2.5 mM, 0.79 ± 0.09; 1.25 mM, 1.19 ± 0.03, n = 8; paired t test, p < 0.01) (Fig. 6C), and in separate voltage-clamp experiments, the AMPA receptor desensitization blocker CTZ (50 μM) did not alter PPR (DMSO vehicle control, 0.99 ± 0.09; CTZ, 0.98 ± 0.07; n = 5; paired t test, p = 0.55), consistent with PPR being primarily an indicator of release probability.
Figure 6.

t-LTD changes paired-pulse ratios. A, Left, Example of change in paired-pulse ratio before (black lines) and after (gray lines) induction of t-LTD. Right, t-LTD and changes in PPR are blocked by postsynaptic BAPTA. B, Top, Closed squares, t-LTD induced with baseline stimulation of five pulses at 25 Hz. Open circles, t-LTD with 10 mM BAPTA in the postsynaptic pipette. Bottom, Same symbols as top. Three cells in which significant t-LTD was not induced are not included. C, Summary of changes in PPR before and after t-LTD induction (open circles), normal to low Ca 2+(open triangles), and t-LTD with postsynaptic BAPTA (open squares). Dashed line, No change in PPR. D, Regression (dashed line) showing the increase in PPR versus the magnitude of t-LTD induction. Symbols 1, 2, and 3 refer to t-LTD induced with 100, 200, and 300 pairings, respectively.
t-LTD (magnitude, 0.78 ± 0.04; n = 16) (Fig. 6B) significantly increased PPR (before LTD, 0.76 ± 0.11; after, 0.91 ± 0.06; paired t test, p < 0.005) (Fig. 6B,C). Postsynaptic BAPTA (10 mM) prevented t-LTD (1.02 ± 0.05; n = 11) and the PPR change (PPR before, 0.80 ± 0.05; after, 0.75 ± 0.05; paired t test, p > 0.05) (Fig. 6A–C). When the number of pairings was varied between cells to enhance the variation in t-LTD magnitude, t-LTD magnitude and the PPR increase were found to be significantly correlated (p < 0.02; R2 = 0.304) (Fig. 6D). This finding, the requirement for retrograde eCB signaling for t-LTD, and the fact that anandamide-induced synaptic depression also increased PPR suggest that both t-LTD and anandamide-induced synaptic depression are expressed, at least in part, by a reduction in presynaptic release.
Coincidence detection windows for t-LTP and t-LTD
The above results suggest that t-LTP involves classical, NMDA receptor-dependent induction mechanisms, whereas t-LTD uses an mGluR-VSCC-IP3R-eCB signaling pathway for induction, leading ultimately to presynaptic expression. To determine how these separate signaling pathways interact to produce the overall STDP rule, we measured the spike-timing dependence of plasticity elicited by each of these pathways separately (Fig. 7). The postsynaptic NMDA receptor pathway was isolated by blocking eCB signaling with bath-applied AM251 and was found to generate only t-LTP and only in response to pre–post spike delays of 5–30 ms. Thus, this pathway required pre-leading-post firing order to drive t-LTP and had a resolution for coincidence detection of ~25 ms. The mGluR-VSCC-IP3R-eCB pathway was isolated by blocking postsynaptic NMDA receptors using internal MK-801 and was found to drive only t-LTD, and only in response to spike delays from −100 ms (post-leading-pre) to +25 ms (pre-leading-post), a coincidence detection resolution of ~125 ms. These findings are consistent with the existence of independent, parallel signaling pathways for t-LTP and t-LTD and demonstrate that short pre-leading-post firing intervals (5–30 ms) trigger both LTP and LTD mechanisms. How t-LTP and t-LTD combine to produce the overall STDP rule (Fig. 7B) was not examined in detail but appears to involve approximately linear summation of LTP and LTD components, except at short pre-leading-post delays (~10 ms), where LTP dominates.
Non-postsynaptic, potentially presynaptic NMDARs are required for t-LTD
Despite the finding that short-duration D-AP5 application did not block t-LTD (Fig. 1), we found that longer D-AP5 application (beginning 20–33 min before the start of pairing) robustly blocked t-LTD (1.08 ± 0.07, n = 5; p < 0.005 vs controls without D-AP5, 0.75 ± 0.03, n = 9) (Fig. 8A). Intermediate D-AP5 application (beginning 10–15 min before pairing), like short-duration application, failed to block t-LTD (0.81 ± 0.04; n = 3) (Fig. 8A). This suggests that although NMDA receptors are not required during pairing, a slow NMDA receptor-dependent process powerfully gates or modulates t-LTD. The NMDA receptor independence during pairing was missed in a previous study that only used long-duration D-AP5 application (Feldman, 2000). To investigate the source of this slow gating, we selectively blocked postsynaptic NMDA receptors with long-duration iMK-801 (iMK-801 introduced 24–26 min before onset of pairing) and found that t-LTD was unimpaired (iMK-801, 0.68 ± 0.04, n = 5; interleaved controls, 0.76 ± 0.06, n = 7; p = 0.29) (Fig. 8A). Thus, the NMDA receptors responsible for slow modulation of t-LTD are unlikely to be postsynaptic.
Figure 8.

Non-postsynaptic NMDA receptors are required for t-LTD and anandamide-induced synaptic depression. A, Intermediate duration (10–15 min) D-AP5 (50 μM) does not block t-LTD, but long-duration (20–33 min) D-AP5 does block t-LTD. Long-duration (24–26 min) internal MK-801 does not block t-LTD. This indicates that postsynaptic NMDA receptors are not the source of slow modulation of t-LTD. B, Bath-applying D-AP5 for 20 min or longer blocks anandamide (AEA)-induced synaptic depression, relative to interleaved control cells with no D-AP5. C, D-AP5 does not block synaptically evoked AMPA receptor currents measured in voltage clamp at −90 mV in single pulses at 0.1 Hz (top) but does block currents when seven pulses are evoked at 50 Hz (bottom). Insets, Single examples of AMPA currents before (black) and after D-AP5 (gray). D, D-AP5 does block AMPA currents measured at −90 mV in trains of two pulses at 30 Hz in the presence of 25 μM TBOA. Inset, Single example of AMPA currents before (black) and after D-AP5 (gray). E, Summary of effects of D-AP5 on the amplitudes (Ampl.) of the first and second EPSCs at 0.1, 30, and 50 Hz and 30 Hz in the presence of TBOA, under normal conditions (control), and in the presence of internal MK-801 (iMK-801). Asterisks indicate significance from baseline using a paired t test.
We considered the possibility that the relevant NMDA receptors may be presynaptic, because presynaptic NMDA receptors exist in the neocortex (Aoki et al., 1994; DeBiasi et al., 1996; Charton et al., 1999) and are required for t-LTD in L5 of V1 (Sjostrom et al., 2003). Consistent with this hypothesis, we found that bath-applied D-AP5 (20–120 min) blocked anandamide-induced synaptic depression, which is likely a presynaptic process (D-AP5 plus anandamide, 1.03 ± 0.10, n = 6; anandamide alone, 0.74 ± 0.03, n = 5; p < 0.03; EPSP amplitude was assessed 50–60 min after anandamide application) (Fig. 8B).
As an additional test for the existence of functional presynaptic NMDA receptors, we tested whether these receptors acutely modulate release probability, as found in L5 of V1 (Sjostrom et al., 2003). To do this, we measured the effect of D-AP5 wash-in on the amplitude and PPR of synaptically evoked AMPA currents, measured in voltage clamp at −90 mV. Under standard conditions, D-AP5 did not significantly alter AMPA-EPSC amplitude when EPSCs were evoked singly at 0.1 Hz or in bursts of five EPSCs at 30 Hz (EPSC amplitude relative to predrug baseline, 0.1 Hz, 1.13 ± 0.07, n = 5, paired t test, p = 0.11; 30 Hz, 0.90 ± 0.05, p = 0.07) (Fig. 8C,E).
However, increasing glutamate levels with the broad-spectrum glutamate transport blocker TBOA (25 μM) (Shigeri et al., 2004) revealed the presence of functional, non-postsynaptic NMDA receptors that regulate release (Lien et al., 2006). TBOA alone evoked a small increase in holding current (62 ± 23 pA at −90 mV, n = 6; p < 0.05), consistent with increased ambient glutamate, but had no significant effect on AMPA-EPSC amplitude (0.91 ± 0.05; n = 7; p = 0.24). Application of D-AP5 (50 μM) in the TBOA background significantly decreased EPSC amplitude, measured using pairs of stimuli at 30 Hz (first EPSC, 0.80 ± 0.04, p < 0.02; second EPSC, 0.90 ± 0.03, p < 0.02; n = 8) (Fig. 8D,E) and increased PPR (before D-AP5, 0.92 ± 0.08; after D-AP5, 1.03 ± 0.08, paired t test, p < 0.03), consistent with a modest decrease in presynaptic release probability. Similar results occurred using higher frequency bursts (seven EPSCs at 50 Hz) without TBOA (first EPSC, 0.74 ± 0.06, p < 0.05; second EPSC, 0.71 ± 0.06, p < 0.01; n = 8) (Fig. 8C,E), although with no change in PPR (before D-AP5, 0.62 ±0.12; after D-AP5, 0.56 ± 0.11, paired t test, p = 0.46). Thus, increasing ambient glutamate either with TBOA or high-frequency bursts revealed the presence of NMDA receptors that regulate release.
These NMDA receptors are likely to be non-postsynaptic, because: (1) the postsynaptic neuron was voltage clamped at −90 mV, which would essentially inactivate postsynaptic NMDA receptors; and (2) the effect on 30 Hz bursts in TBOA persisted when MK-801 was included in the recording pipette to block postsynaptic NMDA receptors (Fig. 8E) (first EPSC amplitude, relative to baseline, 0.62 ± 0.03, p < 0.001; second EPSC, 0.71 ± 0.04, p < 0.001; PPR before D-AP5, 0.96 ± 0.07; after D-AP5, 1.11 ± 0.06; paired t test, p < 0.04; n = 5). Whether these non-postsynaptic, potentially presynaptic NMDA receptors regulate synapse efficacy by directly regulating release probability or by indirectly affecting postsynaptic function (Casado et al., 2000) is unknown.
Discussion
Standard models posit that postsynaptic NMDARs are the sole coincidence detector for LTP and LTD components of STDP (Shouval et al., 2002; Dan and Poo, 2004; Bi and Rubin, 2005; Froemke et al., 2005). Our results support a different model for STDP at L4-L2/3 synapses in which t-LTP induction involves postsynaptic NMDARs, but t-LTD induction instead involves signaling molecules including mGluRs, calcium from IP3R-gated internal stores and VSCCs, retrograde eCB signaling, and non-postsynaptic, potentially presynaptic NMDARs. Thus, LTP and LTD components of STDP involve two distinct sources of calcium and two distinct coincidence detection mechanisms. These mechanisms appear to operate on different time scales and with different firing order dependence and combine to produce overall STDP. Here, we review the evidence for this model and suggest it represents a major class of STDP across cortical synapses.
t-LTP induction mechanisms
t-LTP was blocked by D-AP5, iMK-801, and postsynaptic BAPTA but was unaffected by heparin, indicating that it required postsynaptic NMDARs and postsynaptic calcium but not calcium release from IP3R-gated stores. The narrow coincidence detection window for pharmacologically isolated t-LTP (~25 ms) (Fig. 7) is consistent with NMDAR-based STDP models and known NMDAR kinetics (Shouval et al., 2002; Kampa et al., 2004; Shouval and Kalantzis, 2005). Thus, t-LTP appears to use a standard NMDAR-based coincidence detector, like classical LTP at CA1 hippocampal synapses (Malinow and Malenka, 2002).
t-LTD induction mechanisms
Role of postsynaptic NMDARs
t-LTD is also required postsynaptic calcium, but in contrast to t-LTP, was unaffected by short-duration (5–15 min) D-AP5, postsynaptic hyperpolarization, or iMK-801, which blocked NMDAR currents by 93–99%. Thus, NMDARs are unlikely to be the relevant coincidence detector or a significant source of calcium during spike pairing for t-LTD.
Role of mGluRs, IP3Rs, and VSCCs
t-LTD required mGluR5, the dominant group I mGluR on L2/3 pyramidal cells (Lopez-Bendito et al., 2002). Group I mGluRs activate PLC to generate IP3, which triggers calcium release from IP3R-dependent stores (Berridge, 1998). Calcium stores depletion (using thapsigargin) or IP3R blockade (using heparin) blocked t-LTD, implicating this pathway as a primary source of postsynaptic calcium for t-LTD induction. t-LTD therefore resembles known mGluR-dependent, postsynaptic NMDAR-independent forms of LTD (Anwyl, 1999; Svoboda and Mainen, 1999; Nosyreva and Huber, 2005). These forms of LTD often involve presynaptic expression (Egger et al., 1999; Kemp and Bashir, 2001; Zakharenko et al., 2002) and require calcium through R- and T-type VSCCs, as observed here (Oliet et al., 1997).
Role of eCBs
Blockade of CB1 receptors (by AM251) or postsynaptic synthesis and release of eCBs (by RHC80367 and VDM-11) prevented t-LTD, indicating that retrograde eCB signaling via CB1 receptors is required for t-LTD. eCB signaling must occur downstream of mGluR activation and postsynaptic calcium, because anandamide-induced synaptic depression was unaffected by postsynaptic BAPTA and MPEP (Fig. 5). Thus, we propose that during t-LTD, postsynaptic calcium and/or mGluR signaling drive eCB synthesis and release, which activates CB1 receptors to decrease presynaptic release probability. Although eCBs are best known as mediators of short-term synaptic depression (Wilson and Nicoll, 2002), substantial evidence demonstrates a role for eCBs as retrograde messengers in LTD (Auclair et al., 2000; Huang et al., 2003; Sjostrom et al., 2003), including several forms of mGluR-dependent LTD (Gerdeman et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003; Safo and Regehr, 2005).
Role of non-postsynaptic, potentially presynaptic NMDARs
Long-duration (20–33 min) D-AP5 application prevented t-LTD, whereas brief D-AP5 and long-duration iMK-801, which blocked a similar proportion of NMDAR current, did not (Fig. 8). This implies that a non-postsynaptic, potentially presynaptic, NMDAR-dependent process gates t-LTD induction on a slow, ~20 min time scale. Presynaptic NMDARs exist and modulate transmitter release at several synapses (Berretta and Jones, 1996; Glitsch and Marty, 1999; Casado et al., 2000; Sjostrom et al., 2003; Engelman and MacDermott, 2004; Lien et al., 2006) and are required for eCB-dependent t-LTD in L5 of visual cortex (Sjostrom et al., 2003). At L4-L2/3 synapses, non-postsynaptic NMDARs were found to modulate release during high-frequency burst firing or when glutamate uptake was retarded but not during normal low-frequency transmission (Fig. 8), suggesting these receptors may be perisynaptic. Precisely where these receptors are located, how they are activated under normal conditions to regulate t-LTD induction, and why this regulation is so slow remain unclear. Whether the same receptors regulate both release and t-LTD is also unknown.
Two coincidence detector model for STDP
We propose an overall model for STDP at L4-L2/3 synapses (Fig. 9) in which postsynaptic NMDARs are the coincidence detector and calcium source for t-LTP, but a separate postsynaptic NMDAR-independent pathway performs coincidence detection for, and implements, t-LTD. Postsynaptic NMDARs are proposed to generate strong, brief calcium signals (Lisman, 1989; Hansel et al., 1997) and LTP in response to brief pre-leading-post spike intervals (5–30 ms), as demonstrated by pharmacological isolation of this pathway (Fig. 7) and consistent with known NMDAR kinetics (Kampa et al., 2004). The mGluR-VSCC-IP3R pathway is proposed to generate weaker, slower calcium signals over a broader range of spike intervals (+25 to −100 ms), which either alone or in concert with other mGluR-dependent signals, initiate retrograde eCB signaling, which drives t-LTD. Whether the calcium signals for LTP and LTD merge in a single functional pool (Lisman, 1989; Artola and Singer, 1993; Hansel et al., 1997; Yang et al., 1999) or remain in separate pools (Karmarkar and Buonomano, 2002; Bi and Rubin, 2005) is unknown.
Figure 9.
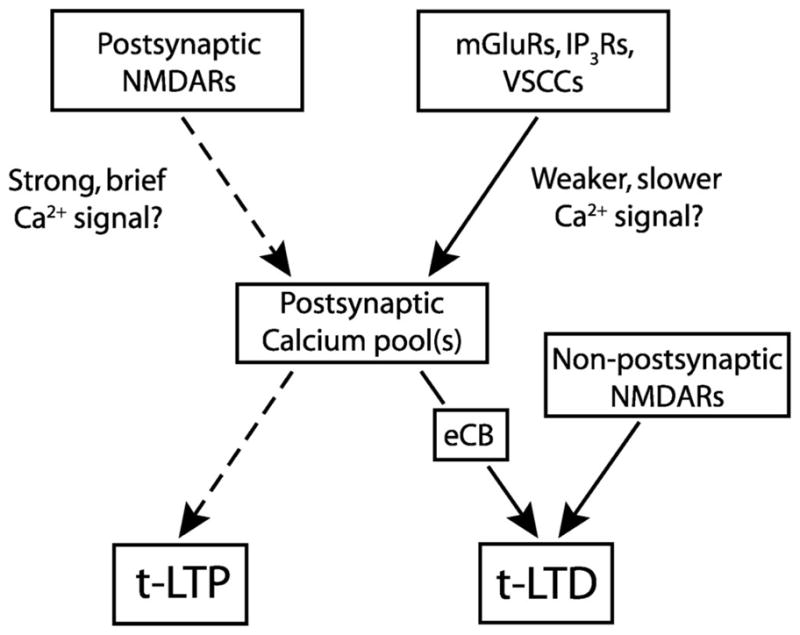
Model for STDP at L4-L2/3 synapses. Separate proposed coincidence detectors for LTP and LTD components of STDP. Postsynaptic NMDA receptors are proposed to be the coincidence detector and calcium source for t-LTP (dashed lines). t-LTD induction protocols are proposed to activate the mGluR-VSCC-IP3R pathway to generate postsynaptic calcium, which drives eCB synthesis, leading to retrograde signaling and presynaptic expression of t-LTD. Non-postsynaptic, potentially presynaptic NMDARs are also required for t-LTD. Whether millisecond scale coincidence detection is performed by the mGluR-VSCC-IP3R module or by eCB- presynaptic NMDAR signaling is unknown. Whether t-LTP and t-LTD share a common or separate pools of dendritic calcium is also unknown.
How mGluRs, VSCCs, IP3Rs, eCBs, and potentially presynaptic NMDARs perform coincidence detection for t-LTD is unknown. One possibility is a postsynaptic coincidence detector model in which mGluR-VSCC-IP3R signaling forms the principal coincidence detector, the output of which is the eCB signal that directly drives t-LTD. Both PLC and IP3Rs are molecular coincidence detectors with strong calcium dependence. Calcium from VSCCs greatly facilitates mGluR-dependent PLC activation and, independently, acts as a coagonist at IP3Rs to promote IP3- mediated calcium release (Berridge, 1998; Finch and Augustine, 1998; Hashimotodani et al., 2005). Thus, in this model, VSCC calcium from each postsynaptic spike (Berridge, 1998; Sabatini et al., 2001) may transiently prime mGluR-IP3 signaling, enabling appropriately timed, single presynaptic spikes to effectively drive mGluR signaling and generate sufficient calcium release for calcium-dependent eCB synthesis and LTD. A similar mechanism has been proposed for short-term synaptic depression in which activation of VSCCs and group I mGluRs, or other Gq/11- coupled receptors, synergistically drive eCB release (Varma et al., 2001; Ohno-Shosaku et al., 2002; Hashimotodani et al., 2005; Kreitzer and Malenka, 2005). This model is essentially identical to the two coincidence detector model for STDP predicted by Karmarkar and Buonomano (2002) and Karmarkar et al. (2002). Interestingly, the mGluR-IP3R pathway is also implicated as a primary coincidence detector in cerebellar LTD, which is also spike timing-dependent (Finch and Augustine, 1998; Svoboda and Mainen, 1999; Wang et al., 2000).
An alternative model is the presynaptic coincidence detector model, which has been proposed by Sjostrom et al. (2003) to explain eCB-dependent t-LTD in layer 5 of V1. Like L4-L2/3 t-LTD, L5 t-LTD is independent of postsynaptic NMDARs but requires presumably presynaptic NMDARs as well as CB1 receptor activation. Sjostrom et al. (2003) proposed that CB1 receptors encode the precise time of postsynaptic spikes (via spike-elicited eCB release), and presynaptic NMDARs encode the time of pre-synaptic spikes (by acting as glutamate autoreceptors), and that coincident activation of these receptors drives t-LTD. A potential difficulty for this model is that eCB synthesis, diffusion, and CB1 receptor signaling may be too slow to allow precise encoding of postsynaptic spike timing (Wilson and Nicoll, 2002; Heinbockel et al., 2005). In the postsynaptic coincidence detector model, eCB signaling represents the output of the coincidence detector, the signal to initiate synaptic weakening, which does not require high temporal precision. The presynaptic coincidence detector model is less likely at L4-L2/3 synapses, where non-postsynaptic, potentially presynaptic NMDARs were not acutely required during t-LTD induction (Fig. 1), but instead seemed to slowly gate t-LTD induction on a ~20 min time scale (Fig. 8). A third model is that both postsynaptic and presynaptic coincidence detectors operate, but at different time scales, to trigger t-LTD.
Two distinct classes of STDP
It now appears that two distinct classes of STDP exist across neocortical and hippocampal synapses. In one class, postsynaptic NMDARs are the primary coincidence detector and calcium source for both t-LTP and t-LTD (Nishiyama et al., 2000; Shouval et al., 2002; Bi and Rubin, 2005; Froemke et al., 2005). The second class of STDP, represented by the present results, incorporates a distinct form of t-LTD that is independent of postsynaptic NMDARs and is presynaptically expressed. Where examined, this form of t-LTD involves postsynaptic mGluRs and retrograde eCB signaling, suggesting a common signaling motif (Egger et al., 1999; Normann et al., 2000; Sjostrom et al., 2003). These contrasting forms of t-LTD are strongly reminiscent of, and may be mechanistically similar to, postsynaptic NMDAR-dependent and mGluR-dependent forms of classical, nontiming-dependent LTD. Because t-LTD at L4-L2/3 synapses are strongly implicated in S1 map plasticity (Feldman and Brecht, 2005; Bender et al., 2006), the mGluR- and eCB-dependent class of STDP may contribute to cortical map plasticity and development.
Acknowledgments
This work was supported by National Institutes of Health Grant RO1 NS046652 and a McKnight Scholars Award to D.E.F. D.J.B. was supported by a predoctoral fellowship from Howard Hughes Medical Institute. We thank Shantanu Jadhav and Jason Wolfe for reading this manuscript.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Allen CB, Celikel T, Feldman DE. Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci. 2003;6:291–299. doi: 10.1038/nn1012. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Aoki C, Venkatesan C, Go CG, Mong JA, Dawson TM. Cellular and subcellular localization of NMDA-R1 subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci. 1994;14:5202–5222. doi: 10.1523/JNEUROSCI.14-09-05202.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol. 2000;83:3287–3293. doi: 10.1152/jn.2000.83.6.3287. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Rangel J, Feldman DE. Development of columnar topography in the excitatory layer 4 to layer 2/3 projection in rat barrel cortex. J Neurosci. 2003;23:8759–8770. doi: 10.1523/JNEUROSCI.23-25-08759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Allen CB, Bender VA, Feldman DE. Synaptic basis for whisker deprivation-induced synaptic depression in rat somatosensory cortex. J Neurosci. 2006;26:4155–4165. doi: 10.1523/JNEUROSCI.0175-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by pre-synaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Rubin J. Timing in synaptic plasticity: from detection to integration. Trends Neurosci. 2005;28:222–228. doi: 10.1016/j.tins.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Blue ME, Martin LJ, Brennan EM, Johnston MV. Ontogeny of non-NMDA glutamate receptors in rat barrel field cortex: I. Metabotropic receptors. J Comp Neurol. 1997;386:16–28. [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci. 2004;24:5623–5631. doi: 10.1523/JNEUROSCI.0918-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Dieudonne S, Ascher P. Presynaptic N-methyl-D-aspartate receptors at the parallel fiber-Purkinje cell synapse. Proc Natl Acad Sci USA. 2000;97:11593–11597. doi: 10.1073/pnas.200354297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW. Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci. 1995;15:5324–5333. doi: 10.1523/JNEUROSCI.15-07-05324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celikel T, Szostak VA, Feldman DE. Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci. 2004;7:534–541. doi: 10.1038/nn1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charton JP, Herkert M, Becker CM, Schroder H. Cellular and subcellular localization of the 2B-subunit of the NMDA receptor in the adult rat telencephalon. Brain Res. 1999;816:609–617. doi: 10.1016/s0006-8993(98)01243-8. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- DeBiasi S, Minelli A, Melone M, Conti F. Presynaptic NMDA receptors in the neocortex are both auto- and heteroreceptors. NeuroReport. 1996;7:2773–2776. doi: 10.1097/00001756-199611040-00073. [DOI] [PubMed] [Google Scholar]
- Egger V, Feldmeyer D, Sakmann B. Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat Neurosci. 1999;2:1098–1105. doi: 10.1038/16026. [DOI] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci. 2004;5:135–145. doi: 10.1038/nrn1297. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Brecht M. Map plasticity in somatosensory cortex. Science. 2005;310:810–815. doi: 10.1126/science.1115807. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Lubke J, Silver RA, Sakmann B. Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signalling within a cortical column. J Physiol (Lond) 2002;538:803–822. doi: 10.1113/jphysiol.2001.012959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Finnerty GT, Roberts LS, Connors BW. Sensory experience modifies the short-term dynamics of neocortical synapses. Nature. 1999;400:367–371. doi: 10.1038/22553. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Dan Y. Spike-timing-dependent synaptic modification induced by natural spike trains. Nature. 2002;416:433–438. doi: 10.1038/416433a. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J Biol Chem. 1988;263:11075–11079. [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Relation between dendritic Ca2+levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. Eur J Neurosci. 1997;9:2309–2322. doi: 10.1111/j.1460-9568.1997.tb01648.x. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Heinbockel T, Brager DH, Reich CG, Zhao J, Muralidharan S, Alger BE, Kao JP. Endocannabinoid signaling dynamics probed with optical tools. J Neurosci. 2005;25:9449–9459. doi: 10.1523/JNEUROSCI.2078-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren CD, Zilberter Y. Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J Neurosci. 2001;21:8270–8277. doi: 10.1523/JNEUROSCI.21-20-08270.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, Wang SJ, Chiou LC, Gean PW. Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. J Neurosci. 2003;23:10311–10320. doi: 10.1523/JNEUROSCI.23-32-10311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissiere S, Luthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL. Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:667–674. doi: 10.1098/rstb.2002.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol (Lond) 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. A model of spike-timing dependent plasticity: one or two coincidence detectors? J Neurophysiol. 2002;88:507–513. doi: 10.1152/jn.2002.88.1.507. [DOI] [PubMed] [Google Scholar]
- Karmarkar UR, Najarian MT, Buonomano DV. Mechanisms and significance of spike-timing dependent plasticity. Biol Cybern. 2002;87:373–382. doi: 10.1007/s00422-002-0351-0. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001;65:339–365. doi: 10.1016/s0301-0082(01)00013-2. [DOI] [PubMed] [Google Scholar]
- Khodakhah K, Armstrong CM. Induction of long-term depression and rebound potentiation by inositol trisphosphate in cerebellar Purkinje neurons. Proc Natl Acad Sci USA. 1997;94:14009–14014. doi: 10.1073/pnas.94.25.14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci USA. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol (Lond) 2000;529:625–646. doi: 10.1111/j.1469-7793.2000.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Mu Y, Vargas-Caballero M, Poo MM. Visual stimuli-induced LTD of GABAergic synapses mediated by presynaptic NMDA receptors. Nat Neurosci. 2006;9:372–380. doi: 10.1038/nn1649. [DOI] [PubMed] [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Spruston N. Postsynaptic depolarization requirements for LTP and LTD: a critique of spike timing-dependent plasticity. Nat Neurosci. 2005;8:839–841. doi: 10.1038/nn0705-839. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man’s land. J Physiol (Lond) 2001;532:285. doi: 10.1111/j.1469-7793.2001.0285f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito G, Shigemoto R, Fairen A, Lujan R. Differential distribution of group I metabotropic glutamate receptors during rat cortical development. Cereb Cortex. 2002;12:625–638. doi: 10.1093/cercor/12.6.625. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol (Lond) 1995;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Malinow R, Miller JP. Postsynaptic hyperpolarization during conditioning reversibly blocks induction of long-term potentiation. Nature. 1986;320:529–530. doi: 10.1038/320529a0. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Normann C, Peckys D, Schulze CH, Walden J, Jonas P, Bischofberger J. Associative long-term depression in the hippocampus is dependent on postsynaptic N-type Ca2+ channels. J Neurosci. 2000;20:8290–8297. doi: 10.1523/JNEUROSCI.20-22-08290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J Physiol (Lond) 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Beltramo M, Giuffrida A, Stella N. Endogenous cannabinoid signaling. Neurobiol Dis. 1998;5:462–473. doi: 10.1006/nbdi.1998.0221. [DOI] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Safo PK, Regehr WG. Endocannabinoids control the induction of cerebellar LTD. Neuron. 2005;48:647–659. doi: 10.1016/j.neuron.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Samson RD, Pare D. Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci. 2005;25:1847–1855. doi: 10.1523/JNEUROSCI.3713-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev. 2004;45:250–265. doi: 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Shouval HZ, Kalantzis G. Stochastic properties of synaptic transmission affect the shape of spike time-dependent plasticity curves. J Neurophysiol. 2005;93:1069–1073. doi: 10.1152/jn.00504.2004. [DOI] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci USA. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. J Neurophysiol. 2004;92:3338–3343. doi: 10.1152/jn.00376.2004. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Mainen ZF. Synaptic [Ca2+]: intracellular stores spill their guts. Neuron. 1999;22:427–430. doi: 10.1016/s0896-6273(00)80698-4. [DOI] [PubMed] [Google Scholar]
- Trettel J, Levine ES. Endocannabinoids mediate rapid retrograde signaling at interneuron right-arrow pyramidal neuron synapses of the neocortex. J Neurophysiol. 2003;89:2334–2338. doi: 10.1152/jn.01037.2002. [DOI] [PubMed] [Google Scholar]
- Trettel J, Fortin DA, Levine ES. Endocannabinoid signalling selectively targets perisomatic inhibitory inputs to pyramidal neurones in juvenile mouse neocortex. J Physiol (Lond) 2004;556:95–107. doi: 10.1113/jphysiol.2003.058198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni VK, Zakharenko SS, Zablow L, DeCostanzo AJ, Siegelbaum SA. Calcium release from presynaptic ryanodine-sensitive stores is required for long-term depression at hippocampal CA3-CA3 pyramidal neuron synapses. J Neurosci. 2004;24:9612–9622. doi: 10.1523/JNEUROSCI.5583-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188, 1–5. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Denk W, Hausser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- Waters J, Larkum M, Sakmann B, Helmchen F. Supralinear Ca2+ influx into dendritic tufts of layer 2/3 neocortical pyramidal neurons in vitro and in vivo. J Neurosci. 2003;23:8558–8567. doi: 10.1523/JNEUROSCI.23-24-08558.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002;35:1099–1110. doi: 10.1016/s0896-6273(02)00898-x. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]