

Abstract
G protein-coupled receptors (GPCRs), which includes the gonadotropin releasing hormone (GnRH) receptor (GnRHR), comprises the largest family of validated drug targets—more than half of all approved drugs derive their benefits by selective targeting of GPCRs. Most drugs in this class are either agonists or antagonists of GPCRs and high throughput screens (HTSs) have typically been designed and performed with a view toward identification of such compounds as lead drug candidates. This manuscript presents the case that valuable drugs which effect the trafficking of GPCRs may have been overlooked because pharmacoperones have been selected from existing screens that identify agonists and antagonists. A “gain of activity assay” is proposed; this assay relies on the expression of a mutant of the GnRHR that is known to be rescuable by pharmacoperone drugs, and which is restored to activity in their presence. Accordingly, “hits” are identified by the appearance of activity. The gene for the mutant is under control of tetracycline and may be prevented from being expressed. This is a valuable feature since it allows false positives to be identified. Such drugs will show apparent activity whether or not the mutant is expressed. This assay will enable identification of these drugs from chemical libraries and does not rely on their activity as agonists or antagonists.
Keywords: G protein-coupled receptors, heritable disease, high throughput screens, pharmacoperones, protein misfolding, protein rescue
Introduction
Mutations in GPCRs are known to be responsible for over 30 disorders, including cancers, heritable obesity and endocrine disease. Normally, nascent GPCRs are subjected to the stringent quality control system (QCS) of the endoplasmic reticulum (ER); this system consists of both protein chaperones that promote correct folding by catalyzing the folding process or may retain misfolded proteins. The QCS ensures that only correctly folded proteins enter the pathway leading to the plasma membrane (PM). Because of this, point mutations may result in the production of misfolded and disease-causing proteins that are unable to reach their functional destinations in the cell since they are retained by the QCS even though they may retain (or regain) function when properly trafficked.
The functional rescue of misfolded mutant receptors by small non-peptide molecules, originally screened from chemical libraries to serve as receptor antagonists, has now been demonstrated (Conn, et al. 2007); these small molecules (pharmacological chaperones or “pharmacoperones”) serve as molecular templates, promoting correct folding and allowing the mutants to pass the scrutiny of the QCS and be expressed at the PM (or, in some cases, other cellular loci) where they may function normally. It is important to note that the QCS is not protein-specific; it recognizes general aspects of misfolding (e.g. exposure of hydrophobic plates in aqueous environments or unpaired sulfhydryl groups in cysteines), frequently with relatively low affinity. Accordingly, GPCRs that retain ligand binding and effector coupling, but are recognized as misfolded by such general criteria, can be misrouted to the ER. Their rescue with pharmacoperones leads to proper folding, passage through the QCS, restoration to the proper site and return of function.
In principle, the pharmacoperone rescue approach may apply to a diverse array of human diseases that result from protein misfolding—among these are cystic fibrosis (misfolding of the cystic fibrosis transmembrane conductance regulator, a transporter class ion channel that transports chloride and thiocyanate ions (Amaral 2006; Dormer, et al. 2001; Galietta, et al. 2001; Zhang, et al. 2003)), hypogonadotropic hypogonadism (HH, The GnRH receptor(Ulloa-Aguirre, et al. 2003)), nephrogenic diabetes insipidus (Bernier, et al. 2004; Bichet 2006; Morello and Bichet 2001), retinitis pigmentosa (rhodopsin (Noorwez, et al. 2004)), hypercholesterolemia (LDL receptor), cataracts (crystallines (Benedek, et al. 1999)), neurodegenerative diseases (Huntington's, Alzheimer's, Parkinson's (Forloni, et al. 2002; Heiser, et al. 2000; Muchowski and Wacker 2005; Permanne, et al. 2002; Soto, et al. 2000)) and particular cancers (Peng, et al. 2003). In the case of certain proteins (e.g. the GnRHR, V2R and rhodopsin), this approach has succeeded with a striking number of different mutants (Conn et al. 2007), supporting the view that pharmacoperones will become powerful weapons in our therapeutic arsenal (Conn et al. 2007).
It has also become clear that variable (but significant) amounts of even some WT GPCRs are misrouted (i.e. frequently retained in the ER), apparently as a result of misfolding (Andersson, et al. 2003; Cook, et al. 2003; Lu, et al. 2003; Lu, et al. 2004; Petaja-Repo, et al. 2001; Petaja-Repo, et al. 2000; Pietila, et al. 2005). This suggests a level of post-translational control that may provide yet another level of potential therapeutic intervention (Ulloa-Aguirre, et al. 2006): increasing the routing of WT GPCRs. In principle, the (endogenous) chaperoning system could mediate the fraction of the newly synthesized receptor that traverses to the PM and may contribute to the physiological changes in levels of GnRHR that are observed in different physiological settings (Marian, et al. 1981).
Physiological significance and rational basis of selection of the hGnRHR model
The gonadotropin-releasing hormone receptor (GnRHR, also known as the luteinizing hormone releasing hormone receptor, LHRHR) resides in the gonadotrope cells of the pituitary and is responsible for producing responses to hypothalamic GnRH, such as the releasing of the gonadotropins, luteinizing hormone (LH) and follicle stimulating hormone (FSH). When the function of this receptor is lost due to mutation, hypogonadotropic hypogonadism (HH) results.
Studies of the hGnRHR have led to useful drugs (agonists and antagonists) for the treatment of reproductive disorders and for cancer. Among the specific reasons for the view that the GnRH is an excellent model for these studies are the following considerations:
GnRHR itself is among the smallest GPCRs (328 amino acids in the human). There are technical advantages to small proteins since these require fewer primers for synthesis and for sequencing than do larger GPCRs (typically at least twice the size of the GnRHR), and there is less sequence length that might lead to random mutation during the PCR process. DNA sequencing of mutants is also cheaper than for a larger protein. The size of the GnRHR means that there are fewer domains to consider in identification of important structural motifs and because the size of hydrophobic domains is relatively constant even in different GPCRs (due to the relatively constant size needed to traverse the membrane), the ratio of these to non-hydrophobic regions is relatively high in the GnRHR due to the short NH2- and COOH-terminals. This promotes the interaction of small hydrophobic molecules that dissolve in the membrane.
The ligand (GnRH) itself and analogs are small and have good thermal and chemical stability which has led to the availability of thousands of agonists and antagonists in the literature; these have been used for I125-radioligand assays and coupling of markers (colloidal gold, fluorescein, Texas Red).
The physiology of the system is well-characterized in many animal models and there is a broad basis for understanding processing differences now known to occur in closely-related species (Knollman, et al. 2005). We have taken advantage of the large number of GnRHR sequences that are available and been able to determine how changes in routing have been impacted by sequence changes (i.e. natural mutation (Janovick, et al. 2006)). Among primate receptors, for example, there are specializations that make these receptors able to decode frequency-modulated, as well as amplitude-modulated, signals (Janovick, et al. 2007b) and note structural changes among particular animals that appear to be explained by reproductive specializations (Janovick et al. 2007b; Janovick et al. 2006; Knollman et al. 2005). The human GnRHR, unlike the rat and mouse counterparts, appears “balanced” in its distribution between the PM and ER (Conn, et al. 2006a; Conn, et al. 2006b), so much that about 50% of the human WT GnRHR (in cells transfected with the corresponding sequence) is retained in the ER and can be “rescued” by the approach described above. At first consideration this appears to be an inefficient use of newly synthesized protein; however, the strong and convergent evolutionary pressure for this suggests a regulatory advantage (Conn et al. 2006a; Conn et al. 2006b; Ulloa-Aguirre et al. 2006). This system offers the ability to examine the evolution of the QCS system since these receptors have been cloned from a wide range of animals (fish, birds, reptiles, many mammals and multiple primates (Janovick, et al. 2003b)).
A great deal of information is also available regarding the cellular mechanism of action and trafficking of the GnRHR (Jennes, et al. 2009) as is substantive information on the mechanism of misfolding (Conn et al. 2006a; Conn et al. 2006b), mutant interactions with pharmacoperones (Ulloa-Aguirre et al. 2003) and the molecular basis of the dominant-negative effect (Conn et al. 2006a; Conn et al. 2006b). Recent studies (Janovick, et al. 2007a) indicate that the mutant receptor that is already trapped in the ER can be freed by pharmacoperones—a surprising result that increases the potential therapeutic reach of this approach since pharmacoperones do not need to be present at the moment of receptor synthesis.
Pharmacoperones
A pharmacoperone (from “pharmacological chaperone”) is a small molecule that enters cells and serves as a “molecular scaffold” to promote correct folding of otherwise-misfolded mutant proteins within the cell (Conn, et al. 2002; Janovick, et al. 2002). Misfolded proteins are often retained by the cellular quality control system (QCS) of the endoplasmic reticulum (ER). For this reason, they do not reach their normal site of function (Bernier et al. 2004; Ulloa-Aguirre, et al. 2004). This event may result in disease (Castro-Fernandez, et al. 2005). Pharmacoperones can rescue misfolded receptor mutants and restore them to function, which is a potentially useful therapeutic approach when the target is a misfolded/misrouted protein. The literature was summarized for the gonadotropin releasing hormone (GnRH) and vasopressin type 2 (V2) receptors pharmacoperones with a view toward moving these compounds in vivo (Conn et al. 2007). Science writers commenting on this approach (Hunter 2004; Kresge 2004) have observed that rescue with pharmacoperones is a viable “alternative (to gene therapy)” since it serves as a means of “skirting gene therapy to correct genetic defects.” This view is supported by the consideration that correction of defective protein folding appears significantly less challenging than replacement of a defective gene (or gene product) by a perfect one. One could, in fact, envision drugs given in a prophylactic manner (in vitamins, for example) that prevent the misfolding that leads to neurodegenerative disorders (Bellotti, et al. 2007; Sun, et al. 2007) and cataracts. In this regard, diseases may be prevented before clinical signs present themselves.
In addition to controlling the trafficking of mutants, this class of drugs may be useful for regulation of the plasma membrane expression of WT proteins since, for some plasma membrane receptors, a percentage of the nascent wild-type (WT) proteins also undergo misfolding (examples in(Conn et al. 2006a)) and are retained; this event may serve a cellular regulatory role (Conn et al. 2006a; Conn et al. 2006b). Pharmacoperones can be used to increase plasma membrane expression of these, as well, and offer a therapeutic means of regulating these GPCRs. Accordingly, pharmacoperones can be viewed as agents that rescue misfolded mutants, but are also potentially able to increase the plasma membrane expression of WT proteins.
1. Problems with existing pharmacoperones and screening techniques
One problem with in vivo use of known pharmacoperones is that almost all existing ones for GPCRs are peptidomimetic antagonists directed against a site near that for the native ligand. This is because existing pharmacoperones, including In3 described below, were selected from assays that identified antagonists with target specificity. In3 is highly specific for the GnRHR and was screened for more than 100 other structures, including α-adrenergics, β-adrenergics, adenosine receptor, bradykinin, CB1 and CB2, dopamine receptors, neurokinins, prostanoid receptors, serotonin receptors, somatostatin, calcium, sodium, potassium channels, monoamine oxidases, and several phosphatases. There are a few exceptions to the use of antagonists-only as pharmacoperones: one agonist has been used for this purpose (Petaja-Repo, et al. 2002) and one molecule has been used that does not appear to compete for the agonist or antagonist binding site (Janovick, et al. 2009b). The latter was identified by our group, based on a fortuitous observation, not a screening process. The “overlap” of the majority of the pharmacoperones with the binding site means that there will likely be competition in vivo that will necessitate episodic administration and washout when used in vivo. From a therapeutic point of view, this presents a hurdle since oral dosing, while desirable (and possible with small molecules), is more difficult (there must be a washout period to prevent the pharmacoperone from competing with the endogenous ligand). It has certainly not been established that binding at or near the binding site of the natural ligand is a necessary pre-requisite for pharmacoperone activity and there is extant information to suggest otherwise (Conn and Janovick 2009; Janovick, et al. 2009a; Janovick et al. 2009b). This would, in fact, be an unexpected requirement since one could imagine pharmacoperones that might stabilize the correctly routed form of the receptor and not show any antagonism. Accordingly, identification of non-antagonistic pharmacoperones is a reasonable goal.
Since most HTS approaches in the pharmaceutical industry revolved around assay models that identified agonists and antagonists, pharmacoperones that were not also agonists or antagonists would have been missed. Accordingly, rethinking the nature of the assay is important both to identify such overlooked structures, but also because they may prove to be more useful and therapeutically facile to use.
It is also worth noting that use of pharmacoperones drugs may unexpectedly alter physiological regulation. As an example, while correction of decreased luteinizing hormone in the male due to the E90K mutations in the GnRHR is likely to be straightforward, such a correction in females may be quite difficult, due to physiological cyclicity and elaborate feedback mechanisms.
2. New screening techniques
We have recently developed a new approach for the identification of pharmacoperones for hGnRHR mutants based on increase of activity of model mutants.
2.1. Primary Screen
The primary screen for pharmacoperones consists of measuring gain of activity in stable HeLa cells expressing the indicated mutant (hGnRHR[E90K]) under control of the tetracycline-controlled transactivator; the parent line was kindly supplied by Dr. Peter Seeburg (Max-Planck-Institut für Medizinische Forschung). Preparation of this mutant has been described (Janovick et al. 2002). The DNA was inserted in the host cell according to published methods (Krestel, et al. 2001).
2.2. Negative Screen
This assay is important in order to preclude moving “false” positives through the process. Stable cell lines which contain the tetracycline-controlled transactivator, which shuts the gene off in the presence of this antibiotic are used (Krestel, et al. 2001). There is literally no measurable expression, as assessed by protein expression or real time PCR in the presence of tetracycline because the mutant GPCRs are under the control of this transactivator. Accordingly, cells cultured in the presence of tetracycline are substantially identical to the primary screen, but lack expression of the target gene and gene product. These cells (in the presence of tetracycline) will serve as an excellent negative control line. In both cases, coupling to second messenger is the measured endpoint.
Candidate compounds could also be screened in a radioligand assay (Janovick et al. 2007b) to determine if they compete with receptor agonists and antagonists. Other routine necessary negative controls, secondary screens, counter-screens and tests for specificity, range, linearity, accuracy, precision, limit of detection and limit of quantitation, robustness, reagent stability and otherwise are not described in this manuscript due to space limitations.
We expect this assay to identify as “specific” other proteins that alter cellular machinery (e.g. ubiquitin ligases, heat shock proteins). It will be necessary to subject hits to a qualifying assay with a non-GnRHR GPCR to exclude such false positives.
3. Selection of the mutants
GnRHR Mutant: The naturally-occurring E90K mutation was selected. Modeling studies (Janovick, et al. 2003a) for the hGnRHR and the data (Janovick et al. 2003a; Janovick et al. 2002; Knollman et al. 2005) support the view that the E90-K121 salt bridge is a fundamental and evolutionarily conserved determinant required for correct protein trafficking to the PM in all mammals examined. This bridge, linking TMS2 to TMS3 is highly conserved in the GnRHR. E90 is conserved in mammals. Even in non-mammals, the TMS2-TMS3 salt bridge is needed and may have been formed by a D98-K121 bridge. D98 is absolutely conserved in all mammalian, reptilian, avian and piscine GnRHRs sequenced to date. K121 is conserved in the same groups; in fruit flies, the residue is also a conservative change R121. A negative amino acid in TMS2 is also found in all other GPCRs (Ni and Lee 2007) of the B-class, except the glucagon receptor. Because this salt bridge is a basic requirement for correct routing, E90K results in a routing defect in both mouse and human GnRHR (Brothers, et al. 2004; Janovick et al. 2007a; Knollman et al. 2005). This leads to full, but rescuable, ER-retention (Brothers et al. 2004) and the predicted phenotype. While it is true that the orthologs of many misrouted human mutants are correctly routed in the mouse (or rat, (Knollman et al. 2005)), E90K behaves similarly in mouse and human sequences (Knollman et al. 2005); the absence of K191 is without impact (Knollman et al. 2005). Further, in cell expression studies, mouse E90K, like the human mutant can be rescued by pharmacoperones (Knollman et al. 2005).
4. Advantages and use of the stably transfected “tTa” cells as an assay for pharmacoperones
We use HeLa cells for the basis of our screen; such cells are human in origin and contain the same major (endogenous) chaperone proteins that are found in most human cells (Feder and Hofmann 1999; Lievremont, et al. 1997; Ravindran, et al. 2005); hGnRHR is processed, expressed and coupled properly in these cells (Knollman and Conn 2008) and they are adherent and well-adapted to cell culture. A special feature of the cell lines used (Krestel et al. 2001) is that the GPCRs or GPCR mutants are expressed under the control of the tTA (tetracycline-controlled transactivator). In the absence of tetracycline, the GPCR is expressed. In the presence of tetracycline, the GPCR is not measurably expressed. This model allows use of the GPCR to measure signal in the HTS (i.e. no tetracycline).
4.1 Cell Culture and RNA Extraction for Real Time PCR
Stably transfected HeLa (tTA) cells were stably transfected with hE90K and then plated at 200,000 cells per well in a 6-well culture plate for extracting RNA. The cells were grown in DMEM/10% fetal calf serum, containing gentamycin, in a humidified atmosphere at 37 C. To measure the time needed to “turn off” the gene, 1μg/ml Tetracycline was added to each well and remained on the indicated cells for a maximum of 12 h. Cells were examined at differing times by dumping off medium, blotting, placing on ice and washing 2 times with 2.5ml 1× PBS (ice cold). Then 1ml TRIZOL reagent is added per well and followed according to the manufacturer's protocol. The cells were pipetted 7 times through an RNase free pipet tip to lyse and transferred to a 1.5ml RNase free centrifuge tube and frozen at –80 C until the total RNA was extracted. After extracting the RNA, it was dissolved in 25μl diethylpyrocarbonate-treated water.
4.2 Real Time PCR
The TaqMan assay was used with the TaqMan PCR Core Reagent Kit with the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). The full length sequence of the human GnRH receptor was used to design the primers and probe for the real-time PCR assays for the mutant E90K. The TaqMan probe for the GnRHR mutant E90K was synthesized by Applied Biosystems ; 5’-FAM-CTCCCTGGCTATCACGAGGCCCCTAG-MGB-3’. For the internal control, 18s RNA, ABI's MGB probe was used in the TaqMan assay.
The following evidence suggests that this screen will be extremely valuable:
4.3. RNA for the GPCR mutant is expressed under the control of tetracycline
Stable cells were cultured in the presence of tetracycline for varying amounts of time in order to show a time course for complete extinction of expression of the E90K mutant. Total RNA was extracted from the cells followed by reverse transcription for obtaining single strand DNA for use in real-time PCR. One microgram of total RNA was used for reverse transcription (RT) to generate single strand DNA. Relative quantitative standard curves for E90K and 18s RNA (the internal housekeeping gene product used to normalize the data or calculate a ratio between the gene of interest and the endogenous and unchanged standard) were generated by serial dilutions of 1:10 of the RT product ranging from 1-10 ng. Ct values (Ct, the cycle threshold, is the cycle at which the fluorescence from a sample crosses the set threshold) were obtained, averaged and converted to (logorithim) nanograms using the straight line extrapolation equation: ([ave Ct value]-b)/m; b = y-intercept of standard curve line, m = slope of standard curve line). The ratio of E90K to 18s RNA was determined. The data (Figure 1) show that by 12 h in the presence of tetracycline, the RNA for hGnRHR[E90K] is no longer detectable.
Figure 1.
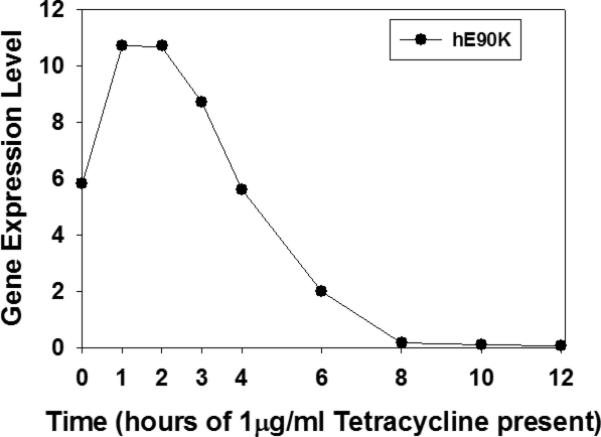
Tetracycline inhibits accumulation of E90K mRNA (normalized with 18S RNA) and assessed by realtime PCR.
4.4. The rescuable GPCR mutant is under the control of tetracycline and rescuable by pharmacoperone, In3
In this study (Figure 2), 20,000 stable cells / 250 μl were cultured in the presence of increasing concentrations of tetracycline, which was continuously present thereafter, for a total of 48 h. Cells (48-well plates) were treated with or without pharmacoperone In3 (“In3” is our lab name for pharmacoperone, [(2S)-2-[5-[2-(2-azabicyclo[2.2.2]oct-2-yl)-1,1-dimethyl-2-oxoethyl]-2-(3,5-dimethylphenyl)-1H-indol-3-yl]-N-(2-pyridin-4-ylethyl)propan-1-amine]), which was originally developed as a specific antagonist of the GnRHR (Ashton, et al. 2001). The pharmacoperone is then removed and cells labeled with 3H-inositol for 18 hours and then stimulated with 10-6 M Buserelin and total inositol phosphate (IP) was then measured by ion exchange chromatography (Huckle and Conn 1987). These studies show that stimulation of IP production in response to the GnRHR agonist, Buserelin, is lost in the presence of tetracycline and that the known pharmacoperone In3 can rescue this mutant with an excellent signal-to-noise ratio (Figure 2). The IP assay, using disposable columns, resin and a skilled operator can conveniently assay 1,000 samples in a day.
Figure 2.
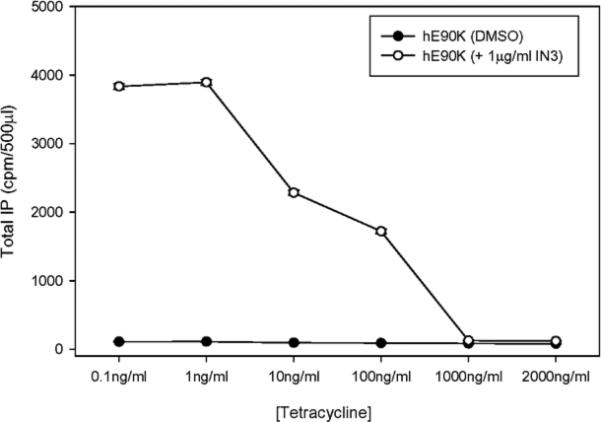
Loss of IP production (in the presence of tetracycline) in stably transfected cells with or without In3 treatment shows that expression of the (rescued) mutant protein is under control of this antibiotic.
4.5
In order to perform (i) a preliminary assessment of the assay performance and (ii) demonstrate the ability to perform the assay in 96-well plates, the study, shown in Figure 3, was performed (all wells in 1% DMSO). This study also (iii) shows the utility of cells treated with tetracycline (which extinguishes expression of the mutant hGnRHR gene) as a negative screen. The upper frame in Figure 3 demonstrates that positives can be identified with a good sensitivity and high signal to noise ratio, with minimal wall effects and good reproducibility.
Figure 3.
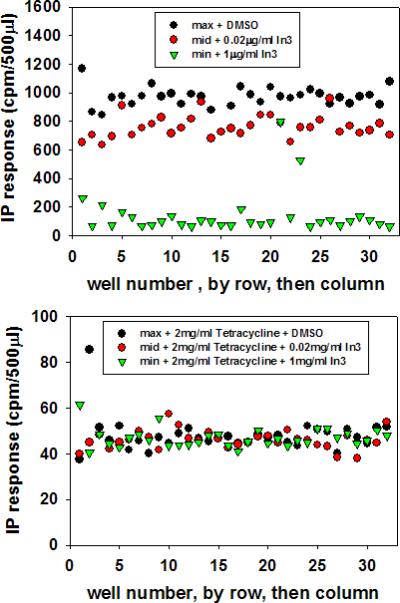
Well-by-well IP stimulation in response to a GnRH agonist, following rescue of mutant E90K by maximum, EC80 or no pharmacoperone (In3). Data is graphed by row, then column. The upper graphic shows the absence of tetracycline; the lower graphic shows the presence of tetracycline. Note that the Y-axis differs.
In this study, stable cells were cultured in the absence (upper frame) or presence (lower frame) of tetracycline which was continuously present thereafter, as described above (“b”). Pharmacoperone was added at the indicated concentrations in 1% DMSO final. Total IP was measured. The x-axis was numerically compressed for this image and is labeled 0-32. There are 32 (interspersed) wells of either DMSO only (DMSO control, green), an EC80 dose of pharmacoperone In3 in 1% DMSO (red) or a saturating dose of In3 in 1% DMSO (black); 3 × 32=96. Every third well contained DMSO, EC80 dose of pharmacoperone In3, or a saturating dose of pharmacoperone In3 so that the next row of wells contained the same treatment diagonally. In Figure 3, rescue was performed on cells in the absence of tetracycline (which allows the receptor mutant to be expressed). This would serve as the primary screen.
In the lower image of Figure 3, tetracycline was present and, accordingly, the gene for the hGnRHR mutant was shut off. This would serve as a negative screen for agents that affect their action by some means other than through the hGnRHR. In that image, there is no significant or reproducible response difference between cells that have been incubated with pharmacoperone In3 because the gene for the mutant receptor is not expressed. Agents that act through a means other than the hGnRHR would be expected to still respond since, other than lacking the hGnRHR (mutant), the cells are otherwise identical.
Figure 4 shows the same data expressed in a “column then row” format instead of the “row then column” format above. This shows, by inspection, the absence of wall effects. These are aberrant effects sometimes observed for cells plated in a well adjacent to an edge or corner of a culture plate.
Figure 4.
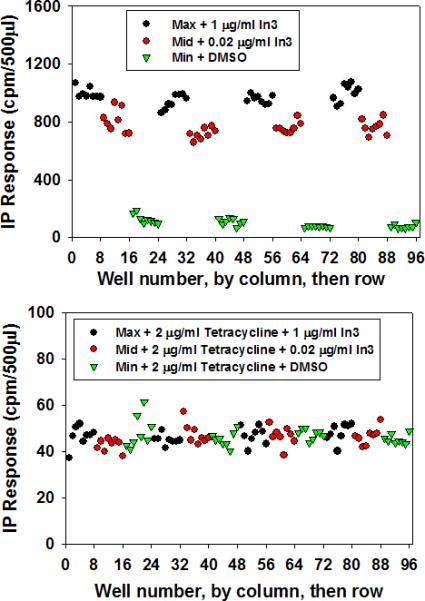
The same data as in Figure 3, but re-graphed by column, then row in order to reveal any “edge” effects or other artifacts due to the plate.
Conclusion
While pharmacoperones may also be agonists or antagonists and are frequently selected from HTS assays for such drugs, this is not a requirement. Pharmacoperones have the potential to override normal physiological levels of receptors and other proteins. This approach may result in altered regulation and may change the number of receptors present at the plasma membrane by rescuing misfolded mutants or increasing plasma membrane expression of fractionally expressed WT receptors. Accordingly, the screening approach we describe is useful for treatment of diseases in which the underlying caused is loss of receptors due to mutation and subsequent misrouting (retention in the ER). This is a novel screen that may lead to identification of pharmacoperones that do not compete with the endogenous ligands, since these agents may have a therapeutic advantage. In principle this approach can form the basis of an HTS for the identification of pharmacoperone drugs for mutant GPCRs, enzymes, or ion channels. Because there are a paucity of such drugs known, the ability to identify them in chemical libraries is one of the keys to moving this approach forward.
Acknowledgements
This work was supported by NIH grants: DK-85040, RR-030229, RR-00163 and HD-18185. We thank Jo Ann Binkerd for formatting the manuscript. We thank Merck and Company for the GnRH peptidomimetic antagonist used.
Footnotes
Conflict of Interest Statement The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Amaral MD. Therapy through chaperones: sense or antisense? Cystic fibrosis as a model disease. J Inherit Metab Dis. 2006;29:477–487. doi: 10.1007/s10545-006-0251-x. [DOI] [PubMed] [Google Scholar]
- Andersson H, D'Antona AM, Kendall DA, Von Heijne G, Chin CN. Membrane assembly of the cannabinoid receptor 1: impact of a long N-terminal tail. Mol Pharmacol. 2003;64:570–577. doi: 10.1124/mol.64.3.570. [DOI] [PubMed] [Google Scholar]
- Ashton WT, Sisco RM, Kieczykowski GR, Yang YT, Yudkovitz JB, Cui J, Mount GR, Ren RN, Wu TJ, Shen X, et al. Orally bioavailable, indole-based nonpeptide GnRH receptor antagonists with high potency and functional activity. Bioorg Med Chem Lett. 2001;11:2597–2602. doi: 10.1016/s0960-894x(01)00512-1. [DOI] [PubMed] [Google Scholar]
- Bellotti V, Nuvolone M, Giorgetti S, Obici L, Palladini G, Russo P, Lavatelli F, Perfetti V, Merlini G. The workings of the amyloid diseases. Ann Med. 2007;39:200–207. doi: 10.1080/07853890701206887. [DOI] [PubMed] [Google Scholar]
- Benedek GB, Pande J, Thurston GM, Clark JI. Theoretical and experimental basis for the inhibition of cataract. Prog Retin Eye Res. 1999;18:391–402. doi: 10.1016/s1350-9462(98)00023-8. [DOI] [PubMed] [Google Scholar]
- Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15:222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Bichet DG. Nephrogenic diabetes insipidus. Adv Chronic Kidney Dis. 2006;13:96–104. doi: 10.1053/j.ackd.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Cornea A, Janovick JA, Conn PM. Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol. 2004;18:1787–1797. doi: 10.1210/me.2004-0091. [DOI] [PubMed] [Google Scholar]
- Castro-Fernandez C, Maya-Nunez G, Conn PM. Beyond the signal sequence: protein routing in health and disease. Endocr Rev. 2005;26:479–503. doi: 10.1210/er.2004-0010. [DOI] [PubMed] [Google Scholar]
- Conn PM, Janovick JA. Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol Cell Endocrinol. 2009;299:137–145. doi: 10.1016/j.mce.2008.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Janovick JA, Brothers SP, Knollman PE. ‘Effective inefficiency’: cellular control of protein trafficking as a mechanism of post-translational regulation. J Endocrinol. 2006a;190:13–16. doi: 10.1677/joe.1.06771. [DOI] [PubMed] [Google Scholar]
- Conn PM, Knollman PE, Brothers SP, Janovick JA. Protein folding as posttranslational regulation: evolution of a mechanism for controlled plasma membrane expression of a G protein-coupled receptor. Mol Endocrinol. 2006b;20:3035–3041. doi: 10.1210/me.2006-0066. [DOI] [PubMed] [Google Scholar]
- Conn PM, Leanos-Miranda A, Janovick JA. Protein origami: therapeutic rescue of misfolded gene products. Mol Interv. 2002;2:308–316. doi: 10.1124/mi.2.5.308. [DOI] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59:225–250. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- Cook LB, Zhu CC, Hinkle PM. Thyrotropin-releasing hormone receptor processing: role of ubiquitination and proteasomal degradation. Mol Endocrinol. 2003;17:1777–1791. doi: 10.1210/me.2003-0073. [DOI] [PubMed] [Google Scholar]
- Dormer RL, Derand R, McNeilly CM, Mettey Y, Bulteau-Pignoux L, Metaye T, Vierfond JM, Gray MA, Galietta LJ, Morris MR, et al. Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci. 2001;114:4073–4081. doi: 10.1242/jcs.114.22.4073. [DOI] [PubMed] [Google Scholar]
- Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol. 1999;61:243–282. doi: 10.1146/annurev.physiol.61.1.243. [DOI] [PubMed] [Google Scholar]
- Forloni G, Terreni L, Bertani I, Fogliarino S, Invernizzi R, Assini A, Ribizzi G, Negro A, Calabrese E, Volonte MA, et al. Protein misfolding in Alzheimer's and Parkinson's disease: genetics and molecular mechanisms. Neurobiol Aging. 2002;23:957–976. doi: 10.1016/s0197-4580(02)00076-3. [DOI] [PubMed] [Google Scholar]
- Galietta LJ, Springsteel MF, Eda M, Niedzinski EJ, By K, Haddadin MJ, Kurth MJ, Nantz MH, Verkman AS. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem. 2001;276:19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, Lehrach H, Wanker EE. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc Natl Acad Sci U S A. 2000;97:6739–6744. doi: 10.1073/pnas.110138997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckle WR, Conn PM. Use of lithium ion in measurement of stimulated pituitary inositol phospholipid turnover. Methods Enzymol. 1987;141:149–155. doi: 10.1016/0076-6879(87)41063-x. [DOI] [PubMed] [Google Scholar]
- Hunter PJ. Receptor redemption: skirting gene therapy to correct genetic defects. The Scientist. 2004;18:30. [Google Scholar]
- Janovick JA, Brothers SP, Cornea A, Bush E, Goulet MT, Ashton WT, Sauer DR, Haviv F, Greer J, Conn PM. Refolding of misfolded mutant GPCR: post-translational pharmacoperone action in vitro. Mol Cell Endocrinol. 2007a;272:77–85. doi: 10.1016/j.mce.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Brothers SP, Knollman PE, Conn PM. Specializations of a G-protein-coupled receptor that appear to aid with detection of frequency-modulated signals from its ligand. FASEB J. 2007b;21:384–392. doi: 10.1096/fj.06-6901com. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003a;305:608–614. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Knollman PE, Brothers SP, Ayala-Yanez R, Aziz AS, Conn PM. Regulation of G protein-coupled receptor trafficking by inefficient plasma membrane expression: molecular basis of an evolved strategy. J Biol Chem. 2006;281:8417–8425. doi: 10.1074/jbc.M510601200. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87:3255–3262. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Maya-Nunez G, Ulloa-Aguirre A, Huhtaniemi IT, Dias JA, Verbost P, Conn PM. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol Cell Endocrinol. 2009a;298:84–88. doi: 10.1016/j.mce.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Patny A, Mosley R, Goulet MT, Altman MD, Rush TS, 3rd, Cornea A, Conn PM. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol. 2009b;23:157–168. doi: 10.1210/me.2008-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Ulloa-Aguirre A, Conn PM. Evolved regulation of gonadotropin-releasing hormone receptor cell surface expression. Endocrine. 2003b;22:317–327. doi: 10.1385/ENDO:22:3:317. [DOI] [PubMed] [Google Scholar]
- Jennes L, Ulloa-Aguirre A, Janovick JA, Adhan V, Conn PM. Gonadotropin releasing hormone and its receptor. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, Brain and Behavior. 2nd Edition Academic Press; New York: 2009. pp. 1645–1668. [Google Scholar]
- Knollman PE, Conn PM. Multiple G proteins compete for binding with the human gonadotropin releasing hormone receptor. Arch Biochem Biophys. 2008;477:92–97. doi: 10.1016/j.abb.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollman PE, Janovick JA, Brothers SP, Conn PM. Parallel regulation of membrane trafficking and dominant-negative effects by misrouted gonadotropin-releasing hormone receptor mutants. J Biol Chem. 2005;280:24506–24514. doi: 10.1074/jbc.M501978200. [DOI] [PubMed] [Google Scholar]
- Kresge N. Pharmacological Chaperones Show. Am Soc for Biochem Molec Bio Today. 2004;3:10–11. [Google Scholar]
- Krestel HE, Mayford M, Seeburg PH, Sprengel R. A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res. 2001;29:E39. doi: 10.1093/nar/29.7.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievremont JP, Rizzuto R, Hendershot L, Meldolesi J. BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+. J Biol Chem. 1997;272:30873–30879. doi: 10.1074/jbc.272.49.30873. [DOI] [PubMed] [Google Scholar]
- Lu M, Echeverri F, Moyer BD. Endoplasmic reticulum retention, degradation, and aggregation of olfactory G-protein coupled receptors. Traffic. 2003;4:416–433. doi: 10.1034/j.1600-0854.2003.00097.x. [DOI] [PubMed] [Google Scholar]
- Lu M, Staszewski L, Echeverri F, Xu H, Moyer BD. Endoplasmic reticulum degradation impedes olfactory G-protein coupled receptor functional expression. BMC Cell Biol. 2004;5:34. doi: 10.1186/1471-2121-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian J, Cooper RL, Conn PM. Regulation of the rat pituitary gonadotropin-releasing hormone receptor. Mol Pharmacol. 1981;19:399–405. [PubMed] [Google Scholar]
- Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607–630. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorwez SM, Malhotra R, McDowell JH, Smith KA, Krebs MP, Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- Peng Y, Li C, Chen L, Sebti S, Chen J. Rescue of mutant p53 transcription function by ellipticine. Oncogene. 2003;22:4478–4487. doi: 10.1038/sj.onc.1206777. [DOI] [PubMed] [Google Scholar]
- Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, Dewachter I, Banks WA, Van Leuven F, Soto C. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a beta-sheet breaker peptide. FASEB J. 2002;16:860–862. doi: 10.1096/fj.01-0841fje. [DOI] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M. Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J. 2002;21:1628–1637. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Laperriere A, Bhalla S, Walker P, Bouvier M. Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J Biol Chem. 2001;276:4416–4423. doi: 10.1074/jbc.M007151200. [DOI] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Laperriere A, Walker P, Bouvier M. Export from the endoplasmic reticulum represents the limiting step in the maturation and cell surface expression of the human delta opioid receptor. J Biol Chem. 2000;275:13727–13736. doi: 10.1074/jbc.275.18.13727. [DOI] [PubMed] [Google Scholar]
- Pietila EM, Tuusa JT, Apaja PM, Aatsinki JT, Hakalahti AE, Rajaniemi HJ, Petaja-Repo UE. Inefficient maturation of the rat luteinizing hormone receptor. A putative way to regulate receptor numbers at the cell surface. J Biol Chem. 2005;280:26622–26629. doi: 10.1074/jbc.M413815200. [DOI] [PubMed] [Google Scholar]
- Ravindran RK, Tablin F, Crowe JH, Oliver AE. Resistance to dehydration damage in HeLa cells correlates with the presence of endogenous heat shock proteins. Cell Preservation Technology. 2005;3:155–164. [Google Scholar]
- Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, Kascsak R, Mendez E, Harris DA, Ironside J, et al. Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet. 2000;355:192–197. doi: 10.1016/s0140-6736(99)11419-3. [DOI] [PubMed] [Google Scholar]
- Sun Y, Breydo L, Makarava N, Yang Q, Bocharova OV, Baskakov IV. Site-specific conformational studies of prion protein (PrP) amyloid fibrils revealed two cooperative folding domains within amyloid structure. J Biol Chem. 2007;282:9090–9097. doi: 10.1074/jbc.M608623200. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM. Misrouted cell surface receptors as a novel disease aetiology and potential therapeutic target: the case of hypogonadotropic hypogonadism due to gonadotropin-releasing hormone resistance. Expert Opin Ther Targets. 2003;7:175–185. doi: 10.1517/14728222.7.2.175. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM. G-protein-coupled receptor trafficking: understanding the chemical basis of health and disease. ACS Chem Biol. 2006;1:631–638. doi: 10.1021/cb600360h. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Wang XT, Yue H, Leung SW, Thibodeau PH, Thomas PJ, Guggino SE. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2003;278:51232–51242. doi: 10.1074/jbc.M309076200. [DOI] [PubMed] [Google Scholar]