

Abstract
Fibroblast growth factors (FGFs) comprise a family of developmental regulators implicated in a wide variety of neurological functions. FGF receptors-1,-2,-3 (Fgfrs) are expressed in the embryonic forebrain, including regions overlapping with ventral sites of oligodendrocyte progenitor (OLP) generation. Although FGF signaling is known to influence the proliferation of OLPs in vitro, functions of different Fgfrs in vivo are lacking. Here, we examined single and double mutants with conditional disruption of Fgfrs, specifically in the embryonic forebrain, to investigate the effect of FGFs on the generation and proliferation of OLPs in vivo. FGF signaling, through cooperation between Fgfr1 and Fgfr2 but not Fgfr3, is required for the initial generation of OLPs in the mouse ventral forebrain, with Fgfr1 being a stronger inducer than Fgfr2. In cultures derived from embryonic mutant forebrains or from normal forebrains grown in the presence of Fgfr inhibitor, a strong attenuation of OLP generation was observed, supporting the role of FGF signaling in vivo. Contrary to in vitro findings, Fgfr1 and Fgfr2 signaling is not required for the proliferation of OLPs in vivo. Finally, failure of OLP generation in the Fgfr mutants occurred without loss of sonic hedgehog (Shh) signaling; and pharmacological inhibition of either Fgfr or hedgehog signaling in parallel cultures strongly inhibited OLP generation, suggesting that Fgfrs cooperate with Shh to generate OLPs. Overall, our results reveal for the first time an essential role of FGF signaling in vivo, where the three Fgfrs differentially control the normal generation of OLPs from the embryonic ventral forebrain.
Keywords: oligodendrocyte, myelin, FGF receptor, Fibroblast Growth Factor, sonic hedgehog
Oligodendrocyte progenitor populations that are embryonically generated originate in the ventral forebrain and then migrate dorsally to their final destinations in the cerebral cortex (Rowitch, 2004; Miller, 2002; Tekki-Kessaris et al., 2001; Kessaris et al., 2008), sequentially maturing through a series of stages (Pfeiffer et al., 1993; Espinosa-Jeffrey et al., 2009). It is generally believed that these cells, expressing the transcription factor Olig2 and platelet derived growth factor receptor alpha (Pdgfra), are generated under the regulation of sonic hedgehog (Shh) (Nery et al., 2001; Tekki-Kessaris et al., 2001; Spassky et al., 2001; Alberta et al., 2001; Rowitch, 2004; Fuccillo et al., 2004). However, evidence has emerged that OLP generation may depend on signals in addition to Shh (Nery et al., 2001; Gabay et al., 2003; Chandran et al., 2003; Kessaris et al., 2004; Cai et al., 2005; Abematsu et al., 2006).
FGFs are a family of signaling molecules that play diverse roles in regulating the development of the nervous system, often in intimate association with other developmental regulators (Ford-Perriss et al., 2001; Hebert, 2005; Aboitiz and Montiel, 2007; Mason, 2007). Fgfrs 1–3 are expressed in the forebrain at embryonic day (E)12.5 in regions that give rise to OLPs, overlapping with Olig2 in the ventral ventricular zone (VZ) (Bansal et al., 2003a). Previous in vitro studies suggest that FGF-2 treatment can induce ventricular cells in culture to acquire an OL fate (Qian et al., 1997), including dorsally derived neural precursors from the embryonic cerebral cortex (Kessaris et al., 2004; Abematsu et al., 2006) and spinal cord in both a Shh dependent (Gabay et al., 2003) and independent (Chandran et al., 2003) manner. We showed that microinjection of FGF-2 into the ventricles of the mouse brain resulted in ectopic induction of OLPs in the embryonic cerebral cortex in a Shh independent manner (Naruse et al., 2006). In the Fgfr1;Fgfr2 double mutants, ventral forebrain precursors were completely absent (Gutin et al., 2006), mimicking the phenotype of mice lacking Shh signaling (Ericson et al., 1995; Fuccillo et al., 2004). In Fgfr1 or Fgfr2 single mutants, ventral precursors were not lost, and while interneurons were induced normally in Fgfr2 mutants they failed to develop in Fgfr1 mutants (Gutin et al., 2006). Taken together, these studies suggest that FGFs are likely candidates to regulate OLP generation, through one or more of the three Fgfrs. However, direct genetic evidence for a role of Fgfr signaling in generating the earliest OLP population from its normal origin in the embryonic ventral forebrain is absent.
Using a combination of loss-of-function approaches to genetically and pharmacologically inactivate Fgfr signaling, specifically in the embryonic forebrain, we investigated the role of FGF and its relationship to Shh in OLP generation. We provide evidence that unlike interneurons, FGF signaling through Fgfr1 as well as Fgfr2 (but not Fgfr3) is required for the generation of OLPs from the ventral forebrain. FGF signaling is not essential for the proliferation or survival of OLPs. Shh signaling remains unaffected in Fgfr deficient forebrains and cooperates with Fgfr signaling to induce the vast majority of the ventrally-derived OLPs in vitro. Thus, the interplay between Fgfr and Shh signaling pathways provides an important mechanism of regulating OLP generation from the embryonic mouse forebrain.
MATERIALS AND METHODS
Generation of mutant mice
Conditional single and double knock-out mice in different combinations, with disrupted FGF receptor signaling were previously generated (Gutin et al., 2006). Briefly, floxed alleles of Fgfr1 and Fgfr2 genes were used to generate telencephalic specific knockouts when crossed to Foxg1-cre mice, as described (Hebert et al., 2003; Yu et al., 2003). The Fgfr3 allele used is a null allele (Deng et al., 1996). Mutant embryos were obtained at the ages indicated in the expected ratios without any signs of necrosis. The transcription factor Foxg1 is expressed from around E8–E9.5 in all cells of the telencephalon, including uncommitted ventral neuroepithelial cells that eventually specify various lineages including the OL-lineage (Hebert and McConnell, 2000). Mutant and control embryos were analyzed from the same litters, facilitating comparisons among the genotypes. Since the majority of Fgfr1 and Fgfr2 mutants rarely survived beyond E17.5, and Fgfr1/Fgfr2 double mutants beyond E14.5, they could not be used to study postnatal OL development.
Tissues Preparation
Dams were deeply anesthetized with halothane and pups removed from the uterus. Brains were fixed by immersion in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) and cryoprotected sequentially in 10% sucrose followed by 30% sucrose, each carried out overnight at 4°C and embedded in OCT compound (Tissue-Teck). Coronal cryostat sections of brains (30 um thick for E12.5 and 15 um for other ages) were used for in situ hybridization and immunohistochemistry.
In situ Hybridization
In situ hybridization was carried out with slight modifications of the procedure previously described (Kaga et al., 2006). Riboprobes specific for Olig2, Pdgfra, Sox10, Shh, Patched1, Foxg1 mRNA were used (gifts from D. Rowitch, W. Richardson, M. Qui, K. Ikenaka, and J.M. Hebert, respectively). Briefly, sections were post-fixed with 4% PFA for 15 min, washed in PBS and incubated in 1 ug/ml proteinase K at 37°C for 30 min. Sections were fixed with 4% PFA and washed with PBS again. Hybridization for each mRNAs was performed overnight at 65°C (Pdgfra) or 70°C (Shh, Patched1, Olig2, Foxg1, and Sox10) by using digoxigenin-labeled antisense riboprobes in a solution containing 50% formamide, 5×SSC (750 mM NaCl, 75 mM Trisodium Citrate) and 1% SDS. After hybridization, the sections were washed in 50% formamide, 2×SSC and 1% SDS at 65 °C or 70°C, followed by rinses in 2×SSC, 0.2×SSC at room temperature and 0.1×SSC at 60°C. After blocking for nonspecific binding in blocking buffer [Tris buffered saline, pH7.4, with 1% Tween-20 and 1% normal goat serum (NGS)] for 1 hr, sections were incubated for 2 hr in alkaline phosphatase-conjugated-antidigoxigenin antibody (1:5,000; Roche Diagnostics, Penzberg, Germany). Color development in the presence of 4-nitroblue tetrazolium chloride, 5-bromo-4-chloro-3-indolylphosphate was performed in the dark at room temperature. The sections were washed in 10 mM Tris and 10 mM EDTA pH8, incubated in Hoechst blue dye 33342 (1 mg/ml; Sigma, St. Louis, MO) to counter stain the nuclei, fixed in 3.7% formaldehyde and mounted with 90% glycerol.
Comparisons between mutants and controls were made within the same litters. Multiple coronal sections were cut from the rostral to caudal regions of the forebrains. Sections from three separate planes from control and mutant mice were matched using anatomical landmarks. All the “scattered” Olig2+ or Pdgfra+ cells in the section were counted from each of the three sections and averaged. Note that Olig2 stains the sections in two distinct patters, (a) the ventricular zone is stained totally in “solid purple” pattern with no clear cellular distinction, while (b) the subventricular region is stained in a “punctate” fashion, which can be clearly distinguished from the solid ventricular zone staining and is defined as “scattered” cells. Similar staining procedure have been employed by other laboratories in previous studies, to reliably quantify these scattered cells as OLPs. The comparison of cell numbers between 3–6 control and mutant mice was determined by an unpaired Student’s t-test.
Cell Culture
Dissociated cultures of whole or ventral portions of forebrains were prepared from E10.5, E11.5 or E12.5 mouse embryos. Briefly, tissue was carefully dissected out from the whole brain and cells were dissociated by trypsinization (0.025% at 37oC for 7 min) and mechanical dissociation. The dissociated cells were plated in 4% fetal calf serum in Dulbeco Modified Eagles medium (FCS/DMEM) at a density of 3×105 cells/cm2 (E12.5) or 1.5×105 cells/cm2 (E10.5, E11.5) into polylysine-D-coated (50 mg/ml; Sigma, St. Louis, MO) 4 well culture plates (Nalge Nunc international, Rochester, NY). Seven hours after plating, the cultures were changed to Bottenstein and Sato defined medium [DMEM with100 ug/ml transferrin, 5.2 ng/ml sodium selenium, 5 ug/ml insulin, 8.8 ug/ml putrescine, 6 ng/ml progesterone, 10,000 units/ml penicillin and 10,000 ug/ml streptomycin (all ingredients from Sigma)] plus 0.5% FCS. In some experiments as indicated, cells were treated with PD173074, a specific inhibitor of Fgfr signaling (Skaper et al., 2000; Bansal et al., 2003b; Kessaris et al., 2004), or cyclopamine, an inhibitor of all hedgehogs including Shh (Enzo Life Science, Plymouth Meeting, PA), at the time when cultures were transferred to defined medium. Since dimethylsulfoxide (DMSO) was used to dissolve the inhibitors, it was added to the control cultures. Every four days the half medium was changed with the re-addition of inhibitors wherever needed.
Immunoflourescence Microscopy
Cells in mixed primary cultures from embryonic forebrains were immunolabelled for successive stage-specific markers of the OL-lineage. Pre/early OLPs were identified with Olig2 and PDGFRa antibodies, late OLPs with O4 antibody, and mature OLs with HPC7 antibody (Pfeiffer et al., 1993; Baas and Barnstable, 1998).
Cells in culture were labeled as described previously (Bansal et al., 1996). Briefly, O4 and HPC7 labeling was done on live cells on ice; and PDGFRa and Olig2 staining was carried out after fixation of cells with 4% PFA for 10 min and permeabilization with 0.1% Triton-X100 in Hepes buffered Earl’s balanced salt solution for 5 min. Cells were blocked with 3% NGS in PBS for 1 hr and incubated with primary antibodies (mouse monoclonal IgM O4 antibody, 1:25; mouse monoclonal IgG HPC7 antibody, 1:25, C.J. Barnstable; rabbit anti-Olig2, 1:50, IBL; rabbit anti-PDGFRa, 1:200, W.B. Stallcup; rat anti-PDGFRa, 1:100, BD Biosciences) for 1 hr, washed and incubated with the appropriate secondary antibodies [mu-chain specific goat anti-mouse IgM-fluorescein, 1:50; gama-chain specific goat anti-mouse IgG-Cy3, 1:600; donkey anti-rabbit IgG-Cy3, 1:600 (all from Jackson Immuno Research, West Grove, PA) or goat anti-rabbit IgG-Alexa-488, 1:200 (Molecular Probes, Eugene, OR, USA)] for 1 hr. Total cells were identified by incubating with the nuclear stain Hoechst Blue 33342 (1:1000), along with secondary antibodies. Cells were washed and mounted in 1,4-diazobicyclo-(2,2,2)-octane in glycerol (DABCO). Immunolabelled cells were counted in 75–150 20X fields to provide an accurate sampling of the positive cell distribution on the whole plate.
Immunohistochemistry
Following antigen retrieval by heat treatment (95oC, 5 min), the E12.5 forebrain sections were incubated in 0.05% H2O2 in PBS for 30 min to inactivate endogenous peroxidase. Next, the sections were blocked in 10% normal goat serum, 0.2% TX100 for 1 hr and then incubated overnight (4oC) in rabbit pan-Erk1/2 antibody (1:500; Promega, Madison, WI). Sections were then incubated in biotinylated anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA) for 1 hr and in ABC reagents (VECTASTAIN Elite ABC kit: Vector Laboratories) for 40 min at room temperature before color development with 0.05% 3-3-diaminobenzidine/0.015% H2O2 (DAB; Research Genetics, Huntsville, AL). Parallel sections processed identically but without the incubation in the primary antibody served as negative controls.
Proliferation Assay
To identify OLPs that were in the S phase of the cell cycle, mice received an intraperitoneal injection of bromodeoxyuridine (BrdU; 100 mg/kg body weight) for incorporation into newly synthesized DNA and sacrificed 1 hr later. Tissue preparation and sectioning was carried out as described above. After detection of Pdgfra by in situ hybridization, the sections were incubated in pre-heated citrate buffer (10mM, pH 6.0) for 5 min at 95oC. Following 1 hr of blocking in 10% NGS and 0.2% Triton X-100 in PBS, the sections were incubated overnight with mouse monoclonal anti-BrdU (1:25; Becton Dickinson, Lincoln Park, NJ), washed three times, and incubated for 1 hr with a biotinylated goat anti-mouse IgG (1:200; Vector Laboratories). The sections were next incubated with avidin-biotin peroxidase complex (VECTASTAIN Elite ABC kit) for 40 min at room temperature and the immune complexes were visualized by treatment with DAB. To identify mitotic cells, the sections were stained with anti-phospho-histone (pH3 ser10) antibody (1:200; Upstate, Lake Placid, NY). Sections were fixed with 4% PFA for 10 min, blocked with 1% NGS in PBS containing 0.1% Triton X-100 for 1 hr, and incubated with the antibody overnight. The sections were then washed and incubated with goat anti-rabbit Alexa-488 (1:200) and Hoechst blue 33342 (1:1000) and washed and mounted in DABCO.
To detect proliferating cells in culture, BrdU was added to the cultures for 3 hr at a final concentration of 50 uM. Following labeling with either rabbit anti-Olig2 or rabbit anti-PDGFRa as described above, the cells were fixed with 4% PFA, washed with PBS, incubated with acid alcohol (95% ethanol/5% acetic acid) for 2 min at −20°C, washed in PBS, denatured with 2N HCL (10 min), neutralized with 0.1 M pH8.5 sodium borate buffer (10 min), blocked with 3% NGS/PBS (1 hr), and incubated in mouse monoclonal anti-BrdU antibody for 30 min. Cells were washed in PBS, incubated in goat anti-mouse IgG conjugated to Cy3 (1:600; Jackson ImmunoResearch, West Grove, PA) and Hoechst Blue 33342 (1:1000), and washed and mounted.
Apoptotic cells assay
Apoptotic cells were detected using terminal deoxynucleotide transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (ApopTag kit; Invitrogen, Purchase, NY) according to the manufacturer protocol. Briefly, brain cryosections were incubated in 4% PFA for 30 min, treated with 0.3% H2O2 to quench endogenous peroxidase, washed in equilibrium buffer, and incubated in reaction buffer containing digoxygenin-dNTP and terminal deoxynucleotidyl transferase (30 min, 37°C). The sections were washed and incubated for 30 min with peroxidase conjugate anti-digoxygenin. The TUNEL+ cells were identified by reaction with DAB and analyzed by epifluorescence microscopy.
Immunoblotting
Dorsal and ventral forebrain tissue from E12.5 normal mice were separated and mechanically dissociated in N2 medium (Fortin et al., 2005). Following 30 min of recovery at 37°C, 0.2 ug/ml heparin was added to both cell suspensions and each divided equally into 6 samples for different treatments as indicated. Cells were incubated in FGF-2 (50 ng/ml) or FGF-8 (50 ng/ml) for 15 min at 37°C. Incubation with Fgfr inhibitor, PD173074 (100 nM), or DMSO was for 1 hr prior to FGF-2 addition. Cells were centrifuged and pellets were lysed in RIPA buffer (10 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 1% deoxycholate and 1% NP40, pH 7.4) with protease and phosphatase inhibitors (2 mM PMSF, 2 ug/ml leupeptin, 2 ug/ml aprotinin, 50 mM NaF, 10 mM NaP2O7, and 1 mM Na o-Vanadate) and briefly sonicated and centrifuged. As described previously (Fortin et al., 2005), total protein was assayed in each sample and equal amounts (2.5 ug) were loaded on SDS-PAGE gel, transferred to PVDF membrane, and immunolabelled with mouse monoclonal anti-phoapho-Erk1/2 (1:1000; Sigma, St. Louis, MO) or rabbit pan-Erk1/2 antibody (1:5,000; Promega, Madison, WI) followed by HRP-conjugated goat-anti-mouse or goat anti-rabbit secondary antibody (1:10,000; Santa Cruz Biotech, Santa Cruz, CA), and developed with ECL plus reagent (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
RESULTS
Fgfr1 and Fgfr2, but not Fgfr3 are required for the generation of OL progenitor from the embryonic mouse forebrain
We asked if the generation of OL progenitors in vivo from the embryonic ventral forebrain was affected by the absence of Fgfr1, Fgfr2 and/or Fgfr3. We examined controls and single mutants of these receptors at E12.5 by in situ hybridization for the expression of Pdgfra and Olig2 to determine the extent of OLP generation (Fig. 1A,B,C). In the normal embryonic forebrains, Pdgfra marks the earlier population of OLPs that appear as an array of scattered cells in the ventral VZ and subventricular zones (SVZ), first localized in the anterior entopeduncular area, later extending into the median and lateral ganglionic eminences (MGE, LGE), and then migrating dorsally to the neocortex (Fig. 1A) (Nery et al., 2001; Tekki-Kessaris et al., 2001). Olig2 is normally expressed strongly in the ventral VZ lining the ventricles terminating sharply at the cortico-striatal boundary and in ‘scattered’ cells outside the VZ, which are presumably OLPs (Fig. 1B) (Tekki-Kessaris et al., 2001; Alberta et al., 2001). Although the VZ expression of Olig2 was unaffected, there was a dramatic decrease in the numbers of scattered Olig2+, Pdgfra+ OLPs that appeared in the ventral forebrain of Foxg1cre/+;Fgfr1lox/lox or Foxg1cre/+;Fgfr2lox/lox mutants (will be referred to as Fgfr1−/− or Fgfr2−/−) compared to littermate controls (Fig. 1A,B,C) or heterozygous receptor mutants (data not shown). In contrast, even though Fgfr3 is expressed in the ventral VZ in a pattern similar to Fgfr1 and Fgfr2 (Bansal et al., 2003a), its loss did not have any effect on OLP generation (Fig. 1A,B,C). Sox10, another marker of OLPs, was also examined showing a reduction of Sox10+ “scattered cells” in the mutants similar to Pdgfra (data not shown). In agreement with the previous finding (Gutin et al., 2006), the morphology of the ventral forebrain was affected in the Fgfr1−/−, but not Fgfr2−/−, mutants (Fig. 1B). Irrespective of this structural defect, OLP generation was equally perturbed in both Fgfr1−/− and Fgfr2−/− mutants suggesting that it occurred independent of morphological defects. This notion is further supported by our observation (presented later, Fig. 3) that OLP production was also deficient in dissociated cell cultures derived from E12.5 Fgfr1−/− forebrains.
Fig.1. Fgfr1 and Fgfr2 but not Fgfr3 are required for the generation of OL progenitor from the embryonic mouse forebrains.

Coronal sections of E12.5 forebrains from control, Fgfr1−/−, Fgfr2−/− or Fgfr3−/− mutants were analyzed for the expression of OLP markers, Pdgfra (A) or Olig2 (B) mRNA by in situ hybridization. Total numbers of “scattered” Pdgfra+ or Olig2+ cells (arrows) were counted in the whole forebrain sections taken caudally (Pdgfra) or rostrally (Olig2). Three to four matched sections from each littermate control and mutant mice were analyzed. Numbers of animals analyzed were six each of control, Fgfr1−/− or Fgfr2−/−, and two of Fgfr3−/−. Data is expressed as percent of control (C). Error Bars represent S.E.M. Scale bars, 200 um. Pdgfra expression is also visible in the meninges (*). Note that compared to controls, decreased numbers of OLPs were found in both Fgfr1−/− and Fgfr2−/− mutants irrespective of abnormal ventral forebrain morphology in the Fgfr1−/− mutants. All the mutants maintained the expression of Olig2 in the ventral ventricular zone (vz) of the median and lateral ganglionic eminences (MGE, LGE), suggesting normal dorsoventral patterning. Dotted line defines the boundary between the dorsal neocortical (d) and the ventral forebrain (v) regions.
Fig.3. Generation of OL progenitors in cultures initiated from E12.5 Fgfr1−/− or Fgfr2−/− mutant forebrains is reduced in a manner similar to that in vivo.

(A) Dissociated cell cultures from E12.5 forebrains of Fgfr1−/− or Fgfr2−/− mutants and littermate controls, were immunolabeled as a function of days in vitro (DIV) for specific markers of pre/early progenitors (Olig2 at 3 DIV and PDGFRa at 6 DIV), late progenitors (O4 at 9 DIV) and OLs (HPC7 at 12 DIV). (B) The numbers of Olig2+, PDGFRa+ and O4+ cells were counted from each culture and expressed as a percent of control showing significantly reduced numbers of these cells in both mutants compared to littermate controls. Scale bar, 50 um. Error Bars represent S.E.M. (P<0.01*, P<0.02**). Cultures from 3–6 pups were analyzed for each group.
In order to investigate if the reduction of OLP progenitors in the mutant embryos observed at E12.5 reflected a developmental delay, we examined forebrains of Fgfr1−/− or Fgfr2−/− mutants at E14.5 and E16.5 for the expression of Pdgfra, Sox10 or Olig2 (Fig. 2). Normally by this time, a significant proportion of OLPs progressively migrate from their ventral sites of origin dorsally to the cortical regions. As expected, the numbers of OLPs at these ages were increased in all the groups relative to E12.5, but the reduction of OLPs was maintained in Fgfr1−/− or Fgfr2−/− embryos compared to controls both in the cortical regions (Fig. 2) and the ventral regions (data not shown), ruling out any significant developmental delay.
Fig.2. The decrease in OL progenitors in Fgfr1−/− or Fgfr2−/− mice is maintained even at E14.5 and E16.5.

Coronal sections of E14.5 (A) or E16.5 (B) forebrains from control, Fgfr1−/− or Fgfr2−/− mutants were analyzed for the expression of Pdgfra, Sox10 or Olig2 mRNA by in situ hybridization. Diagram shows the plane in which the sections were cut, and the boxed area depicts the neocortical (ncx) region of the forebrain that is shown in the figures. Scale bars, 200 um.
It is important to note that, unlike the double Fgfr1−/−;Fgfr2−/− mutant, in the single Fgfr1−/− or Fgfr2−/− mutants the ventral neuroepithelial precursor cells are not lost but clear differences are observed in how the interneuron (Gutin et al., 2006) or OLPs (present study) are generated from these cells at E12.5. Specifically, (a) in the Fgfr1 single mutants 100% of the interneurons failed to develop from the neuroepithelial precursors (Gutin et al. 2006); in contrast, 40% of OLPs were able to develop in these mutants (present study). (b) In Fgfr2 single mutants interneuron generation was completely unaffected (Gutin et al. 2006); in contrast, OLP generation was significantly inhibited in these mutants (by 60%) (present study).
We conclude that signaling by both Fgfr1 and Fgfr2, but not Fgfr3, is needed to generate the full numbers of ventrally-derived OLPs, since loss of either one attenuated the generation of the majority, but not all of the OLPs. Further, comparison with a previous study (Gutin et al., 2006) demonstrates that there are important differences in the requirements for FGF signaling by ventral neuroepithelial precursors for generating OLPs or interneurons.
Generation of OL progenitors in cultures initiated from E12.5 Fgfr1−/− or Fgfr2−/− mutant forebrains is reduced in a manner similar to that in vivo
Lack of Fgfr signaling could potentially disrupt migration of cells out of the germinal zone resulting in reduced numbers of “scattered” OLPs observed outside the VZ in the Fgfr1−/− and Fgfr2−/− mutants (Fig. 1). To test this possibility and to substantiate the in vivo findings, OL progenitor development was analyzed in dissociated cell cultures initiated from E12.5 forebrains of Fgfr1−/− or Fgfr2−/− mutants and their littermate controls (Fig. 3). Cells were examined as a function of time by immunofluorescence microscopic labeling for early OLP markers, Olig2, PDGFRa. As in vivo, there were reduced numbers of Olig2+ and PDGFRa+ OLP that developed in cultures from each of the two mutant forebrains compared to controls. Since the PDGFRa+ OLPs mature into O4+ late OLPs (Pfeiffer et al., 1993), we also examined the expression of the O4-antigen and found considerably reduced numbers of these cells correlating with the reduced numbers of early OLP-marker expressing cells. Hoescht staining of cell nuclei showed a similar distribution of total cells in cultures of mutant and control forebrains (Supplementary Fig. 1). Thus, since reduced numbers of OLPs are also observed in cultures of mutant forebrains, the reduction of OLPs observed in vivo cannot be explained by potential migration defects in the mutants.
Since the mutant mice die before OLPs differentiate into OLs, precluding the in vivo analysis of Fgfr function in the maturation of OLs, we also utilized these mutant forebrain cultures to analyze OLP terminal differentiation by examining OL marker, HPC7, whose expression overlaps with galactocerebroside, a well-recognized marker of OLs (Pfeiffer et al., 1993; Baas and Barnstable, 1998). HPC7 immunolabeling was detected in the cultures of Fgfr1−/− and Fgfr2−/− mutants similar to controls, demonstrating that the small numbers of OLPs that were produced in these mutant cultures were able to differentiate into OLs. However, the proportion of late progenitors that differentiated into OLs was not determined here and is being examined in another study that addresses the role of FGF signaling at later stages of OLP maturation.
Loss of Fgfr1 or Fgfr2 does not affect proliferation or cell death of OL progenitors
The reduction in the numbers of OLPs observed in the Fgfr1−/− and Fgfr2−/−mutants could, in principle, be due to a decreased efficiency of specification by the neuroepithelial cells towards an OL progenitor fate, reduced proliferation, and/or increased death of specified cells. To explore these possibilities, we examined the proliferation and cell death in the E12.5 forebrains of mutants and their littermate controls. The proliferative capacity of OLPs was studied in vivo by injecting BrdU intraperitonially into mothers, 1 hr before harvesting brains from the embryos, for the analysis of Olig2 or Pdgfra mRNA positive cells double labeled with anti-BrdU (Fig. 4A). Proliferation of OLPs was also studied in cultures initiated from E12.5 control and mutant forebrains (Fig. 4B). No differences were observed in the numbers of OLPs incorporating BrdU in control and Fgfr2−/− or Fgfr1−/− mutants either in vivo or in vitro. Similarly, immunolabeling of mutant and control forebrains with anti-phospho-histone 3 (pH3, a marker for mitotic cells) also did not show any significant differences in the numbers of pH3+ cells (Fig. 4C). These data suggest that the proliferation of OLPs is not affected in the forebrains of Fgfr1−/− and Fgfr2−/− mutants and therefore cannot account for the reduced numbers of OLPs.
Fig.4. Loss of Fgfr1 or Fgfr2 does not affect proliferation of OL progenitors.

(A) Mothers were injected intraperitoneally with BrdU (100 mg/kg body wt), 1 hr before harvesting the embryos. To identify proliferating cells, coronal sections of E12.5 forebrain from Fgfr1−/−, or Fgfr2−/− mutants and littermate control were double labeled by combining immunohistochemistry for BrdU (brown) and in situ hybridization for Olig2 or Pdgfra mRNA expression (purple). Total numbers of BrdU+ cells double labeled for Pdgfra or Olig2 mRNA (arrows) were counted in the entire section as in Fig.1 and expressed as percent of Olig2 or Pdgfra positive cells. No significant differences were observed in the numbers of proliferating OLPs between controls and either of the mutants. Error bars represent S.E.M (N=3–4). (B) Dissociated cell cultures initiated from E12.5 forebrains of control, Fgfr1−/− or Fgfr2−/− embryos were treated with BrdU (50 uM) for 3 hr and cells were double immunolabelled with anti-BrdU and either anti-Olig2 (3DIV) or anti-PDGFRa (6DIV). No differences were observed in the numbers of Olig2+ cells incorporating BrdU in control and Fgfr2−/− (quantified) or Fgfr1−/− mutant cultures. Arrows show examples of double-labeled cells. Error bar represents S.E.M. for control (N=3), and means of the difference for Fgfr2−/− (N=2). (C) Coronal sections of E12.5 forebrain from control, Fgfr1−/− or Fgfr2−/− mice, were immunostained with anti-phospho-histone 3 (pH3, a marker for mitotic cells). Total numbers of pH3 positive scattered cells were counted in the entire section as for Fig.1 [representative area with pH3+ cells (arrowheads) is shown in insert], and the numbers were expressed as a percent of the control. No differences in the numbers of pH3+ cells were observed between the control and mutants. Error bars represent mean of duplicates. Scale bars A, 50 um; B, 20 um; C, 20 um.
The reduced numbers of OLPs observed in the Fgfr1−/− and Fgfr2−/− mutants can also be attributed to a localized increase in ventral forebrain cell death. It has been previously reported that in the Fgfr1−/−;Fgfr2−/− double mutants at E10.5, there is increased cell death in the dorsal midline compared to control, and in the Foxg1cre/+,Fgfr1lox/lox;Fgfr3null/null mutants (will be referred to as Fgfr1−/−;Fgfr3−/−) at E11.5, there was a slightly increased percentage of TUNEL-labeled cells in the ventral medial area (0.8% versus 0.2%; Gutin et al., 2006). When we examined Fgfr1−/− and Fgfr2−/−mutants at E12.5, we observed a slight increase in TUNEL+ cells in the mutants compared to controls, which was fairly widespread over the whole forebrain, including the dorsal regions (data not shown). Taken together, there was no evidence of massive cell death specifically in the ventral regions that could have ablated the majority of the OLP population, accounting for the dramatically reduced numbers of OLPs in the ventral forebrains of these mutants.
In the absence of evidence for either major alterations in OLP proliferation or regionally localized massive cell death in the ventral forebrain, we conclude that the dramatic decrease in the numbers of OLPs in the Fgfr1−/− or Fgfr2 −/− mice is due to a decreased efficiency of their induction from the neuroepithelial cells of the forebrain.
Fgfr1 and Fgfr2 gene dosages govern the extent of OL progenitor induction
Analysis of Fgfr1 and Fgfr2 single mutants showed that loss of either Fgfr1 or Fgfr2 was sufficient to attenuate the generation of the majority of OLPs from the ventral neuroepithelial precursors (Fig. 1). Nevertheless, a small population of OLPs appeared in these single mutants suggesting that neither of the receptors could fully compensate for the other. To test this hypothesis, we examined mutants with deletion of different combinations of Fgfr1–3 genes. We also compared Fgfr1−/−;Fgfr2−/− with Fgfr1−/−;Fgfr3−/−double mutants to determine if deletion of any of the two receptors together would have the same effect on OLP generation. E12.5 forebrains were analyzed by in situ hybridization for the expression of Pdgfra (Fig. 5A). Combined loss of both alleles of Fgfr1 and Fgfr3 (Fgfr1−/−;Fgfr3−/−) or one allele of Fgfr1 and both alleles of Fgfr2 (Fgfr1+/−;Fgfr2−/−) were not sufficient to cause a significant additional reduction of OLPs over single mutants of Fgfr1 or Fgfr2. In contrast, elimination of Fgfr1 in combination with one or both alleles of Fgfr2 (Fgfr1−/−;Fgfr2+/− or Fgfr1−/−;Fgfr2−/−) led to a significant reduction or complete failure of OLP generation, respectively. It is important to note that in the Fgfr1−/−;Fgfr2−/− double mutants, the telencephalon is dorsalized [evident by the absence of Olig2 expression in the ventral VZ (Fig. 5B) and other ventral markers (Gutin et al., 2006)], suggesting a loss of all the ventral precursor cells. Thus, it is likely that the complete absence of OLPs (and interneurons, Gutin et al., 2006) in the double mutants (Fgfr1−/−;Fgfr2−/−) could be secondary to a loss of all ventral precursor cells. However, the almost complete failure of OLP development in Fgfr1−/−;Fgfr2+/− cannot be attributed to this fact, since ventral precursors were apparently present in these mutants, evident by normal Olig2 expression in the ventral VZ (Fig. 5B), and therefore normal dorsoventral patterning. It is also interesting to note that one allele of Fgfr2 was largely sufficient for maintaining normal dorsoventral patterning (Fig. 5B).
Fig.5. Fgfr1 and Fgfr2 gene dosages govern the extent of OL progenitor induction.
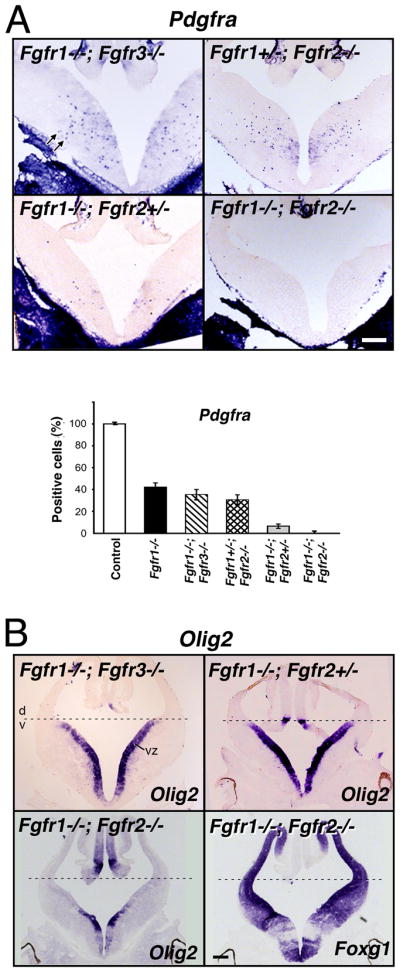
Coronal sections of E12.5 forebrain from control or mutants with different combinations of the three receptors deleted (Fgfr1−/−;Fgfr3−/−, Fgfr1+/−;Fgfr2−/−, Fgfr1−/−;Fgfr2+/−, Fgfr1−/−;Fgfr2−/−) were analyzed by in situ hybridization for the expression of Pdgfra (A), Olig2 and Foxg1 (B) mRNA. Three to four matched sections, each from three to four mice, were analyzed from each group. (A) Compared to the controls in mice lacking Fgfr1 in combination with one or both alleles of Fgfr2, but not of Fgfr3, the numbers of Pdgfra+ OLPs (arrows) were dramatically reduced (Fgfr1−/−;Fgfr2+/−) or totally failed to develop (Fgfr1−/−;Fgfr2−/−). Error bars represent S.E.M (N=3–4). (B) Dorsal-ventral (d,v) patterning, shown by the expression of Olig2 in the ventral ventricular zone (VZ), remained intact in all mutants except in the Fgfr1−/−;Fgfr2−/− double mutant. Foxg1 and Olig2 expression patterns are shown in serial sections in these mutants to demonstrate the loss of Olig2 expression from the regions of the ventricular zone corresponding to regions of strong Foxg1 expression (in which floxed Fgfr1 and Fgfr2 genes should be deleted by Foxg1-cre), but as expected, Olig2 remained in the regions largely negative for Foxg1. Scale bars, 200 um. Note the severe failure of Pdgfra+ OLPs to develop in Fgfr1−/−;Fgfr2+/− mutants without disruption of d-v patterning.
We conclude that reducing the combined gene dosage of Fgfr1 and Fgfr2 (but not Fgfr3), significantly influences the extent of OLP induction, with Fgfr1 playing a more dominant role than Fgfr2. These data emphasize the important difference between the three FGF receptors in influencing OLP generation in vivo.
Shh expression and signaling is unaffected in the ventral forebrains lacking individual or combined alleles of Fgfr1 and Fgfr2
Shh is a potent inducer of OLPs, therefore it is plausible that the reduction of OLPs in the conditional Fgfr mutants could be indirect i.e., loss of Fgfr signaling may down-regulate Shh signaling, which in turn could adversely affect OLP generation. Previous studies addressed this question related to dorsoventral patterning in the Fgfr1−/−;Fgfr2−/− double mutants at E10.5 and showed that Shh and its dependent gene Gli1 were unaffected in these mutants (Gutin et al., 2006). However, Fgfr2−/− and Fgfr1−/−;Fgfr2+/− mutants were not examined in this study. Given that, in the single Fgfr2−/− mutant, unlike interneurons, OLP generation was significantly affected and that in the Fgfr1−/−;Fgfr2+/− mutants it was virtually abolished (even when ventral precursors were present), it became essential to determine if the expression and signaling potential of Shh in these mutants was compromised. We, therefore, examined these mutants for the first time and also extended previous studies and examined E12.5 embryos of single and double mutants of Fgfr1 and Fgfr2 for the expression of Shh and its receptor Patched1, as a readout gene of Shh signaling. Coronal sections from control and mutants taken from rostral (Fig. 6A) or caudal regions (Fig. 6B,C) of the forebrain were analyzed for Shh, Patched1 and Foxg1 mRNA expression by in situ hybridization of adjacent serial sections. Expression of Shh and Patched1 was not significantly altered in Foxg1+ region of the mutants in which floxed Fgfr genes should be deleted by Foxg1-cre (Foxg1 expression shown only for Fgfr1−/− and Fgfr1−/−;Fgfr2−/− sections).
Fig.6. Shh and Patched1 expression is unaffected in the ventral forebrains lacking individual or both of Fgfr1 and Fgfr2.

Coronal sections of E12.5 forebrain from single and double mutants of Fgfr1 and Fgfr2 and littermate controls taken from rostral (A) or caudal (B and C) regions of the forebrains were analyzed by in situ hybridization for the expression of Shh and its receptor Patched1 (as a readout of Shh signaling). Expression of Shh and Patched1 were not significantly affected in any of the mutants: Fgfr2−/− (A–C), Fgfr1−/− (B), Fgfr1−/−;Fgfr2+/− (C) or Fgfr1−/−;Fgfr2−/− (B). In situ hybridization for Foxg1 in adjacent sections of Fgfr1−/− or Fgfr1−/−; Fgfr2−/− mutants, show that Shh continues to be expressed in Foxg1+ region (brackets) in which floxed Fgfr1/2 genes should be deleted by Foxg1-cre. The same image of Foxg1 was used in Fig. 5B. Scale bar, 200 um.
Thus, in the absence of a significant affect on Shh signaling in any of the mutants of Fgfr1 and Fgfr2, we conclude that FGF receptor signaling acts either independently or downstream of Shh to control OLP generation.
Cooperation between sonic hedgehog and FGF receptor signaling is required for the generation of ventrally derived OL progenitor populations in vitro
It has been shown that attenuation of Shh signaling in vivo and in cultures of embryonic forebrains results in a strong inhibition of OLP generation (Tekki-Kessaris et al., 2001; Nery et al., 2001; Fuccillo et al., 2004; Kessaris et al., 2004). In the present study we showed that, like Shh, attenuation of Fgfr signaling in vivo also inhibits OLP generation not only in the double Fgfr1−/−;Fgfr2−/− mutants but also in the single Fgfr1−/−and Fgfr2−/− mutants (Figs. 1,5). To substantiate our in vivo loss-of-function genetic approach, we asked if inhibition of OLP generation observed in vivo and in cultures of Fgfr1−/− or Fgfr2−/− mutants could be mimicked in cultures of normal forebrains grown in the presence of a specific chemical inhibitor of Fgfr signaling (PD173074). The specificity of this inhibitor for Fgfrs has been established previously where PD173074, at similar doses used in this experiment, specifically inhibited FGF-2-mediated but not PDGF-mediated MAPK activation and proliferation of isolated OLPs (Bansal et al., 2003b). Dissociated cell cultures derived from whole forebrains of wild type E12.5 mice were cultured in defined media in the presence or absence of PD173074 (Fig. 7A,B). OLP development was analyzed by immunolabeling cells with specific markers of early and late OLPs (PDGFRa, O4). Hoescht staining of cell nuclei showed a similar distribution of total cells in cultures grown in the absence or presence of the inhibitor (Fig. 7A and Supplementary Fig. 2). There was a dramatic reduction in the numbers of PDGFRa+ and O4+ OLPs that develop in these cultures in the presence of the Fgfr inhibitor. These data are consistant with our in vivo genetic loss-of-function results, but differ somewhat from a previous in vitro report that did not observe a decrease in the numbers of ventrally-derived OLPs, defined by the expression of NG2, by PD173074 treatment (Kessaris et al., 2004). The difference could partly be due to the examination of a different OLP marker, NG2, that identifies a little different stage of OLP development than PDGFRa or O4. We were unable to duplicate these studies as in our cultures of embryonic forebrain; the NG2 antibody immunostained a much larger population of cells (many with a fibroblast-like morphology) than PDGFRa (data not shown). Given the current debate about the identity and fate of NG2+ cells (Zhu et al., 2008; Nishiyama et al., 2009; Trotter et al., 2010) and the difficulty we encountered in definitively identifying OLPs among the mixed population of cells immunostained by the NG2 antibody, we preferred to use PDGFRa and O4 to reliably mark OLPs in the cultures initiated from very early embryonic brains.
Fig.7. Cooperation between sonic hedgehog and FGF receptor signaling is required for the generation of ventrally derived OL progenitor population in vitro.

Dissociated cell cultures initiated from whole forebrains of E12.5 wild-type mice (A,B,C) or from the ventral portion of the forebrain from E11.5 wild-type mice (D) were grown in defined media in the presence or absence of a chemical inhibitor of Fgfrs [PD173074; 150 nM (A–C), 100 nM (D) or 300 nM (B)], inhibitor of all hedgehogs (cyclopamine; 1uM) or both. OLP development was analyzed by immunolabeling with specific marker of early OLPs (PDGFRa) at 6 DIV (A,B,C) or 9 DIV (D), late OLPs (O4) at 9 DIV (A,B) double labeled with the nuclear marker Hoechst (blue). The total numbers of PDGFRa- or O4-positive cells were counted and expressed as a percent of control. Error bars represent S.E.M. (N=3). Each experiment was performed at least two to three times. Scale bars, 50 um. Note that OLP generation is strongly inhibited by either of the inhibitors.
To investigate the relationship of FGF and Shh signaling in OLP generation, we next examined the effects of cyclopamine (hedgehog inhibitor) and PD173074, individually and in combination, in parallel dissociated cultures of not only whole E12.5 forebrains (Fig. 7C) but also of ventral E11.5 (Fig. 7D) and E10.5 (data not shown) forebrains for OLP generation. We found that the appearance of PDGFRa+ OLPs in these cultures was strongly inhibited by either of the two inhibitors, individually or in combination, at each of the ages or regions examined.
From these data, we conclude that a cooperation between sonic hedgehog and FGF receptor signaling is required for the generation of the vast majority of ventrally derived OLPs, since loss of either one resulted in a severe attenuation of OLP induction. Importantly, these data also indicate that E12.5 forebrain cells, grown in defined media, must normally produce FGF (and Shh) endogenously at concentrations sufficient for the development of the vast majority of OLPs in these cultures.
Erk1/2 MAPKs are expressed in the ventral forebrain at the time of OL progenitor generation and can be activated by FGF receptor stimulation
Since the Ras/mitogen activated protein kinase (MAPK) pathway is one of the major signaling pathways downstream of Fgfrs and, at least in the dorsal forebrain, has been implicated in the OLP-inducing activity of both Shh and FGF-2 (Kessaris et al., 2004), we next asked if the ventral forebrain cells also expressed Erk1/2 MAPK which could be activated by stimulating, and inhibited by attenuating Fgfr signaling. Immunolabeling of coronal sections of E12.5 forebrains with anti-pan-Erk1/2 showed a clear expression of Erk1/2 MAPK, both in the dorsal and ventral forebrain cells (Fig. 8A). The specificity of the Erk1/2 staining was confirmed by the absence of a positive signal in (a) non-neuronal tissue (Fig. 8A, asterisk), (b) in control serial section with no primary antibody (Fig. 8B) and (c) in brain sections from Erk1/2 knock-out mice (part of another study; data not shown). In addition, immunoblotting of tissue from E12.5 ventral and dorsal forebrains, with anti-pan-Erk1/2, further demonstrated the presence of Erk1/2 in these regions (Fig. 8C).
Fig.8. Erk1/2 MAPKs are expressed in the ventral forebrain at the time of OL progenitor generation that can be activated by FGF receptor stimulation.

Coronal sections of normal E12.5 forebrains were immunolabelled with anti-pan-Erk1/2 (A). Negative controls did not receive the primary antibody (B). (C) Dorsal (d) or ventral (v) forebrains were separated and mechanically dissociated. The cell suspensions were incubated with FGF-2, in the absence or presence of the Fgfr inhibitor (PD173074, 100 nM), FGF-8, PD173074 alone or DMSO. Equal amounts of total cellular protein were analyzed by immunoblotting with anti-phospho-Erk1/2 or anti-pan Erk1/2. Note the positive immunostaining signal of Erk1/2 in the forebrain over the non-neural tissue (*) and its activation by FGF-2 and FGF-8 in both the dorsal and ventral forebrain. Both the basal and FGF-2 stimulated activity of Erk1/2 MAPK was abolished by PD173074. Scale bar, 100 um.
To examine the effect of Fgfr stimulation on Erk1/2 MAPK activation, dorsal and ventral forebrain cells were freshly dissociated separately and incubated with FGF-2 or FGF-8. These FGFs are normally expressed in the embryonic forebrains and are considered physiological ligands for the three Fgfrs (Ford-Perriss et al., 2001). Immunoblot analysis of the cells showed that these FGFs dramatically increased Erk1/2 MAPK phosphorylation over basal levels both in the dorsal and ventral region cells (Fig. 8C). Inhibition of Fgfr signaling by PD173074 completely abolished not only the FGF-induced Erk1/2 MAPK phosphorylation but importantly, the basal level as well. These data suggest that Fgfrs are clearly a major (if not the only) upstream regulator of Erk1/2 MAPK activity in the E12.5 forebrain.
We conclude that activation of the Erk1/2 MAPK pathway in the embryonic forebrain is very likely to play a primary role in the FGF-mediated induction of ventrally derived OLPs and perhaps of other lineages that emerge in a similar spatial and temporal manner.
DISCUSSION
By conditionally deleting Fgfr1–3 in different combinations from the embryonic forebrain, we demonstrate that differential loss of Fgfr1 and Fgfr2 but not Fgfr3 genes results in a partial to complete failure of ventrally-derived OLPs to develop, without a loss of Shh signaling, providing strong evidence for a direct role of Fgfr signaling in OLP generation. Furthermore, since OLP generation was strongly inhibited in vitro by attenuating either FGF or Shh signaling, we suggest that Fgfr cooperates with Shh to control OLP generation.
Dorsal forebrain and spinal cord neuroepithelial cells have the potential to generate OLPs under the influence of FGF-2 in vitro (Chandran et al., 2003; Gabey et al., 2003; Kessaris et al., 2004; Abematsu et al., 2006). However, the relevance of these studies in the context of the in vivo generation of OLPs has been questioned (Gabey et al., 2003). Furthermore, since the normal origin of the embryonically derived OLP population is the ventral and not the dorsal forebrain (Tekki-Kessaris et al., 2001; Nery et al., 2001; Spassky et al., 1998; Rowitch et al., 2004; Kessaris et al., 2008), these approaches, although informative about the potential of dorsal forebrain cells to generate OLPs, suffer from possible in vitro deregulatory effects and fail to directly address the role of FGF signaling in the normal generation of the embryonically derived OLP populations. Thus, the present study not only directly demonstrates the in vivo consequence of eliminating FGF signaling on OLP generation, but also reveals the differential signaling potential of the three FGF receptors.
Exposure of isolated OLPs to FGF-2 in vitro leads to continuous proliferation, predicting a prominent role of FGF signaling in OLP proliferation (McKinnon et al., 1990; Bansal, 2002; Fortin et al., 2005). However, contrary to this notion, the present study shows that in vivo, the loss of FGF signaling in Fgfr1−/− or Fgfr2−/− mutants does not affect OLP proliferation in the embryonic forebrain or in dissociated cultures derived from the embryonic mutant forebrains. Thus, the severe reduction of OLP population in the Fgfr1−/− and Fgfr2−/− mutants cannot be accounted for simply by inhibition of proliferation (or increased cell death) of specified cells. Further, a potential inhibition of the migration of cells from the germinal zone is unlikely to account for the reduced numbers of “scattered” OLPs observed in the mutants, since similar reduction was observed in cultures of E12.5 mutant forebrains and of normal brains grown in the presence of a chemical inhibitor of FGF receptors. We, therefore, favor the hypothesis that like Shh, FGF signaling generates ventral OLPs by directly promoting their specification. Consistent with the inductive role of FGF signaling in specifying ventral cells, FGF8-soaked beads ectopically induced ventral telencephalic cells (Kuschel et al., 2003); and FGF-2 infused into the ventricles induced OLPs in the neocortex and potentially in the ventral forebrain, judged by the increased numbers of OLPs in the cortical intermediate zone, the accepted migration route of ventrally-derived OLPs (Naruse et al., 2006). Thus, although FGF signaling effects proliferation of OLPs in vitro, it is not essential for the proliferation of specified OLPs in vivo, but is required for their induction from the neuroepithelial cells.
In addition to FGF, as shown here, it is well accepted that Shh induces the generation of OLPs from the ventral forebrain (Tekki-Kessaris et al., 2001; Alberta et al., 2001; Nery et al., 2001; Spassky et al., 2001; Rowitch et al., 2004; Fuccillo et al., 2004). This raises the question whether these molecules operate entirely independently or cooperatively, perhaps in a sequential manner. Our in vivo results, together with previous findings (Gutin et al., 2006), show that loss of Fgfr signaling in the mutant forebrains did not adversely effect Shh signaling, which suggests that attenuated OLP generation in these mice is not simply due to down-regulation of Shh signaling. To address this further, when we attenuated either Shh or Fgfr signaling in ventral forebrain cells by cyclopamine or PD173074 respectively, we found a strong inhibition of OLP induction by either of the inhibitors, suggesting that cooperation between FGF and Shh signaling must exist to generate the majority of ventrally-derived embryonic populations of OLPs. Although the mechanism is currently unclear, given our present data and previous studies showing that several Fgf genes are down-regulated in mice lacking Shh (Rash and Grove, 2007), and that Shh-induced OLP induction from the dorsal forebrain is blocked by attenuating FGF signaling in vitro (Kasseris et al., 2004), we favor the model that FGF functions downstream of Shh in the induction of ventrally-derived OLPs. Our results from mouse forebrain analysis are consistent with the studies in zebrafish hindbrain, where loss of FGF receptor signaling resulted in the loss of OLP but not of Shh (Esain et al., 2010). The authors also suggest cooperation between FGF and Shh; specifically, Shh signaling establishes a progenitor domain that is competent to express Olig2, and Fgfr signaling permits or promotes Olig2 gene transcription (Esain et al., 2010). Another model proposed by Kassaris et al. (2004) for cooperative signaling between FGF and Shh, specifically for the dorsal forebrain cells, is that FGF receptor signaling maintains a basal level of MAPK phosphorylation, which is essential for Olig2 expression and the OLP-inducing activities of Shh. Taken together, it appears that Shh activity is not sufficient to generate OLPs and depends on FGF signaling for the generation of the vast majority of ventrally derived OLPs.
What are the mechanisms by which FGF receptor activation leads to OLP generation? Fgfrs are known to signal via two major phosphorylation cascades: the Ras/MAPK and the phosphotidylinositol 3-kinase/AKT pathway (Turner and Grose, 2010). In vitro evidence suggests that Erk1/2 MAPK pathway is a major contributor leading to the induction of OLPs, at least in the dorsal forebrain and spinal cord, since inhibition of Erk1/2 MAPK by a chemical inhibitor attenuated FGF-2-mediated induction of Olig2 cells in cultures of dorsal spinal cord or neocortex (Chandran et al., 2003; Kessaris et al., 2004). Our data show that the embryonic ventral forebrain cells also express Erk1/2 MAPK at the time of OLP generation, which is active at a basal level and can be further activated by stimulation of the Fgfrs by their physiological ligands, FGF-2 and FGF-8. Importantly, abolition of the basal level of active Erk1/2 by Fgfr specific inhibitor suggests that Erk1/2 MAPK are exclusively controlled by Fgfr signaling in the E12.5 forebrain. Thus, it is plausible that activation of Erk1/2 MAPK downstream of Fgfrs plays a primary role in the FGF-mediated induction of OLPs from the embryonic ventral forebrain.
Previous studies showed that in the Fgfr1−/−;Fgfr2−/− double mutants, ventral precursor cells were lost and as a result interneurons failed to develop from the ventromedial telencephalon (Gutin et al., 2006), mimicking the phenotype of mice lacking Shh signaling (Ericson et al., 1995; Fuccillo et al., 2004; Xu et al., 2010). The present study shows that in the double mutants OLPs also failed to develop, as was shown for mice deficient in Shh signaling (Nery et al., 2001; Tekki-Kessaris et al., 2001; Alberta et al., 2001; Rowitch, 2004; Fuccillo et al., 2004), thereby providing strong genetic evidence that like Shh, generation of both cell types is under the regulation of the same molecules. However, in the single Fgfr1 and Fgfr2 mutants, where the ventral precursor cells are generated normally, some remarkable differences exist between interneuron and OLP generation. Specifically, in Fgfr1 single mutants the generation of both interneurons (Gutin et al., 2006) and OLPs (present study) was inhibited but to different extents; and in Fgfr2 single mutants generation of OLPs was strongly inhibited but interneurons developed normally. This suggests that the nature of FGF signaling required for neuron or OLP generation from the neuroepithelial precursors may be somewhat different. The difference can be qualitative, i.e., Fgfr2 may activate certain pathways conducive for OLPs but not neurons. This connection of Fgfr2 with the OL-lineage cells is also maintained in the adult brain, where Fgfr2 is localized primarily to OLs and myelin and not neurons (Miyake et al., 1996; Fortin et al., 2005). Another possible difference can be quantitative, where the generation of neurons and OLPs may depend on achieving different strengths of downstream signaling by the activated receptors. For example, it is possible that the strength or duration of Erk1/2 MAPK phosphorylation induced by Fgfr2 may be sufficient to promote OLP generation but not strong enough for interneuron generation, where as the Fgfr1 induced phosphorylation of these molecules may be robust enough to acquire a critical signaling threshold needed for the generation of both cell types. This suggests differences in the signaling potential of individual Fgfrs. Consistent with this notion, the differential level of Erk1/2 MAPK phosphorylation was observed following the activation of distinct Fgfrs in the OL-lineage cells (Fortin et al., 2005).
In summary, we provide novel evidence that Fgfr1 and Fgfr2 but not Fgfr3 have critical functions in the regulation of OLP generation from the embryonic mouse forebrain operating in cooperation with Shh.
Supplementary Material
Acknowledgments
We would like to thank Dr. D. Ornitz for providing the Fgfr3-null mice; Drs. D. Rowitch, W.D. Richardson, and K. Ikenaka for riboprobes; and Dr. W.B. Stallcup for the PDGFRa antibody. This work was supported by NIH Grant NS38878 and in part by grants from the National Multiple Sclerosis Society, RG 4087-A-3 and NIH, NS41078 to RB and NIH grant MH070596 (JH).
References
- Aboitiz F, Montiel J. Co-option of signaling mechanisms from neural induction to telencephalic patterning. Rev Neurosci. 2007;18:311–42. doi: 10.1515/revneuro.2007.18.3-4.311. [DOI] [PubMed] [Google Scholar]
- Abematsu M, Kagawa T, Fukuda S, Inoue T, Takebayashi H, Komiya S, Taga T. Basic fibroblast growth factor endows dorsal telencephalic neural progenitors with the ability to differentiate into oligodendrocytes but not gamma-aminobutyric acidergic neurons. J Neurosci Res. 2006;83:731–743. doi: 10.1002/jnr.20762. [DOI] [PubMed] [Google Scholar]
- Alberta JA, Park SK, Mora J, Yuk D, Pawlitzky I, Iannarelli P, Vartanian T, Stiles CD, Rowitch DH. Sonic hedgehog is required during an early phase of oligodendrocyte development in mammalian brain. Mol Cell Neurosci. 2001;18:434–41. doi: 10.1006/mcne.2001.1026. [DOI] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: Implications for demyelination and remyelination. Dev Neurosci. 2002;46:24–35. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfr alpha, Olig2 and Plp/dm20: implications for oligodendrocyte development. Dev Neurosci. 2003a;25:83–95. doi: 10.1159/000072258. [DOI] [PubMed] [Google Scholar]
- Bansal R, Magge S, Winkler S. Specific Inhibitor of FGF receptor signaling: FGF-2-mediated effects on proliferation, differentiation and MAPK activation are inhibited by PD173074 in oligodendrocyte-lineage cells. J Neurosci Res. 2003b;74:486–493. doi: 10.1002/jnr.10773. [DOI] [PubMed] [Google Scholar]
- Baas D, Barnstable CJ. HPC-7: A novel oligodendrocyte lineage protein, which appears prior to galactcerebroside. Glia. 1998;23:169–179. [PubMed] [Google Scholar]
- Cai J, Qi Y, Hu X, Tan M, Liu Z, Zhang J, Li Q, Sander M, Qiu M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron. 2005;45:41–53. doi: 10.1016/j.neuron.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Chandran S, Kato H, Gerreli D, Compston A, Svendsen CN, Allen ND. FGF-dependent generation of oligodendrocytes by a hedgehog-independent pathway. Development. 2003;130:6599–6609. doi: 10.1242/dev.00871. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Ericson J, Muhr J, Placzek M, Lints T, Jessell TM, Edlund T. Sonic hedgehog induces the differentiation of ventral forebrain neurons: a common signal for ventral patterning within the neural tube. Cell. 1995;81:747–756. doi: 10.1016/0092-8674(95)90536-7. [DOI] [PubMed] [Google Scholar]
- Esain V, Postlethwait JH, Charnay P, Ghislain J. FGF-receptor signalling controls neural cell diversity in the zebrafish hindbrain by regulating Olig2 and sox9. Development. 2010;137:33–42. doi: 10.1242/dev.038026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Jeffrey A, Wakeman DR, Kim SU, Snyder EY, de Vellis J. Culture system for rodent and human oligodendrocyte specification, lineage progression, and maturation. Curr Protoc Stem Cell Biol. 2009;Chapter 2 doi: 10.1002/9780470151808.sc02d04s10. [DOI] [PubMed] [Google Scholar]
- Ford-Perriss M, Abud H, Murphy M. Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol. 2001;28:493–503. doi: 10.1046/j.1440-1681.2001.03477.x. [DOI] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M, Rallu M, McMahon AP, Fishell G. Temporal requirement for hedgehog signaling in ventral telencephalic patterning. Development. 2004;131:5031–5040. doi: 10.1242/dev.01349. [DOI] [PubMed] [Google Scholar]
- Gabay L, Lowell S, Rubin LL, Anderson DJ. Deregulation of dorsoventral patterning by FGF confers trilineage differentiation capacity on CNS stem cells in vitro. Neuron. 2003;40:485–499. doi: 10.1016/s0896-6273(03)00637-8. [DOI] [PubMed] [Google Scholar]
- Gutin G, Fernandes M, Palazzolo L, Paek H, Yu K, Ornitz DM, McConnell SK, Hebert JM. FGF signalling generates ventral telencephalic cells independently of SHH. Development. 2006;133:2937–2946. doi: 10.1242/dev.02465. [DOI] [PubMed] [Google Scholar]
- Hébert JM, Lin M, Partanen J, Rossant J, McConnell SK. FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development. 2003;130:1101–1111. doi: 10.1242/dev.00334. [DOI] [PubMed] [Google Scholar]
- Hébert JM. Unraveling the molecular pathways that regulate early telencephalon development. Curr Top Dev Biol. 2005;69:17–37. doi: 10.1016/S0070-2153(05)69002-3. [DOI] [PubMed] [Google Scholar]
- Hébert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Pringle N, Richardson WD. Specification of CNS glia from neural stem cells in the embryonic neuroepithelium. Philos Trans R Soc Lond B Biol Sci. 2008;363:71–85. doi: 10.1098/rstb.2006.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Jamen F, Rubin L, Richardson WD. Cooperation between sonic hedgehog and fibroblast growth factor/MAPK signalling pathways in neocortical precursors. Development. 2004;131:1289–1298. doi: 10.1242/dev.01027. [DOI] [PubMed] [Google Scholar]
- Kaga Y, Shoemaker WJ, Furusho M, Bryant M, Rosenbluth J, Pfeiffer SE, Oh L, Rasband M, Lappe-Siefke C, Yu K, Ornitz DM, Nave KA, Bansal R. Mice with conditional inactivation of FGF receptor-2 signaling in oligodendrocytes have normal myelin but display dramatic hyperactivity when combined with Cnp1 inactivation. J Neurosci. 2006;26:12339–12350. doi: 10.1523/JNEUROSCI.3573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschel S, Ruther U, Theil T. A disrupted balance between Bmp/Wnt and Fgf signaling underlies the ventralization of the Gli3 mutant telencephalon. Dev Biol. 2003;260:484–495. doi: 10.1016/s0012-1606(03)00252-5. [DOI] [PubMed] [Google Scholar]
- Mason I. Initiation to endpoint:the multiple roles of fibroblast growth factors in neural development. Nature Rev Neurosci. 2007;8:583–596. doi: 10.1038/nrn2189. [DOI] [PubMed] [Google Scholar]
- Miyake A, Hattori Y, Ohta M, Itoh N. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor-2 and -3 mRNAs. J Neurosci Res. 1996;45:534–541. doi: 10.1002/(SICI)1097-4547(19960901)45:5<534::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Naruse M, Nakahira E, Miyata T, Hitoshi S, Ikenaka K, Bansal R. Induction of oligodendrocyte progenitors in dorsal forebrain by intraventricular microinjection of FGF-2. Dev Biol. 2006;297:262–273. doi: 10.1016/j.ydbio.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Nery S, Wichterle H, Fishell G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development. 2001;128:527–540. doi: 10.1242/dev.128.4.527. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- Qian X, Davis AA, Goderie SK, Temple S. FGF2 concentration regulates the generation of neurons and glia from multipotent cortical stem cells. Neuron. 1997;18:81–93. doi: 10.1016/s0896-6273(01)80048-9. [DOI] [PubMed] [Google Scholar]
- Rash BG, Grove EA. Patterning the dorsal telencephalon: a role for sonic hedgehog? J Neurosci. 2007;27:11595–603. doi: 10.1523/JNEUROSCI.3204-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH. Glial specification in the vertebrate neural tube. Nat Rev Neurosci. 2004;5:409–19. doi: 10.1038/nrn1389. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Kee WJ, Facci L, Macdonald G, Doherty P, Walsh FS. The FGFR1 inhibitor PD 173074 selectively and potently antagonizes FGF-2 neurotrophic and neurotropic effects. J Neurochem. 2000;75:1520–1527. doi: 10.1046/j.1471-4159.2000.0751520.x. [DOI] [PubMed] [Google Scholar]
- Spassky N, Goujet-Zalc C, Parmantier E, Olivier C, Martinez S, Ivanova A, Ikenaka K, Macklin W, Cerruti I, Zalc B, Thomas JL. Multiple restricted origin of oligodendrocytes. J Neurosci. 1998;18:8331–8343. doi: 10.1523/JNEUROSCI.18-20-08331.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Heydon K, Mangatal A, Jankovski A, Olivier C, Queraud-Lesaux F, Goujet-Zalc C, Thomas JL, Zalc B. Sonic hedgehog-dependent emergence of oligodendrocytes in the telencephalon: evidence for a source of oligodendrocytes in the olfactory bulb that is independent of PDGFRalpha signaling. Development. 2001;128:4993–5004. doi: 10.1242/dev.128.24.4993. [DOI] [PubMed] [Google Scholar]
- Tekki-Kessaris N, Woodruff R, Hall AC, Gaffield W, Kimura S, Stiles CD, Rowitch DH, Richardson WD. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development. 2001;128:2545–2554. doi: 10.1242/dev.128.13.2545. [DOI] [PubMed] [Google Scholar]
- Trotter J, Karram K, Nishiyama A. NG2 cells: Properties, progeny and origin. Brain Res Rev. 2010;63:72–82. doi: 10.1016/j.brainresrev.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to Cancer. Nature Neuroscience. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Xu Q, Guo L, Moore H, Waclaw RR, Campbell K, Anderson SA. Sonic hedgehog signaling confers ventral telencephalic progenitors with distinct cortical interneuron fates. Neuron. 2010;65:328–40. doi: 10.1016/j.neuron.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–57. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.