

Abstract
Mutations in human PRPF31 gene have been identified in patients with autosomal dominant retinitis pigmentosa (adRP). To begin to understand mechanisms by which defects in this general splicing factor cause retinal degeneration, we examined the relationship between PRPF31 and pre-mRNA splicing of photoreceptor-specific genes. We used a specific anti-PRPF31 antibody to immunoprecipitate splicing complexes from retinal cells and identified the transcript of rhodopsin gene (RHO) among RNA species associated with PRPF31-containing complexes. Mutant PRPF31 proteins significantly inhibited pre-mRNA splicing of intron 3 in RHO gene. In primary retinal cell cultures, expression of the mutant PRPF31 proteins reduced rhodopsin expression and caused apoptosis of rhodopsin-positive retinal cells. This primary retinal culture assay provides an in vitro model to study photoreceptor cell death caused by PRPF31 mutations. Our results demonstrate that mutations in PRPF31 gene affect RHO pre-mRNA splicing and reveal a link between PRPF31 and RHO, two major adRP genes.
Keywords: autosomal dominant retinitis pigmentosa (adRP), PRPF31, pre-mRNA splicing, retinal cells, apoptosis, rhodopsin
Introduction
Retinitis pigmentosa (RP) is a common form of retinal degeneration. RP is characterized by progressive degeneration of the peripheral retina, leading to night blindness, loss of the peripheral visual field and an abnormal electroretinogram. The most prominent pathological feature of RP is the loss of photoreceptor, often followed by alterations in the retinal pigment epithelium and retinal glia with the appearance of bone spicule-like pigment deposits. RP is clinically and genetically heterogeneous, displaying all three modes of Mendelian inheritance: autosomal dominant RP (adRP), autosomal recessive RP (arRP), and X-linked RP (xlRP) (Inglehearn, 1998; Phelan and Bok, 2000; Dejneka and Bennett, 2001; Farrar et al., 2002; Rivolta et al., 2002; Hims et al., 2003; Pacione et al., 2003) (www.sph.uth.tmc.edu/RetNet).
Among ∼130 genetic loci associated with retinal degeneration, >50% have been cloned (Baehr and Chen, 2001; Dejneka and Bennett, 2001; Swaroop and Zack, 2002; Pacione et al., 2003). Although most adRP genes are expressed predominantly in the retina, three genes have been identified that are ubiquitously expressed in different tissues. These non-retina-specific adRP genes encode proteins essential for pre-mRNA splicing, including HPRP3 [for RP18 (Anthony et al., 1997; Chakarova et al., 2002)], PRPC8 [for RP13 (McKie et al., 2001; van Lith-Verhoeven et al., 2002)], and PRPF31 [for RP11 (Moore et al., 1993; Evans et al., 1995; Al-Maghtheh et al., 1996; Vithana et al., 2001)]. In fact, RP11 has been reported as the second most common locus for adRP (Vithana et al., 1998). However, mechanisms underlying the pathogenesis of RP caused by mutations in these ubiquitously expressed splicing factors are not understood.
Pre-mRNA splicing is a critical step in eukaryotic gene expression. The vast majority of mammalian transcription units contain introns that must be accurately excised to form functional mRNA. Pre-mRNA splicing reaction occurs in spliceosomes, the large RNA–protein complexes containing pre-mRNA, five small nuclear ribonucleoprotein (snRNP) particles, U1, U2, U4/U6, and U5, as well as a large number of accessory protein factors (Burge et al., 1999; Hastings and Krainer 2001; Zhou et al., 2002; Wu et al., 2004). Proteins required for the formation of stable U4/U6 snRNPs and for assembly of the U4/U6.U5 tri-snRNP have been identified, including HPRP3, PRPC8, and PRPF31. Yeast prp31 gene was identified in a screen of for splicing defects after a shift to nonpermissive temperatures. It encodes a 60 kDa protein, PRP31 also named as PRPF31, essential for yeast cell viability and critical for pre-mRNA splicing. PRPF31 is important for recruiting the U4/U6-U5 tri-snRNP to prespliceosome complexes or stabilizing these interactions (Maddock et al., 1996; Weidenhammer et al., 1996, 1997). It is not known how its mutations lead to retinal degeneration.
In this study, we examined the role of PRPF31 in the pre-mRNA splicing of photoreceptor-specific genes. We began by searching for potential splicing substrate genes for PRPF31 using an immunoprecipitation-coupled microarray assay. This experiment suggests that rhodopsin (RHO) is among the target splicing substrate genes for PRPF31. Cotransfection of adRP mutants of PRPF31 with a rhodopsin minigene inhibited rhodopsin pre-mRNA splicing. Expression of these mutant PRPF31 proteins significantly reduced rhodopsin expression in cultured retinal cells and induced apoptosis of retinal cells. These experiments establish a link between the general splicing factor PRPF31 and the expression of a critical retina-specific gene, rhodopsin.
Materials and Methods
Plasmid, oligonucleotides, and antibodies
The mammalian expression plasmids carrying either wild-type or mutant PRPF31 were constructed by inserting the corresponding cDNA fragments into pCS2 vector (Wu et al., 1999). PCR-amplified full-length PRPF31 open reading frame or fragments containing N-terminal 371 or 256 aa residues (N371 and N256 mutants, respectively) were inserted at the BamHI site downstream of the cytomegalovirus (CMV) promoter in the pCS2 vector. The expression of wild-type or mutant PRPF31 proteins was under the control of CMV promoter (Wu et al., 1999). The corresponding cDNA inserts as well as junction sequences were confirmed by using PRISM Ready reaction DyeDeoxy Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA). Oligonucleotides were purchased from IDT Integrated DNA Technologies (Coralville, IA). nti-PRPF31 polyclonal antibodies were prepared in chicks using synthetic peptides (Aves Laboratories, Tigard, OR). Other antibodies including anti-rhodopsin (Chemicon, Temecula, CA), anti-synapsin (Chemicon), and anti-green fluorescent protein (GFP) (Roche Applied Sciences, Welwyn Garden City, UK) were purchased from commercial suppliers.
Primary retinal neuronal culture, transfection, and neuronal cell death assays
Interspecific consomic strain (ICS) mice at postnatal day 0 (P0) to P5 were purchased from Charles River Laboratories (Wilmington, MA). Primary retinal cell culture was performed by modifying a published protocol (Luo et al., 2001). Briefly, the enucleation procedure was performed within minutes after killing. Ca2+−Mg2+-free Hank's medium (Invitrogen, Gaithersburg, MD) containing 20% glucose was used for retina dissection. The dissected retinal tissue was treated with 0.25% trypsin for 15 min at 37°C, and the digestion was terminated by adding heat-inactivated fetal bovine serum (HiFBS) to a final concentration of 20%. DNase (10 mg/ml; Sigma, St. Louis, MO) was added, and polished Pasteur pipettes were used to triturate dissected retinal tissue three times. The cell suspension was washed with culture medium three times. Dissociated cells were counted and plated on precoated coverslips (poly-l-lysine and laminin) at 4 × 105 cells per coverslip in six-well tissue culture dishes. Cells were allowed to attach to coverslips for at least 6 h before adding the DMEM/F-12 media containing 10% HiFBS.
Transfection was performed 24–48 h after the cultures were established, using 5 μg of DNA per well in six-well dishes with a modified calcium precipitation method as described previously (Jiang et al., 2000). When boc-aspartyl(OMe)-fluoromethylketone (BAF) (50–100 μm; Calbiochem, La Jolla, CA) was used, it was added to the culture medium 12 h before transfection. From >50 independent transfection experiments, the transfection efficiency was 3–7%, as detected by fluorescent microscopy of cells expressing GFP-tagged proteins. Data shown in this paper were derived from those experiments with comparable transfection efficiency (∼5%) among control and different experimental groups.
Cell morphology and viability were monitored by microscopic examination after staining with bis-benzimide (Sigma) and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay using tetra-methyl-rhodamine red staining kit (Roche). Immunofluorescent staining was performed to examine the expression of rhodopsin, synapsin, and PRPF31 in cultured cells using corresponding specific primary antibodies following secondary antibodies conjugated with either cyanine 3 (Cy3) or Cy2. Microscopic images were taken under an inverted microscope using an Axiocam digital camera (Zeiss, Oberkochen, Germany). In corresponding panels of each figure, the images were captured with the same exposure time and displayed with the same parameters so that the fluorescent intensity can be compared among the corresponding groups.
Immunoprecipitation microarray analysis
The detailed protocol will be described in a separate publication (D. Mordes and J. Y. Wu, unpublished observations), and a brief description is provided below. Affinity-purified polyclonal anti-PRPF31 against a synthetic peptide (corresponding to amino acid residue 416–432) was prepared by Aves Laboratories. Mouse retinas were dissected from 8-week-old ICS mice (Charles River Laboratories). Retinal cells were dissociated by gentle treatment with trypsin and lysed in cell lysis buffer (20 mm HEPES, pH 7.4, 10 mm NaCl, 5 mm MgCl2, and 0.1% NP-40). After a 10 min incubation on ice, cell nuclei were collected by centrifugation at 2000 rpm for 10 min and resuspended in cell lysis buffer containing 1% paraformaldehyde for 30 min at room temperature in the presence of protease inhibitors (Roche). The cell nuclei were pelleted by centrifugation as described above and resuspended in dilution buffer [20 mm HEPES, pH 7.4, 167 mm NaCl, 1.2 mm EDTA, 0.01% SDS, 0.1% Triton X-100, 1 mm DTT, 1× protease inhibitor (Roche), and 40 U/ml RNasin]. The nuclear lysate was sonicated for 10 × 30 s, and insoluble particles were removed by centrifugation at 13,000 rpm for 10 min at 4°C. Protein A/G-Sepharose beads (Sigma) were added to the supernatant to preclear the nuclear lysate. The nuclear lysate was precleared using protein A/G-Sepharose beads and was then incubated with the affinity-purified anti-PRPF31 antibody for 4 h at 4°C. The preimmune Ig preparation was used as a control. Pelleted beads were then sequentially washed with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm HEPES, pH 7.5, and 150 mm NaCl) and high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm HEPES, pH 7.5, and 500 mm NaCl). The enriched protein–RNA complex was eluted twice using freshly prepared elution buffer (1% SDS, 0.1 m NaHCO3) and centrifuged. DNA contaminants were removed by DNase I treatment, and proteins were digested by proteinase K treatment for 30 min at 45°C. Six volumes of Trizol (Invitrogen, San Diego, CA) were added, and RNA was precipitated in the presence of glycogen (Roche). The RNA samples immunoprecipitated with anti-PRPF31 or control preimmune Ig preparation were amplified by reverse transcription (RT)-PCR for 23–25 cycles using the SMART amplification kit according to the manufacturer's instructions (Clontech, Palo Alto, CA), except that the last five cycles of PCR were performed in the presence of a primer containing oligo-dT coupled to T7 promoter sequence. The amplified samples (∼5 μg) were labeled with biotinylated CTP and UTP in in vitro transcription using T7 RNA polymerase. The biotinylated cRNA samples were then fragmented and hybridized to Affymetrix (Santa Clara, CA) mouse genome chips (430 array, version 2.0) and further processed at Vanderbilt microarray core facility.
In situ hybridization assay
To examine the expression of PRPF31 in the retina, in situ hybridization was performed using an antisense RNA probe of PRPF31 with the sense probe as a control. The in situ hybridization was performed using a protocol as described previously (Li et al., 1999). Briefly, digoxygenin (DIG)-labeled sense and antisense probes for PRPF31 were prepared from linearized PRPF31 cDNA plasmids in in vitro transcription reactions using a DIG–RNA labeling kit (Roche). The retinal sections were deparaffinized, hydrated, treated with proteinase K (10 mg/ml), and fixed in 4% paraformaldehyde in PBS, pH 7.6. After treatment with triethanolamine and acetic anhydride, the slides were prehybridized for 1 h at 60° in hybridization solution (50% formamide, 5× SSC, 1× Denhardt's solution, 0.1% Tween 20, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 5 mm EDTA, 0.3 mg/ml yeast tRNA, and 0.1 mg/ml heparin) and then hybridized in the probe-containing hybridization solution overnight at 60°. After washing in SSC buffers (2× to 0.1× SSC), slides were incubated with preabsorbed anti-digoxygenin–alkaline phosphatase–Fab fragment in 20% sheep serum in PBT (PBS containing 2 mg/ml BSA and 0.1% Triton X-100) (1:2000 dilution) overnight at 4°C. After washing, color was developed with alkaline phosphatase substrates (nitroblue-tetrazolium-chloride/5-bromo-4-chloro-3-indolyl-phosphate) in the dark. Slides were mounted and photographed.
Results
Expression of PRPF31 gene in the retina and other tissues
Although mutations in PRPF31 gene have been identified in adRP patients, it is not clear whether the PRPF31 gene is differentially expressed in different types of retinal cells. We examined the expression of the PRPF31 gene in the retina. In situ hybridization was performed using an antisense PRPF31 RNA probe, as described previously (Li et al., 1999). The expression of PRPF31 was detected in the cells in several layers of the retina, with strong signals detected in outer nuclear layer (the nuclei of photoreceptor cells) and inner nuclear layer. The majority of photoreceptor cells express a high level of PRPF31 mRNA, and a fraction of retinal ganglion cells also show the expression of PRPF31 (Fig. 1A, part I). Such in situ signals are specific because they were not detectable when the sense control probe was used (Fig. 1B, part I). This PRPF31 expression pattern is consistent with the role of PRPF31 acting as a general pre-mRNA splicing factor important for photoreceptor cells.
Figure 1.
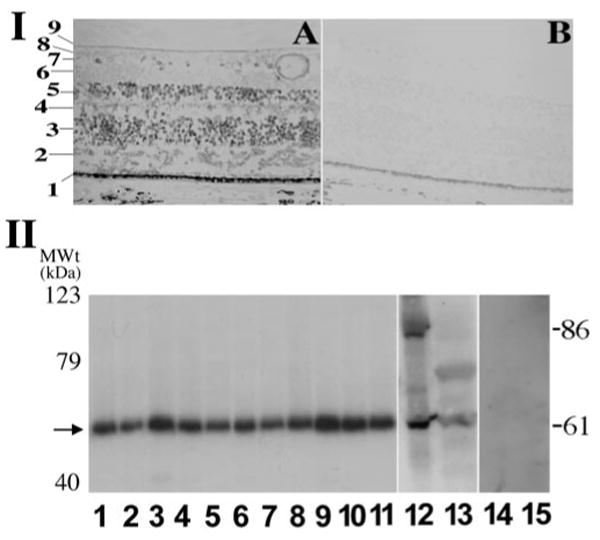
Detection of human PRPF31 gene expression by in situ hybridization and by Western blotting assays. I, Expression of PRPF31 gene in the retina, as detected by in situ hybridization. Cells in different layers of the retina express PRPF31, as detected by the PRPF31 antisense probe (A). The sense control probe does not show detectable signal (B). Distinct histologic layers of the retina are marked as follows: 1, retinal pigment epithelium; 2, photoreceptor cell layer; 3, outer nuclear layer, nuclei of photoreceptor cells; 4, outer plexiform layer; 5, inner nuclear layer; 6, inner plexiform layer; 7, ganglion cell layer; 8, retinal nerve fiber layer; 9, internal limiting membrane. II, Western blotting results showing a prominent band of 61 kDa in a range of adult murine tissues (lanes 1–9; 1, brain; 2, heart; 3, kidney; 4, spleen; 5, lung; 6, thymus; 7, muscle; 8, lymph node; 9, retina), in postnatal retina (from P2 mice, lane 10), in embryonic retina (from embryonic day 18 mice, lane 11), and in human HEK cells as well as stable HEK cells expressing a PREF31–GFP fusion protein (lanes 13 and 12, respectively). Protein concentrations of the cell lysates were measured, and equal amounts were loaded in each lane. These bands represent specific PRPF31 signals (61 kDa for the endogenous protein and 86 kDa for PRPF31–GFP fusion protein), because they are not detectable when preimmune antibody preparation was used (lanes 14 and 15). There is no significant change in the gel mobility of PRPF31 protein detected in different tissues. MWt, Molecular weight.
To test whether PRPF31 protein is expressed as unique isoforms or at particularly high levels in the retina compared with other tissues, we examined the PRPF31 protein in different tissues using Western blotting assay. Polyclonal anti-PRPF31 antibodies were raised in chicks against synthetic peptides corresponding to different regions in the human PRPF31 gene. The affinity-purified antibodies against the peptide corresponding to amino acid residues 416–432 gave consistent and specific signals. Western blotting experiments using murine tissue samples demonstrate that the antibodies raised against human PRPF31 peptides specifically recognize the murine PRPF31 protein as one prominent band of 61 kDa in size, similar to that in human cells (Fig. 1, part II). No significant difference was detected in either the level or pattern of PRPF31 protein among different tissues. PRPF31 protein was expressed in the retinas from embryonic throughout adult stages in mice (Fig. 1, part II, lanes 9–11).
The size of the PRPF31 protein detected is consistent with a previous report (Makarova et al., 2002) and with the predicted protein products of human and murine PRPF31 cDNA sequences obtained from GenBank. The amino acid sequences of predicted peptide sequences corresponding to PRPF31 cDNAs from Homo sapiens and Mus musculus (accession numbers NM_015629 and NM_027328, respectively) are almost identical except for a single amino acid at the position 496. The mammalian PRPF31 sequences are homologous to those of Drosophila as well as yeast (Vithana et al., 2001; Makarova et al., 2002). The central domain of PRPF31 shows homology to nucleolar proteins NOP56 and NOP58 from the box C/D snoRNPs, hence named NOP domain. In particular, the human PRPF31 protein shows ∼36% and 29% homology to PRPF31 protein sequence of Saccharomyces pombe and Saccharomyces cerevisiae (Weidenhammer et al., 1997; Bishop et al., 2001), respectively, indicating a high degree of conservation of PRPF31 genes through evolution. Such sequence similarity is not restricted to the NOP domain but distributed throughout the entire protein. For example, in the C terminal region, outside of NOP domain and nuclear localization signal, from amino acid residue 364–499, the amino acid identity and similarity of PRPF31 proteins between human and S. pombe is 31 and 41%, respectively (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Construction of mammalian expression plasmids for wild-type and RP mutant forms of PRPF31
Murine PRPF31 protein is nearly identical to human PRPF31, and the murine retina is highly similar to the human retina both in structure and in function. We established primary retinal cell cultures using mouse retinal tissue to investigate the potential role of PRPF31 mutations in retinal degeneration. We engineered mammalian expression plasmids for wild-type and mutant PRPF31 proteins. Two PRPF31 mutants were constructed to mimic two frame-shift mutations found in either sporadic (SP117) or familial (AD5) forms of adRP. As a result of either a single base insertion (SP117) or an 11 bp deletion (AD5), these mutations lead to truncations of the PRPF31 peptide after the residue 256 or 371, respectively. Thus, we named these mutants N256 and N371 (Fig. 2). The wild-type or mutant PRPF31 proteins were expressed as either nontagged or GFP-tagged proteins. The GFP-tagged proteins behave in a similar manner to the non-tagged proteins in transfection and splicing assays. We explored different methods to express these proteins in primary retinal cells and established a modified transfection protocol using calcium phosphate precipitation method similar to the one described previously (Jiang et al., 1998). Compared with other protocols, this method allowed us to achieve a moderate level of GFP fusion protein expression in retinal cells without detectable non-specific cytotoxicity.
Figure 2.
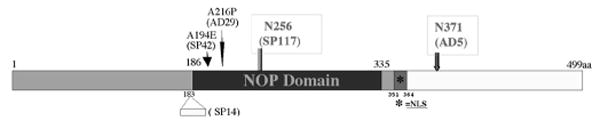
Peptide sequence features of PRPF31 protein. Positions of reported mutations in human PRPF31 gene and domain features of predicted PRPF31 protein are shown. The position of a predicted nuclear localization signal (NSL) is marked by an asterisk. Two RP mutations, N256 (SP117) and N371 (AD5), are depicted in red.
Transfection of plasmids encoding GFP-tagged forms of PRPF31 into either human embryonic kidney (HEK) cells or primary retinal cells led to the expression of PRPF31 proteins with similar intracellular distributions. Both wild-type and the N371 mutant proteins showed predominantly nuclear expression. The N256 mutant, however, was primarily detected in the cytoplasm, and only a low level of this mutant protein was detectable in the nucleus. This observation is consistent with the prediction that the nuclear localization signal (Fig. 2, NLS) of PRPF31 was retained in N371 mutant but deleted in the N256 mutant (data not shown) (see Figs. 5, 7).
Figure 5.
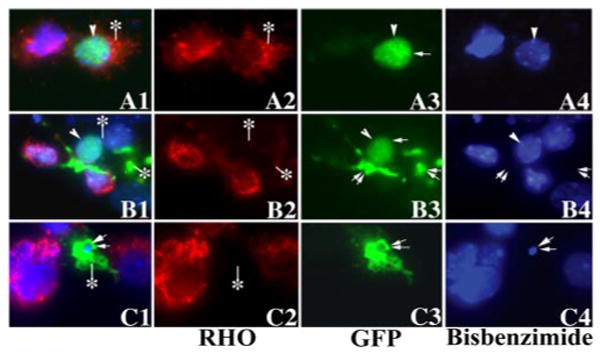
The expression of mutant PRPF31 significantly inhibited the expression of rhodopsin protein. After the transfection of murine primary retinal cells with plasmids expressing either wild-type or mutant PRPF31 as GFP-fusion proteins, wild-type (A1–A4), N371 (B1–B4), or N256 (C1–C4) PRPF31 proteins were detected in the transfected primary retinal cells by monitoring GFP fluorescence (A3–C3), and rhodopsin expression was demonstrated by immunofluorescent staining (red fluorescence) using anti-rhodopsin antibody followed by staining with the secondary antibody conjugated with Cy3 (A2–C2). The nuclear morphology was revealed by staining with bis-benzamide (A4 –C4). A1–C1 show the superimposed images in columns 2– 4. Single arrowheads or arrows indicate nuclei with normal morphology. The cells marked with double arrows show abnormally condensed nuclei or dissolution of nuclei, as shown by bis-benzimide staining. Rhodopsin staining signal shows a significant reduction in cells expressing mutant PRPF31 proteins compared with those expressing the wild-type PRPF31 (compare cells marked by an asterisk in A with those in B and C; also compare transfected cells with neighboring nontransfected cells).
Figure 7.
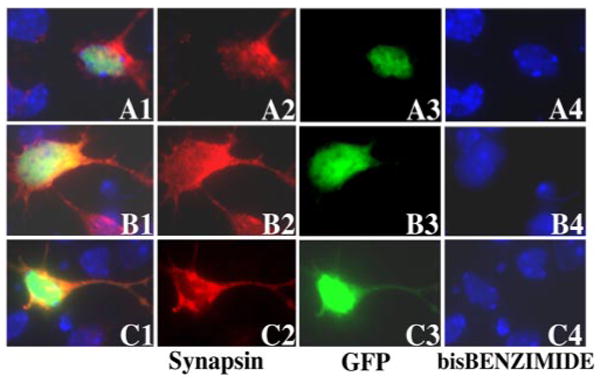
Mutant PRPF31 proteins do not affect synapsin expression. Primary retinal cells were transfected with plasmids expressing either wild-type or mutant PRPF31 as GFP-fusion proteins; wild-type (A1–A4), N371 (B1–B4), or N256 (C1–C4) PRPF31 proteins were detected in the transfected primary retinal cells by monitoring GFP fluorescence (A3–C3). Synapsin expression was detected by immunofluorescent staining (red fluorescence) using a monoclonal anti-synapsin antibody followed by the staining with the secondary antibody conjugated with Cy3 (A2–C2). The nuclear staining was performed with bis-benzamide (A4 –C4). A1–C1 show the superimposed images in columns 2– 4. The synapsin expression in cells expressing mutant PRPF31–GFP proteins shows no significant difference from those expressing the wild-type PRPF31–GFP protein (compare B2 and C2, with A2).
Searching for potential target genes for PRPF31
PRPF31 mutations cause adRP, suggesting that PRPF31 may have retina-specific splicing substrate genes. We searched for such potential target genes using a combined immunoprecipitation and microarray approach. First, we established a stable cell line expressing PRPF31 protein as a GFP-tagged protein in HEK cells. Using a monoclonal anti-GFP antibody and the affinity-purified polyclonal anti-PRPF31 antibody as well as preimmune Ig preparation for immunoprecipitation, we optimized conditions for immunoprecipitating PRPF31-containing splicing complexes. RNA species associated with the immunoprecipitated complexes were extracted and followed by microarray. Having established conditions for such immunoprecipitation microarray analysis using the HEK cell lines, we performed immunoprecipitation experiments using mouse retinal cells with the specific anti-PRPF31 antibody and the preimmune Ig control antibody (see Materials and Methods). RNA species associated with the PRPF31-containing splicing complexes were examined using microarray analysis. We detected 146 genes in the samples immunoprecipitated using PRPF31 antibody, but not in those prepared using the control preimmune antibodies. Among the potential target genes identified by this approach are a number of retina-specific genes. Results on other candidate target genes for PRPF31 will be described elsewhere. In this report, we will focus on these two adRP genes relevant to this study, RHO and rod cell outer membrane protein 1 (ROM1) genes. We confirmed the identity of RHO and ROM1 RNA species by using RT-PCR with specific primers and by sequencing the PCR products. The observation that RHO and ROM1 transcripts were detected among RNA species associated with splicing complexes containing PRPF31 protein suggests that these genes may be splicing substrates for PRPF31 protein and that they may act as “downstream target genes” for PRPF31.
Previous studies show that PRPF31 is a nuclear protein involved in pre-mRNA splicing (Weidenhammer et al., 1996, 1997; Makarova et al., 2002). Because immunoprecipitation microarray experiments suggest that RHO and ROM1 may be among splicing target genes for PRPF31, we examined the effects of PRPF31 on pre-mRNA splicing of these candidate target genes. Specific primers were synthesized to construct minigenes containing individual introns in RHO and ROM1 genes, especially in the peptide coding regions where adRP mutations have been identified. RHO or ROM1 minigenes were constructed covering different intronic regions with the corresponding flanking exonic sequences (Fig. 3A). In particular, RHO intron 3 and ROM1 intron 2 captured our attention. Prp31 gene in yeast, the ortholog for human PRPF31, is essential for pre-mRNA splicing. Most yeast introns are short, with their average size being 270 nt (Burge et al., 1999). Therefore, we decided to focus on small introns in human RHO and ROM1 genes. The removal of these small introns may be relatively inefficient, and failure to remove of these two introns would be predicted to cause a frame-shift in their peptide translation. We tested effects of mutant PRPF31 on the pre-mRNA splicing of these minigenes in HEK cells using co-transfection coupled with RT-PCR assay as described previously (Jiang et al., 1998; Cote et al., 2001). The splicing of RHO intron 3 was examined using the minigene containing intron 3 with flanking exon 3 and exon 4. After cotransfection of this RHO intron 3 minigene with a plasmid expressing either wild-type or adRP mutant forms of PRPF31 into HEK cells, the pre-mRNA splicing of the RHO intron 3 was examined using a semiquantitative RT-PCR assay. Such an RT-PCR assay is specific for the cotransfected minigene, because in the cells with an empty vector that does not contain the RHO minigene insert, there was no PCR band detectable (Fig. 3B, lane 1). When this RHO minigene was transfected into HEK cells, intron 3 underwent splicing at a moderate efficiency. In the cells expressing wild-type PRPF31 protein, the splicing of RHO intron 3 was more efficient than that in cells transfected with the expression vector control, as shown by the appearance of the spliced product (Fig. 3B, compare lanes 2 and 3). However, in the cells cotransfected with either N371 or N256 mutant forms of PRPF31 (lanes 4 and 5, respectively), splicing of intron 3 of RHO gene was inhibited, as demonstrated by the reduction in the spliced product with concomitant accumulation of the unspliced pre-mRNA transcript (Fig. 3B, compare lanes 4 and 5 with lanes 2 and 3). We also examined ROM1 intron 2, another small intron similar in size to RHO intron 3. When the ROM1 intron 2 minigene was cotransfected with a PRPF31-expressing plasmid, the wild-type PRPF31 or N371 or N256 mutant PRPF31 expression did not affect the ROM1 intron 2 splicing, because the ratio of pre-mRNA to splicing products showed no detectable changes (Fig. 3B, lanes 7–10). These splicing events occurred in the presence of the endogenous PRPF31 protein in the HEK cells, as shown by the Western blotting assay (Fig. 1, part II, lane 12). The levels of wild-type and mutant PRPF31 proteins in transfected cells were similar (Fig. 3C). These results indicate that the processing of RHO intron 3 is affected by PRPF31 mutations and suggest that the mutant PRPF31 proteins may be capable of blocking the activity of the endogenous wild-type PRPF31 protein in a dominant-negative manner. The removal of these introns in RHO and ROM1 genes are not equally sensitive to PRPF31 mutations, suggesting that mutations in PRPF31 gene may preferentially affect the pre-mRNA splicing of a subset of retinal genes.
Figure 3.
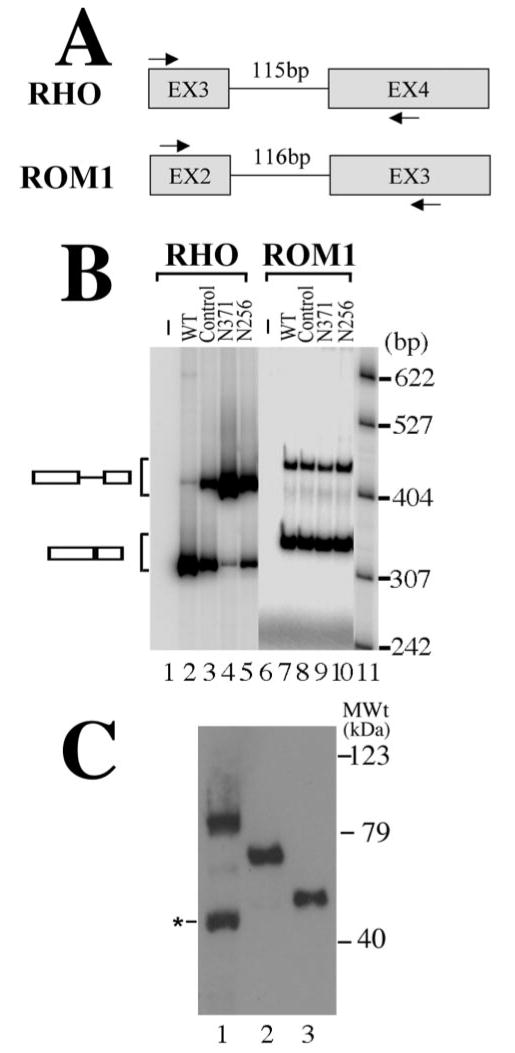
Mutant PRPF31 expression inhibits the splicing of RHO intron 3 but not ROM1 intron 2. A, A diagram is shown for RHO and ROM1 minigenes. The arrows mark the positions of specific primers used in RT-PCR assay to detect the corresponding splicing products. B, Effects of PRPF31 mutant proteins on the pre-mRNA splicing of RHO intron 3 and ROM1 intron 2 in transfected cells. After cotransfection of plasmids expressing wild-type (lanes 2 and 7), GFP vector control (lanes 3 and 8), or mutant [N371 (lanes 4 and 9) or N256 (lanes 5 and 10)] proteins together with either the RHO intron 3 minigene or the ROM1 intron 2 minigene into HEK cells, the corresponding pre-mRNA and splicing products are detected using RT-PCR with specific primers in the corresponding regions as shown in A. Lanes 1 and 6 contain the RT-PCR products prepared from cells transfected with the empty vector not containing either RHO or ROM1 minigene inserts. Lanes 2–5 (RHO minigene) and lanes 7–10 (ROM1 minigene) contain the RT-PCR products from cells transfected with the corresponding minigenes. DNA size markers are in lane 11, with sizes indicated on the right. C, The expression levels of wild-type and mutant PRPF31 proteins are equivalent in transfected HEK cells. The expression plasmids that were used for transfecting retinal cells (Figs. 4, 5) were transfected into HEK cells, and the GFP fusion proteins were detected by Western blotting using an anti-GFP antibody. Lane 1, Wild type; lane 2, N371; lane 3, N256. A degradation band of wild-type PRPF31–GFP fusion protein (marked by the asterisk) was sometimes detectable. MWt, Molecular weight.
Expression of mutant PRPF31 leads to a significant reduction in rhodopsin protein expression
To investigate effects of PRPF31 expression on RHO gene expression in retinal cells, we established primary cultures using dissociated retinal tissues from early postnatal mice. We used an immunofluorescent staining assay to examine rhodopsin protein expression in the primary retinal cells. Under our culture conditions, ∼70% retinal cells were rhodopsin-positive when cultured without transfection or transfected with either wild-type or vector control (see Fig. 6). With our optimized calcium phosphate method, we were able to achieve ∼5% transfection efficiency without causing nonspecific cytotoxicity (Fig. 4). After the transfection of the plasmid expressing the GFP vector or GFP-tagged wild-type PRPF31, N371, or N256 mutants, we monitored cells expressing these GFP-tagged proteins by fluorescent microscopy (Fig. 5). Similar to what was detected in HEK cells, wild-type PRPF31 and N371 mutant were primarily expressed in the nucleus, whereas N256 mutant was distributed throughout the cytoplasm (Fig. 5A1,A3,B1,B3,C1,C3). This was consistent with the prediction that the nuclear localization signal sequence was deleted in the N256 mutant. Interestingly, there was a marked reduction in the rhodopsin immunostaining in cells expressing mutant PRPF31, either N371 or N256, compared with cells expressing wild-type PRPF31 (Fig. 5A1–C1, compare the red immunofluorescent staining signals for rhodopsin in the transfected cells expressing PRPF31–GFP fusion proteins) (Fig. 5A2–C2, see cells marked with an asterisk). Similarly, comparison of the rhodopsin-immunofluorescent staining signals in cells transfected with mutant PRPF31–GFP plasmid with those in surrounding nontransfected cells in the same tissue culture dishes demonstrates that expression of either the N371 or the N256 mutant PRPF31 proteins reduced the expression of rhodopsin in these primary retinal cells. In cells expressing mutant PRPF31 (Fig. 5B1,B4, arrowheads), the reduction of rhodopsin expression appeared to be an early event, before there was any detectable change in nuclear morphology. Rhodopsin-positive cells were quantified at different time points after transfection. Consistently, the percentage of rhodopsin-positive cells was significantly reduced in cells expressing mutant (either N371 or N256) PRPF31 protein compared with cells expressing the vector or wild-type PRPF31 by 24 h after transfection (Fig. 6). At the same time frame, there was no detectable change in the expression of another neuronal marker, synapsin (Fig. 7). The percentage of synapsin-positive cells in these cultures was 70–75% without detectable differences among cells expressing either wild-type or mutant PRPF31–GFP proteins, suggesting that the reduction in rhodopsin expression observed is not a result of general shutdown of protein synthesis after cell death. These results show that mutant PRPF31 proteins inhibit the expression of RHO gene in primary retinal cells.
Figure 6.
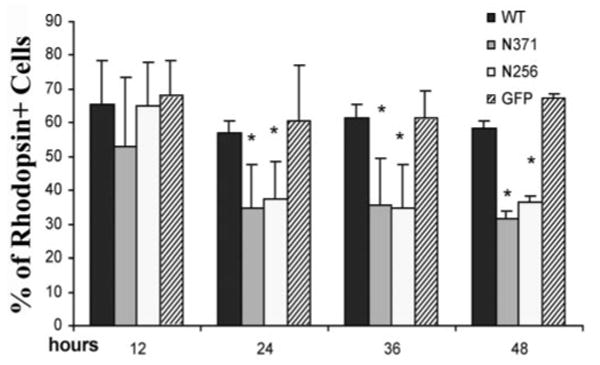
Quantification of rhodopsin-expressing cells after transfection of primary retinal cultures. Transfection and immunostaining were performed as described for Figure 4. The percentage of rhodopsin-expressing cells was scored at different time points. By 24 h after transfection, cells expressing mutant PRPF31 (either N371 or N256) show a significantly lower percentage of rhodopsin expression compared with cells expressing either GFP vector control or wild-type (WT) PRPF31–GFP fusion protein. The differences between the mutant group (either N371 or N256) and wild-type group (or GFP vector control) are statistically significant (*p<0.005). The data were collected from three independent experiments.
Figure 4.
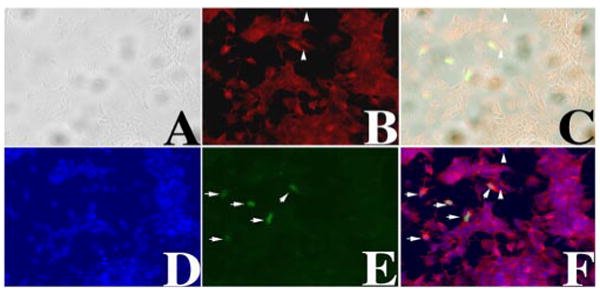
Expressing PRPF31–GFP protein in primary retinal cells. Primary retinal cell cultures were established as described in Materials and Methods. A calcium-phosphate method was established to transfect primary retinal cells. Twelve hours after transfection, cells were fixed and immunostained using anti-rhodopsin antibody, and images were captured. A phase-contrast image (A), rhodopsin immunostaining signal (B), a superimposed A–B image (C), nuclear staining (D), green fluorescence (E), and a superimposed B–D–E image (F) are shown. The arrowheads mark two cells that are rhodopsin negative. PRPF31–GFP-expressing cells are marked by white arrows (E, F). Nuclear morphology as detected by bis-benzimide (D), GFP fluorescence (E), and superimposition of B, D, and E (F) demonstrate that our transfection method did not cause nonspecific cytotoxicity and allowed us to express PRPF31–GFP in rhodopsin-positive retinal cells.
PRPF31 mutants cause retinal cell death in primary cultures
During the transfection experiments, we noticed that cells expressing mutant PRPF31 proteins began to show signs of cell death after transfection. We examined cell death and compared cells expressing wild-type PRPF31 with those expressing mutant PRPF31 proteins in primary cultures. After the transfection of the plasmid expressing the GFP vector, GFP-tagged wild-type PRPF31, or mutant forms (N371 or N256), we monitored cells expressing these GFP-tagged proteins. Significant differences in cell viability were observed between cells expressing the wild-type PRPF31 and those expressing mutant forms of PRPF31 protein. Nuclear staining using bis-benzimide demonstrated that the majority of cells expressing the wild-type PRPF31 showed normal healthy nuclei (Fig. 8A1–A3), whereas a significant fraction of cells expressing the mutant PRPF31 proteins showed the nuclear morphology characteristic of apoptosis, with fragmented or condensed nuclei (Fig. 8B,C). The retinal cell loss after the expression of mutant PRPF31 proteins was further investigated using TUNEL assay to determine whether such neuronal death is apoptotic in nature. A significant fraction of cells expressing the mutant PRPF31 proteins, either N371 or N256, exhibited positive TUNEL staining, compared with surrounding cells not expressing mutant PRPF31 proteins or with cells expressing wild-type PRPF31–GFP (Fig. 9A). The quantification of TUNEL-positive cells among the transfected cells demonstrated that cells expressing N371 or N256 mutant forms of PRPF31 showed significantly increased TUNEL staining by 24 h after transfection. The percentage of TUNEL-positive cells in the cells expressing these mutant PRPF31 proteins was more than doubled compared with cells expressing wild-type PRPF31 by 36 h after transfection (Fig. 9B). In the same time period, cells expressing wild-type PRPF31 or GFP vector control showed only a slight increase, similar to the basal level of TUNEL staining in the surrounding nontransfected cells. These results suggest that PRPF31 mutations led to apoptosis of the retinal neurons.
Figure 8.
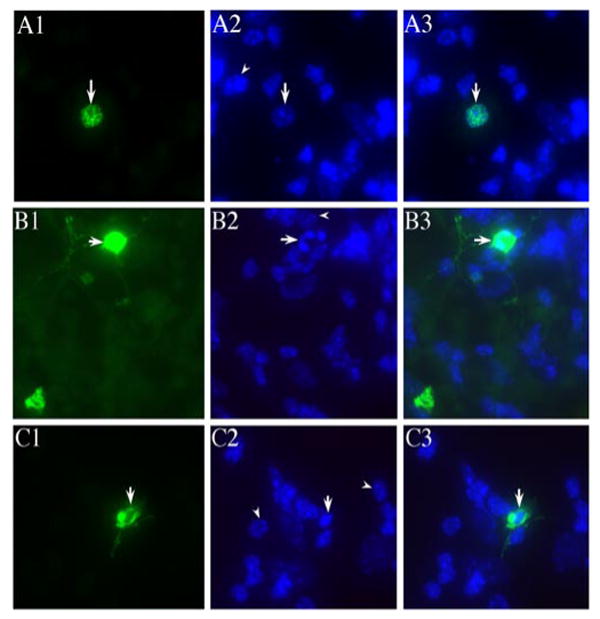
Expression of mutant PRPF31 proteins leads to retinal cell death. Primary murine retinal cells were transfected to express PRPF31 wild-type (A1–A3), N371 (B1–B3), or N256 (C1–C3) as GFP-fusion proteins, with green fluorescence showing the nuclear distribution of wild-type or N371 mutant PRPF31 (A1 and B1, respectively) and predominantly cytoplasmic localization of N256 mutant PRPF31 protein (C1). Nuclear staining was performed using bis-benzamide (A2–C2). The arrowheads in A2–C2 mark healthy nuclei in the neighboring cells in the same dishes. The arrows in B2 and C2 show a fragmented nucleus (B) and condensed nucleus (C), respectively. A3–C3 are superimposed images from corresponding ones in columns 1 and 2. The nuclear morphology of cells expressing mutant PRPF31 proteins suggests that these cells are undergoing apoptosis.
Figure 9.
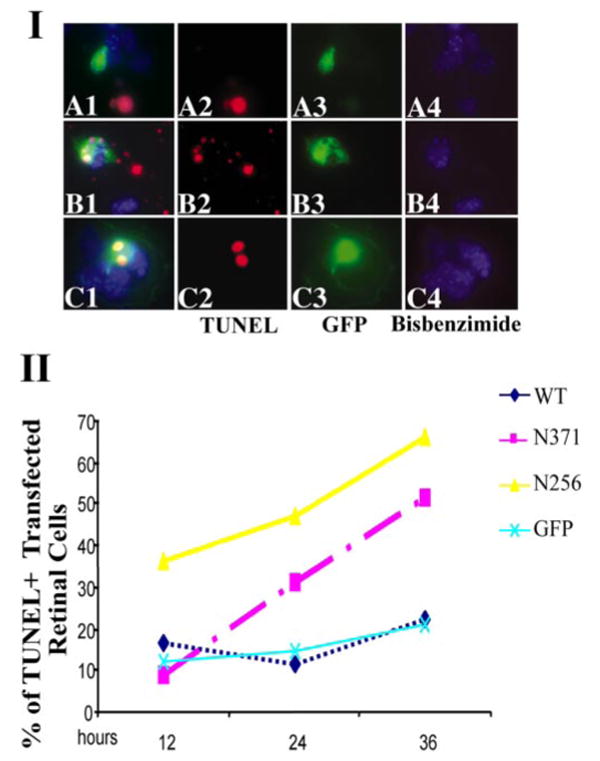
The expression of mutant PRPF31 causes a significant increase in retinal cell death as detected by TUNEL staining. I, TUNEL staining shows that N371 and N256 mutant PRPF31 expression significantly increased DNA fragmentation in retinal cells. In the primary retinal cultures transfected with either GFP vector control or wild-type PRPF31–GFP, only a small percentage of cells were TUNEL positive, and a majority of these TUNEL-positive cells were not expressing wild-type PRPF31–GFP protein. A significantly increased percentage of cells expressing PRPF31 mutant proteins were TUNEL positive (red signals). II, Quantification of TUNEL-positive cells at different time points after transfection of primary retinal cells with plasmids expressing either wild-type (WT) or mutant PRPF31–GFP fusion proteins. By 24 h after transfection, cells expressing either N371 or N256 mutant PRPF31 protein showed more than a twofold increase in the number of TUNEL-positive cells. The data shown are representative of three independent experiments.
Retinal cell death caused by the expression of mutant PRPF31 was blocked by the treatment with a caspase inhibitor, BAF
A critical feature of apoptosis is the activation of caspases, which is the convergent point for signal transduction pathways downstream of a variety of cell death signals. We, therefore, tested whether the retinal neuronal death induced by expression of these mutant PRPF31 proteins was caspase dependent. A broad-spectrum caspase inhibitor, BAF, was used in the transfected retinal neurons. When BAF was added in the culture media, retinal neurons expressing mutant PRPF31 proteins (either N371 or N256) showed significantly improved viability, as shown by both cell morphology and nuclear staining with bis-benzimide. Consistently, monitoring retinal cell death using TUNEL staining at different time points after transfection of PRPF31 expression constructs showed that BAF treatment blocked the cell death induced by expression of mutant PRPF31, N371, or N256, bringing the percentage of TUNEL-positive cells to the basal level (Fig. 10). These results indicate that the expression of mutant PRPF31 proteins induces retinal cell death in a caspase-dependent manner.
Figure 10.
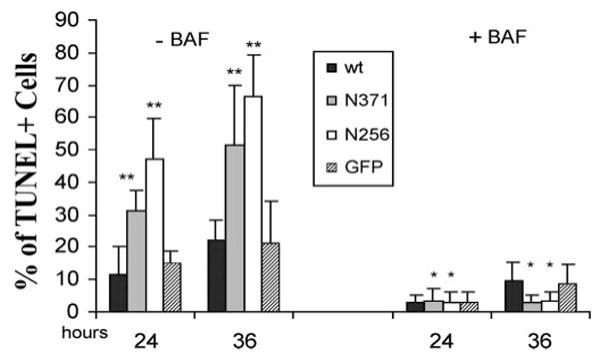
The cell death induced by the expression of mutant PRPF31 proteins was significantly reduced by the treatment with BAF, a caspase inhibitor. Transfection and TUNEL staining were performed as described for Figure 9, except BAF or vehicle control was added to the cultured retinal cells. In the absence of BAF, cells expressing mutant PRPF31, either N371 or N256, showed a significantly increased percentage of TUNEL-positive cells compared with cells expressing GFP alone or wild-type (wt) PRPF31 (**p<0.01). Treatment with BAF blocked cell death and eliminated the differences between the mutant groups (*) and control groups.
Discussion
Our study using primary retinal cultures indicates that mutant PRP31 proteins cause apoptosis of retinal cells. This suggests a mechanism for retinal degeneration caused by PRPF31 mutations. Transfection experiments with RHO minigene show that the wild-type PRPF31 protein stimulates RHO intron 3 splicing, whereas mutant PRPF31 proteins inhibit this splicing event. Our findings suggest a functional link between ubiquitously expressed PRPF31 protein and the expression of the retina-specific gene, rhodopsin. Our experiments reveal a previously unknown relationship between two major players in the pathogenesis of adRP, rhodopsin and PRPF31.
Rhodopsin is a primary visual pigment of the rod photoreceptor cells. It is a G-protein-coupled receptor activated by light that initiates the phototransduction cascade converting light signals to electrophysiological signals in retinal neurons. This photo-activated signal transduction process is essential for vision. A prominent early clinical feature of retinitis pigmentosa is the loss of night vision as a result of death of rod photoreceptor cells. Proper expression of the wild-type rhodopsin gene is essential for the development and sustained function of photoreceptor cells. Mutations in the RHO gene account for a significant proportion (∼25%) of all adRP cases in North America (Dryja et al., 2000; Dejneka and Bennett, 2001). More than 100 mutations in RHO gene have been reported (www.sph.uth.tmc.edu/RetNet). Most of these mutations are missense mutations affecting single amino acid residues in the rhodopsin protein (Vaithinathan et al., 1994). Photoreceptor cell death in RP is believed to occur by programmed cell death or apoptosis (Chang et al., 1993) (for review, see Baehr and Chen, 2001; Dejneka and Bennett, 2001). Mice carrying a targeted disruption of rhodopsin gene showed rapid and early-onset photoreceptor degeneration (Humphries et al., 1997). A similar phenotype was seen in transgenic rats carrying P23H rhodopsin mutation (for review, see Dejneka and Bennett, 2001). Our observation that PRPF31 mutations block RHO intron 3 splicing provides a new mechanism for generating defective RHO gene products, either as an aberrant RNA or as a truncated rhodopsin protein. Such defective RHO gene products may contribute to photoreceptor cell death.
Genetic studies suggest that mutations in PRPF31 gene may be a major cause of adRP (Vithana et al., 1998, 2001). In this report, we investigated the expression pattern, potential target genes, and functional activities of the PRPF31 gene. Indeed, PRPF31 gene expression is detected at a high level in the photoreceptors, although this gene is also expressed in other cell layers of the retina as well as in other tissues. The immunoprecipitation microarray approach led to the identification of candidate retina-specific target genes for PRPF31 protein, such as the rhodopsin gene. Cotransfection experiments using a RHO minigene with wild-type or RP mutant forms of PRPF31 expression constructs in HEK cells indicated the functional relationship between the PRPF31 and RHO genes. These experiments revealed the dominant-negative effects of RP mutants of PRPF31 on RHO pre-mRNA splicing. We also examined effects of PRPF31 mutations in primary retinal cultures. Consistently, rhodopsin protein expression was markedly decreased in retinal cells transfected with the RP mutant forms of PRPF31. Retinal cells expressing mutant PRPF31 proteins showed significantly reduced viability, providing an explanation for the retina-specific manifestation of mutations in the non-retina-specific PRPF31 gene. It is conceivable that modifier genes that could remove or functionally antagonize mutant PRPF31 proteins could affect the penetrance or expressivity of PRPF31 mutations. Together, these results show that the rhodopsin transcript is a pre-mRNA splicing substrate affected by PRPF31 protein.
It is likely that RHO is not the only gene affected by PRPF31 mutations. Our immunoprecipitation microarray experiments revealed a number of RNA transcripts associated with PRPF31-containing splicing complex or complexes. It will be interesting to examine whether pre-mRNA splicing of other retina-specific genes, especially those implicated in adRP, are affected by PRPF31 mutations. Our results show that not all splicing events are equally sensitive to PRPF31 mutations. PRPF31 mutants may inhibit pre-mRNA splicing of a subset of photoreceptor genes, such as RHO intron 3, but not other splicing events such as ROM1 intron 2. It is likely that the photoreceptor cell death caused by PRPF31 mutants is the result of combinatorial effects on a range of PRPF31 splicing substrate genes. Such collective effects on these PRPF31-sensitive photoreceptor genes may lead to the photoreceptor loss in RP11 patients.
We engineered PRPF31 mutants mimicking mutations identified in patients with either familial or sporadic retinitis pigmentosa, N371 and N256. In primary retinal culture, these PRPF31 mutants caused a reduction in rhodopsin expression at an early stage, before there was obvious changes nuclear morphology. Using the RHO intron 3 minigene in transfected HEK cells, we showed that both N371 and N256 mutant PRPF31 inhibited the pre-mRNA splicing of RHO intron 3 in the presence of endogenous wild-type PRPF31 protein. The failure in removing intron 3 during pre-mRNA splicing of RHO gene is predicted to produce an aberrant RHO transcript that contains a premature stop codon. Such an aberrant RHO transcript may be subjected to nonsense-mediated mRNA degradation. Alternatively, the retention of RHO intron 3 may cause the formation of a defective rhodopsin peptide truncated after the codon 232. The fact that a number of RHO mutations have been identified downstream of its exon 3 in adRP patients suggests that the retention of intron 3 and the formation of the truncated peptide would have deleterious effects on rhodopsin function. Similar to other adRP mutations in RHO gene, RHO intron 3 retention caused by PRPF31 mutations likely leads to retinal degeneration. These observations suggest that effects on RHO pre-mRNA splicing exerted by PRPF31 mutants may contribute to photoreceptor cell death in adRP.
Our results indicate that intron 3 removal in RHO pre-mRNA splicing is inhibited by the presence of RP mutant forms of PRPF31 protein. However, not all introns in RHO gene are equally sensitive to PRPF31 mutations. Similar to a previous report that intron 2 removal in RHO pre-mRNA splicing was not affected by a point mutation in PRPF31 gene (Deery et al., 2002), RHO intron 2 removal was not affected by N371 or N256 truncation mutations of PRPF31 in our study (data not shown). The small size of RHO intron 3 is not the only factor that makes the removal of this intron sensitive to PRPF31 mutations, because splicing of another small intron similar in size (i.e., ROM1 intron 2) is not inhibited by PRPF31 mutations (Fig. 3). It remains to be investigated what factors determine the sensitivity of different introns to mutations in PRPF31 gene.
Retinal cell death was blocked by treatment with the caspase inhibitor BAF, suggesting a caspase-dependent mechanism underlying the retinal cell loss caused by mutations in human PRPF31 gene. We noticed that it was necessary to apply BAF early in retinal cell culture and that delayed application of BAF (for example, 24 h after transfection of PRPF31 mutant plasmids) would not efficiently rescue the retinal cell death. This observation suggests that early application of caspase inhibitors might have therapeutic benefits in patients with adRP caused by mutations in PRPF31 gene.
The goal of therapeutic approaches to retinal degeneration is to restore vision or increase the residual functional visual field. Experimental approaches to retinitis pigmentosa include transplantation of photoreceptor cells and retinal pigment epithelium (RPE) cells using normal fetal retinal tissues (Mohand-Said et al., 2000; Radtke et al., 2002), delivery of genes to produce photoreceptor trophic factors, application of ribozymes to suppress the expression of disease-causing or disease-inducing genes, and therapies aimed at reducing photoreceptor cell death (LaVail et al., 2000; Bennett, 2002; Chaum and Hatton, 2002). In addition, electronic photoreceptor prosthesis is under development to restore vision (Margalit et al., 2002). Our results on PRPF31 mutations suggest that the loss of photoreceptor cells in adRP patients carrying PRPF31 mutations may be caused by the gain-of-function toxicity of mutant PRPF31 proteins. Simply providing wild-type PRPF31 protein may not be sufficient for preserving or restoring retinal neuronal function, and removing the neurotoxic mutant PRPF31 proteins may be essential for expression of functional rhodopsin protein. Our observations may have implications for understanding pathogenesis and designing new therapeutic interventions for neurodegenerative disorders caused by defects in general regulatory genes, such as those encoding pre-mRNA splicing factors or other gene expression regulatory proteins.
Aberrant splicing has been implicated in the pathogenesis of several neurodegenerative disorders, including amyotrophic lateral sclerosis, spinal muscular atrophy, and dementia (Grabowski and Black, 2001; Dredge et al., 2001; Faustino and Cooper, 2003; Wu et al., 2003). Mutations have been identified in intronic regions in addition to the ones at the exon–intron junctions, leading to perturbation of alternative splicing of the genes involved. However, genetic defects have been identified that directly affect the pre-mRNA splicing machinery and yet cause specific neurological manifestations. For example, spinal muscular atrophy, a motor neuron degenerative disorder can be caused by deletion of or mutations in survival of motor neurons, a ubiquitously expressed gene critical for biogenesis of spliceosomes (Paushkin et al., 2002). Our results on PRPF31 provide an explanation why such genetic defects in the seemingly ubiquitously expressed general splicing factors can cause neuron-specific diseases.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grants RO1 EY014576 and R01 AG17518 to J.Y.W., by the Vanderbilt Vision Center core facility (NIH Grant EY08126), and by the Muscular Dystrophy Association. J.Y.W. was also supported by a scholarship from the Leukemia Society of America. We are grateful to Drs. David Beebe, Eugene M. Johnson Jr, Pat Levitt, David Ornitz, and Arnold Strauss, and also to members of Wu laboratory for generous help, insightful suggestions, and critical reading of this manuscript.
References
- Al-Maghtheh M, Vithana E, Tarttelin E, Jay M, Evans K, Moore T, Bhattacharya S, Inglehearn CF. Evidence for a major retinitis pigmentosa locus on 19q13.4 (RP11) and association with a unique bimodal expressivity phenotype. Am J Hum Genet. 1996;59:864–871. [PMC free article] [PubMed] [Google Scholar]
- Anthony JG, Weidenhammer EM, Woolford JL., Jr The yeast Prp3 protein is a U4/U6 snRNP protein necessary for integrity of the U4/U6 snRNP and the U4/U6.U5 tri-snRNP. RNA. 1997;3:1143–1152. [PMC free article] [PubMed] [Google Scholar]
- Baehr W, Chen CK. RP11 and RP13: unexpected gene loci. Trends Mol Med. 2001;7:484–486. doi: 10.1016/s1471-4914(01)02177-3. [DOI] [PubMed] [Google Scholar]
- Bennett J. Gene therapy for retinitis pigmentosa. Curr Opin Mol Ther. 2002;2:420–425. [PubMed] [Google Scholar]
- Bishop DT, McDonald WH, Gould KL, Forsburg SL. Isolation of an essential Schizosaccharomyces pombe gene, prp31(+), that links splicing and meiosis. Nucleic Acids Res. 2001;28:2214–2220. doi: 10.1093/nar/28.11.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge CB, Tuschl TH, Sharp PA. In: The RNA world. Gesteland RF, Cech TR, Atkins JF, editors. New York: Cold Spring Harbor Laboratory; 1999. pp. 525–560. [Google Scholar]
- Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, Rosenberg T, Webster AR, Bird AC, Gal A, Hunt D, Vithana EN, Bhattacharya SS. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- Chaum E, Hatton MP. Gene therapy for genetic and acquired retinal diseases. Surv Ophthalmol. 2002;47:449–469. doi: 10.1016/s0039-6257(02)00336-3. [DOI] [PubMed] [Google Scholar]
- Cote J, Dupuis S, Jiang Z, Wu JY. Caspase-2 pre-mRNA alternative splicing: identification of an intronic element containing a decoy 3′ acceptor site. Proc Natl Acad Sci USA. 2001;98:938–943. doi: 10.1073/pnas.031564098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deery EC, Vithana EN, Newbold RJ, Gallon VA, Bhattacharya SS, Warren MJ, Hunt DM, Wilkie SE. Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet. 2002;11:3209–3219. doi: 10.1093/hmg/11.25.3209. [DOI] [PubMed] [Google Scholar]
- Dejneka NS, Bennett J. Gene therapy and retinitis pigmentosa: advances and future challenges. BioEssays. 2001;23:662–668. doi: 10.1002/bies.1092. [DOI] [PubMed] [Google Scholar]
- Dredge BK, Polydorides A, Darnell RB. The splice of life: alternative splicing and neurological disease. Nat Neurosci Rev. 2001;2:43–50. doi: 10.1038/35049061. [DOI] [PubMed] [Google Scholar]
- Dryja TP, McEvoy JA, McGee TL, Berson EL. Novel rhodopsin mutations Gly114Val and Gln184Pro in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:3124–3127. [PubMed] [Google Scholar]
- Evans K, al-Maghtheh M, Fitzke FW, Moore AT, Jay M, Inglehearn CF, Arden GB, Bird AC. Bimodal expressivity in dominant retinitis pigmentosa genetically linked to chromosome 19q. Br J Ophthalmol. 1995;79:841–846. doi: 10.1136/bjo.79.9.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar GJ, Kenna PF, Humphries P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 2002;21:857–864. doi: 10.1093/emboj/21.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- Grabowski PJ, Black DL. Alternative RNA splicing in the nervous system. Prog Neurobiol. 2001;65:289–308. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Hastings ML, Krainer AR. Pre-mRNA splicing in the new millennium. Curr Opin Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- Hims MM, Diager SP, Inglehearn CF. Retinitis pigmentosa: genes, proteins and prospects. Dev Ophthalmol. 2003;37:109–125. doi: 10.1159/000072042. [DOI] [PubMed] [Google Scholar]
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- Inglehearn CF. Molecular genetics of human retinal dystrophies. Eye. 1998;12:571–579. doi: 10.1038/eye.1998.147. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol Cell Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZH, Zhang WJ, Rao Y, Wu JY. Regulation of Ich-1 pre-mRNA alternative splicing and apoptosis by mammalian splicing factors. Proc Natl Acad Sci USA. 1998;95:9155–9160. doi: 10.1073/pnas.95.16.9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Drenser KA, Flannery JG, Lewin AS, Hauswirth WW. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci USA. 2000;97:11488–11493. doi: 10.1073/pnas.210319397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Chen JH, Wu W, Fagaly T, Zhou L, Dupuis S, Jiang ZH, Nash W, Gick C, Wu JY, Rao Y. Vertebrate Slit, a secreted ligand for the transmembrane protein Roundabout, is a repellent for olfactory bulb axons. Cell. 1999;96:807–818. doi: 10.1016/s0092-8674(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Luo X, Heidinger V, Picaud S, Lambrou G, Dreyfus H, Sahel J, Hicks D. Selective excitotoxic degeneration of adult pig retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci. 2001;42:1096–1106. [PubMed] [Google Scholar]
- Maddock JR, Roy J, Woolford JL., Jr Six novel genes necessary for pre-mRNA splicing in Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:1037–1044. doi: 10.1093/nar/24.6.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov EM, Makarova OV, Urlaub H, Gentzel M, Will CL, Wilm M, Luhrmann R. Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science. 2002;298:2205–2208. doi: 10.1126/science.1077783. [DOI] [PubMed] [Google Scholar]
- Makarova OV, Makarov EM, Liu S, Vornlocher HP, Luhrmann R. Protein 61K, encoded by a gene (PRPF31) linked to autosomal dominant retinitis pigmentosa, is required for U4/U6*U5 tri-snRNP formation and pre-mRNA splicing. EMBO J. 2002;21:1148–1157. doi: 10.1093/emboj/21.5.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margalit E, Maia M, Weiland JD, Greenberg RJ, Fujii GY, Torres G, Piyathaisere DV, O'Hearn TM, Liu W, Lazzi G, Dagnelie G, Scribner DA, de Juan E, Jr, Humayun MS. Retinal prosthesis for the blind. Surv Ophthalmol. 2002;47:335–356. doi: 10.1016/s0039-6257(02)00311-9. [DOI] [PubMed] [Google Scholar]
- McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, Mackey DA, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- Mohand-Said S, Hicks D, Dreyfus H, Sahel JA. Selective transplantation of rods delays cone loss in a retinitis pigmentosa model. Arch Ophthalmol. 2000;118:807–811. doi: 10.1001/archopht.118.6.807. [DOI] [PubMed] [Google Scholar]
- Moore AT, Fitzke F, Jay M, Arden GB, Inglehearn CF, Keen TJ, Bhattacharya SS, Bird AC. Autosomal dominant retinitis pigmentosa with apparent incomplete penetrance: a clinical, electrophysiological, psychophysical, and molecular genetic study. Br J Ophthalmol. 1993;77:473–479. doi: 10.1136/bjo.77.8.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacione LR, Szego MJ, Ikeda S, Nishina PM, McInnes RR. Progress toward understanding the genetic and biochemical mechanisms of inherited photoreceptor degenerations. Annu Rev Neurosci. 2003;26:657–700. doi: 10.1146/annurev.neuro.26.041002.131416. [DOI] [PubMed] [Google Scholar]
- Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol. 2002;14:305–312. doi: 10.1016/s0955-0674(02)00332-0. [DOI] [PubMed] [Google Scholar]
- Phelan JK, Bok D. A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Mol Vis. 2000;6:116–124. [PubMed] [Google Scholar]
- Radtke ND, Seiler MJ, Aramant RB, Petry HM, Pidwell DJ. Transplantation of intact sheets of fetal neural retina with its retinal pigment epithelium in retinitis pigmentosa patients. Am J Ophthalmol. 2002;133:544–550. doi: 10.1016/s0002-9394(02)01322-3. [DOI] [PubMed] [Google Scholar]
- Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002;11:1219–1227. doi: 10.1093/hmg/11.10.1219. [DOI] [PubMed] [Google Scholar]
- Swaroop A, Zack DJ. Transcriptome analysis of the retina. Genome Biol. 2002;3:1022.1–1022.4. doi: 10.1186/gb-2002-3-8-reviews1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaithinathan R, Berson EL, Dryja TP. Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics. 1994;21:461–463. doi: 10.1006/geno.1994.1301. [DOI] [PubMed] [Google Scholar]
- van Lith-Verhoeven JJ, van der Velde-Visser SD, Sohocki MM, Deutman AF, Brink HM, Cremers FP, Hoyng CB. Clinical characterization, linkage analysis, and PRPC8 mutation analysis of a family with autosomal dominant retinitis pigmentosa type 13 (RP13) Ophthalmic Genet. 2002;23:1–12. doi: 10.1076/opge.23.1.1.2206. [DOI] [PubMed] [Google Scholar]
- Vithana E, Al-Maghtheh M, Bhattacharya SS, Inglehearn CF. RP11 is the second most common locus for dominant retinitis pigmentosa. J Med Genet. 1998;35:174–175. doi: 10.1136/jmg.35.2.174-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–381. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- Weidenhammer EM, Singh M, Ruiz-Noriega M, Woolford JL., Jr The PRP31 gene encodes a novel protein required for pre-mRNA splicing in Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:164–170. doi: 10.1093/nar/24.6.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenhammer EM, Ruiz-Noriega M, Woolford JL., Jr Prp31p promotes the association of the U4/U6 x U5 tri-snRNP with prespliceosomes to form spliceosomes in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:3580–3588. doi: 10.1128/mcb.17.7.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Tang H, Havlioglu N. Alternative pre-mRNA splicing and regulation of programmed cell death. In: Jeanteur P, editor. Progress in molecular and sub-cellular biology. Vol. 31. New York: Springer; 2003. pp. 153–185. [DOI] [PubMed] [Google Scholar]
- Wu JY, Yuan L, Havlioglu N. Alternatively spliced genes. In: Meyers RA, editor. Encyclopedia of molecular cell biology and molecular medicine. 2. Vol. 1. Weinheim, Germany: Wiley-VCH; 2004. pp. 125–177. [Google Scholar]
- Wu W, Wong K, Chen JH, Jiang ZH, Dupuis S, Wu JY, Rao Y. Directional guidance of neuronal migration in the olfactory system by the secreted protein Slit. Nature. 1999;400:331–336. doi: 10.1038/22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human sliceosome. Nature. 2002;419:182–185. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.