

Abstract
The deubiquitinase HAUSP (herpesvirus-associated ubiquitin-specific protease; also called USP7) has a critical role in regulating the p53-Mdm2 (murine double minute 2) pathway. By using the conventional knockout approach, we previously showed that hausp inactivation leads to early embryonic lethality. To fully understand the physiological functions of hausp, we have generated mice lacking hausp specifically in the brain and examined the impacts of this manipulation on brain development. We found that deletion of hausp in neural cells resulted in neonatal lethality. The brains from these mice displayed hypoplasia and deficiencies in development, which were mainly caused by p53-mediated apoptosis. Detailed analysis also showed an increase of both p53 levels and p53-dependent transcriptional activation in hausp knockout brains. Notably, neural cell survival and brain development of hausp-mutant mice can largely be restored in the p53-null background. Nevertheless, in contrast to the case of mdm2- and mdm4 (murine double minute 4)-mutant mice, inactivation of p53 failed to completely rescue the neonatal lethality of these hausp-mutant mice. These results indicate that HAUSP-mediated p53 regulation is crucial for brain development, and also suggest that both the p53-dependent and the p53-independent functions of HAUSP contribute to the neonatal lethality of hausp-mutant mice.
Keywords: HAUSP, p53, Mdm2, deubiquitination, gene conditional knockout, mouse
The p53 tumor suppressor has been extensively studied because of its critical functions in maintaining cellular home-ostasis and in tumor suppression.1–4 Activation of p53 triggers changes in a host of downstream targets and causes cell growth arrest, senescence, and apoptosis.5–7 Numerous studies suggest that ubiquitination has a critical role in modulating stability and activity of p53.8–12 It has been well established that Mdm2 (murine double minute 2) is the major E3 ubiquitin ligase for p53.9,13 Knockout of mdm2 in mice causes embryonic lethality due to p53 activation, whereas knockout of both mdm2 and p53 completely rescues the lethality.14,15 In addition, the lethality of the knockout mice of mdm4 (murine double minute 4), which encodes an Mdm2-related p53-binding protein, can also be rescued by deletion of p53, highlighting the complexity in inhibiting p53.9,16 Furthermore, other ubiquitin ligases, such as Pirh2, COP1, and Arf-BP1, have been shown to ubiquitinate p53, underscoring the importance of ubiquitination in p53 regulation.17–19 Adding to the complexity, HAUSP (herpesvirus-associated ubiquitin-specific protease), a deubiquitinase, has been shown to stabilize p53 by reducing p53 ubiquitination.20 Interestingly, HAUSP was later shown to deubiquitinate Mdm2 as well.21,22 Because of this dual substrate specificity, moderate knockdown of hausp decreases p53 stability, whereas complete knockdown of hausp leads to loss of Mdm2 and stabilization of p53.22 Moreover, HAUSP has also been shown to deubiquitinate Mdm4, whose functions are implicated in radiosensitivity and tumorigenesis, further establishing the critical role of HAUSP in regulating p53.23–26
Deletion of hausp in mice causes embryonic lethality between embryonic day 6.5 (E6.5) and E7.5, due to increased p53 levels and cell growth arrest.27 Deletion of p53 in hausp knockout mice extends the embryonic development, but fails to rescue the lethality, suggesting that both p53-dependent and p53-independent functions of HAUSP contribute to the lethality. Indeed, several studies have implicated the roles of HAUSP in p53-independent functions of HAUSP, that is, regulation of gene transcription by deubiquitination of histone proteins.28 HAUSP has been shown to bind and regulate PML and its associated proteins.29–31 Additional targets of HAUSP have also emerged, such as ataxin-1, Daxx, and FoxO4a, which are critical for normal cellular functions.32–35 To extend the study of HAUSP functions in vivo, hausp conditional knockout mice were generated in this study. To validate this mouse model, transgene nes-cre (loxP site-specific recombinase gene cre driven by nestin promoter) was chosen to generate deletion of hausp in neural cells.36 In previous studies, the regulation of p53 by Mdm2 and Mdm4 has been studied using nes-cre-mediated mdm2 and mdm4 conditional knockout mice,37 as well as using nes-cre-mediated brain-specific expression of p53 in mdm2 and mdm4 knockout mice.38 Loss of Mdm2 causes apoptosis, whereas loss of Mdm4 leads to apoptosis and cell cycle arrest, due to activation of p53.37 As expected, concomitant deletion of p53 rescues the lethality of mdm2 nes-cre knockout mice and mdm4 nes-cre knockout mice. More importantly, by combining mdm2 and mdm4 deletions, the study reveals unique and synergistic roles of Mdm2 and Mdm4 for p53 inhibition in neural cells. These findings are reinforced using nes-cre-mediated brain-specific expression of p53 in mdm2 and mdm4 knockout mice,38 suggesting that mouse brain is an excellent model to study regulations of p53. Therefore, we chose to study brain-specific knockout of hausp mediated by nes-cre to elucidate HAUSP functions in regulating p53 during brain development.
Results
Generation of hausp conditional knockout mice
The conditional knockout of hausp was designed to flox exon 6 of hausp and to allow deletion of exon 6 upon expression of Cre recombinase. Deletion of exon 6 removes the conserved catalytic site (cysteine 206), and causes truncation of the rest of the protein because of reading frame shift to ensure loss of functions of HAUSP (Figure 1a). The gene targeting in mouse embryonic stem (ES) cells yielded 33 correctly targeted clones out of 192 G418-resistant clones. Two of the targeted ES cell clones were injected into blastocyst to generate chimeras, from which germline transmission of the exon-6-floxed allele (FL) of hausp (hauspFL) was achieved. Through intercross of hausp conditional knockout heterozygote (hauspFL/+) mice, hausp conditional knockout homozygote (hauspFL/FL) mice were obtained at the expected Mendelian ratio. Both hauspFL/+ and hauspFL/FL mice were fertile and indistinguishable from the wild-type littermates, suggesting that floxing exon 6 of hausp did not cause discernible changes in hausp gene expression. The mice were maintained in a 129 and C57BL/6J mix background. Initially, mice were genotyped by Southern blot (Figure 1b). Subsequently, offsprings were genotyped by PCR (Figure 1c).
Figure 1.
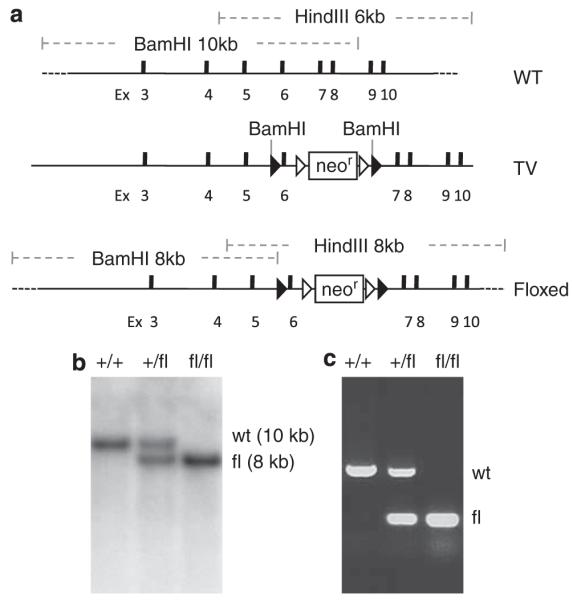
Generation of the conditional knockout allele of mouse hausp gene. (a) Diagram of the hausp genomic region centering on exon 6 is shown, with restriction fragments of BamHI and HindIII indicated. WT, wild-type locus. Targeting vector contains exon 6 flanking with loxP sites and 7 kb 5′ homology and 3 kb 3′ homology. TV, targeting vector. Diagram of the conditional knockout allele shows the altered restriction fragments of BamHI and HindIII after homologous recombination. Floxed, exon 6-floxed conditional knockout allele. (b) Genotyping was determined by Southern blot using genomic DNA digested with BamHI. Hybridization with 5′ external probe detected a 10 kb band in wild-type mice (+/+) and an additional 8 kb band for the FL in heterozygote mice (+/fl). Only the 8 kb band for the FL was detected in hausp conditional knockout homozygote mice (fl/fl). (c) Genotyping was determined using three mixed primers to detect wild-type allele and the FL simultaneously to identify wild-type mice (+/+), heterozygote mice (+/fl), and homozygote mice (fl/fl)
Deletion of exon 6 of hausp in mice resulted in embryonic lethality
To determine whether deletion of exon 6 of hausp produces similar phenotypes to the previous hausp knockout mice, generated by deletion of exon 14, hence hauspΔEx14/ΔEx14 mice,27 hauspFL/+ mice were crossed with ROSA26-cre mice to generate exon 6-deleted hausp heterozygote (hauspΔEx6/+) mice. Intercross of hauspΔEx6/+ mice did not yield hauspΔEx6/ΔEx6 mice, suggesting that deletion of exon 6 of hausp caused embryonic lethality. Analysis of the embryos collected at embryonic day E7.5 revealed the presence of severely abnormal embryos (Figure 2b). The normal developing embryos displayed formation of amniotic cavity, exocoelom, and ectoplacental cavity (Figure 2a), whereas the abnormal embryos showed no recognizable structures and contained only disorganized cells (Figure 2b). All of the abnormal embryos showed no detectable HAUSP protein, suggesting that they were knockouts of hausp (hauspΔEx6/ΔEx6 ; Figure 2d). Furthermore, the abnormal embryos showed p53 staining in the remaining cells, suggesting loss of HAUSP caused accumulation of p53 (Figure 2f). In contrast, normal embryos showed HAUSP staining (Figure 2c), little or no p53 staining (Figure 2e). The nearly identical phenotypes between hauspΔEx6/ΔEx6 and hauspΔEx14/ΔEx14 mice suggested that deletion of exon 6 of hausp also created a loss of function allele of hausp, and that hauspFL/FL mice were suitable to study tissue-specific loss of functions of HAUSP.
Figure 2.

Analysis of embryos of day E7.5 from hauspΔEx6/+ intercross. Two representative embryos are shown: an HAUSP staining positive embryo (hausp+) in a, c, and e; and an HAUSP staining negative embryo (hauspΔEx6/ΔEx6) in b, d, and f. The hausp+ embryo showed formation of germinal layers and characteristic structures of exocoelom (EC) and ectoplacental cavity (EP) for normal embryos at day E7.5 (a), positive staining for HAUSP (c), but little or no p53 staining (e), whereas the hauspΔEx6/ΔEx6 embryo showed no recognizable structures (b), no HAUSP staining in the remaining embryonic cells (d), and detectable p53 staining in all remaining embryonic cells (f)
Deletion of hausp in mouse brain during embryonic development caused neonatal lethality and defects in brain development
Previous studies have shown that activation of p53 results in abnormalities in mouse brain during embryonic development, suggesting development of mouse brain is sensitive to p53 regulations.37,38 Breeding between hauspFL/FL female mice and hauspFL/+; nes-cre male mice was set up to obtain hauspFL/FL; nes-cre mice. Among 75 newborn pups, 21 of them were hauspFL/FL; nes-cre mice, 15 of them died soon after birth, and 6 of them died the next day. The surviving hauspFL/FL; nes-cre mice at birth showed uncoordinated body movements and no milk in their stomach, suggesting lack of feeding caused their death. There were 15 hauspFL/+, 17 hauspFL/FL, and 22 hauspFL/+; nes-cre mice, which were all alive at weaning age without obvious defects. These results suggested that although hauspFL/FL; nes-cre mice were born at the expected Mendelian ratio, deletion of hausp in neural cells during development caused neonatal lethality. The heads from hauspFL/FL; nes-cre mice displayed distinct appearances with a flat forebrain (Figure 3a, arrow head), forming almost a right angle at the midbrain junction (Figure 3a, asterisk). Upon dissection of the brains at day E18.5, the mutant brains (Figures 3b and c, arrow head) had much smaller forebrain than those from the control mice (Figures 3b and c). Dramatically, cerebellum was mostly missing (Figure 3b and c, asterisk) in hauspFL/FL; nes-cre mice, but was visible in the brains of the control mice (Figures 3b and c, arrow). These developmental defects of the mutant brains contributed to their abnormal appearance of the heads.
Figure 3.

Developmental defects in brains from hauspFL/FL; nes-cre knockout mice. (a) Embryos collected at day E18.5 from breeding between hauspFL/FL mice and hauspFL/+; nes-cre mice are shown. The control hauspFL/FL embryo is on the left (arrow). The hauspFL/FL; nes-cre embryo (right) showed flattened forebrain (arrow head), forming almost a right angle at the midbrain junction (asterisk). (b) Dorsal and (c) lateral views of dissected brains from the control embryo (arrow) and the hauspFL/FL; nes-cre embryo (arrowhead) at day E18.5. The cortex of hauspFL/FL; nes-cre brain was much smaller than that of the control brain. Development of cerebellum was largely missing in brains from the hauspFL/FL; nes-cre mice (asterisk) compared with the cerebellum in the control brain (arrow). (d–g) Sagittal sections of the control (d and f) and the hauspFL/FL; nes-cre (e and g) brains are shown. The brain from hauspFL/FL; nes-cre showed reduced cell density in cortex and formation of large cavity (triangle; e) compared with the control brain (d). In addition, the brain from the hauspFL/FL; nes-cre mouse showed no development of cerebellum at the midline (asterisk; g) compared with the control brain (arrow; f)
To further understand the abnormalities, sagittal sections were collected from the midline of the brains of day E18.5. The brains from hauspFL/FL; nes-cre mice had dramatically reduced size and thickness in cortex, because of a severe reduction of neural cells (Figure 3e versus 3d). As a result, the third ventricle appeared to be greatly enlarged in hauspFL/FL; nes-cre brain (Figure 3e, open triangle). Strikingly, cerebellum was missing at the midline of the hauspFL/FL; nes-cre brain (Figure 3g, asterisk). In contrast, the brains from the control mice showed primitive cerebellum and formation of external germinal layer at E18.5 (Figure 3f, arrow).
Neural cells from hausp nes-cre knockout brain showed accumulation of p53 protein
To study the molecular events that preceded these structural changes, immunostaining was performed using antibodies against HAUSP, p53, and Cleaved Caspase-3. Immunostaining of the brains from day E12.5 showed that ~50% of the cells had lost HAUSP staining in hauspFL/FL; nes-cre brains, most of which were in the subventricular zone and the ventricular zone (data not shown). The timing and location of the loss of HAUSP staining was consistent with the pattern of nes-cre expression (The Jackson Laboratory, Bar Harbor, ME, USA). The neurons in the marginal zone at day E12.5, which were generated earlier, still retained HAUSP staining.36,39 The p53 staining was weak but was detectable in some of the cells in hauspFL/FL; nes-cre cortex at day E12.5 (data not shown), indicating the onset of the accumulation of p53 following depletion of HAUSP protein.
In the cortex of hauspFL/FL; nes-cre brains from day E14.5, HAUSP staining was lost in most of the cells (Figures 4c and d). Dramatically, number of p53 staining positive neural cells, as well as the intensity of the staining, was increased (Figures 4g and h). Moreover, immunostaining revealed the widespread presence of Cleaved Caspase-3 in the neural cells in the intermediate zone and the subventricular zone, accompanied by the abundant appearance of cells with pyknotic nuclei (Figures 4k and l). In contrast, staining for HAUSP protein was present in all neural cells in the control brains (Figures 4a and b), in which cells positive for p53 staining were rarely observed (Figures 4e and f). Consistently, no Cleaved Caspase-3 staining was detected in the control brains (Figures 4i and j). The highly temporal- and spatial-restricted events were consistent with the expression pattern of nes-cre during neurogenesis in the cortex. These results suggested that deletion of hausp resulted in depletion of HAUSP protein, followed by accumulation of p53 protein and potential activation of p53 downstream targets, which resulted in apoptosis.
Figure 4.

Immunostaining of the sagittal sections from the control and hauspFL/FL; nes-cre brains at day E14.5. HAUSP was detected using a polyclonal antibody anti-HAUSP C terminus (a–d). HAUSP was ubiquitously expressed in all cells indicated by the presence of brown staining in nuclei of the control brain (a and b), whereas most of the cells from ventricular zone and intermediate zone of the hauspFL/FL; nes-cre brain showed no staining of HAUSP protein (c and d). Immunostaining of p53 was done using an anti-p53 polyclonal antibody (CM5) (e–h). Dramatic p53 activation was detected in the hauspFL/FL; nes-cre brain (g and h), whereas no p53 staining was detected in the control brain (e and f). Apoptosis was determined by the presence of Cleaved Caspase-3 (i–l). Consistent with the abundant appearance of pyknotic nuclei, Cleaved Caspase-3 was detected in large number of neural cells in the hauspFL/FL; nes-cre brain by immunostaining (k and l). No Cleaved Caspase-3 was detected in the control brain (i and j). b, d, f, h, j, and l are the higher magnified pictures of the insects in a, c, e, g, i, and k, respectively. IZ, intermediate zone; MZ, marginal zone; V, ventricle; VZ, ventricular zone
Similarly, the brains from hauspFL/FL; nes-cre mice and the control mice at day E16.5 and E18.5 were also studied. Most of the remaining neural cells in hauspFL/FL; nes-cre brains showed no staining for HAUSP protein. Although only some of the cells in the cortex still retained p53 staining, the cells displaying strong p53 staining were mostly located in the ventricular zone, suggesting accumulation of p53 at these stages was restricted to the cells in the areas of active neurogenesis (data not shown).
Deletion of hausp in neural cells led to accumulation of p53 and activation of p53 downstream targets
To uncover the molecular events following accumulation of p53 protein, protein extracts were prepared from cortex at E13.5 and E14.5. As shown by western blot, HAUSP protein was mostly depleted in hauspFL/FL; nes-cre brains (Figure 5a, lanes 3–4 and 7–8 versus lanes 1–2 and 5–6). Significant accumulation of p53 was detected in protein extracts from hauspFL/FL; nes-cre brains (Figure 5b, lanes 3–4 and 7–8). Importantly, p21 (cyclin-dependent kinase inhibitor 1) and PUMA (p53 upregulated modulator of apoptosis), two of the signature p53 targets, were also increased in protein extracts from hauspFL/FL; nes-cre brains (Figures 5c and d, lanes 3–4 and 7–8), compared with the controls (Figures 5c and d, lanes 1–2 and 5–6), suggesting that accumulation of p53 protein resulted in transcriptional activation of its downstream targets. Additionally, the level of Daxx protein, a reported substrate of HAUSP,33 showed slight reduction after deletion of hausp (Figure 5e, lanes 3–4 and 7–8), suggesting reduced stability of Daxx in HAUSP-depleted cells.
Figure 5.

Depletion of HAUSP in neural cells led to accumulation of p53 and activation of p53 target genes. Protein extracts were prepared from cortex of the brains collected at days E13.5 and E14.5. The protein levels of HAUSP (a), p53 (b), p53 downstream target p21 (c), PUMA (d), Daxx (e), and total proteins represented by β-actin (f) were determined by western blot. Protein extracts from two individual hauspFL/FL brains at E13.5 (lanes 1 and 2) and at E14.5 (lanes 5 and 6), along with protein extracts from two individual hauspFL/FL; nes-cre brains at E13.5 (lanes 3 and 4) and at E14.5 (lanes 7 and 8), were analyzed. HAUSP protein was shown to be nearly depleted in hauspFL/FL; nes-cre brains (a; lanes 3, 4, 7, and 8) compared with HAUSP protein in the control brains (a; lanes 1, 2, 5, and 6). In contrast, the protein level of p53 was elevated in hauspFL/FL; nes-cre brains (b; lanes 3, 4, 7, and 8 versus lanes 1, 2, 5, and 6) and so were the p53 downstream targets, p21 (c) and PUMA (d). Daxx, a reported HAUSP substrate, was slightly reduced after knockout of hausp (e; lanes 3, 4, 7, and 8 versus lanes 1, 2, 5, and 6). Total proteins were comparable, indicated by the protein levels of β-actin in each lane (f). (g) Quantitative PCR analysis of transcriptional levels of p53, mdm2, p21, bax, and puma in hauspFL/FL; nes-cre brains collected at days E13.5 and E14.5. The hauspFL/+; nes-cre brains were used as controls. Whereas little or no change of transcription was observed for p53 and mdm2, the transcription levels of p53 target genes, p21, bax, and puma were significantly increased in hauspFL/FL; nes-cre brains. The error bars represent S.D.
To confirm transcriptional activation by p53, total RNA was prepared from both hauspFL/FL; nes-cre brains and the control, hauspFL/+; nes-cre brains, from which the transcriptional levels of p53, mdm2, and p53 downstream targets were determined using quantitative PCR. The results showed significant increases in transcription of p21, bax (BCL2-associated X protein), and puma in hauspFL/FL; nes-cre brains from both E13.5 and E14.5 compared with that in the control brains (Figure 5g). There was little or no change of transcription for p53 and mdm2, suggesting that accumulation of p53 was potentially not because of increase of p53 transcription, but resulted from stabilization of p53 protein. Together, these data indicated that accumulation of p53 in hauspFL/FL; nes-cre brains caused transcriptional activation of downstream targets, particularly, apoptotic genes, such as bax and puma, which led to apoptosis in neural cells.
Deletion of hausp in mouse embryonic fibroblasts resulted in destabilization of Mdm2 and activation of p53
To elucidate potential mechanisms that led to the accumulation of p53, deletion of hausp was carried out in hauspFL/FL mouse embryonic fibroblasts (MEFs) by infecting them with adenovirus expressing Cre. After infection for 3 days, HAUSP protein was nearly depleted (Figure 6a, lane 6). Consistent with the results in the hausp nes-cre knockout brains, the p53 level increased in hausp knockout MEFs (Figure 6b, lane 6) compared with that in MEFs infected with the control adenovirus expressing GFP (Figure 6b, lane 5). No obvious increase of p53 levels was observed in wild-type MEFs infected with either control adenovirus (Figure 6b, lane 3) or Cre-expressing adenovirus (Figure 6b, lane 4). Interestingly, the Mdm2 level decreased after depletion of HAUSP (Figure 6c, lane 6 versus lane 5), consistent with the previous finding that Mdm2 is a substrate of HAUSP, and Mdm2 is destabilized in hausp knockout cells.21,22,40 In contrast, the level of Mdm2 dramatically increased in the control MEFs (hauspFL/FL) after induction of DNA damage (Figure 6c, lane 2), because of increase of mdm2 transcription mediated by p53 activation (Figure 6b, lane 2). Consistent with the previous studies,23–25 the protein level of Mdm4 in the control MEFs reduced dramatically in response to DNA damage (Figure 6d, lane 2). Interestingly, the level of Mdm4 showed slight increase in hausp knockout MEFs (Figure 6d, lane 6 versus lane 5), suggesting p53 activation in hausp knockout MEFs was mainly because of downregulation of Mdm2. In addition, accumulation of p53 in control MEFs after DNA damage was accompanied by increase of phosphorylation on Ser15 of p53 (Figure 6e, lane 2). No increase of phosphorylation on Ser15 of p53 was detected in HAUSP-depleted MEFs (Figure 6e, lane 6). Together, these results suggested that accumulation of p53 was potentially caused by destabilization of Mdm2, not by DNA damage-induced phosphorylation of Ser15 of p53, which stabilizes p53 through inhibiting ubiquitination of p53 by Mdm2. Moreover, accumulation of p53, regardless whether it was due to DNA damage or due to destabilization of Mdm2, resulted in activation of p21 (Figure 6f, lanes 2 and 6).
Figure 6.
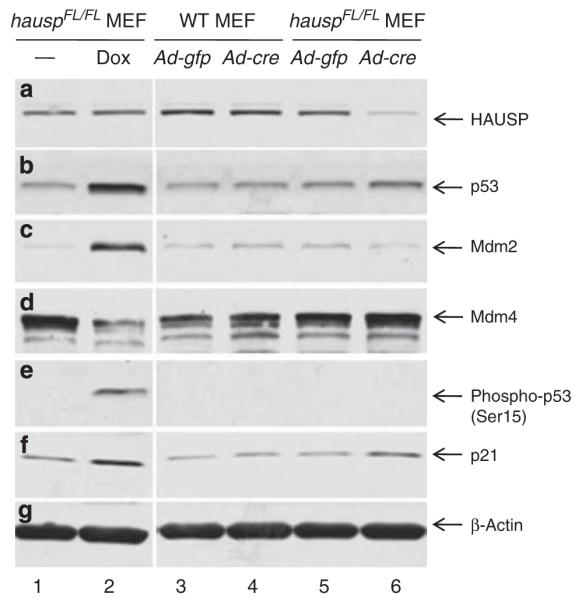
Deletion of hausp in MEFs resulted in destabilization of Mdm2 and stabilization of p53. Protein extracts were prepared from hauspFL/FL MEFs infected with control virus (Ad-CMV-GFP; lane 5) and Cre-expressing adenovirus (Ad-CMV-Cre; lane 6), as well as from wild-type MEFs infected with control virus (lane 3) and Cre-expressing adenovirus (lane 4). Protein extracts were also prepared from hauspFL/FL MEFs treated without (lane 1) and with doxorubicin (lane 2) for 6 h to induce DNA damage response. The protein levels of HAUSP (a), total p53 (CM5) (b), Mdm2 (c), Mdm4 (d), Phospho-p53 (Ser15) (e), p21 (f), and protein loading control β-actin (g) were determined by western blot. Deletion of hausp resulted in depletion of HAUSP (a; lane 6 versus 5), accumulation of p53 (b; lane 6 versus 5), destabilization of Mdm2 (c;lane 6 versus 5), and activation of p21 (f; lane 6 versus 5). There was slight increase of Mdm4 level in hausp knockout MEFs (d; lane 6 versus 5). No change in phosphorylation on Ser15 of p53 was observed in hausp knockout cells (e; lane 6 versus 5). In contrast, DNA damage caused activation of p53 (b; lane 2 versus 1), increase of Mdm2 (c; lane 2 versus 1), decrease of Mdm4 (d; lane 2 versus 1), phosphorylation on Ser15 of p53 (e; lane 2 versus 1), and activation of p21 (f; lane 2 versus 1). There was no obvious change of protein levels for Hausp, p53, Mdm2, Mdm4, and p21 in wild-type MEFs infected with either control adenovirus or Cre-expressing adenovirus (lanes 3 and 4). The protein levels of β-actin indicated comparable levels of total proteins in each lane (g)
Hausp nes-cre knockout was partially rescued by p53 knockout
As p53 activation and ensuing apoptosis were observed in neural cells of hauspFL/FL; nes-cre brains, crosses between hauspFL/FL; p53+/− mice, and hauspFL/+; p53+/−; nes-cre mice were set up to generate hauspFL/FL; p53−/−; nes-cre mice to determine whether neonatal lethality in hauspFL/FL; nes-cre mice could be rescued by concomitant deletion of p53. Among the newborns of hauspFL/FL; nes-cre mice, 8 of them were wild type for p53, 11 of them were heterozygote for p53, and 3 of them were null for p53. None of these mice survived more than 1 day, suggesting that the neonatal lethality of hauspFL/FL; nes-cre mice cannot be rescued by deletion of p53. Those p53-null mice with open neural tube defect were excluded, which could have resulted from p53 deficiencies during embryonic development and would not have survived regardless of the genotypes for hausp or nes-cre.41 However, upon dissection of the brains, the hauspFL/FL; p53−/−; nes-cre brain showed similar size in forebrain, midbrain, and cerebellum (Figures 6a and b, star) compared with the hauspFL/FL; p53+/− brain (Figures 7a and b, arrow), suggesting that the developmental defects in hauspFL/FL; nes-cre brain (Figures 7a and b, arrow head) were partially rescued by concomitant deletion of p53. Furthermore, sagittal sections of the cortical layers also showed comparable thickness and density of neural cells in the hauspFL/FL; p53−/−; nes-cre brain (Figure 7e) compared with that in the control hauspFL/FL; p53+/− brain (Figure 7c). In contrast, hauspFL/FL; nes-cre cortical layers were dramatically smaller and contained less cells than the control (Figure 7d). These results suggested that although deletion of p53 failed to rescue the neonatal lethality of hauspFL/FL; nes-cre mice, survival of neural cells and brain development were largely restored in the absence of p53.
Figure 7.

Deletion of p53 in hauspFL/FL; nes-cre mice restored the cell density in the cortex and the size of the brain. Dorsal (a) and lateral (b) views of the brains collected at day E18.5 from hauspFL/FL; p53+/− (arrow), hauspFL/FL; nes-cre (arrowhead), and hauspFL/FL; p53−/−; nes-cre (star) are shown. The hauspFL/FL; nes-cre brain (arrow head) was much smaller than the control brain (arrow), displaying little or no development of cerebellum (asterisk), whereas the hauspFL/FL; p53−/−; nes-cre brain (star) showed comparable size to that of the control brain (arrow), with similar development of cerebellum. (c–e) The sagittal sections of the brains were stained with hematoxylin and eosin. The thickness and cell density of the cortex of the hauspFL/FL; p53−/−; nes-cre brain (e) was comparable to that of the control hauspFL/FL; p53+/− brain (c), whereas the hauspFL/FL; nes-cre brain (d) showed much reduced thickness and much lower cell density than that of the control brain (c). These results suggested that concomitant deletion of p53 partially rescued the developmental defects in hauspFL/FL; nes-cre brains
Ectopic expression of nes-cre caused p53 stabilization in epithelial cells of the lens from hausp nes-cre knockout mice
It has been shown that ectopic expression of nes-cre occurs in the epithelial cells of the mouse lens, which causes eye abnormalities due to nes-cre-mediated gene inactivation.42 Similarly, HAUSP protein was depleted in the epithelial cells in the lens of hauspFL/FL; nes-cre mouse at day E18.5 (Figure 8b versus 8a), accompanied by accumulation of p53 in those cells (Figure 8d and f). No p53 staining was detected in the lens cells from the control mouse (Figures 8c and e). The previous study has shown that conditional deletion of DDB1 mediated by nes-cre caused lens degeneration due to DNA damage-induced p53 activation.42 However, accumulation of p53 in lens epithelial cells in hausp nes-cre knockout mice had little or no effects on cell survival and morphology of the eyes (Figure 8b versus 8a). The reasons for lack of abnormalities in the eyes from hausp nes-cre mice could be because of the timing and the cause of p53 activation associated with individual gene inactivation, and could also be because of differences in p53-independent functions associated with individual gene inactivation.
Figure 8.

Accumulation of p53 in the epithelial cells of the lens from hauspFL/FL; nes-cre knockout mice. HAUSP staining was detected in the epithelial cells of the lens (a; arrows) and surrounding cells from the hauspFL/FL mice, which was missing in the epithelial cells of lens (b; arrowheads), but not in the surrounding cells from the hauspFL/FL; nes-cre mice, because of ectopic expression of nes-cre in the epithelial cells of mouse lens. The stabilization of p53 was detected in the epithelial cells of lens from the hauspFL/FL; nes-cre mice (d; arrowheads), but not in the epithelial cells of lens from the control mice (c; arrows). The higher magnified pictures of the insects showed nuclear staining of accumulated p53 protein in the epithelial cells of the lens from the hauspFL/FL; nes-cre mice (f; arrowheads), but not in the epithelial cells of the lens from the control mice (e; arrows)
Discussion
In this study, hausp conditional knockout mice were generated to circumvent the early embryonic lethality in hausp conventional knockout mice. Analysis of hausp nes-cre conditional knockout mice showed dramatic accumulation of p53 protein and activation of its downstream targets in neural cells after HAUSP protein was depleted, which led to loss of neural cells and neonatal lethality. Concomitant deletion of p53 partially rescued the developmental defects in hausp nes-cre conditional knockout mice, providing direct evidences for p53-dependent functions of HAUSP. Moreover, deletion of hausp in MEFs showed reduced level of Mdm2, which supports our major conclusion that destabilization of Mdm2 in hausp knockout cells leads to p53 activation in vivo. Although previous studies indicated that hausp knockdown destabilizes Mdm4 in cancer cell lines,25 our data derived from the same experiments revealed that the levels of Mdm4 were slightly increased in hausp knockout MEFs (Figure 6d, lane 6). Thus, the precise role of HAUSP in modulating Mdm4 stability needs to be further elucidated.
Activation of p53 in hausp knockout mouse brains resulted in apoptosis in neural cells and developmental defects in brains, which at least, in part, led to neonatal lethality in mice. These findings displayed significant similarities to those of nes-cre-mediated mdm2 conditional knockout mice, in which the accumulation of p53 is because of inability to inhibit p53. However, the severity of the defects resulted from p53 activation differ dramatically between these mutant mice. The newborn mdm2 nes-cre conditional knockout mice (mdm2FM/FM; nes-cre) have almost no brain tissues as a result of p53 activation at as early as day E10.5, demonstrating the immediate and complete loss of inhibition of p53. Interestingly, although stabilization of p53 occurred in hausp nes-cre knockout mouse brains, it appeared at a much later time than that in mdm2 nes-cre knockout mouse brains. The differences were potentially because of the fact that HAUSP is a much more stable protein than Mdm2, evidenced by the lingering presence of HAUSP protein in neural cells up to day E13.5. Second, regulation of p53 by Mdm2 is direct, whereas regulation of p53 by HAUSP is complicated, in part, because of regulation of Mdm2 by HAUSP. The greater loss of neural tissues in mdm2 nes-cre knockout brains than hausp nes-cre knockout brains was consistent with the earlier presence of p53 activation in mdm2 nes-cre knockout brains.
In conclusion, hausp conditional knockout mice provide a powerful tool in understanding tissue-specific regulations of p53 by HAUSP. Once again, the current study demonstrated the essential roles of p53-independent functions of HAUSP, indicated by the inability to rescue the neonatal lethality of hauspFL/FL; nes-cre mice by concomitant deletion of p53. Specifically, the protein level of Daxx was slightly decreased in hausp knockout brains in this study. In addition, the potential effects on p63/p73 in hausp knockout cells cannot be excluded, as well as a host of other potential HAUSP targets. Although future investigations are required to identify the key substrates for HAUSP-mediated p53-independent function, our study provides strong evidence that the HAUSP/Mdm2/p53 interplay represents the major role for HAUSP in vivo.
Materials and Methods
Gene targeting and generation of hausp conditional knockout mice
The hausp conditional knockout-targeting vector contained floxed exon 6 and neighboring genomic DNA of mouse hausp gene. Briefly, an 10 kb genomic DNA was cloned from a 129 Sv/Ev BAC clone using recombineering technology, into which a loxP site was inserted between exons 5 and 6, and an frt-neo-frt-loxP cassette was inserted between exons 6 and 7 (Figure 1a).43 The targeting vector was electroporated into mouse ES cells W4/129S6 cells (Taconic, Hudson, NY, USA). The correctly targeted ES cell clones were screened by Southern blot using 5′ external probes. The mutated ES cells were injected into blastocyst to generate chimeras, from which germline transmission of the mutated allele of hausp gene was accomplished. Genotyping was determined initially by Southern blot (Figure 1b). Subsequently, PCR assay was designed to genotype hausp FL using hausp-specific primers 5′-GAAGCATACTGGC-TATGTCGGCTT-3′ and 5′-GCCTGTCAGCTACAAGAGAGCCAG-3′ in combination with a neo-specific primer 5′-CATCAGTCAGGTACATAATATAAC-3′ to detect wild-type allele and mutant allele simultaneously (Figure 1c). The hausp-floxed mice were maintained on a mixed background of 129Sv and C57BL/6J through breeding between littermates. Maintenance and handling protocols were approved by the Institutional Animal Care and Use Committee of Columbia University.
Embryo collection and histology
Embryos were collected from timed pregnancy at various stages of gestation for histological analysis. Heads were removed from embryos and fixed in PBS-buffered 4% paraformaldehyde overnight. Serial 5-μm sagittal sections were collected and stained by hematoxylin and eosin according to standard procedures. The sections were immunostained using antibodies against HAUSP C terminus at 1.1 μg/ml,20 p53 (CM5, Novocastra, Newcastle upon Tyne, UK) and Cleaved Caspase-3(Asp175) (#9661, Cell Signaling, Danvers, MA, USA), followed by counter staining with hematoxylin.
Western blot analysis of the protein extracts from brain cortex
The brains from mouse embryos at days E13.5 and E14.5 were collected. The proteins extracts were prepared from cortex individually using RIPA buffer and analyzed by western blot. The western blot was probed with anti-HAUSP, anti-p53 (CM5, Novocastra), anti-p21 (SX118, Santa Cruz, Santa Cruz, CA, USA), anti-PUMA (Sigma, St. Louis, MO, USA), anti-Daxx (H-7, Santa Cruz), and anti-β-actin (Sigma)) antibodies.
Deletion of hausp in MEFs using adenovirus expressing Cre recombinase
MEFs of hauspFL/FL were prepared from E13.5 embryos, and cultured in DMEM containing 10% FBS. To delete hausp, 2.5×105 of cells were mixed with either control adenovirus expressing GFP (Ad-CMV-GFP, Vector Biolabs, Philadelphia, PA, USA) or adenovirus expressing Cre (Ad-CMV-Cre, Vector Biolabs) at an MOI of 200 before being plated onto a 6-well plate. Cells were harvested after 3 days, and protein extracts were prepared in RIPA buffer and analyzed by western blot. The western blot was probed with anti-Mdm2 (AB5, EMD, Gibbstown, NJ, USA), anti-Mdm4 (MX-82, Sigma), and anti-Phospho-p53(Ser15) (#9284, Cell Signaling) antibodies, in addition to antibodies against HAUSP, p53, p21, and β-actin. The hauspFL/FL MEFs treated with doxorubicin (0.2 μg/ml) for 6 h were used as control to show changes of p53, Mdm2, Mdm4, phosphorylation of p53 on Ser15, and p21 after DNA damage.
Acknowledgements
We thank Dr. Neal Copeland for recombineering reagents; Qiong Li and Xi Sun for expert assistance with histology. This study was supported, in part, by grants from the NIH/NCI and by grants from the Leukemia and Lymphoma Society and Ellison Medical Foundation to WG.
Abbreviations
- HAUSP
herpesvirus-associated ubiquitin-specific protease
- Mdm2
murine double minute 2
- Mdm4
murine double minute 4
- nes-cre
loxP site-specific recombinase gene cre driven by nestin promoter
- FL
floxed allele
- p21
cyclin-dependent kinase inhibitor 1
- BAX
BCL2-associated X protein
- PUMA
p53 upregulated modulator of apoptosis
- MEF
mouse embryonic fibroblast
Footnotes
Conflict of interest The authors declare no conflict of interest.
References
- 1.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 2.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 5.Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol. 2000;20:3224–3233. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marchenko ND, Moll UM. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle. 2007;6:1718–1723. doi: 10.4161/cc.6.14.4503. [DOI] [PubMed] [Google Scholar]
- 7.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 8.Stommel JM, Wahl GM. A new twist in the feedback loop: stress-activated MDM2 destabilization is required for p53 activation. Cell Cycle. 2005;4:411–417. doi: 10.4161/cc.4.3.1522. [DOI] [PubMed] [Google Scholar]
- 9.Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927–934. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- 10.Wahl GM. Mouse bites dogma: how mouse models are changing our views of how P53 is regulated in vivo. Cell Death Differ. 2006;13:973–983. doi: 10.1038/sj.cdd.4401911. [DOI] [PubMed] [Google Scholar]
- 11.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shan J, Brooks C, Kon N, Li M, Gu W. Dissecting roles of ubiquitination in the p53 pathway. Ernst Schering Found Symp Proc. 2008;1:127–136. doi: 10.1007/2789_2008_105. [DOI] [PubMed] [Google Scholar]
- 13.de Rozieres S, Maya R, Oren M, Lozano G. The loss of mdm2 induces p53-mediated apoptosis. Oncogene. 2000;19:1691–1697. doi: 10.1038/sj.onc.1203468. [DOI] [PubMed] [Google Scholar]
- 14.de Oca Luna R Montes, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 15.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 16.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 17.Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 18.Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 19.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121:1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 20.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 21.Cummins JM, Rago C, Kohli M, Kinzler KW, Lengauer C, Vogelstein B. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428:1. doi: 10.1038/nature02501. following 486. [DOI] [PubMed] [Google Scholar]
- 22.Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang YV, Leblanc M, Wade M, Jochemsen AG, Wahl GM. Increased radioresistance and accelerated B cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell. 2009;16:33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci USA. 2005;102:5056–5061. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, et al. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell. 2005;18:565–576. doi: 10.1016/j.molcel.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 26.Meulmeester E, Pereg Y, Shiloh Y, Jochemsen AG. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle. 2005;4:1166–1170. doi: 10.4161/cc.4.9.1981. [DOI] [PubMed] [Google Scholar]
- 27.Kon N, Kobayashi Y, Li M, Brooks CL, Ludwig T, Gu W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene. 2010;29:1270–1279. doi: 10.1038/onc.2009.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Knaap JA, Kumar BR, Moshkin YM, Langenberg K, Krijgsveld J, Heck AJ, et al. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol Cell. 2005;17:695–707. doi: 10.1016/j.molcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997;16:1519–1530. doi: 10.1093/emboj/16.7.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sivachandran N, Sarkari F, Frappier L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 2008;4:e1000170. doi: 10.1371/journal.ppat.1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong S, Kim SJ, Ka S, Choi I, Kang S. USP7, a ubiquitin-specific protease, interacts with ataxin-1, the SCA1 gene product. Mol Cell Neurosci. 2002;20:298–306. doi: 10.1006/mcne.2002.1103. [DOI] [PubMed] [Google Scholar]
- 33.Tang J, Qu L, Pang M, Yang X. Daxx is reciprocally regulated by Mdm2 and Hausp. Biochem Biophys Res Commun. 2010;393:542–545. doi: 10.1016/j.bbrc.2010.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 35.Tang J, Qu LK, Zhang J, Wang W, Michaelson JS, Degenhardt YY, et al. Critical role for Daxx in regulating Mdm2. Nat Cell Biol. 2006;8:855–862. doi: 10.1038/ncb1442. [DOI] [PubMed] [Google Scholar]
- 36.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 37.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Liu G, Lozano G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci USA. 2006;103:3226–3231. doi: 10.1073/pnas.0508500103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Francoz S, Froment P, Bogaerts S, De Clercq S, Maetens M, Doumont G, et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc Natl Acad Sci USA. 2006;103:3232–3237. doi: 10.1073/pnas.0508476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- 40.Hu M, Gu L, Li M, Jeffrey PD, Gu W, Shi Y. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 2006;4:e27. doi: 10.1371/journal.pbio.0040027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- 42.Cang Y, Zhang J, Nicholas SA, Bastien J, Li B, Zhou P, et al. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell. 2006;127:929–940. doi: 10.1016/j.cell.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 43.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]