

Abstract
Huntington's disease (HD) is an autosomal-dominant neurological disorder caused by expanded CAG repeats in the Huntingtin (Htt) gene but it is not known how this mutation causes neurodegeneration. Herein, we found that dysfunction of upstream binding factor-1 (UBF-1) is linked to reduced ribosomal DNA (rDNA) transcription in HD. We identified that UBF1 acetylation at Lys (K) 352 by CBP is crucial for the transcriptional activity of rDNA. UBF1 mutation (K352A, K352Q, and K352R) decreased rDNA transcriptional activity. Moreover, both CBP-ΔHAT mutant and knock-down of CBP by siRNA reduced acetylation of UBF1 and resulted in the decreased transcription of rDNA into rRNA. ChIP analysis showed a significant reduction of UBF1 occupancy in the promoter of rDNA in STHdhQ111 cell line model of HD. These results demonstrate that abnormal activity of UBF1 and its acetylation by CBP are linked to impaired rDNA transcription in HD. This novel mechanism suggests that modulation of UBF-mediated rDNA synthesis by CBP may be a therapeutic target for improving neuronal rDNA transcription in HD.
INTRODUCTION
Huntington’s disease (HD) is an incurable and fatal autosomal dominant neurodegenerative disorder, typically of mid-life onset, characterized by a triad of chorea, cognitive abnormalities and progressive dementia. The motor symptoms of HD are largely a consequence of profound neurodegeneration affecting the striatum1. HD is caused by an expansion of CAG repeats coding for glutamine (Q) in exon 1 of the Huntingtin (Htt) gene. One mechanism by which mutant Htt (mHtt) contributes to neurodegeneration is through transcriptional dysregulation and chromatin remodeling.2 Indeed, polyQ stretches in mutant htt interact physically with CBP (CREB binding protein), a transcriptional coactivator, and block the intrinsic histone acetyltransferase (HAT) activity of CBP.3 These specific interactions show that mHtt modulates transcriptional signaling cascades that may initiate a number of downstream pathophysiological mechanisms relevant to HD.
The nucleolus is an important subnuclear component of the transcription machinery of ribosomal genes.4 The ribosomal DNA (rDNA) encoding ribosomal RNA (rRNA) is organized as tandem repeats and is transcribed into 47S precursor rRNA by a specialized transcription complex, consisting of RNA polymerase I and other co-regulatory factors.5 Neurons are postmitotic cells that have prominent nucleoli but the role of this structure and the regulatory mechanism of rDNA remain unknown.6 RNA polymerase I (RNA pol I) and upstream binding factor (UBF) play critical roles in the formation of active nucleolar organizer regions (NORs) and maintenance of rRNA transcriptional activity.7 UBF is a member of the high mobility group (HMG) protein family and contains six HMG box DNA binding motifs. It is highly conserved protein and there is UBF1 and UBF2 polypeptides 97 and 94 kDa, respectively.8 Not only UBF is a RNA pol I factor essential for rDNA (genes encoding ribosomal RNAs in eukaryotes) transcription by inducing remodeling of ribosomal gene chromatin, but it also serves a structural role by binding to other sequences across the entire rDNA repeat4. Interestingly, nucleolar accessory bodies [Cajal bodies (CBs)] are associated with disorders caused by expansions of CAG repeats within genes, including Huntington’s disease and the neuronal nuclear inclusions characteristic of polyQ diseases show a marked association with CBs.9 In our study, we show for the first time that transcriptional modulation of rDNA, a key process in ribosome biogenesis, is altered in HD and that UBF and CBP play a role in altered rRNA expression. We found that UBF1 protein levels and UBF-mediated transcriptional activity of rDNA are impaired in cellular and animal model of HD. In parallel, we determined that CBP interacts with UBF1, acetylates UBF1 at Lys (K) 352, and modulates rDNA transcription.
RESULTS
UBF1 is dysregulated in an animal model of HD
We examined UBF1 immunoreactivity, protein, and mRNA levels in the R6/2 transgenic mouse model of HD (Fig. 1). The R6/2 line is a transgenic mouse line expressing exon 1 of the human HD gene with an expanded CAG repeat.9 R6/2 mice develop brain and body weight loss at 6 weeks of age and a gait and movement disorder by 9–11 weeks. These clinical features, along with striatal atrophy and neuronal intranuclear inclusions, are similar to changes found in human’s with HD. At 10 weeks of age, UBF1 levels in the striatum of R6/2 mice were found to be significantly decreased compared with littermate controls as measured by immunofluorescence staining and confocal microscopy (Fig. 1a,b) and Western blot analysis (Fig. 1c). In addition, UBF1 levels in the striatum of human HD victims were markedly reduced compared to controls (Supplementary Fig. 1a). The level of 18S RNA correlatively reduced in HD (Supplementary Fig. 1b and c). In contrast to protein level (Fig. 1d), UBF1 mRNA levels in striatum were not significantly decreased in R6/2 mice compared to WT controls (Fig. 1e). We also found that 48S levels were down regulated in R6/2 mice in compared to controls using quantitative real-time PCR (Fig. 1f). These results indicate that the UBF1 protein levels are reduced in HD mice and are linked to reduced ribosomal transcription in vivo.
Figure 1.
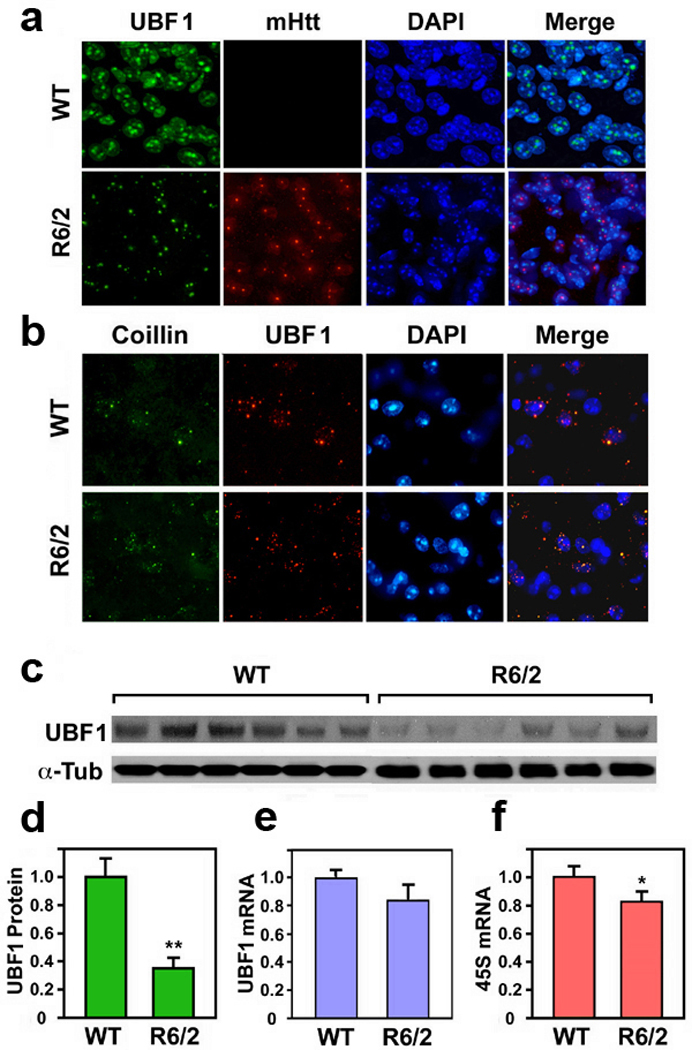
Upstream binding factor 1 (UBF1) is decreased in an animal model of HD (R6/2 line). (a) The immunoreactivity of UBF1 is markedly reduced in striatal neurons of R6/2 mice in comparison to wild type littermate control mice at 9 weeks of age. UBF1 and mtHtt are not co-localized. Scale bar: 10 µm. (b) Confocal microscopic analysis for UBF1 and coillin co-localization in the striatal tissue. Colocalization of coilin with UBF is decreased in striatal neurons of R6/2 mice in comparison to control mice. (c) Immunoblot assay for UBF-1 in the striatal tissue of WT and R6/2 mice. (d) Densitometry analysis of UBF1 protein level normalized to the levels of α–tubulin (α–Tub). (e) Quantitative RT-PCR analysis of UBF1 mRNA shows no significant difference between WT and R6/2 mice. Densitometry analysis from WT (n=6) and R6/2 (n=6). UBF1 mRNA is normalized to GAPDH mRNA. (f) Quantitative RT-PCR analysis of 45S mRNA shows a difference between WT and R6/2 mice. *, Significantly from wild type mice at p < 0.05; ** at p < 0.01. Scale bar: 10 µm.
UBF1-induced rDNA transcription is impaired in a cellular model of HD
In order to examine how the transcriptional activity of ribosomal DNA (rDNA) is modulated by UBF1, we transiently transfected luciferase reporter vectors containing pIRES (internal ribosome entry segment) or pHrD (human rDNA)-IRES with UBF1 and measured the change of luciferase activity (Fig. 2a). As expected, UBF1 increased in pHrD-IRES promoter activity in a dose dependent manner in STHdhQ7/7 (Q7) cells (Fig. 2b). Because CBP can modulate UBF1 activity, we further elucidated how the interplay between UBF1 and CBP affects the regulation of rDNA transcription. We found that CBP enhanced rDNA transcription in a dose dependent manner (Fig. 2c). When UBF and CBP constructs were co-transfected, a synergistic effect on rDNA transcription was found (Fig. 2d). To confirm whether CBP-dependent and UBF1-induced rDNA transcription was due to the HAT activity of CBP, we used a CBP mutant construct with a mutant HAT site (CBP-dHAT). As we predicted, co-transfection of UBF1 and mutant CBP- dHAT lead to the loss of the synergistic effect on the rDNA transcription (Fig. 2e). The loss of CBP HAT activity reduced the promoter activity of pHrD-IRES to basal levels. UBF knock-down by shRNA reduced CBP-induced rDNA transcription (Supplementary Fig. 2). We then examined transcriptional activity of rDNA in a cell line model of HD using wild type (STHdhQ7/7) and mutant (STHdh Q111/111) huntingtin expressing cells of striatal origin (Fig. 2f). Basal rDNA transcriptional activity was higher in cells expressing STHdhQ7/7 (Q7) and downregulated in STHdh Q111/111 (Q111) cells. UBF1-dependent rDNA transcription was dramatically impaired in mutant Q111 cells in contrast to Q7 cells where UBF1 led to increased pHrD-IRES promoter activity in a dose dependent manner. Using a dual luciferase assay (HrD-IRES fire fly luciferase versus Renilla luciferase), we found that the transcriptional system is dysregulated in Q111 cells compared to Q7 cells (Supplementary Fig. 3). UBF1 knock-down using shRNA only reduced basal pHrD-IRES promoter activity in WT cells.
Figure 2.
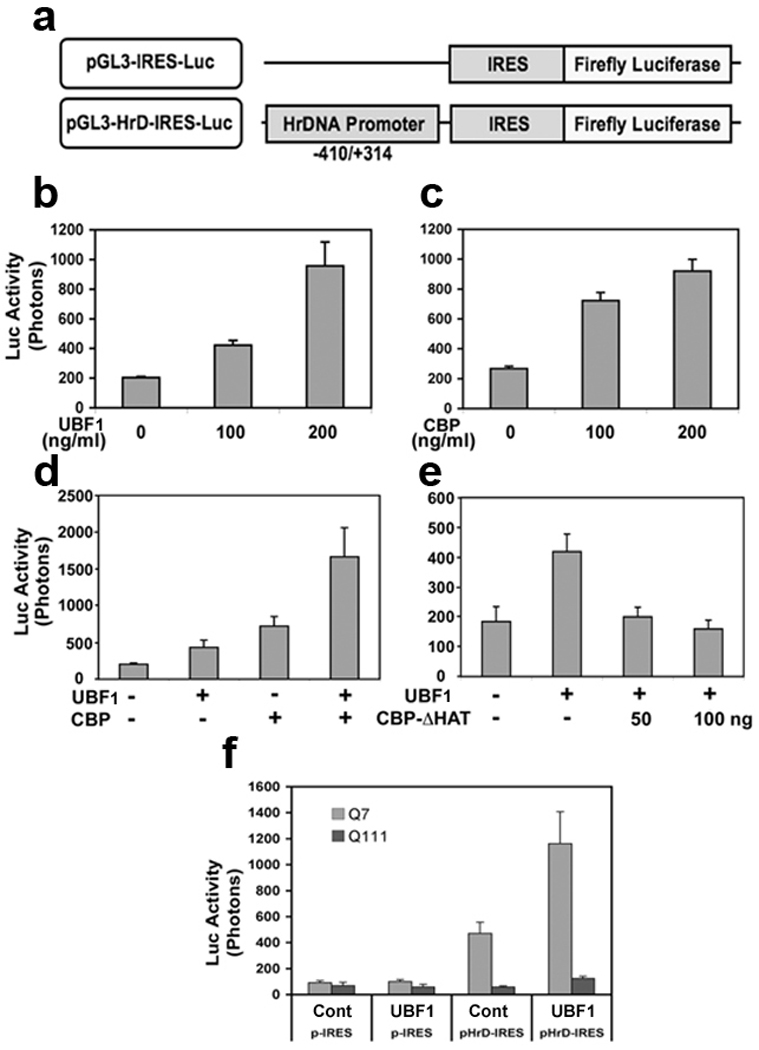
UBF1-dependent rDNA transcription is impaired in mutant HD cells. (a) The scheme of HrDNA IRES-reporter constructs. (b) UBF1 increases pHrD-IRES promoter activity in a dose dependent manner. STHdhQ7/7 (Q7) cells were transiently cotransfected with 400 ng of pHrD-IRES luciferase reporter plasmid and the other indicated constructs. Cells were incubated for 48 hr and luciferase activity was measured. Transactivation of pHrD-IRES by UBF1 is increased in a dose dependent manner. (c) CBP increases pHrD-IRES promoter activity in a dose dependent manner. (d) UBF1 and CBP synergistically stimulate pHrD-IRES promoter activity. (e) The loss of acetyltransferase activity of CBP reduces UBF1-dependent transactivation of pHrD-IRES promoter. UBF1 was cotransfected with a CBP mutant with HAT domain deletion (CBP-ΔHAT). (f) The basal rDNA transcriptional activity was down regulated in STHdhQ111/111 (Q111) cells. The data represent an average of three independent experiments.
CBP dysregulation is linked to UBF1 dysfunction in HD
Since CBP is known to modulate UBF1 and CBP and is dysregulated in HD,10 we sought to determine the relationship between CBP and UBF1 activity in HD. First, we confirmed that CBP levels are affected in HD mice and HD cell lines. CBP levels in the striatum and hippocampus of R6/2 mice were significantly reduced compared with littermate controls at 10 weeks of age, as monitored by Western blot analysis (Fig. 3a). Punctate CBP immunoreactivity colocalized with mutant Htt in neuronal nuclear inclusions in R6/2 mice (Fig. 3b,c). CBP and UBF1 protein levels were reduced in mutant Q111 cells compared to WT Q7 cells (Fig. 3d,e) paralleling our in vivo findings. We examined the basal stability of UBF1 and CBP in intact cells and determined whether CBP modulates UBF1 stability or not (Supplementary Fig. 4). There was no difference in the stability of UBF1 and CBP in Q7 and Q111 cells. We did find, however, that ectopic expression of CBP increases the stability of UBF1. qRT-PCR analysis showed decreased 45S mRNA in mutant Q111 cells (Fig. 3f), indicating impairment of ribosomal biosynthesis similar to that we found in R6/2 mice.
Figure 3.
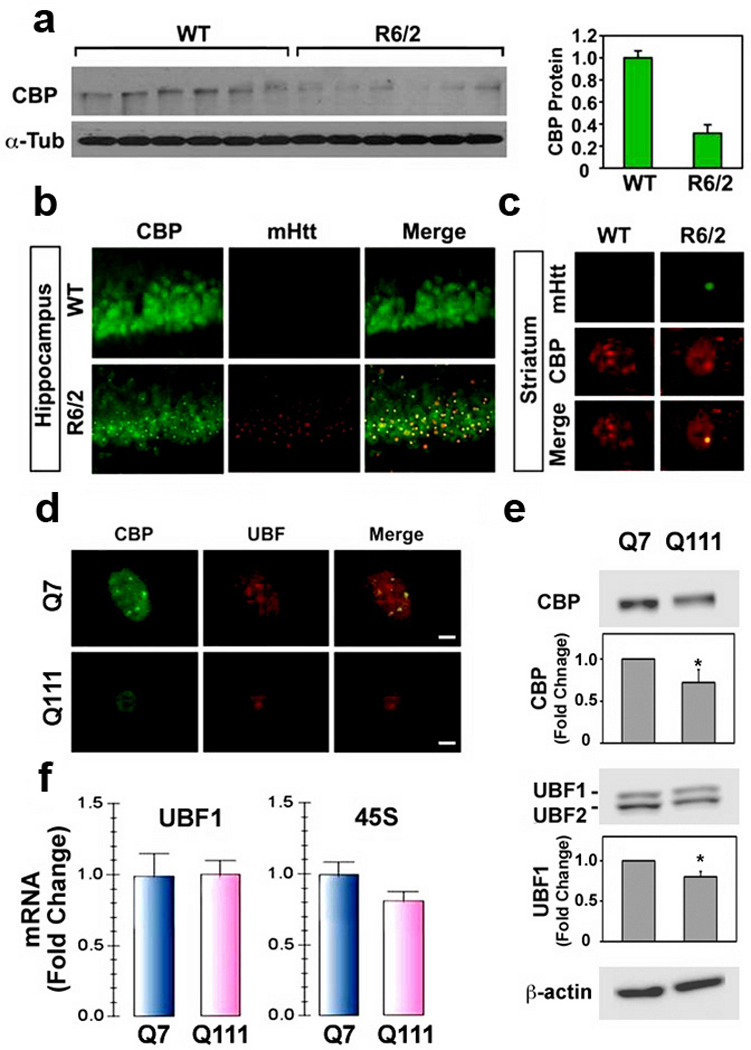
CBP is impaired in HD mice and cell line (Q111). (a) The protein level of CBP is markedly reduced in striatal neurons of R6/2 mice compared to wild type littermate control mice at 9 weeks of age. (b) CBP and mtHtt are co-localized in hippocampal neurons of R6/2 mice but not in WT mice. (c) Confocal microscopic analysis for mtHtt and CBP co-localization in striatal neurons of R6/2 mice. Colocalization of mtHtt with CBP is increased in striatal neurons of R6/2 mice in comparison to control mice. (d) CBP and mtHtt are co-localized in Q7 cells and its clocalization is decreased in Q111 cells. (e) Immunoblot assay for CBP, UBF1 and β-actin in Q7 and Q111 cells. The data represent an average of ten independent experiments. *, Significantly from the control at p < 0.05. (f) Quantitative RT-PCR analysis of UBF1 mRNA and 45S in Q7 and Q111 cells. UBF1 mRNA and 45S were normalized to GAPDH mRNA. The data represent an average of three independent experiments.
Next, we further characterized the in vivo association of UBF1 and CBP by performing immunoprecipitations (IPs) on neuronal lysates using either anti-UBF1 or anti-CBP antibodies. A prominent 300 kDa band of CBP protein was present in UBF1 IPs (Fig. 4a). The association of UBF1 and CBP was apparent in both Q7 and Q111 cells (Fig. 4a). We also confirmed the association between CBP and UBF1 using reverse IP with CBP antibody (Fig. 4b). These results indicate that both UBF and CBP constitutively interact to generate rDNA transcription complex formation in neurons. Immunoblotting with anti-UBF1 antibody of anti-UBF1 IP verified that the same amounts of UBF1 were recovered by IP under different conditions. To test whether the acetylation status of UBF1 was altered in HD cells, we detected acetylated UBF1 (Ac-UBF1) using an anti-Ac-Lys antibody on UBF1 IPs. Interestingly, we found that Ac-UBF1 levels were reduced (>40%) in mutant Q111 cells compared to WT Q7 cells (Fig. 4c). We then addressed whether acetylation of UBF1 by CBP-HAT activity is responsible for CBP-dependent UBF1 transcriptional activation. Indeed, UBF1 acetylation was significantly augmented by WT-CBP in Q7 cells but not by the CBP-dHAT mutant, as determined by immunoprecipitation using an UBF1 antibody followed by immunoblotting using acetyl lysine antibody (Ac-UBF1) or UBF1 antibody alone (Fig. 4d). CBP knock down using siRNA CBP reduced UBF1 acetylation levels but the basal acetylation levels of UBF1 was not changed by siRNA Control treatment (Fig. 4e). In addition, CBP knock down by siRNA markedly decreased UBF1-induced transcriptional activity of rDNA compared to the siRNA Control (Fig. 4f).
Figure 4.

UBF1 interacts with CBP and its acetylation is modulated by CBP-HAT activity. (a) UBF1 interacts with CBP in intact cells. Cell lysates were immunoprecipitated with UBF1 and subsequently blots were probed with anti-CBP antibody. The same blots were then stripped and reprobed with anti-UBF1 antibody. (b) CBP is also immunoprecipiated with UBF1. (c)The acetylated level of UBF1 is reduced in Q111 cells in comparison to Q7 cells. (d) HAT deletion in CBP abrogates the acetylation of UBF1. Cells were transiently transfected with pCMV-WT-CBP or pCMV-CBP-dHAT mutant. The UBF1 was immunoprecipitaed and the blot was probed with anti-Ac-Lys antibody. (e) Knock down of CBP by siRNA reduces the acetylation of UBF1. (f) Knock down of CBP reduces UBF1-induced transcriptional activity of rDNA. *, Significantly from the control at p < 0.05.
UBF1 is acetylated at K352 by CBP
To identify which lysine (Lys) residue of UBF1 is directly acetylated by CBP, we performed an in vitro acetylation assay using GST-UBF1-HMG1-6 proteins and GST-CBP protein. Since we found that GST-UBF1-HMG3 domain is specifically acetylated by CBP (Supplementary Fig. 5), acetylated GST-UBF1-HMG3 protein was pooled and cut by Prescission enzyme for LC-MS/MS analysis. As shown in the Figure 5, we identified that the Lys (K) 352 of UBF1 was specifically acetylated by CBP (Fig. 5a,b). To further confirm whether the acetylation of UBF1 at K352 is critical for the transcriptional activation of rDNA, we generated K352 acetylation site mutants of UBF1 using site-directed mutagenesis. We co-transfected UBF1 acetylation site mutants (K352A, K352Q, K352R) with or without CBP and checked the acetylation status of UBF1 using immunoprecipitation and Ac-lys blot analysis. Three acetylation site mutants of UBF1 (K352A, K352Q, K352R) showed a marked reduction of acetylation by CBP (Fig. 5c). These result suggest that UBF1 can be directly acetylated at K352 by CBP in intact neuronal cells. Concurrent with the reduced level of UBF1 acetylation in Western blot analysis (Fig. 5c), acetylation site mutants (K352A, K352Q, K352R) decreased the transcriptional activity of rDNA as determined by reporter assay (Fig. 5d).
Figure 5.
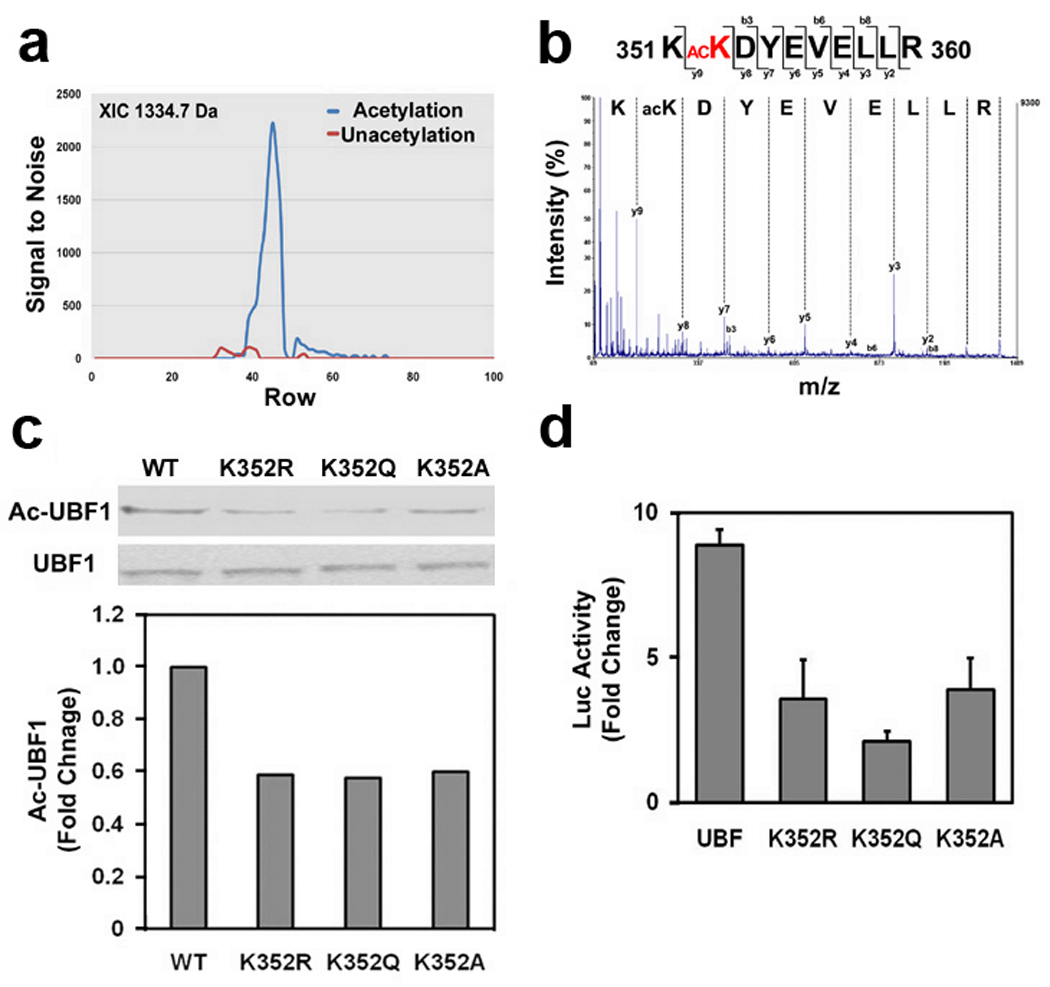
UBF1 is acetylated at K352 residue by CBP and UBF1 acetylation site mutants (K352A, K352Q, and K352R) decreases the transcriptional activity of rDNA. (a) Comparison of LC-MALDI-MS/MS extracted ion chromatogram between in vitro acetylated UBF1-HMG3 domain peptide (acetylation) and untreated control UBF1-HMG3 domain peptide (unacetylation). The extracted ion chromatogram (XIC) of the precursor ion at m/z 1334.7 Da was plotted as signal to nose (vertical) versus spot number (horizontal). (b) MALDI-TOF/TOF MS/MS spectrum of acetylated peptide KacKDYEVELLR (1334.7 m/z) from human UBF1 HMG3 domain (accession number: P17480). The chart represents m/z (horizontal) versus intensity (vertical). (c) Mutation of UBF1 at K352 to A, Q, and R reduced its acetylation level in neuronal cells. pCMV-Flag-UBF1s (WT, K352A, K352Q, and K352R) were transiently transfected, immunoprecipitaed with anti-Flag antibody and blotted with anti-Ac-Lys antibody. (d) UBF1 acetylation site mutants (K352A, K352Q, and K352R) decrease the transcriptional activity of rDNA in response to CBP.
The occupancy of UBF1 to the rDNA and ribosomal biosynthesis is altered in HD
ChIP assays were performed to detect whether the UBF1 occupancy of DNA was altered within the rDNA gene promoter.11 Q7 and Q111 cells were cross-linked with formaldehyde, sonicated, and immunoprecipitated with anti-UBF1 and normal rabbit IgG (as a control for normalization) antibodies. Eluted DNAs were amplified with specific primer sets for mouse rDNA promoter region such as ENH, UCE, CORE, and ETS3 (Fig. 6a and Supplementary Table 1). As we expected, the occupancy of UBF1 to ENH, UCE, CORE, and ETS3 regions of the rDNA promoter was decreased (>40%) in Q111 cells. In contrast, little or no PCR product was generated in Q7 cells or with normal rabbit IgG as negative control (Fig. 6b). In order to examine whether ribosomal biosynthesis is altered in HD, Q7 and Q111 cells were pulsing labeled for 30 min with 5- fluorouracil (FU) and immunolabeled with a mouse anti-BrdU antibody to visualize nascent transcripts of nucleolus. 5-FU incorporation was significantly decreased in mutant HD Q111 cells compared to WT Q7 cells (Fig. 6c,d). These results confirm that the occupancy of UBF1 within rDNA gene loci is significantly reduced, which subsequently contributes to the dysregulation of rDNA transcription in HD. Figure 6e summarizes how CBP interacts and acetylates UBF1 which is important in basal constitutive and inducible expression of rDNA. In the context of HD, mtHtt interferes with the ability of CBP to inhibit HAT activity and co-factor function. This in turn disrupts the interaction between UBF1 and CBP and decreases the acetylation of UBF1 at the proximal promoter of rDNA to repress rDNA transcription.
Figure 6.
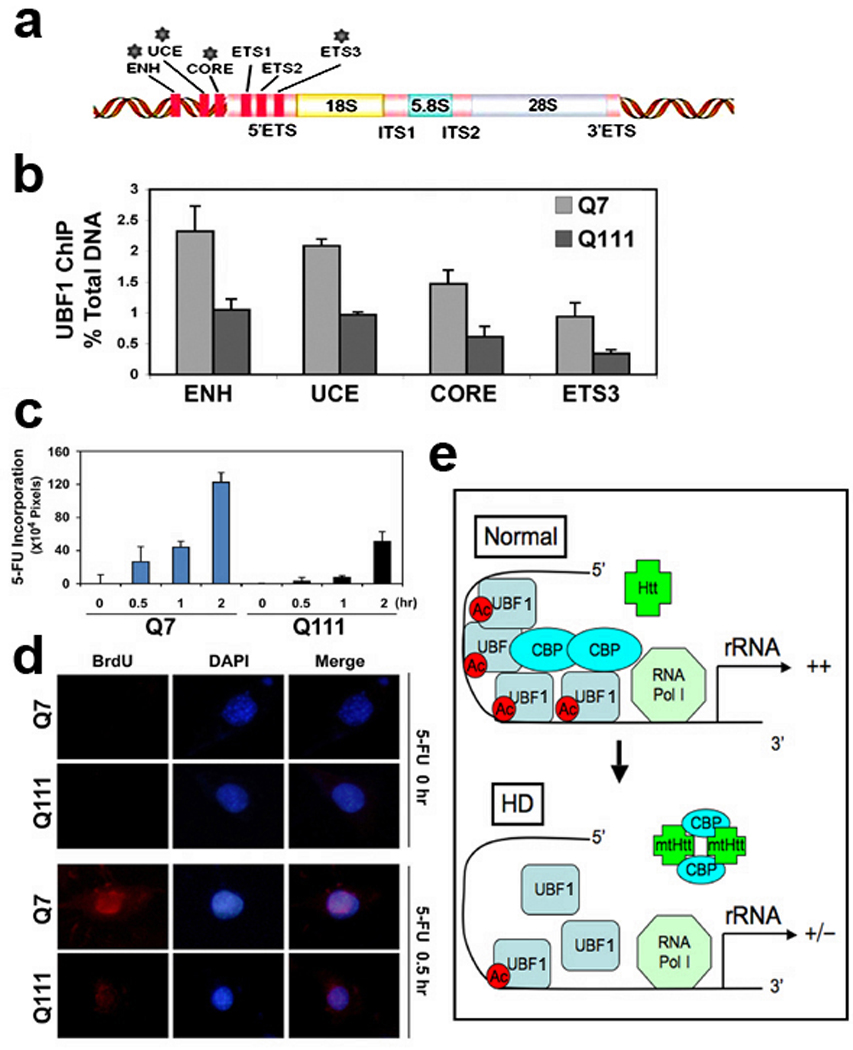
The occupancy of UBF1 to rDNA is changed in HD cell lines. (a) Positions of qRT-PCR primer sets for detecting UBF1 occupancy in the promoter of mouse rDNA. (b) Quantitative ChIP analysis of UBF1 occupancy in the mouse rRNA gene. The UBF1 binding to rDNA was decreased in Q111 HD cells. Quantitative ChIP analysis of UBF1 binding to the rDNA (ENH, UCE, CORE, and ETS3) was performed as described in Materials and Methods. (c) In situ transcription assay of 5’-FU incorporation into nucleolar rRNA in Q7 and Q111 cells. The level of 5’-FU incorporation was lowered in Q111 cells in comparison to Q7 cells. The time-dependent (0.5, 1, and 2 hr) incorporation of halogenated nucleotide was detected by anti-BrdU antibody. The density (pixels) was averaged by counting 5 areas of each 60 cells. (d) Immunofluorescence staining images of Q7 and Q111 after 5’-FU incorporation. The nucleus was counterstained with DAPI. (e) A scheme of UBF1-dependent rDNA transcription in normal and HD condition. Normal condition (top): CBP interacts and acetylates UBF1 that is important in basal and constitutive transcription of rDNA (top). HD condition (bottom): mtHtt disrupts UBF1 and CBP complexes by sequestering CBP and decreases the acetylation of UBF1 at the proximal promoter of rDNA, subsequently leading to the repression of rDNA transcription.
DISCUSSION
UBF is intimately involved in rDNA transcription as a RNA polymerase I specific transcription factor and is required for maintenance of rRNA genes to be transcriptionally active.12 UBF appears to be transcriptionally more active in specific neuronal cell type such as sensory ganglia neurons. UBF consists of two polypeptides (UBF1 and 2), which form hetero-and homodimers and arise from alternative splicing of a single transcript.13UBF2 is five-fold less active that UBF1,14 and reasons why UBF2 has such poor transcriptional activity are unclear. UBF1 targeting to regions of heterochromatin is sufficient to induce large-scale chromatin decondensation.15 UBF1 binding throughout the rDNA gene repeat might therefore contribute to the formation of the active chromatin state of rDNA genes.16 In the current study, we found that UBF1 protein levels in the striatum were significantly decreased in R6/2 transgenic HD mice. We also found that the 45S was down regulated in R6/2 mice in comparison to WT controls. These data indicate that abnormal protein level of UBF1 protein levels correlate with impairmed ribosomal transcription in HD mice.
Post-translational modifications of UBF, such as acetylation and phosphorylation, play an important role in the control of rDNA transcription.17 Transcription factor acetylation is responsible for both activation and subnuclear localization.18 It has been well established that CBP has histone acetyl transferase (HAT) activity that contributes to transcriptional activation by nuclear receptors. UBF acetylation is also important for cell cycle-dependent regulation of rRNA expression. It has been proposed that the transcription of rDNA is regulated upon the influence of two opposite processes through UBF acetylation by CBP and deacetylation by HDAC.10 In this context, CBP-dependent acetylation of UBF is linked to the transcription activation of rDNA.17 It is noteworthy that reduced HAT activity of CBP is thought to play a role in the toxic, transcriptional, and repressive effects of mutated proteins with expanded polyglutamine repeats such as Htt and atrophin.19–22 These findings, along with the established ability of UBF1-mediated rDNA transcription to be activated by acetyl transferases,23 raises the possibility that the neurodegenerative process in HD might be associated with changes in adaptive rDNA transcription caused by altered CBP acetyl transferase that affects the balance of acetylated and deacetylated UBF1. Indeed, we found that the acetylation levels of UBF1 are significantly lowered due to dysregulation of CBP function in both HD cells and HD mice. Our data shows that UBF1 and CBP interact in intact neurons. Reduced levels of acetylated UBF1 correlate with the reduced CBP level and activity in HD. We also directly demonstrated that the HAT activity of CBP controls UBF1 acetylation using a HAT domain deletion mutant of CBP that caused a marked reduction of UBF1 acetylation.
In order to identify the specific acetylation site of UBF1 by CBP acetyl transferase, we performed LC-MALDI-MS/MS analysis for the in vitro acetylated UBF-HMG protein by CBP. We confirmed the ability of CBP to acetylate UBF1 at K352 that resides in the HMG3 domain. Mutations at K352 to Aalanine (A), glutamine (Q), and arginine (R) abrogated the acetylation of UBF1 as well as the transcriptional activation of UBF1 in response to CBP. These data prove that the CBP-dependent acetylation of UBF1 plays a pivotal role in rDNA gene transcription17,24 and that CBP-dependent UBF1-induced rDNA expression is disrupted in HD models. It is unclear, however, whether mtHtt and HD-related cellular changes modulate the association of HDAC to the UBF1 complex. It will be important to define precisely how UBF1 protein is deacetylated in HD in order to more fully understand the role of neuronal rDNA transcription and ribosomal synthesis in HD pathogenesis.
Deacetylase inhibitors, such as trichostatin A (TSA), that enhance UBF acetylation have been show to enhance rDNA gene expression.7,10 In the present study, we found that sodium butyrate triggered dose dependent stimulation of rDNA transcription in mutant HD Q111 cells and WT Q7 cells in the presence of UBF1 (Supplementary Fig. 6). We previously showed that enhanced transcription factors acetylation using three structurally distinct HDAC inhibitors improves neuronal survival in response to oxidative stress 25 The results of these studies are consistent with a model in which altered rDNA transcription is an additional mechanism contributing to the protective effects of HDAC inhibitors in neurons. Several groups have now reported the neuroprotective effects of HDAC inhibitors in combating the toxicity of mutant huntingtin or mutant androgen receptors, proteins with expanded polyglutamine repeats in vitro26,27 and in vivo.3 Future studies will be needed, however, to clarify the contribution of UBF1 acetylation and rDNA transcription to the neuroprotective effects of HDAC inhibitors.
In summary, our in vitro and in vivo findings show that CBP acetylates UBF1 at K352 to enhance the transcriptional activity of rDNA. Our results also indicate that dysregulation of UBF1 and CBP is linked to impaired rDNA transcription in HD. A HDAC inhibitor enhanced rDNA transcription in mutant HD cells. These data suggest that modulation of CBP-dependent UBF1-mediated rDNA synthesis may be a therapeutic target to improve neuronal rDNA transcription in HD by blunting the deleterious effects of expanded polyglutamine repeats.
MATERIALS AND METHODS
Plasmid constructs
pIRES-Luc and Human rRNA-luciferase Vector (pHrD-IRES-Luc) was generously provided from Dr. Samson T. Jacob (Ohio State University).28 UBF1 acetylation mutants (K352A, K352Q, and K352R) were generated from pCMV-Flag-UBF1 using site-directed mutagenesis kit (TOYOBO) (Supplementary Table 1). pCMV-WT-CBP and pCMV-CBP-dHAT were previously described.11
In Vitro Protein Acetylation Assay
GST, GST-CBP, and GST-UBF1 HMG1-6 proteins were synthesized in Escherichia coli and purified with glutathione beads. Fusion proteins were eluted by using 20 mM glutathione in 1×HAT buffer [50 mM Tris-HCl, pH 8.0, 10% glycerol,1 mM PMSF, 0.1 mM EDTA, 1 mM DTT, 0.05 M NaCl, 0.01 M sodium butyrate (Calbiochem)]. The acetylation assay was performed as previously described.25
Cell Culture and Transfection Assays
STHdhQ7/7 (wild type) and STHdhQ111/111 (HD knock-in striatal cell line expresses mutant huntingtin at endogenous level), were generously provided from Dr. Marcy MacDonald (Harvard Medical School).29 For transfection assays, 2.5×105 cells were plated onto 48-well cell culture plates 24 h prior to transfection. DMRIE-C reagent (Invitrogen) has been used as a transfection reagent and transfections were performed according to the manufacturer`s protocol. When NaB was used in the experiments, the calculated amount of the HDAC inhibitor was added directly to the medium 24h after transfection. The luciferase activity was measured and normalized to protein concentration. The Dual Luciferase Assay kit (Promega) was used to determine the difference of internal transcriptional activity between Q7 and Q111 cells. Data represent the mean of three or more independent experiments.
Animals
Male R6/2 mice were bred with females from their background strain (B6CBAFI/J), and offspring were genotyped using PCR.9,30,31 CAG repeat length remained stable within a 147–153 range. Female mice were used in the experimental paradigms.
Quantitative Real Time-PCR (qRT-PCR)
Total RNA was isolated from cells using a commercial extraction system (Qiagen). 1µg total RNA has been used for cDNA preparation with iScript cDNA Synthesis Kit (BioRad) according to manufacturer’s protocols. cDNA from each sample was amplified by real-time PCR using iQ SYBR Green Supermix (BioRad). RNA quantities were normalized using GAPDH mRNA as a reference11. PCR cycling conditions were: denaturation for 3 min at 95 °C; then 40 cycles of amplification for 15 sec at 95 °C, 15 sec at 60 °C, 20 sec at 70 °C; followed with 30 sec at 72 °C. For melt curve data collection has been used 33 cycles, 6 sec each, with the temperature increased from 60 °C to 92 °C (increase set point temperature after cycle 2 by 1 °C).
Chromatin immunoprecipitation (ChIP)
ChIP for UBF1 binding to rDNA was performed using a CHIP assay kit (Upstate Biotech.) as described previously.11,25,32 Q7 and Q111 cells were crossed-linked with 1% formaldehyde for 20 min at room temperature. The lysates were sonicated six times with each time for 30 sec using Bioruptor (Diogenode). After centrifugation, the supernatant was diluted in ChIP dilution buffer and then incubated overnight at 4 °C with anti-UBF1 antibody. qPCR primers were shown on the Supplementary Table 2.
In-gel trypsin digestion and Off-line 2 dimensional LC-MALDI-MS/MS analysis
In-gel trypsin digestion and Nano LC separations were performed as previously reported with some minor modifications.32,33 The details of method are on line Supplementary Materials and Methods.
Confocal Microscopy
Indirect labeling methods were used to determine the UBF-1 (Santa Cruz Biotech) (1:200), coillin (Chemicon) (1:200), mtHtt (Chemicon) (1:1,000), and CBP (Santa Cruz Biotech) (1:200). The procedures were performed as previously described.31 Images were analyzed using a spinning disk confocal microscope (Olympus DSU, Tokyo, Japan). Deconvolution and 3-dimentainal construction of the confocal image was performed by AQI-X-COMBO-CWF program (Media cybernetics Inc. Bethesda, MD). Control experiments were performed in the absence of primary antibody or in the presence of blocking peptide.31
Western blot analysis
Western blot was performed as previously described.30,35 30 microgram of protein was subjected to SDS-PAGE (10%) and blotted with anti-UBF1 (Upstate Biotech.), anti-CBP (A-22: sc-369 and C-20: sc-583) (Santa Cruz Biotech.), and anti-huntingtin (C-20: sc-351) (Santa Cruz Biotech.) antibody. Protein loading was controlled by probing for alpha-tubulin (Sigma) or beta-actin (Santa Cruz Bitotech) on the same membrane.
RNA interference experiment
Q7 and Q111 (1 × 105 cells/ml) or SH-SY5Y (2 × 105 cells/ml) were transiently transfected with 100 nM to 400 nM of Stealth control RNAi and CBP RNAi (Invitrogen Life Tech., Carlsbad, CA) and control shRNA and UBF1 shRNA using DMRIE-C transfection reagent (Invitrogen Life Tech., Carlsbad, CA) in the presence or absence of HrD-IRES reporter for 48hr.11
Run-on transcription assay and immunofluorescence
Nascent RNA was detected using a method previously described with slight modifications.4 Briefly, cells were incubated with 5 mM 5-FU (Sigma) for indicated time and fixed with 4% paraformaldehyde (PFA) for 30 min at 4 °C. Incorporated 5-FU was labeled with anti-BrdU antibody (Serotec) for overnight at 4 °C and with DyLight-594 conjugated anti-mouse IgG antibody (Jackson Laboratories) for 1 hr. The nuclei were counterstained with DAPI and labeled signals were visualized using fluorescent microscopy (Olympus, Japan). For the quantitative analysis, emitted signals for 5-FU was captured and their intensities were analyzed using multigauge program (Fuji, Japan).
Supplementary Material
ACKNOWLEDGEMENT
We thank to Dr. Samson T. Jacob for pIRES-Luc and Human rRNA-luciferase Vector and Dr. Dr. Marcy MacDonald for STHdhQ7/7 and STHdhQ111/111 cells. We thank Dr. Byoung Yul Soh for his technical assistance. This study was supported by WCU Neurocytomics Program Grant (800-20080848) (H.R.) and SRC Grant (2010-0029-403) (H.R.) from KOSEF and NIH NS52724 (H.R.).
REFERENCES
- 1.Vonsattel JP, DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Sadri-Vakili G, Cha JH. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006;2:330–338. doi: 10.1038/ncpneuro0199. [DOI] [PubMed] [Google Scholar]
- 3.Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 4.Casafont I, et al. The giant fibrillar center: A nucleolar structure enriched in upstream binding factor (UBF) that appears in transcriptionally more active sensory ganglia neurons. J. Struct. Biol. 2007;159:451–461. doi: 10.1016/j.jsb.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Lafontaine DL, Tollervey D. The function and synthesis of ribosomes. Nat Rev Mol Cell Biol. 2001;2:514–520. doi: 10.1038/35080045. [DOI] [PubMed] [Google Scholar]
- 6.Akhmanova A, Verkerk T, Langeveld A, Grosveld F, Galjart N. Characterisation of transcriptionally active and inactive chromatin domains in neurons. J. Cell. Sci. 2000;113:4463–4474. doi: 10.1242/jcs.113.24.4463. [DOI] [PubMed] [Google Scholar]
- 7.McStay B, Grummt I. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu. Rev. Cell. Dev. Biol. 2008;24:131–157. doi: 10.1146/annurev.cellbio.24.110707.175259. [DOI] [PubMed] [Google Scholar]
- 8.Hirschler-Laszkiewicz I, et al. The role of acetylation in rDNA transcription. Nucleic Acids Res. 2001;29:4114–4124. doi: 10.1093/nar/29.20.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada M, et al. Interaction between neuronal intranuclear inclusions and promyelocytic leukemia protein nuclear and coiled bodies in CAG repeat diseases. Am. J. Pathol. 2001;159:1785–1795. doi: 10.1016/S0002-9440(10)63025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangiarini L, et al. Exon1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 11.Pelletier G, et al. Competitive recruitment of CBP and Rb-HDAC regulates UBF acetylation and ribosomal transcription. Mol. Cell. 2000;6:1059–1066. doi: 10.1016/s1097-2765(00)00104-0. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, et al. Monoallele deletion of CBP leads to pericentromeric heterochromatin condensation through ESET expression and histone H3 (K9) methylation. Hum. Mol. Genet. 2008;17:1774–1782. doi: 10.1093/hmg/ddn067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stefanovsky VY, Moss T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res. 2008;36:5093–5101. doi: 10.1093/nar/gkn484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Mahony DJ, Rothblum LI. Identification of two forms of the RNA polymerase I transcription factor UBF. Proc. Natl. Acad. Sci. USA. 1991;88:3180–3184. doi: 10.1073/pnas.88.8.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannan RD, et al. Overexpression of the transcription factor UBF1 is sufficient to increase ribosomal DNA transcription in neonatal cardiomyocyte: implications for cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 1996;93:8750–8755. doi: 10.1073/pnas.93.16.8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen D, Belmont AS, Huang S. Upstream binding factor association induces large-scale chromatin decondensation. Proc. Natl. Acad. Sci. USA. 2004;101:15106–15111. doi: 10.1073/pnas.0404767101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanij E, et al. UBF levels determine the number of active ribosomal RNA genes in mammals. J. Cell Biol. 2008;183:1259–1274. doi: 10.1083/jcb.200805146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meraner J, et al. Acetylation of UBF changes during the cell cycle and regulates the interaction of UBF with RNA polymerase I. Nucleic Acids Res. 2006;34:1798–1806. doi: 10.1093/nar/gkl101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin CH, et al. Mass spectrometric identification of phosphorylation sites of rRNA transcription factor upstream binding factor. Am. J. Physiol. Cell Physiol. 2007;292:1617–1624. doi: 10.1152/ajpcell.00176.2006. [DOI] [PubMed] [Google Scholar]
- 20.Steffan JS, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 22.McCampbell A, Fischbeck KH. Polyglutamine and CBP: Fatal attraction? Nat. Med. 2001;7:528–530. doi: 10.1038/87842. [DOI] [PubMed] [Google Scholar]
- 23.Nucifora FC, et al. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 24.Doetzlhofer A, et al. Histone deacetylase 1 can repress transcription by binding to SP1. Mol. Cell. Biol. 1999;19:5504–5511. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stefanovsky VY, Moss T. Regulation of rRNA synthesis in human and mouse cells is not determined by changes in active gene count. Cell Cycle. 2006;5:735–739. doi: 10.4161/cc.5.7.2633. [DOI] [PubMed] [Google Scholar]
- 26.Ryu H, et al. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. USA. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCampbell A, et al. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc. Natl. Acad. Sci. USA. 2001;98:15179–15184. doi: 10.1073/pnas.261400698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes RE, Olson JM. Therapeutic opportunities in polyglutamine disease. Nat. Med. 2001;7:419–423. doi: 10.1038/86486. [DOI] [PubMed] [Google Scholar]
- 29.Ghoshal K, et al. Role of human ribosomal RNA (rRNA) promoter methylation and of methyl-CpG-binding protein MBD2 in the suppression of rRNA gene expression. J. Biol. Chem. 2004;279:6783–6793. doi: 10.1074/jbc.M309393200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trettel F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 31.Ryu H, et al. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc. Natl. Acad. Sci. USA. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryu H, et al. Sp1 and Sp3 are oxidative stress-inducible, anti-death transcription factors in cortical neurons. J. Neurosci. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ. Antioxidants modulate mitochondrial protein kinase A and increase CREB binding to D-Loop DNA of the mitochondrial genome in neurons. Proc. Natl. Acad. Sci. USA. 2005;102:13915–13920. doi: 10.1073/pnas.0502878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim T, et al. Profiling of vitreous proteomes from proliferative diabetic retinopathy and nondiabetic patients. Proteomics. 2007;7:4203–4215. doi: 10.1002/pmic.200700745. [DOI] [PubMed] [Google Scholar]
- 35.Garaguso I, Borlak J. Matrix layer sample preparation: an improved MALDI-MS peptide analysis method for proteomic studies. Proteomics. 2008;8:2583–2595. doi: 10.1002/pmic.200701147. [DOI] [PubMed] [Google Scholar]
- 36.Ryu H, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.