

Abstract
The visual system can adjust its sensitivity over a wide range of light intensities. Photoreceptors account for some of this adjustment, but there is evidence that postreceptoral processes also exist. To investigate the latter, we pharmacologically mimicked the effects of light stimulation on mouse On bipolar cells, thus avoiding confounding effects of receptoral mechanisms. Here, we report that cGMP selectively enhances responses to dim, but not bright, stimuli through a purely postsynaptic mechanism. This action of cGMP was completely blocked by inhibitors of cGMP-dependent kinase. We propose that cGMP-dependent kinase decreases coupling of the On bipolar cell glutamate receptor to the downstream cascade, thus amplifying small decreases in photoreceptor transmitter levels that would otherwise go undetected by the visual system.
Keywords: retina, On bipolar cell, G-protein-coupled receptor, cGK, mGluR6, PKG
Introduction
On bipolar cells are designed to detect the onset of light. They express an unusual sign-inverting receptor designated mGluR6 (Nakajima et al., 1993), which couples via Go (Vardi, 1998; Nawy, 1999) to a nonspecific cation channel. In darkness, when gluta-mate is released by photoreceptors, activation of the receptor leads to the closing of the cation-selective transduction channel, thereby hyperpolarizing the cell. Conversely, when synaptic glutamate is decreased during light, the transduction channel opens, and the cell depolarizes.
The human eye can distinguish light levels spanning 10 log units (Dowling, 1987) because of the exquisite sensitivity and large adaptive capacity of the retina. Many adaptive processes occur on the photoreceptor level, but electrophysiological and psychophysical studies have shown that, in addition, a significant portion of adjustment to illumination occurs at the network level.
The On bipolar cell relays signals from photoreceptors to ganglion cells, making it strategically positioned to modify visual input, and there is strong evidence that the On bipolar cell is involved in adjusting the sensitivity of the retina to light. The electroretinogram b-wave, which is thought to reflect mainly On bipolar cell activity (Miller and Dowling, 1970; Dong and Hare, 2000), is sensitive enough to signal the capture of single photons (Cone, 1963) and displays an absolute sensitivity far greater than that of photoreceptors (Witkovsky, 1980). In addition, changes in b-wave sensitivity occur at light levels too dim to adapt photoreceptors (Green and Powers, 1982; Frishman and Sieving, 1995). Pharmacological interruption of synaptic transmission suggests that these changes most likely occur within the On bipolar cell (Gurevich and Slaughter, 1993), and although the cellular mechanisms involved remain unknown, several research groups have found that mGluR6-mediated responses to glutamate and light are potentiated by intracellular addition of cGMP (Nawy and Jahr, 1990; Shiells and Falk, 1990, 2002; de la Villa et al., 1995), making it well suited as a modulator of sensitivity.
To directly investigate the role of cGMP within the On bipolar cell transduction pathway, without presynaptic and lateral interference from photoreceptors and horizontal cells, we developed a protocol for pharmacological simulation of light flashes in complete darkness. To mimic darkness, we bath applied saturating concentrations of an mGluR6 agonist, and simulated light flashes were elicited by controlled applications of a competitive antagonist of the receptor. In this way, we were able to vary the stimulus intensity while only affecting the On bipolar cell, thereby obtaining a readout of the purely postsynaptic processes involved in adjusting stimulus sensitivity.
We report that cGMP, rather than gate the transduction channel as previously proposed, potentiates On bipolar cell responses by activating cGMP-dependent kinase (cGK). Our findings suggest that activation of cGK uncouples mGluR6 from its transduction channel, thus reducing the ability of glutamate to keep the channel closed. This results in an enhanced ability of the On bipolar cell to sense small changes in synaptic glutamate levels, thereby increasing its sensitivity to changes in light that may otherwise go undetected.
Materials and Methods
Electrophysiology
Tissue preparation
Retinal slices from 4- to 6-week-old C57BL/6 mice (Charles River, Cambridge, MA) were prepared as described previously for salamander (Walters et al., 1998; Nawy, 1999). Briefly, mice were anesthetized with halothane (Sigma, St. Louis, MO), killed by cervical dislocation, and their eyes were removed and enucleated. Whole retinas were isolated and placed on a 0.65 μm cellulose acetate/nitrate membrane filter (Millipore, Bedford, MA), which was secured with vacuum grease to a glass slide adjacent to the recording chamber. Slices were then cut to a thickness of 100 μm using a tissue slicer (Stoelting, Wood Lane, IL), transferred to the recording chamber while remaining submerged, and viewed with an Axioskop (Zeiss, Thornwood, NY) equipped with a water-immersion 40× objective with Hoffman modulation contrast (Modulation Contrast, Greenvale, NY). Slices were bathed in Ames medium (Sigma), supplemented at all times with 4 μm l-AP-4 and 95% O2 and 5% CO2. The solution was perfused continuously through the recording chamber at a rate of ∼4 ml/min.
Solutions
The pipette solution was composed of (in mm) 108 K+gluconate, 20 TEA, 10 HEPES, 20 BAPTA, 4 MgATP, and 1 LiGTP, and the pH was adjusted to 7.4 with KOH. Alexa Fluor 488 was added to the pipette solution at a concentration of 14 μg/ml. All drugs, except S-nitroso-N-acetylpenicillamine (SNAP), were added in the recording pipette at the following concentrations (in μm): 500 and 1000 IBMX, 1 DT-3, 1 KT5823, 1000 cGMP, and 100 Sp-8-Br-PET-cGMPS. SNAP was dissolved in DMSO and added to the bath solution through a separate gravity perfusion system, with a final concentration of 500 μm (DMSO concentration, 0.05%). (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG; 600 μm) was delivered to the cell by applying positive pressure to a fine pipette (2–3 psi using Picospritzer III; Parker Instrumentation, Fairfield, NJ). All chemicals were obtained from Sigma, except CPPG and l-AP-4 (Tocris, Ballwin, MO), Alexa Fluor 488 (Molecular Probes, Eugene, OR), DT-3 (Biolog, Bremen, Germany), and KT5823, SNAP, and Sp-8-Br-PET-cGMPS (Calbiochem, La Jolla, CA). All drugs, except CPPG and l-AP-4, were aliquoted, stored at −20°C, and dissolved in pipette solution immediately before use.
Electrophysiology
Patch pipettes of resistance 7–9 MΩ were fabricated from borosilicate glass (World Precision Instruments, Sarasota, FL) using a two-stage vertical puller (Narishige, Sea Cliff, NY). Whole-cell recordings were obtained with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) and had input and series resistances of ∼1 GΩ and 10–19 MΩ, respectively. Cells were discarded if the series resistance exceeded 20 MΩ, the holding current changed suddenly, or if the holding current during the first application of agonist exceeded 150 pA at +50 mV. Holding potentials were corrected for the liquid junction potential, which was measured to be −10 mV with the K+ gluconate pipette solution. On bipolar cells were identified by their position in the slice, by their characteristic outward responses to CPPG at positive holding potentials, and by fluorescence imaging. Fluorescence imaging was performed using a CCD camera (COHU Electronics, San Diego, CA) and digitizing with a Sicon frame grabber. Data were acquired with Axograph software and the Digidata 1200 interface (Axon Instruments) and analyzed with Axograph and Kaleidagraph (Synergy Software, Reading, PA).
Statistics
Results are represented as the mean ±SEM. Significance was determined with the unpaired Student t test, set to 0.001, and is illustrated with asterisks in the figures.
Immuocytochemistry
Antibodies and chemicals
We used the following primary antibodies: PKC-MC5, mouse monoclonal, targeted against aa 292–317 within the hinge region of PKC; cGKIα/β (T-19), polyclonal, targeted against a 19 amino acid sequence within aa 600–700 of the C terminus; cGK1α (N-16), polyclonal, targeted against 16 amino acids within the first 100 amino acids of the N terminus; cGKIβ (E-20), polyclonal, epitope defined as 20 amino acids within aa 50–150 of the N terminus. All antibodies and blocking peptides were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). We used the following secondary antibodies: Alexa Fluor 488 donkey anti-mouse (Molecular Probes); Cy3 donkey anti-goat (Jackson Immunoresearch, West Grove, PA). All other chemicals were obtained from Sigma.
Immunolabeling
Eyeballs were fixed in 0.1% PBS with 4% paraformaldahyde for 2 hr at 4°C, rinsed in PBS overnight, and then infiltrated with 20% sucrose for 2–3 d. Eyecups were embedded in 7.5% gelatin with 15% sucrose, flash-frozen, and cryosectioned into 15 μm slices. Auto-flourescence was reduced by a 10 min infiltration with 50 mm NH4Cl, followed by a 15 min wash in 0.1 m PBS. Slices were then incubated for 1.5 hr in 0.1 m PBS containing 0.1% BSA, 0.1% Triton-X, and 10% natural donkey serum. The primary antibody (1:100) was applied and allowed to incubate overnight. For control staining, blocking peptides specific for each of the antibodies were included during incubation in the same ratios as corresponding antibodies. After 24 hr and a 15 min wash, slices were incubated for 1 hr with secondary antibodies (1:100 donkey anti-mouse Alexa 488, 1:300 donkey anti-goat Cy3). Flourescence was visualized using an Fluoview laser-scanning microscope (Olympus, Melville, NY) with Melles Giot ion laser power supply. Image acquisition and analysis was performed using Fluoview 500 (Olympus) and MetaMorph (Universal Imaging, Downingtown, PA) software.
Results
Light and dark conditions can be mimicked using mGluR6 analogs
We obtained whole-cell patch-clamp recordings from On bipolar cells in slices of mouse retina. All cells in this study were filled with Alexa Fluor 488 and visualized under fluorescent light for morphological identification. Data from both rod- and cone-driven On bipolar cells were combined because no differences relating to this study were found between the groups. Figure 1A shows an example of a rod bipolar cell (left) and a cone On bipolar cell (right) visualized with Alexa Fluor 488. Cells were voltage clamped to +50 mV and dialyzed with 20 mM BAPTA to minimize the influx of calcium and reduce response depression (Nawy, 2000) as well as voltage-dependent desensitization (Nawy, 2004). Slices were bathed continuously in a saturating concentration (4 μm) of the group III mGluR agonist l-AP-4. This treatment mimics darkness by activating the mGluR6 cascade, resulting in closure of the transduction channel and hyperpolarization of the cell, as would occur when glutamate is released by photoreceptors in the dark. Responses were elicited by puffs of CPPG, an antagonist of group III metabotropic glutamate receptors. CPPG simulates light stimulation by preventing mGluR6 agonist from binding, thereby opening the mGluR6-regulated transduction channel. A concentration of 600 μm is sufficient to fully antagonize bath l-AP-4 and turn on the maximum transduction current. However, the volumes of solution ejected by these brief puffs were probably diluted quickly, and as a result, only a fraction of the current was activated. Brief picospritzer puffs of CPPG are referred to as simulated flashes (sim-flashes). Puffs of saline alone produced smaller responses than CPPG, presumably because inclusion of the antagonist in the puff solution decreased rebinding of bath-applied l-AP-4 (data not shown). Applications longer than 1500 msec did not further increase the size of the response, suggesting that the concentration of CPPG reached saturating levels and therefore was sufficient to fully compete off the agonist.
Figure 1.
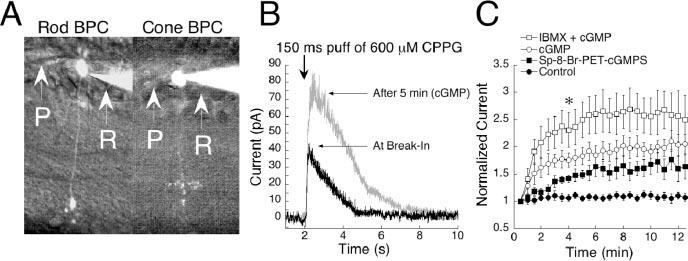
cGMP potentiates On bipolar cell responses in mouse. A, Light microscope images of a rod bipolar cell (left) and a cone On bipolar cell (right) filled with Alexa Fluor 488 and visualized with fluorescence. The two cell types can be distinguished by the position of their terminals in the inner plexiform layer. P, Puffer pipette; R, Recording pipette. B, Raw traces from a cell filled with 1 mm cGMP and stimulated with a 150 msec sim-flash at the 2 sec mark. As cGMP diffused into the cell, the amplitude was potentiated by approximately twofold. C, During 15 min of recording, cells dialyzed with cGMP potentiated by 180% (n = 12). Responses of control cells remained stable in amplitude throughout the session (n = 11). When IBMX was included in the pipette together with cGMP (n = 7), responses potentiated more than with cGMP alone. Inclusion in the pipette of the non-hydrolyzable cGMP analog sp-8-Br-PET-cGMPS (n = 3) caused an amplification of 60%. Data from each condition were individually normalized to the initial response, and the results were averaged. The plot represents the mean ± SEM for each time point. *p < 0.001, significance at 4 min for all conditions compared with baseline.
cGMP potentiates mouse On bipolar cell responses
Amplification of On bipolar cell responses by cGMP has been reported in salamander (Nawy and Jahr, 1990), dogfish (Shiells and Falk, 1990), and cat (de la Villa et al., 1995). In this study, amplification was also observed in mouse. When 1 mm cGMP was added to the recording pipette and cells were stimulated every 30 sec with 150 msec sim-flashes, which open ∼50% of the transduction channels, responses were enhanced by twofold within 15 min of recording (Fig. 1C, open circles). Control responses remained stable throughout the experimental session (Fig. 1C, closed circles). Figure 1B shows superimposed examples of raw traces from an On bipolar cell before (at break-in) and after (at 5 min) dialysis with cGMP.
Hydrolysis of cGMP is not necessary for gating of the transduction channel
It has been hypothesized that the mGluR6-coupled transduction channel operates in a manner similar to that of photoreceptors, in which cGMP gates the channel open, and hydrolysis through phosphodiesterases (PDEs) is required for channel closure (Nawy and Jahr, 1990; Shiells and Falk, 1990). If this is true, the addition of a nonhydrolizable analog of cGMP, or inhibition of endogenous PDEs, should increase channel opening but prevent closure of the channel by PDE activity, resulting in diminished response amplitudes. This hypothesis has previously been investigated in salamander (Nawy, 1999), in which it was reported that the addition of either constitutively active cGMP or IBMX, a broad spectrum PDE inhibitor, actually increased amplitudes of On bipolar cell responses. To investigate the possibility of a cGMP/PDE mechanism for gating the channel in mouse, we dialyzed cells with 100 μm Sp-8-Br-PET-cGMPS, a nonhydrolyzable analog of cGMP. This potentiated responses by an average of 60% compared with control, suggesting that hydrolysis of cGMP is not necessary for channel closure (Fig. 1 C, closed squares). We next inhibited endogenous PDEs by the addition of 500 μm or 1 mm IBMX and cGMP, to the recording pipette. IBMX and cGMP produced a 60% increase in response amplitudes compared with cGMP alone (Fig. 1 C, open squares), suggesting that On bipolar cells contain endogenous PDEs that continuously hydrolyze cGMP, but that PDEs are not directly involved in the transduction pathway. The finding that IBMX and nonhydrolizable analogs of cGMP do not interfere with cGMP-dependent potentiation rules out cGMP hydrolysis as an obligatory step for channel closure.
Phosphorylation by cGK is necessary for cGMP enhancement of On bipolar cell responses
We next investigated whether responses may be amplified through activation of cGK, a serine/threonine kinase specifically activated by cGMP. There are three known isoforms of cGK: splice variants cGK1α and cGK1β (Wernet et al., 1989), as well as cGKII (Uhler, 1993). To block the action of all isoforms, we included the broad spectrum cGK antagonist KT5823 (1 μm) in the pipette solution together with cGMP. Under these conditions, amplitudes initially increased by an average of 15% during the first 2 min of recording, then steadily decreased until, after 15 min, the average amplitude was 45% of the size observed at break-in (Fig. 2A). The initial potentiation of responses is presumably attributable to activation of cGK before the effect of the antagonist can be observed. This result was replicated with DT-3 (2.5 μm), a highly specific peptide inhibitor of the cGK1α isoform. Within the first 3 min of recording, DT-3 caused a 30% amplification of peak amplitudes, similar to the early amplitude increase observed with KT5823. After this initial potentiation, the amplitudes depressed until, after 15 min of recording, they reached levels equivalent to those at break-in (Fig. 2B). The observation that inhibitors of cGK abolish amplification of sim-flashes implies that activation of this kinase is a necessary step in cGMP-induced potentiation.
Figure 2.
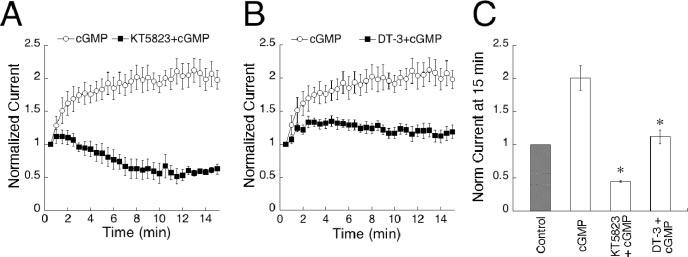
cGMP potentiates On bipolar cell responses through a pathway involving phosphorylation by cGK. A, Results from cells dialyzed with KT5823 and cGMP (n =5) or cGMP alone (n = 12) are plotted over 15 min. Inclusion of KT5823 in the pipette abolished potentiation of responses to a 150 msec sim-flash and induced a depression. B, The cGK inhibitory peptide DT-3 (n = 10) prevented potentiation of cGMP dialyzed cells when included in the internal solution. C, Summary of the results shown in A and B. Data points at 14–15 min of recording were averaged and are plotted for each condition. *p < 0.001, significance compared with cGMP.
On bipolar cells express both isoforms of cGK1
Expression of all three cGK isoforms has been reported in the retina (Gamm et al., 2000), including the outer plexiform layer. To verify this, and to specifically characterize cGK expression in On bipolar cells, we performed immunocytochemistry in mouse retinal slices. Immunolabeling for cGK1α was dense in the outer plexiform layer and the inner nuclear layer where On bipolar cells are situated (Fig. 3A, left). Staining was also present in the photoreceptor layer and the ganglion cell layer and was somewhat weaker in the outer nuclear layer. Staining for cGK1β showed similar intensity in the outer plexiform layer and inner nuclear layer but was also evident in the inner plexiform layer (Fig. 3B, left). Preabsorption controls for the two antibodies eliminated the staining (Fig. 3A,B, right). To verify that On bipolar cells express cGK1, we stained for cGK1α/β together with an antibody against PKC, a marker for rod-driven On bipolar cells (Negishi et al., 1988; Greferath et al., 1990). We found that 86% of PKC-positive rod bipolar cells expressed cGKIα or cGKIβ (Fig. 3C). PKC-positive On bipolar cells (red) show colocalization with cGK1 (green) in dendrites, somas, along the axon, and in the terminals. High-resolution confocal imaging of 0.5-μm-thick sections immunolabeled with antibodies against the separate isoforms of cGK1 revealed staining in the plasma membrane, the cytosol, and within and around the dendritic arbors of the On bipolar cells, as well as generally throughout the outer plexiform layer (Fig. 3D, cGK1α, 3E cGK1β). Neighboring cells in the rod bipolar layer also stained positive for cGK1. These cells displayed typical On bipolar cell morphology and may belong to other classes of On bipolar cells (Tsukamoto et al., 2001; Ghosh et al., 2004). Our results were similar to those of Gamm et al. (2000), indicating that cGK1 is expressed in most cell types in the retina, suggesting a global function for this kinase in regulating light responses. We conclude that cGK1α and cGK1β are both expressed in On bipolar cells, and in light of the electrophysiological data showing abolished potentiation by cGMP in the presence of DT-3, we believe that cGK1α is the primary isoform mediating cGMP-dependent potentiation of On bipolar cell responses.
Figure 3.
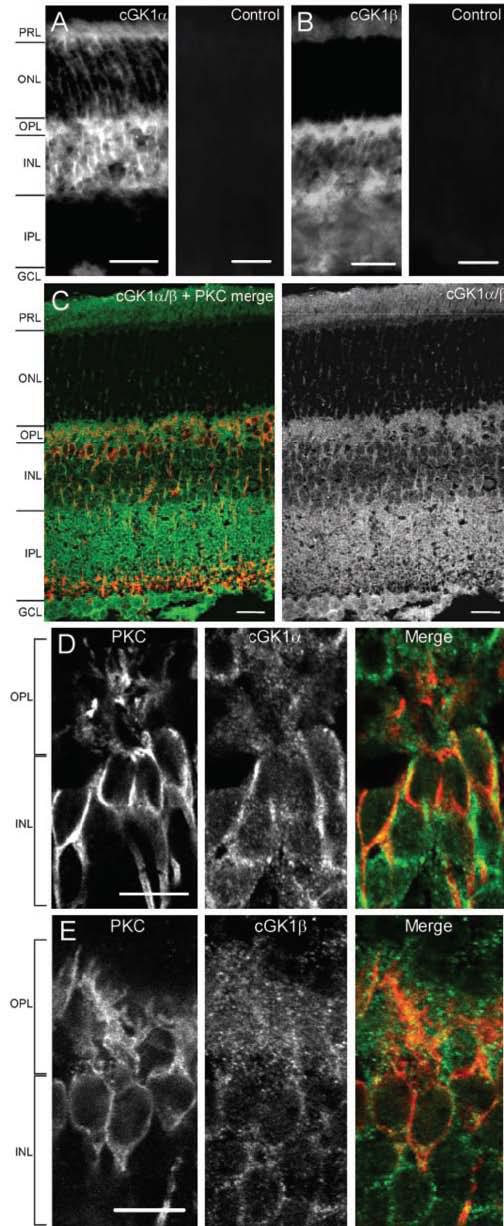
Immunocytochemistry showing the localization of cGK1 in PKC-positive On bipolar cells. PRL, Photoreceptor layer; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. A, Left, Expression of cGK1α in the layers of a 15-μm-thick retinal section. Right, Preincubation with an antibody-specific peptide blocks cGK1α staining. Scale bars, 25 μm. B, Left, Staining pattern of cGK1β in a 15-μm-thick retinal section. cGK1β shows an expression pattern similar to cGK1α but is also present in the inner plexiform layer. Right, Preincubation with an antibody-specific peptide blocks cGK1β staining. Scale bars, 25 μm. C, Left, Immunocytochemistry in a 15-μm-thick section of mouse retina, visualized with confocal microscopy. cGK1α or -β (green) are expressed in 86% of PKC-positive cells (red) (n = 71). Right, cGK1α/β, single channel. Staining is evident in the outer plexiform layer and in the inner nuclear layer, as well as in the inner plexiform layer, the ganglion cell layer, and the photoreceptor layer. Scale bars, 20 μm. D, Confocal imaging of On bipolar cells in a 0.5 μm section of mouse retina. Cells are labeled for PKC (left), cGK1α (middle), and merged (right; PKC, red; cGK1α, green). cGK1α is expressed in the plasma membrane, the cytosol of the cell body, in the immediate dendritic area, and along the axon. Scale bar, 10μm. E, Confocal imaging of a 0.5 μm section. cGK1β is expressed in the same compartments as cGKα. Scale bar, 10 μm.
Activation of endogenous guanylyl cyclase by SNAP potentiates On bipolar cell responses
Most studies involving cGMP modulation of On bipolar cell responses have used exogenous cGMP, although modulation of dogfish On bipolar cell responses by nitroprusside has been reported previously (Shiells and Falk, 1992). On bipolar cells express a nitric oxide (NO)-sensitive form of guanylyl cyclase (Ahmad and Barnstable, 1993; Haberecht et al., 1998; Spreca et al., 1999), and there is evidence that NO, acting as a messenger molecule in the retina (Goldstein et al., 1996; Cudeiro and Rivadulla, 1999), can increase cGMP levels in On bipolar cells (Gotzes et al., 1998; Baldridge and Fischer, 2001). We examined the effect of endogenous cGMP on On bipolar cell responses by adding SNAP, an agonist of NO-activated soluble guanylyl cyclase (sGC), to the external bath solution. SNAP application potentiated responses to sim-flashes by 60% (Fig. 4A), indicating that endogenous production of cGMP can be sufficient to potentiate On bipolar cell responses. Figure 4B shows examples of raw traces obtained before and during bath application of SNAP. The potentiation was reversible within minutes of washout, with a decrease in amplitude to near control levels (12% above baseline), suggesting a high rate of endogenous PDE activity. In cases in which application and washout of SNAP had not lasted for >20 min, a second application of SNAP could once again produce an enhancement of the response (data not shown).
Figure 4.
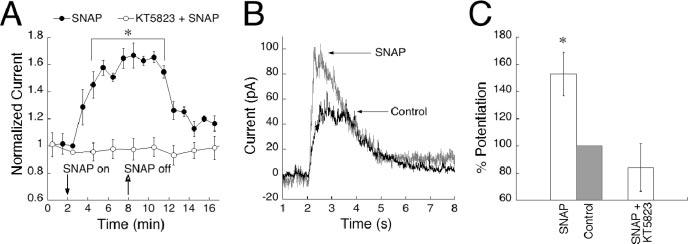
Bath application of the NO analog SNAP results in a reversible, KT5823-sensitive potentiation of On bipolar cell responses. A, On bipolar cells with stabilized responses were bathed in SNAP (closed symbols) for 6 min (time on and off are indicated by arrows) and stimulated with 150 msec sim-flashes every 30 sec (n=7). Data points were binned into 1 min intervals, and averages were plotted over 16 min. Responses started to potentiate 1 min after SNAP application and lasted for ∼3 min after removal (p < 0.001 at 4 min, compared with baseline and KT5823). The amplification was reversible, returning to amplitudes between 10 and 20% above baseline. The addition of KT5823 to the recording pipette (n = 7) completely blocked the SNAP-induced potentiation (open symbols). B, Example of single-cell traces illustrating the potentiating effect of SNAP compared with a control trace recorded before SNAP was applied. C, Data summarized in histogram form. The bar labeled “Control” represents the normalized response after SNAP has been washed out.
SNAP-induced potentiation was abolished by inclusion of KT5823 in the recording pipette, indicating that amplification of responses by SNAP acts via the same cGK pathway as exogenously applied cGMP (Fig. 4A). Figure 4C shows a histogram summary of the percentage of potentiation caused by SNAP and the blocking effect of KT5823. These results suggest that NO activation of sGC has the capacity to produce sufficient endogenous levels of cGMP to potentiate responses through cGK and that a physiological pathway for modulation of the transduction cascade is present in On bipolar cells.
cGMP reduces efficacy of the mGluR6 transduction cascade
cGMP-mediated amplification of sim-flash responses is not equal for all flash strengths. We applied a paradigm of interleaved sim-flashes lasting 15, 150, and 1500 msec. Briefer sim-flashes displace a smaller amount of l-AP-4 from the receptor, opening a submaximal fraction of channels, as would occur during dim light flashes, whereas longer sim-flashes mimic brighter light by displacing more of the agonist. When 1 mm cGMP was included in the pipette, responses to brief sim-flashes (15 and 150 msec duration) potentiated greatly, whereas responses elicited by the 1500 msec sim-flash remained close to their initial size. Figure 5A shows raw traces from a cell subjected to the three sim-flash paradigm, recorded before (left) and after (right) dialysis of cGMP. Because the long sim-flash caused only marginal potentiation, the plots have been scaled to the largest amplitudes to clearly illustrate the potentiation of the smaller responses. In Figure 5B, responses have been divided into groups according to flash strength and plotted versus time to demonstrate the difference in cGMP potentiation between the sim-flash durations. After 15 min of recording, responses to the briefest sim-flash were amplified by 308.01 ± 13.1%, twofold that of responses to the medium sim-flash, which potentiated by 140.78 ± 5.1%. The longest sim-flashes remained close to their initial amplitude with an average amplification of 26.14 ± 2.48%.
Figure 5.
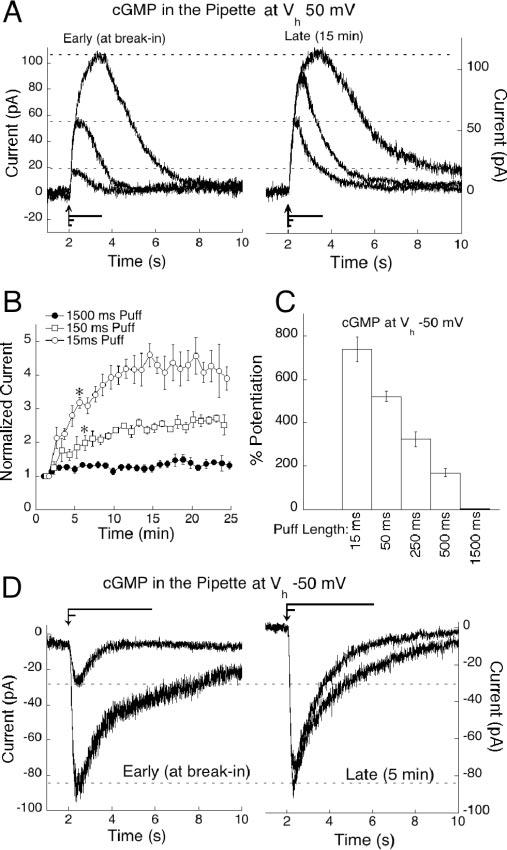
cGMP preferentially potentiates small On bipolar cell responses. A, On bipolar cells were stimulated by interleaved sim-flashes of 1500, 150, and 15 msec. cGMP potentiation of each stimulation is illustrated by raw traces from an On bipolar cell at break-in (left) and after 15 min (right). Responses have been scaled to the largest amplitude. Flash lengths are represented by lines positioned above the x-axis, with the arrow indicating stimulus onset. B, Responses from six cells dialyzed with cGMP were sorted into groups corresponding to sim-flash duration, pooled and plotted over 25 min. cGMP amplified responses differentially according to flash strength (p < 0.001 at 6 min for 150 msec sim-flashes compared with 1500 msec sim-flashes; p < 0.001 at 6 min for 15 msec sim-flashes compared with 150 msec sim-flashes). C, Histogram representing the average potentiation for each of five sim-flash durations from six cells voltage clamped at −50 mV. Amplitudes at 5 min were normalized to the amplitude at break-in and plotted as percentage of potentiation for each puff length. D, Size-dependent cGMP potentiation at Vh −50 mV illustrated by raw traces from an On bipolar cell stimulated with a 50 and a 1500 msec sim-flash, at break-in (left) and after 4 min (right). Puff lengths are represented by lines positioned above the traces, with the arrow indicating stimulus onset.
Amplitude-dependent potentiation was even more dramatic at physiological potentials. To avoid the use-dependent depression at negative potentials, a series of five sim-flashes ranging from 15 to 1500 msec were applied only twice: at break-in and again after 4 min, to allow cGMP to diffuse into the cell. Examples of raw traces from a cell subjected to this paradigm can be seen in Figure 5D. The average percentage of potentiation for each puff length at a holding potential of −50 mV is plotted in Figure 5C. Thus, cGMP also preferentially amplifies dim sim-flashes at physiologically relevant membrane potentials.
To further investigate the relationship between amplitude and cGMP-induced potentiation, a more complete dose–response function was obtained by applying a series of stimulations lasting 5, 15, 150, 1500, and 5000 msec. The two longest applications resulted in similar response amplitudes, suggesting that they delivered enough CPPG to completely displace l-AP-4, whereas the briefest application resulted in responses that were barely discernable above the noise. For the dose–response analysis, we obtained a series of responses at break-in and after 5 min to allow for dialysis of cGMP into the cell. Results were plotted on a semi-log scale and fitted with the Hill equation to obtain a PL50 (the puff length required to reach 50% of response maximum) value for CPPG in the absence and presence of cGMP. At break-in, a puff length of 85.6 msec was required to produce a half-maximal response in control cells that were not dialyzed with cGMP (Fig. 6A), and this value did not change significantly after 5 min (77.0 msec). In contrast, dialysis with cGMP caused a dramatic leftward shift of the dose–response function, decreasing the PL50 by fourfold from 88.9 msec at break-in to 20.3 msec after 5 min (Fig. 6B).
Figure 6.
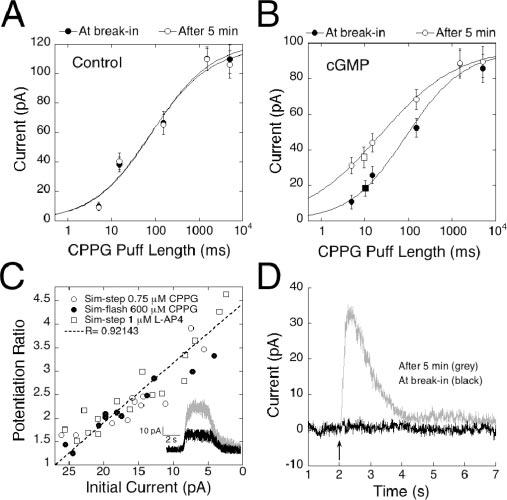
cGMP induces a shift in efficacy of the mGluR6 signal transduction cascade. A, Plot of the mean responses to a series of five sim-flashes/sim-steps of 5, 15, 150, 1500, and 5000 msec. Dose–response series from control cells voltage clamped at +50 mV were obtained at break-in and after 5 min of recording (n = 4). Results are plotted on a semi-log scale. Data were fitted with the equation 1/(1 + (P/PL50 ))n, where P is the puff length, PL50 is the length of the puff producing a half-maximal response, and n is the Hill coefficient. The PL50 value was 85.67 msec at break-in and 77.02 after 5 min. B, Dose–response series obtained immediately after break-in and 5 min later from cells dialyzed with cGMP and voltage clamped to +50 mV (n = 4). The PL50 value decreased by fourfold from 88.94 msec at break-in to 20.27 msec after 5 min. Superimposed on the intensity response curve are data points representing the average amplitudes of dim sim-steps (n = 8). Steady-state responses obtained with a 5 sec puff of 1 μm l-AP-4 or 0.75 μm CPPG, before (filled square) and after (open square) dialysis with cGMP, indicate that responses reaching steady state potentiate to the same degree as sim-flash responses. C, Scatter plot illustrating the relationship between initial amplitude and amount of potentiation for responses smaller than 25 pA. Cells were dialyzed with cGMP, and responses wereelicitedwitha5or15msecCPPGpuff(closedcircles; n=8). To simulate steps of dim light, cells were stimulated with a 5 sec puff of either 0.75 μm CPPG (open circles; n = 8) or 1 μm l-AP-4 (open squares; n = 9). The potentiation ratio, Ap /AI (I, initial; P, potentiation) is plotted on the y-axis, whereas the x-axis corresponds to AI. cGMP potentiated responses to dim sim-steps and sim-flashes equally. Inset, Raw traces at break-in (black) and after 5 min (gray) from a cell dialyzed with 1 mm cGMP and stimulated with a dim sim-flash produced by a 5 sec puff of 1 μm l-AP-4 given at the 2 sec mark. D, An average of raw traces illustrating that cGMP can increase the functional intensity of a stimulus. Responses from five cells, in which a 15 msec puff initially caused no detectable response, were averaged at break-in (black trace) and after 5 min (gray). Dialysis with cGMP either shifted the threshold for response initiation or potentiated an existing, undetectable response, allowing a 30 pA current to rise out of the baseline.
Sim-flashes were too brief to allow responses to reach steady state. We therefore considered the possibility that cGMP potentiated sim-flash responses by accelerating the channel opening rate. This seems unlikely for two reasons. First, the rising phases of pre- and post-potentiated responses appeared virtually identical. Second, we mimicked dim light steps (sim-steps) with a 5 sec puff of solution containing a low concentration of CPPG (0.75 μm) or a nonsaturating concentration of l-AP-4 (1 μm). These sim-steps elicited responses that reached steady state but were submaximal (Fig. 6C, inset). The mean amplitude of the response to sim-steps obtained at break-in is indicated by the filled square in Figure 6B, which has been positioned according to size on the y-axis. Note that the size of the averaged sim-step response after 5 min of recording (Fig. 6B, open square) is in good agreement with the size predicted from analysis of the sim-flash responses (Fig. 6B, open circles). Thus, cGMP potentiates steady-state and flash responses to the same degree, and its effects cannot be explained by acceleration of the rising phase of the response. To further verify that short sim-flashes amplified within the same size-dependent range as dim sim-steps, which reach steady state, we plotted the potentiation ratio of all cells with responses <25 pA. The y-axis in Figure 6C represents the potentiation ratio, AP/AI (A, amplitude; P, potentiation; I, initial), whereas the x-axis corresponds to AI. Regardless of whether the puff was generated by a brief puff of CPPG or a long application of a low concentration of l-AP-4 or CPPG, all responses fell along a straight line, suggesting that the primary parameter determining the amount of potentiation was the amplitude of the initial response.
The lateral shift in the puff length–response curve, without a change in maximum amplitude, suggests that cGMP does not change the number of available channels or substantially alter single-channel properties such as conductance. Instead, the affinity shift indicates that cGMP, by activation cGK, decreases the ability of the agonist to close the mGluR6-coupled transduction channel. The inability to close the channel in the presence of agonist changes the apparent level of stimulus intensity, allowing small decreases in agonist to have larger effects. Physiologically, an increase in intracellular cGMP may allow the On bipolar cell to respond to small changes in illumination across a large spectrum of ambient light levels. Furthermore, it may permit for detection of events hidden in the baseline noise. Figure 6D shows the averaged traces of five cells subjected to subthreshold sim-flashes, before and after cGMP dialyzed into the cell. Initially, responses could not be detected, but after 4 min, a robust response developed as a result of cGMP.
Discussion
In this study, we used a technique to pharmacologically mimic synaptic transmission from photoreceptors to On bipolar cells to study purely postsynaptic contributions to sensitivity adjustments within the On bipolar cell. Photoreceptors release gluta-mate continuously in darkness, and we simulated this by adding a saturating concentration of the mGluR6 agonist l-AP-4 to the bath solution. To simulate the synaptic action of dim light flashes, we displaced the agonist locally by applying brief puffs (sim-flashes) of the mGluR6 antagonist CPPG. Lengthening the flash resulted in the opening of more cation channels, analogous to brighter flashes, which suppress a greater fraction of transmitter release. Lengthening the puff still further did not increase the size of the cation current, suggesting that we were able to completely antagonize bath L-AP-4 and open all available postsynaptic channels.
Currents elicited by sim-flashes were markedly potentiated by cGMP. By measuring the effect of cGMP on a series of sim-flash intensities, we found that cGMP selectively potentiated responses to brief stimuli but had little or no effect on saturating stimuli that were long enough to displace nearly all of the agonist. Functionally, this is equivalent to selective amplification by cGMP of responses to dim, but not bright, light stimulation. A selective enhancement of responses to dim light by cGMP has previously been reported in the dogfish retina (Shiells and Falk, 2002).
The observed potentiating effects of cGMP could be fully accounted for by the activation of cGK. Taken together with the evidence that hydrolysis of cGMP is not an obligatory step in channel function, this suggests that cGK is the main effector of cGMP. The cGK inhibitors KT5823 and DT-3 effectively blocked amplitude potentiation in response to both exogenously administered cGMP and to application of SNAP. The cGK antagonist DT-3 is selective for the cGK1α isoform, and immunocytochemical localization by confocal microscopy revealed that cGK1α was the most prevalent isoform expressed in rod bipolar cells, closely followed by cGK1β. We conclude that cGMP acts via cGK1 to modulate the mGluR6 cascade and that the α splice variant is the most likely effector because pharmacologically blocking this isoform abolishes potentiation.
Mechanistically, the potentiation of responses to simulated puffs causing submaximal channel opening implies that cGMP reduces the ability of the agonist to close the channel, thereby decreasing the efficacy of the mGluR6 pathway. One way this could be achieved is if cGMP decreases functional coupling of the receptor to its G-protein. There is abundant evidence that the interactions of G-protein-coupled receptors with their target G-proteins are strongly regulated by phosphorylation of well defined sites within the intracellular loops and the C terminal of the receptors (Dale et al., 2002). A typical cGK phosphorylation site, RKRS, is present in the C terminus of mGluR6 (National Center for Biotechnology Information accession number NM_173372). Furthermore, several variations of the standard cGK consensus sequence, RKXS/T (Kennelly and Krebs, 1991), have been identified as important for regulation of signaling in various systems (Yuasa et al., 2001; Zhou et al., 2001; He and Weber, 2003) and in the modulation of several receptors (Wang and Robinson, 1997), including modulation of group III mGluRs (Sorensen et al., 2002). Although it is tempting to speculate that cGK1 phosphorylation of the mGluR6 terminal might promote receptor uncoupling, it could also lower the affinity of the agonist for the receptor. Alternatively, the target for cGK1 phosphorylation could be the channel (Kwan et al., 2004), in which case phosphorylation would make channel closure more difficult after binding an effector.
In the intact retina, changes in cGMP levels are most likely mediated by NO. Several lines of evidence suggest that NO is an important modulator in the retina. For example, NO has been implicated in closing gap junctions between horizontal cells (DeVries and Schwartz, 1989), and between amacrine and cone bipolar cells (Mills and Massey, 1995)]. It has also been shown to modulate a variety of channels and receptors, including Ca2+ channels (Hirooka et al., 2000), GABAA receptors (Wexler et al., 1998), and AMPA/kainite receptors (McMahon and Ponomareva, 1996; McMahon and Schmidt, 1999). On bipolar cells express a NO-activated guanylyl cyclase (Ahmad and Barnstable, 1993; Haberecht et al., 1998; Spreca et al., 1999), and stimulation with NO upregulates cGMP levels in On bipolar cells (Koistinaho et al., 1993; Gotzes et al., 1998; Baldridge and Fischer, 2001). Similar to findings by Shiells and Falk (1992), we observed potentiation of sim-flash responses by SNAP, consistent with the idea that NO analogs trigger sufficient production of cGMP to stimulate cGK. Furthermore, we found this potentiation to be readily reversible, suggesting that On bipolar cells contain an endogenous PDE capable of restricting the duration of the signal.
Production of NO may occur in several retinal cell types. Neuronal and epithelial forms of NO synthase (cNOS) are expressed in amacrine and ganglion cells of virtually every species examined. In mammals, cNOS is also expressed in bipolar cells, and in some species in horizontal cells and photoreceptor outer segments as well (Koistinaho et al., 1993; Haberecht et al., 1998; Kim et al., 2000). Importantly, there is evidence that NO release is regulated by adaptation state (Zemel et al., 1996; Neal et al., 1998; Wexler et al., 1998). One group, using NADPH staining as a measure of NO production, found that NO was preferentially produced by horizontal cells in dark-adapted retinas and by amacrine cells during light adaptation (McMahon and Ponomareva, 1996). These studies provide support for the hypothesis that NO contributes to postsynaptic sensitivity of On bipolar cells in response to changes in ambient light conditions. However, a more specific model addressing how the mGluR6 cascade is affected by the adaptation state requires additional information about NO levels in the outer retina in response to ambient light intensity.
We propose that activation of the NO–cGMP–cGK1 pathway in mouse On bipolar cells decreases the coupling strength of mGluR6 to downstream elements in the transduction cascade. Evidence suggests that the mGluR6 pathway is saturated in darkness because transmitter is sufficient to close all of the transduction channels (Sampath and Rieke, 2004). In this model of postsynaptic saturation, small synaptic events fail to lower transmitter levels sufficiently to relieve saturation, and there is no increase in channel opening. Thus, the mGluR6 cascade imposes an intrinsic threshold on the size of detectable events. A decrease in mGluR6 coupling caused by cGK1 may constitute a mechanism for shifting the threshold to allow small, but relevant, events to be detected. In support of this idea, we observed that in cells in which a brief sim-flash initially failed to cause a detectable event, dialysis with cGMP facilitated the rise of distinct responses from the noise (Fig. 6D). Presumably, the unbinding of the agonist resulting from these brief sim-flashes was sufficient to open channels only after cGMP reduced the effective coupling of mGluR6 to closure of the transduction channel. The ability of cGMP to elevate subthreshold events to threshold may be viewed as a special case of a more general mechanism of signal amplification, because it also potentiated responses that were initially well above threshold.
Acknowledgments
This work was supported by the National Eye Institute. We thank Reed Carroll for helpful discussions and Karen Cusato for advice on immunocytochemistry.
References
- Ahmad I, Barnstable CJ. Differential laminar expression of particulate and soluble guanylate cyclase genes in rat retina. Exp Eye Res. 1993;56:51–62. doi: 10.1006/exer.1993.1008. [DOI] [PubMed] [Google Scholar]
- Baldridge WH, Fischer AJ. Nitric oxide donor stimulated increase of cyclic GMP in the goldfish retina. Vis Neurosci. 2001;18:849–856. [PubMed] [Google Scholar]
- Cone RA. Quantum relations of the rat electroretinogram. J Gen Physiol. 1963;46:1267–1286. doi: 10.1085/jgp.46.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudeiro J, Rivadulla C. Sight and insight—on the physiological role of nitric oxide in the visual system. Trends Neurosci. 1999;22:109–116. doi: 10.1016/s0166-2236(98)01299-5. [DOI] [PubMed] [Google Scholar]
- Dale LB, Babwah AV, Ferguson SS. Mechanisms of metabotropic glutamate receptor desensitization: role in the patterning of effector enzyme activation. Neurochem Int. 2002;41:319–326. doi: 10.1016/s0197-0186(02)00073-6. [DOI] [PubMed] [Google Scholar]
- de la Villa P, Kurahashi T, Kaneko A. l-Glutamate-induced responses and cGMP-activated channels in three subtypes of retinal bipolar cells dissociated from the cat. J Neurosci. 1995;15:3571–3582. doi: 10.1523/JNEUROSCI.15-05-03571.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH, Schwartz EA. Modulation of an electrical synapse between solitary pairs of catfish horizontal cells by dopamine and second messengers. J Physiol (Lond) 1989;414:351–375. doi: 10.1113/jphysiol.1989.sp017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CJ, Hare WA. Contribution to the kinetics and amplitude of the electroretinogram b-wave by third-order retinal neurons in the rabbit retina. Vision Res. 2000;40:579–589. doi: 10.1016/s0042-6989(99)00203-5. [DOI] [PubMed] [Google Scholar]
- Dowling JE. The retina, an approachable part of the brain. Belknap; Cambridge, MA: 1987. [Google Scholar]
- Frishman LJ, Sieving PA. Evidence for two sites of adaptation affecting the dark-adapted ERG of cats and primates. Vision Res. 1995;35:435–442. doi: 10.1016/0042-6989(94)00165-i. [DOI] [PubMed] [Google Scholar]
- Gamm DM, Barthel LK, Raymond PA, Uhler MD. Localization of cGMP-dependent protein kinase isoforms in mouse eye. Invest Ophthalmol Vis Sci. 2000;41:2766–2773. [PubMed] [Google Scholar]
- Ghosh KK, Bujan S, Haverkamp S, Feigenspan A, Wassle H. Types of bipolar cells in the mouse retina. J Comp Neurol. 2004;469:70–82. doi: 10.1002/cne.10985. [DOI] [PubMed] [Google Scholar]
- Goldstein IM, Ostwald P, Roth S. Nitric oxide: a review of its role in retinal function and disease. Vision Res. 1996;36:2979–2994. doi: 10.1016/0042-6989(96)00017-x. [DOI] [PubMed] [Google Scholar]
- Gotzes S, de Vente J, Muller F. Nitric oxide modulates cGMP levels in neurons of the inner and outer retina in opposite ways. Vis Neurosci. 1998;15:945–955. doi: 10.1017/s0952523898155141. [DOI] [PubMed] [Google Scholar]
- Green DG, Powers MK. Mechanisms of light adaptation in rat retina. Vision Res. 1982;22:209–216. doi: 10.1016/0042-6989(82)90120-1. [DOI] [PubMed] [Google Scholar]
- Greferath U, Grunert U, Wassle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- Gurevich L, Slaughter MM. Comparison of the waveforms of the ON bipolar neuron and the b-wave of the electroretinogram. Vision Res. 1993;33:2431–2435. doi: 10.1016/0042-6989(93)90122-d. [DOI] [PubMed] [Google Scholar]
- Haberecht MF, Schmidt HH, Mills SL, Massey SC, Nakane M, Redburn-Johnson DA. Localization of nitric oxide synthase, NADPH diaphorase and soluble guanylyl cyclase in adult rabbit retina. Vis Neurosci. 1998;15:881–890. doi: 10.1017/s0952523898155104. [DOI] [PubMed] [Google Scholar]
- He B, Weber GF. Phosphorylation of NF-kappaB proteins by cyclic GMP-dependent kinase. A noncanonical pathway to NF-kappaB activation. Eur J Biochem. 2003;270:2174–2185. doi: 10.1046/j.1432-1033.2003.03574.x. [DOI] [PubMed] [Google Scholar]
- Hirooka K, Kourennyi DE, Barnes S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J Neurophysiol. 2000;83:198–206. doi: 10.1152/jn.2000.83.1.198. [DOI] [PubMed] [Google Scholar]
- Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- Kim IB, Oh SJ, Chun MH. Neuronal nitric oxide synthase immuno-reactive neurons in the mammalian retina. Microsc Res Tech. 2000;50:112–123. doi: 10.1002/1097-0029(20000715)50:2<112::AID-JEMT3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Koistinaho J, Swanson RA, de Vente J, Sagar SM. NADPHdiaphorase (nitric oxide synthase)-reactive amacrine cells of rabbit retina: putative target cells and stimulation by light. Neuroscience. 1;57:587–597. doi: 10.1016/0306-4522(93)90008-4. [DOI] [PubMed] [Google Scholar]
- Kwan HY, Huang Y, Yao X. Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Natl Acad Sci USA. 2004;101:2625–2630. doi: 10.1073/pnas.0304471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon DG, Ponomareva LV. Nitric oxide and cGMP modulate retinal glutamate receptors. J Neurophysiol. 1996;76:2307–2315. doi: 10.1152/jn.1996.76.4.2307. [DOI] [PubMed] [Google Scholar]
- McMahon DG, Schmidt KF. Horizontal cell glutamate receptor modulation by NO: mechanisms and functional implications for the first visual synapse. Vis Neurosci. 1999;16:425–433. doi: 10.1017/s0952523899163041. [DOI] [PubMed] [Google Scholar]
- Miller RF, Dowling JE. Intracellular responses of the Muller (glial) cells of mudpuppy retina: their relation to b-wave of the electroretinogram. J Neurophysiol. 1970;33:323–341. doi: 10.1152/jn.1970.33.3.323. [DOI] [PubMed] [Google Scholar]
- Mills SL, Massey SC. Differential properties of two gap junctional pathways made by AII amacrine cells. Nature. 1995;377:734–737. doi: 10.1038/377734a0. [Comment (1995) 377:676] [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Iwakabe H, Akazawa C, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. J Biol Chem. 1993;268:11868–11873. [PubMed] [Google Scholar]
- Nawy S. The metabotropic receptor mGluR6 may signal through Go, but not phosphodiesterase, in retinal bipolar cells. J Neurosci. 1999;19:2938–2944. doi: 10.1523/JNEUROSCI.19-08-02938.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawy S. Regulation of the On bipolar cell mGluR6 pathway by Ca2+ J Neurosci. 2000;20:4471–4479. doi: 10.1523/JNEUROSCI.20-12-04471.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawy S. Desensitization of the mGluR6 transduction current in tiger salamander On bipolar cells. J Physiol (Lond) 2004;558:137–146. doi: 10.1113/jphysiol.2004.064980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawy S, Jahr CE. Suppression by glutamate of cGMP-activated conductance in retinal bipolar cells. Nature. 1990;346:269–271. doi: 10.1038/346269a0. [DOI] [PubMed] [Google Scholar]
- Neal M, Cunningham J, Matthews K. Selective release of nitric oxide from retinal amacrine and bipolar cells. Invest Ophthalmol Vis Sci. 1998;39:850–853. [PubMed] [Google Scholar]
- Negishi K, Kato S, Teranishi T. Dopamine cells and rod bipolar cells contain protein kinase C-like immunoreactivity in some vertebrate retinas. Neurosci Lett. 1988;94:247–252. doi: 10.1016/0304-3940(88)90025-0. [DOI] [PubMed] [Google Scholar]
- Sampath AP, Rieke F. Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse. Neuron. 2004;41:431–443. doi: 10.1016/s0896-6273(04)00005-4. [DOI] [PubMed] [Google Scholar]
- Shiells R, Falk G. Retinal on-bipolar cells contain a nitric oxide-sensitive guanylate cyclase. NeuroReport. 1992;3:845–848. doi: 10.1097/00001756-199210000-00006. [DOI] [PubMed] [Google Scholar]
- Shiells RA, Falk G. Glutamate receptors of rod bipolar cells are linked to a cyclic GMP cascade via a G-protein. Proc R Soc Lond B Biol Sci. 1990;242:91–94. doi: 10.1098/rspb.1990.0109. [DOI] [PubMed] [Google Scholar]
- Shiells RA, Falk G. Potentiation of “on” bipolar cell flash responses by dim background light and cGMP in dogfish retinal slices. J Physiol (Lond) 2002;542:211–220. doi: 10.1113/jphysiol.2002.019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen SD, Macek TA, Cai Z, Saugstad JA, Conn PJ. Dissociation of protein kinase-mediated regulation of metabotropic glutamate receptor 7 (mGluR7) interactions with calmodulin and regulation of mGluR7 function. Mol Pharmacol. 2002;61:1303–1312. doi: 10.1124/mol.61.6.1303. [DOI] [PubMed] [Google Scholar]
- Spreca A, Giambanco I, Rambotti MG. Ultracytochemical study of guanylate cyclases A and B in light- and dark-adapted retinas. Histochem J. 1999;31:477–483. doi: 10.1023/a:1003712110751. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Morigiwa K, Ueda M, Sterling P. Microcircuits for night vision in mouse retina. J Neurosci. 2001;21:8616–8623. doi: 10.1523/JNEUROSCI.21-21-08616.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler MD. Cloning and expression of a novel cyclic GMP-dependent protein kinase from mouse brain. J Biol Chem. 1993;268:13586–13591. [PubMed] [Google Scholar]
- Vardi N. Alpha subunit of Go localizes in the dendritic tips of ON bipolar cells. J Comp Neurol. 1998;395:43–52. [PubMed] [Google Scholar]
- Walters RJ, Kramer RH, Nawy S. Regulation of cGMP-dependent current in On bipolar cells by calcium/calmodulin-dependent kinase. Vis Neurosci. 1998;15:257–261. doi: 10.1017/s0952523898152057. [DOI] [PubMed] [Google Scholar]
- Wang X, Robinson PJ. Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system. J Neurochem. 1997;68:443–456. doi: 10.1046/j.1471-4159.1997.68020443.x. [DOI] [PubMed] [Google Scholar]
- Wernet W, Flockerzi V, Hofmann F. The cDNA of the two isoforms of bovine cGMP-dependent protein kinase. FEBS Lett. 1989;251:191–196. doi: 10.1016/0014-5793(89)81453-x. [DOI] [PubMed] [Google Scholar]
- Wexler EM, Stanton PK, Nawy S. Nitric oxide depresses GABAA receptor function via coactivation of cGMP-dependent kinase and phosphodiesterase. J Neurosci. 1998;18:2342–2349. doi: 10.1523/JNEUROSCI.18-07-02342.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkovsky P. Excitation and adaptation in the vertebrate retina. Curr Top Eye Res. 1980;2:1–66. [PubMed] [Google Scholar]
- Yuasa K, Ohgaru T, Asahina M, Omori K. Identification of rat cyclic nucleotide phosphodiesterase 11A (PDE11A): comparison of rat and human PDE11A splicing variants. Eur J Biochem. 2001;268:4440–4448. doi: 10.1046/j.1432-1327.2001.02366.x. [DOI] [PubMed] [Google Scholar]
- Zemel E, Eyal O, Lei B, Perlman I. NADPH diaphorase activity in mammalian retinas is modulated by the state of visual adaptation. Vis Neurosci. 1996;13:863–871. doi: 10.1017/s0952523800009111. [DOI] [PubMed] [Google Scholar]
- Zhou XB, Arntz C, Kamm S, Motejlek K, Sausbier U, Wang GX, Ruth P, Korth M. A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J Biol Chem. 2001;276:43239–43245. doi: 10.1074/jbc.M104202200. [DOI] [PubMed] [Google Scholar]