

Abstract
Mouse models with conditional activation of K-ras (K-rasG12D) are used widely to investigate the role of oncogenic K-ras in a tissue-specific manner. However, the effect of ubiquitous activation of K-ras in adult mice has not been well studied. Herein, we report that systemic activation of K-ras in mice leads to rapid changes in gastric cellular homeostasis. Conditional activation of K-ras results in activation of the MAPK pathway and hyperproliferation of squamous epithelium in the forestomach and metaplasia in the glandular stomach. Parietal cells almost completely disappear from the upper part of the stomach adjacent to forestomach of K-ras activated mice. CDX2, a caudal-related homeobox transcription factor normally expressed in the intestine, is upregulated in parts of the stomach, following activation of K-ras in mice. Cyclooxygenase 2 (COX-2), a mediator of inflammation, is also upregulated in parts of the stomach of the K-ras activated mice with concomitant infiltration of hematopoietic cells in the hyperplastic tissue. Moreover, in K-ras activated mice, the expression of putative progenitor cell marker Dcamkl1 is upregulated in the glandular stomach. Expression of CD44, a candidate stomach cancer stem cell marker, is also increased in forestomach and the glandular stomach. These results suggest that cells of the stomach, potentially stem or progenitor cells, are highly susceptible to K-ras activation-induced initiation of gastric precancerous lesions. The histological changes in the K-ras activated mice resemble the pre-neoplastic changes that take place during gastric carcinogenesis in humans. Thus, a mouse model with systemic K-rasG12D activation could be useful for studying the early molecular events leading to gastric carcinogenesis.
Keywords: K-ras, intestinal metaplasia, gastric stem cells, Dcamkl1, CD44
Introduction
Gastric cancer, which can be divided into two major histologic sub-types, intestinal and diffuse, is the second leading cause of cancer-related deaths worldwide [1]. According to the Lauren classification, the intestinal-type gastric cancer is well- differentiated and is often accompanied with intestinal metaplasia (IM), whereas the diffuse-type cancers are poorly differentiated [2]. IM of the stomach is a precursor legion for intestinal-type gastric cancer [3]. K-ras, a member of the ras gene family that encode small GTPases, plays an important role in cell differentiation, growth, and apoptosis through multiple effector proteins including MAPKs (mitogen activated protein kinases)/ERKs (extracellular signal-regulated kinases) [4]. K-ras mutation has been frequently detected in a number of human cancers such as pancreatic, colonic, and lung, amongst others [5]. Activating K-ras mutations are also found in approximately 5-20% of gastric cancers [6]. Mutations of K-ras are more prevalent in intestinal-type gastric cancers [7]. K-ras mutations are also detected frequently in squamous cell carcinoma caused by carcinogens from betel quid [8], as well as in a subset of Barrett’s esophagus and adenocarcinoma at the gastroesophageal junction [9].
Several K-ras mouse models of human cancer are available to study human cancers in a tissue-specific manner. The mouse model with knock-in of inducible K-rasG12D (LSL-K-rasG12D) has the advantage of expressing endogenous levels of oncogenic K-rasG12D protein following removal of the Stop element by activated Cre [10]. The mouse model that ubiquitously expresses a conditional knock-in K-ras, using a CMV promoter-driven Cre recombinase, does not show any effect on major organs except for the lungs after seven months [11], perhaps due to a low percentage (5-15%) of the cells expressing the activated K-ras in adult mice. By contrast, conditional expression of the K-rasG12D transgene from the keratin 5 (K5) promoter causes squamous cell carcinoma in the skin, the oral cavity, the esophagus and the forestomach in mice with high penetrance within 3 to 4 weeks [12]. Thus, it is possible that the effect of K-ras activation may depend on which population of cells (such as stem/progenitor cells) is targeted. As the K-ras transgene was driven by the exogenous K5 promoter, the activated K-ras might be expressed at a higher level than that from its endogenous promoter. Hence, we decided to ubiquitously express activated K-ras from its endogenous promoter in all cellular populations to study whether the endogenous levels of activated K-ras have any early impact on tumor initiation from the digestive system.
Lgr5, an orphan G protein coupled receptor, a marker of stem cells in the small intestine, colon, and hair follicles [13,14], is also a bona fide marker for gastric epithelial stem cells for the squamous forestomach and the pylorus [15], but not for the corpus of the stomach in adult mice. In this regard, Doublecortin and calcium/calmodulin- dependent protein kinase-like1 (Dcamkl1) is expressed in the neck/isthmus region of the glandular stomach where the progenitor cells are believed to reside, and may serve as a candidate marker for gastric progenitor cells in the corpus [16,17]. It is also thought that gastric cancer stem cells can contribute to gastric carcinogenesis [18]. In the case of gastric cancer, CD44 is a transmembrane glycoprotein, and CD44(+) cells have been shown to possess the properties of cancer stem cells [19].
In the current study, we show that after systemic and conditional K-ras activation in adult mice, histologic changes that occur in the stomach of the mice resemble the events that occur during the early stages of gastric neoplasia in humans.
Materials and methods
Mice
All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and were performed according to institutional and national guidelines. The KrasG12D/+ mice (C57BL/6 and 129/Sv mixed background) were obtained from Dr. Tyler Jack’s group via Dr. Ben Stanger, and crossed with Ubc9 promoter driven Cre-ERT2 mice (C57B6 background) that were provided by Dr. Eric Brown [20]. Animal genotyping was done by PCR according to published methods using DNA extracted from tails, as well as stomach tissues [10]. PCR primers used were as follows P1 5′ gtc ttt ccc cag cac agt gc 3′, P2 5′ ctc ttg cct acg cca cca gct c 3′, P3 5′ agc tag cca cca tgg ctt gag taa gtc tgc a 3′.
Activation of K-ras by tamoxifen
KrasG12D/+;Ubc9 Cre-ER and their littermate controls,Ubc9 Cre-ER around six weeks old, were fed tamoxifen (TAM) at 200mg/kg.bw/day by gavaging for two consecutive days, followed by one day off and then, for two more consecutive days.
Histopathology
The mice were monitored closely and ill mice were sacrificed when they exhibited signs of strong distress. For histological examination, tissues were fixed in 10% neutral buffered formalin followed by dehydration in graded solutions of alcohol, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H & E).
Measurement of blood glucose
Blood glucose levels were measured before the animals were sacrificed, using One Touch Ultra test strips (Lifescan, Milpitas) on venous blood from the tail vein.
Immunohistochemical (IHC) / immunofluorescence (IF) staining
Mice were injected with BrdU at 100mg/kg in phosphate buffered saline, 2 hrs before collecting tissue samples to study BrdU incorporation. Stomach sections (5 μm) were stained with antibodies against BrdU (1:250, Accurate Chemical and Scientific Corporation), H+ K+ ATPase (1:1000, Medical and Biological Laboratories Co.), CD44 (1:100, B.D. Biosciences), CD45 (1:25, B.D. Biosciences), CD68 (1:100, Abcam), COX-2 (1:100, B.D. Biosciences), CDX2 (1:200 Biogenex), phospho- ERK1/2 (1:250, Cell Signaling Technologies) phospho p38 (1:50 Cell Signaling Technologies), chromogranin A (1:2000), and Dcamkl1 (Abjent, 1:100), as per manufacturer’s instructions. The signal for immunohistochemical analysis was visualized using the Vectorstain Elite Kit (Vector Laboratories). For immunofluorescence, alexafluor labeled secondary antibodies were used. The number of BrdU positive cells were counted in three microscopic fields per tissue sample (three tissue samples per condition) and a mean was recorded.
Western blot analysis
The whole stomach tissue samples were homogenized in RIPA buffer containing protease inhibitor cocktail (Sigma), 0.2 M sodium fluoride and 0.2 M β-phosphoglycerate. Protein supernatants (50μg) were separated on 4-12% SDS gradient gels (Invitrogen) and transferred to nitrocellulose membranes. The primary antibodies used were Phospho Mek, Mek, phospho Erk1/2, Erk 1/2, phospho p38, p38 (1:1000, Cell Signaling Technologies), CD44, Lgr5 (1:1000 Abcam) and CDX2 (1:1000 Biogenex). ECL detection system (Amersham Biosciences) was used for visualization of the bands.
Alcian blue staining
Paraffin sections were deparaffinized and placed in 3% acetic acid for 3 min, incubated in 1% Alcian blue in 3% acetic acid, pH 2.5, for 30 min, and washed in water. The slides were then incubated in 0.1% nuclear fast red for 5 min as a counter stain, washed in water, dehydrated, and mounted with Cytoseal. Alcian blue stains-weakly acidic sulfated mucosubstances, hyaluronic acid and sialomucins- dark blue.
Statistical analysis
Microsoft Excel and GraphPad Prism software was used for statistical analysis. Student’s t-test was used to determine the significance of the results. Kaplan-Meier statistical analysis was performed with the log-rank test.
Results
Systemic K-ras activation causes hyperplasia of the forestomach and the glandular stomach
To determine the effect of systemic activation of K-ras on mice, we bred LSL-K-rasG12D/+ mice with mice expressing the Ubc9 Cre-ERT2 transgene (referred to as control mice hereafter) that was driven by the pan-active Ubc9 promoter, to generate LSL-K-rasG12D/+;Ubc9 Cre-ERT2 (referred to as K-rasG12D/+ mice hereafter). These mice allow inducible and conditional activation of K-rasG12D/+ ubiquitously after tamoxifen (TAM) feeding (Figure 1A). Control and K-rasG12D/+ mice were fed with TAM to activate the Cre recombinase. K-rasG12D/+ mice did not express K-rasG12D allele before treatment with TAM (data not shown). To examine the efficiency of Cre-mediated recombination, PCR was performed with mouse tail and stomach DNA samples using specific primers for genotyping. The results showed complete excision of the LSL stop sequence upstream of K-rasG12D allele in the stomachs of the K-rasG12D/+ mice (Figure 1B, lane 2, P2/P3). Genotyping of lung, pancreas, intestine and colon tissue samples showed similar levels of K-ras activation in these tissues (data not shown). Notably, K-rasG12D/+ mice started dying 13-18 days after TAM treatment (Figure 1C). The ill mice did not show any obvious tumor formation in the pancreas, lung, liver, kidney, small intestine and colon (Figure 2 and data not shown), except for one mouse showing oral papilloma.
Figure 1.
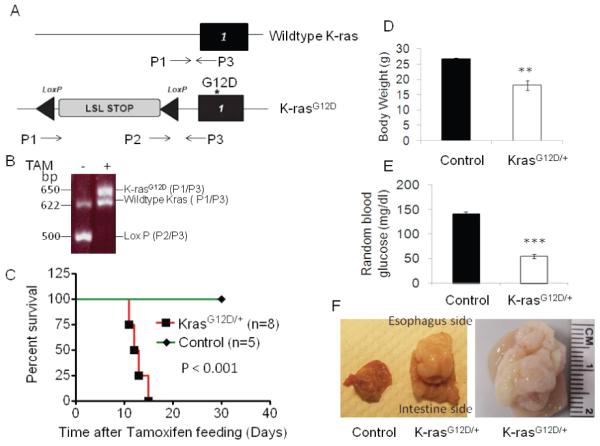
Activation of K-ras leads to quick death of mice. A) Schematic of LSL-K-rasG12D locus. K-rasG12D with an upstream LoxP-STOP-LoxP cassette that can be excised by Cre recombinase. P1, P2 and P3, primers for genotyping. B) Genotyping products, before and 15 days after TAM treatment. 500 bp LSL cassette, 625 bp wild-type and 650 bp recombined fragments. TAM-induced activation of K-ras decreases survival of mice. C) Kaplan-Meier curve of control Ubc9 CreER mice and LSL-K-rasG12D/+ Ubc9 Cre ER mice after TAM feeding. Mice with K-ras activation had D) decreased body weights and E) decreased random blood glucose levels. F) Tumor in stomach of K-ras Ubc9 Cre-ER mice.
Figure. 2.
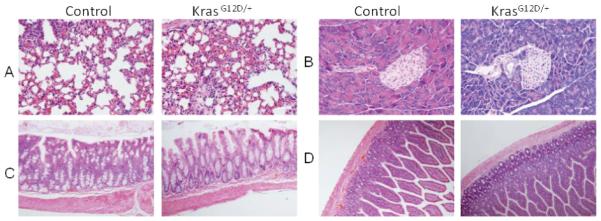
Histology of tissues from mice two weeks after tamoxifen treatment. H & E staining of A) lung, B) Pancreas, C) Colon and D) Small intestine.
However, all ill mice had enlarged stomachs and a large mass was found along the lesser curvature, near the gastroesophageal junction spreading throughout the stomach in advanced cases (Figure 1F). The stomachs of K-rasG12D/+ mice were devoid of any food, perhaps partly due to very limited space within the thickened wall of the stomach. The body weights of K-ras activated mice were significantly reduced (Figure 1D), and blood glucose levels were also substantially decreased (without obviously affecting pancreatic beta cells) in these mice as compared to the control mice (Figure 1E), suggesting that death may be attributed to malnourishment and hypoglycemia.
For histological sections, stomachs were cut open from the larger curvature, and were sectioned along longitudinal axis. Histological examination showed that K-rasG12D/+ mice har-bored hyperplasia of squamous epithelium of forestomach (Figure 3A and B) and mucus gland metaplasia of the glandular stomach (more prominent at the junction of forestomach and glandular stomach) (Figure 3C, D and E). The cross section of the stomach shows that the enlarged mass was composed of hyperplastic glands (Figure 3F).
Figure 3.
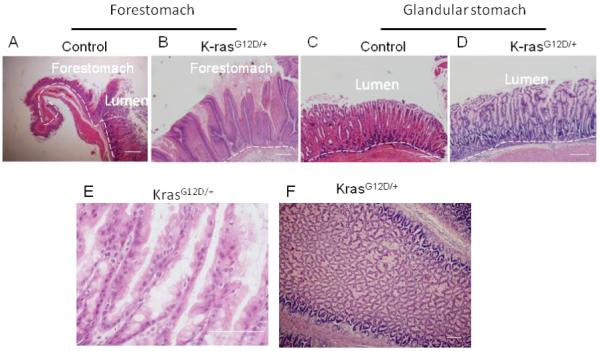
Activation of K-ras causes hyperplasia of squamous epithelium and glandular epithelium within 15 days after TAM treatment. A) Control and B) K-rasG12D/+ H and E staining of gastric squamous epithelium. C) Control and D) KrasG12D/+ H and E staining of glandular epithelium. E) Glandular stomach in KrasG12D/+ mice in higher magnification (100X). F) Cross section of tumor in KrasG12D/+ mice.
To examine the effect(s) of K-ras activation on cell proliferation, K-rasG12D/+ and the control mice were injected with BrdU to detect rapidly cycling cells. Mouse forestomach, which is comparable to distal esophagus of humans in physiology and cellular composition, is composed of keratinized stratified squamous epithelium. The immunofluorescence staining showed that the BrdU positive cells in the squamous epithelium of control mice were sparse among the basal cells (Figure 4A); whereas their percentage was significantly increased in the K-ras activated mice (Figure 4B) indicating enhanced cell proliferation in the K-ras activated mice (Figure 4C).
Figure 4.
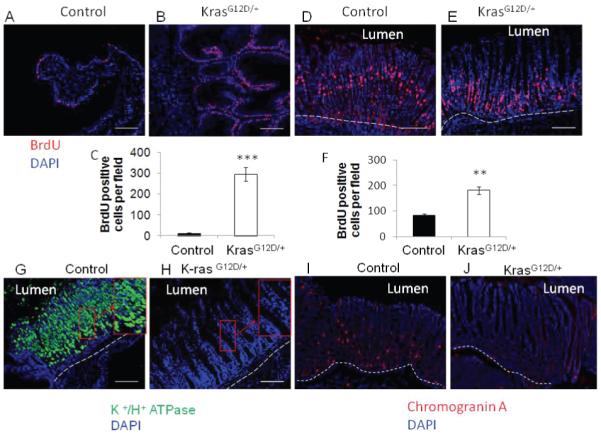
A) Control and B) K-rasG12D/+ BrdU incorporation of sqamous epithelium and C) Quantitation of BrdU positive cells in squamous epithelium. D) Control and E) K-rasG12D/+ BrdU incorporation in glandular epithelium. F) Quantitation of BrdU positive cells in glandular epithelium. Immunofluorescence staining with H+/ K+ ATPase antibody to detect parietal cells in G) control and H) K-ras activated mice. Immunofluorescence staining with Chromogranin A antibody to detect enteroendocrine cells in I) control and J) K-ras activated mice. Scale bar 200μM.
In the glandular part of the stomach, the normal cell proliferation zone is in the isthmus region [21]. In the control mice, the immunofluorescence staining clearly showed the BrdU-labeled cells in the isthmus (Figure 4D). However in the K-ras activated mice, the BrdU-labeled cells were shifted closer to the crypts (Figure 4E). The H & E staining showed that the glandular region of the stomach in K-ras activated mice was metaplastic . These metaplastic glands were highly hyperproliferative, as indicated by the quantitation of the BrdU positive cells in the stomachs of K-ras activated mice as compared to the control mice (Figure 4F).
Activation of K-ras leads to depletion of parietal cells in the glandular part of the stomach
Parietal or oxyntic cells of the stomach are acid-secreting cells and play an important role in the digestion of food. H+/K+ ATPase is an enzyme specifically expressed in parietal cells, serving as a reliable marker for parietal cells [22]. To determine whether K-ras activation affects the identity or fate of these cells, we performed immunofluorescence staining for H+/K+ ATPase. TAM-induced K-ras activation led to almost complete depletion of parietal cells in the glandular stomach close to the forestomach (Figure 4H), and also dramatic reduction of parietal cells in the body of the stomach (data not shown) as compared to the control mice (Figure 4G and data not shown). Mechanisms for rapid loss of parietal cells in K-ras activated mice remain to be further investigated. We further examined whether K-ras activation also affects the enteroendocrine cells. Immunofluorescence staining for chromogranin A, a marker of enteroendocrine cells, showed that enteroendocrine cells were barely detectable in the stomachs of K-ras activated mice (Figure 4I and J).
Gastric cancer develops in a multi-step process, and intestine metaplasia (IM) is considered a premalignant lesion of tumorigenesis in which the gastric mucosa is replaced by mucussecreting intestinal mucosa [23]. To determine whether K-ras activation caused IM in gastric mucosa, the gastric tissues were stained with Alcian blue to identify acidic mucins that are normally found only in the goblet cells of the small intestine ,colon and pylorus part of stomach. The mucins are not found in the part of stomach adjacent to forestomach, unless there is intestinal metaplasia [24]. Although the gastric mucosa of control mice contained no Alcian blue-stained cells (Figure 5A), a significant number of positively stained cells were found in the K-ras activated transgenic mice (Figure 5B), indicating that the glandular epithelium was replaced with the intestinal-type columnar epithelium (the figure shows the part of glandular epithelium next to forstomach region).
Figure 5.
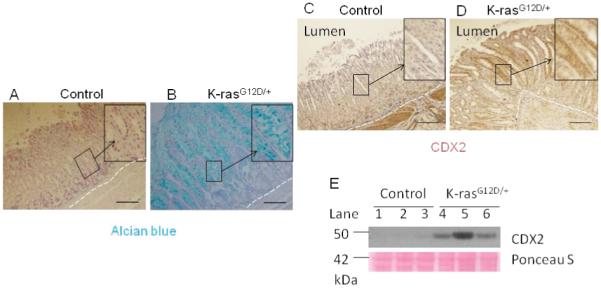
K-ras activation causes intestinal metaplasia in 13-16 days after TAM treatment. Alcian blue staining for A) Control and B) K-ras activated mice. Immunohistochemistry staining for CDX2 level in C) Control and D) K-ras activated mice. Scale bar 200μM. E) Western blot for CDX2.
Intestine-specific gene expression is increased in K-ras activated mice
CDX2, a transcription factor involved in intestinal cell proliferation and differentiation, is often upregulated in IM [25]. Immunohistochemical analysis showed that CDX2 was upregulated in K-ras activated mice as compared to the control (Figure 5C, D). The results were confirmed by Western blot (Figure 5E, lanes 4-6 vs. 1-3). Since ectopic expression of Cdx2 in the stomach of transgenic mouse induces intestinal metaplasia [24], our results strongly suggest that activation of K-ras-induced expression of CDX2 may play an important role in induction of IM in the stomach, promoting development of gastric pre-neoplasia. However, other pathways may be involved and need to be further investigated.
MAPK pathway is activated in the stomach of mice carrying a K-ras mutation
To test whether downstream targets of the K-ras signaling pathway were upregulated in K-ras activated mice, we examined the expression of downstream effectors, phosphorylated MEK, phosphorylated ERK1/2, and phosphorylated p38 by Western blot analysis. We found that expression of phosphorylated MEK, phosphorylated ERK1/2, and phosphorylated p38 were upregulated in K-ras activated mice (Figure 6A, lanes 4-6) as compared to the control (Figure 6A, lanes 1-3). Western blotting showed that total p38 was also upregulated in the K-ras activated mice, consistent with an earlier report that total p38 is upregulated in some gastric tumors [26]. Immunohistochemical staining showed that phosphorylated ERK1/2 was upregulated mostly at the top of the crypts in K-ras activated mice (Figure 6C), as compared to the control (Figure 6B), while expression of phosphorylated p38 was upregulated at the top and the bottom of the crypts in K-ras activated mouse (Figure 6E) as compared to the control (Figure 6D). These results suggest that the classical MAPK pathway is activated in the stomachs of mice upon K-ras activation.
Figure 6.
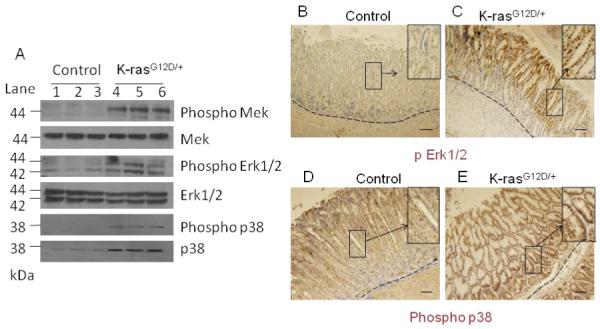
Activation of downstream targets of the MAPK pathway within 13-16 days after TAM treatment. A) Western blot for phospho MEK, phospho Erk1/2, and phosphor p38 in K-ras activated mice and control mice. Immunohistochemical staining for phospho Erk1/2 in the stomach of the B) control and C) K-ras activated mice, or for phospho p38 (D and E), respectively, 15 days after tamoxifen feeding. Scale bar 200μM.
K-ras activation induces chronic inflammatory response in the stomach of mice
Chronic gastritis is an important risk factor for the development of IM and gastric cancer [27]. The presence of macrophages and lymphocytes indicates a chronic inflammatory state. H & E staining of stomachs of K-ras activated mice showed infiltration of cells with small nuclei in the submucosal layer of the foresomatch squamous epithelium (Figure 7B), while the control mice did not show such staining (Figure 7A). To determine whether K-ras activation in mice induced inflammation in the stomach, we performed immunohistochemical staining for CD45, a marker of hematopoietic cells and observed infiltration of hematopoietic cells throughout the squamous epithelium of the forestomachs of K-ras activated mice (Figure 7D), but not in the control mice (Figure 7C). We also observed the presence of macrophages (stained with CD68) in the areas of chronic inflammation (Figure 7F), but not in the control mice (Figure 7E). The inflammatory response was observed also in the glandular stomach of K-ras activated mice (data not shown).
Figure 7.
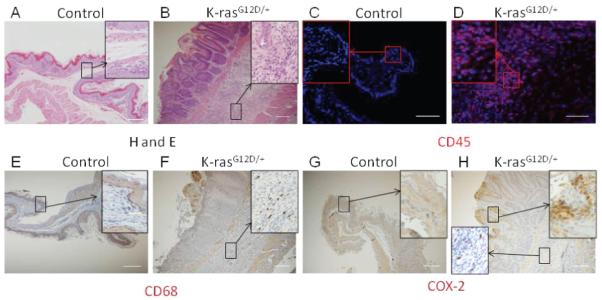
Activation of K-ras induces inflammatory response within 15 days after TAM treatment. H and E staining for stomach of A) control and B) K-ras activated mice, Immunohistochemistry for CD45 in stomach of C) control and D) K-ras activated mice. Immunohistochemistry for CD68, a macrophage marker, in E) control and F) K-ras activated mice. Immunohistochemical staining for COX-2 from G) control and H) K-ras activated mice. Scale bar 200μM.
Cyclo-oxygenase 2 (COX-2) is an inducible enzyme found in activated macrophages and other cells at the site of inflammation. COX-2 expression is significantly upregulated in gastric cancer[28-30]. Recent studies have suggested a strong link between K-ras activation and COX-2 expression[31,32]. Immunohistochemical staining of stomach tissues in K-ras activated mice showed COX-2 expression in a subset of hematopoietic cells at the site of inflammation and also, in the surrounding forestomach squamous epithelial cells (Figure 7H), but not in the control mice (Figure 7G). Together, these results suggest that K-ras activation may induce an inflammatory response in the stomach tissues.
K-ras activation results in expansion of cells expressing putative progenitor cell marker Dcamkl1and also candidate gastric cancer stem cell marker CD44
Rapid induction of hyperplasia of the forestomach squamous epithelium and hyperplasia and IM of the glandular stomach after K-ras activation prompted us to investigate the potential alteration of stem/progenitor cell markers in the stomach. Lgr5, a marker for adult stomach stem cells, is expressed at the base of pylorus in gastric antrum and squamous forestomach of the adult mouse [15]. Notably, the expression of Lgr5 was found to be reduced drastically in K-ras activated mice compared to control mice, as shown by Western blotting (Figure 8A, lanes 4-6). However, we could not perform immunohistochemical staining to detect the precise location of Lgr5 expression because of the unavailability of a suitable antibody.
Figure 8.
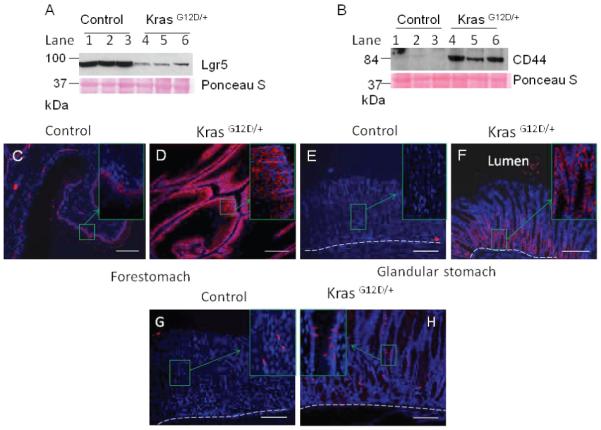
Change in stem cell markers in K-ras activated mouse within 15 days after TAM treatment. Western blot for A) LGR5 or B) CD44 from the lysates of stomach from control or K-ras activated mice 13-16 days after TAM treatment. Immunofluorescence staining for CD44 in the forestomach of the C) control and D) K-ras activated mice, or in the glandular stomach (E and F), 15 days after TAM feeding. Immunofluorescence staining for Dcamkl1 in stomach of G) control and H) K-ras activated mice. Scale bar 200μM.
CD44 is a candidate for gastric cancer stem cells [19]. The expression of CD44 was dramatically increased in the stomachs of K-ras activated mice compared to the control, as shown by Western blotting (Figure 8B, lanes 4-6). Immunofluorescence staining showed that the expression of CD44 was particularly high in the basal cells of the K-ras activated mice and appeared to be present in multiple layers of forestomach squamous epithelium compared to the control mice, which show CD44 (+) cells only at the base (Figure 8C and D). This region coincides with the highly proliferative BrdU positive region.
In the glandular part of the stomach, the CD44 (+) cells were especially located at the bottom of the crypts. Although cells in the large intestine express CD44 at the basolateral region of the crypt [33], the CD44-positive cells in the Kras activated mice were present in a considerately larger part of the crypts (Figure 8F), while the control did not show any CD44 positive cells (Figure 8E) except at the base of the pylorus region (data not shown). Thus, the highly proliferative region of the stomachs in K-ras activated mice was also found to express CD44. It has been reported that in intestinal metaplasia, v5 and v6 exons of CD44 are expressed similar to intestinaltype gastric cancer[34]. Thus, these results suggest that elevated expression of CD44 might be an early, important event in stomach pre-carcinogenesis. Dcamkl1, a putative progenitor cell marker in glandular stomach is normally expressed in the isthmus/neck region of stomach. The control mouse showed a few Dcamkl1 expressing cells in isthmus region of stomach (Figure 8G). Notably, Dcamkl1 expressing cells were increased and detectable in a larger part of glandular stomach of K-ras activated mice (Figure 8H). Collectively, our results indicate that K-ras activation altered the expression of gastric stem cell markers or fate of potential gastric progenitor cells.
Discussion
We found that acute and ubiquitous K-ras activation in adult mice had dramatic and rapid effects on the stomach, namely hyperplasia, IM, an inflammatory response, and changes in the expression of candidate progenitor cell markers, while we did not detect obvious tumors in other organs examined up to 18 days after K-ras activation. Moreover, the forestomach epithelium and the adjacent glandular stomach epithelium are more dramatically affected by K-ras mutation. These results indicate that amongst all the tissues in which K-ras is activated, the stomach appears to be most susceptible to K-ras mutation in an earliest window of time, suggesting a crucial role of K-ras activation in initiation of IM.
Thus far, reported K-ras models that show a gastric phenotype include the K-rasG12V expressed from the K19 promoter [35] and tetracycline-inducible expression of the K-rasG12D transgene from the K5 promoter [12]. K-rasG12V expressed from the K19 promoter causes hyperproliferation of mucus neck cells with an expansion of this compartment in 6-12 months, and the mice are viable; it is conceivable that stem cells or progenitor cells might reside also in this compartment. Another mouse model conditionally expresses the K-rasG12D transgene from the K5 promoter leads to squamous cell hyperplasia within 3-4 weeks [12]. Knock-in KrasG12D induced by the expression of Mx-cre induces hyperplasia in the forestomach along with myeloproliferative disorder with mean survival of 35 days [36]. However, the reported phenotype and/or the K-ras expression systems used in these studies were different from our studies. In our mouse model, K-rasG12D/+ was expressed from the endogenous K-ras promoter and only one allele of the knock-in K-rasG12D was activated in mice induced by the expression of Ubc9-cre. We did not observe any skin, oral cavity and esophageal lesions [23]. Ubiquitous induction of mutant K-ras in our mice rapidly resulted in forestomach squamous cell hyperplasia and dysplasia, as well as hyperplasia and intestinal metaplasia of the glandular stomach within two weeks, the latter of which resemble the precancerous stages of intestinal-type human gastric cancer [25]. Therefore, the conditional and systemic activation of K-ras in the adult mouse may be very useful to study the precancerous legions which are the early events in human gastric carcinogenesis.
Overexpression of EGFR(HER1), an upstream activator of K-ras, has been detected in more than 70% of gastro-esophageal cancers [37]. While K-ras mutations have been detected in gastric cancers, the significance of K-ras activation in the pre-neoplastic legions of gastric cancer is not well studied. A clinical study performed on a large number of patients with atrophic gastritis suggests that K-ras mutation is a good predictor of progression of pre-neoplastic legions [38], indicating that activation of the K-ras pathway can be an early event in gastric tumorigenesis. Furthermore, it is possible that activation of this pathway, even in the absence of a K-ras mutation in certain cell populations, can drive cells to proliferate faster, facilitating neoplastic transformation.
K-ras can activate multiple downstream signaling proteins, and typical effectors are MAPK/MEK/ERKs [4], which also have pleiotropic roles including induction of cell proliferation. We observed that K-ras activation increased phosphorylation and activation of MEK1 and ERK1/2 in the forestomach and the glandular stomach and also enhanced cell proliferation. Notably, in the gastric epithelium of K-ras activated mice that underwent IM in the stomach, expression of CDX2 was markedly increased. These findings are consistent with the notion that K-ras activated MEK/ERKs increases cell proliferation of the gastric epithelium leading to IM. The mechanisms involved in the intestinalization of gastric epithelium need to be further investigated.
Clinical data suggest that adenocarcinoma of the cardia region is closely associated with inflammation and IM [39]. We found that K-ras activation resulted in an inflammatory response and enhanced expression of COX-2 in certain areas of the forestomach and the glandular stomach. COX2 is upregulated in the gastric epithelium, and in the infiltrating inflammatory cells in the stomach with gastritis [40,41], and sulindac suppresses the progression of gastric cancer in mice [42]. Hence, a K-ras activation-induced inflammatory response may facilitate formation of IM and promote progression of gastric cancer. TGF-β signaling in fibroblasts can affect epithelial growth and induce oncogenesis [43]. As gastric cancer can originate from bone marrow derived cells [44], K-ras activated hematopoietic cells or stromal cells in stomach may also contribute to the phenotype. The bone marrow transplant from K-ras activated mice to the donor mice leads to hematological malignancies in mice with C57BL/6J genetic background in three months, with no abnormality reported in other organs [45]. A similar bone marrow transplant study in mice with 129Sv and C57BL/6 mixed background (same as in this study) did not show any phenotype and the mice were healthy for at least 120 days [36]. Thus, it is less likely that the phenotype is entirely a result of recruitment of K-ras activated hematopoietic cells to the stomach. Nevertheless, it remains possible that a combination of K-ras activation in gastric epithelial cells along with activation in hematopoietic system contribute to the phenotype.
Deletion of Apc in the stem cell population of intestine, leads to their transformation within days, whereas the same gene deleted in the cells of the transit amplifying region has little impact on tumorigenesis [46], suggesting that targeting the stem cell population enhances carcinogenesis. In our mouse model, K-ras was activated systemically in almost all cellular compartments, leading to the rapid initiation of IM in the stomach, perhaps via targeting a stem/progenitor cell population. K-ras activation induced IM and expression of CD44, a putative marker for gastric cancer stem cells [19], but did not lead to development of gastric cancer within two weeks, suggesting that CD44 may act as an oncogenic factor rather than a cancer stem marker at an early stage.
We found that the gastric epithelial stem cell marker Lgr5, which only marks the stem cells for the forestomach, at the junction of forestomach and glandular stomach and pylorus was decreased substantially in the K-ras activated mice. It is possible that K-ras-induced proliferation of the normal stem cell population led to exhaustion of this population. Alternatively, it is also possible that the stomach stem cell population in the forestomach and at the junction of forestomach and glandular stomach are more susceptible to K-ras activation-induced change of fate. In the glandular stomach, the cells expressing a putative progenitor cell marker, Dcamkl1, was increased in the K-ras activated mice. Thus, there appears to be differential regulation of stem/progenitor cell population in the forestomach and glandular stomach of K-ras activated mice. The molecular mechanisms underlying the link between K-ras activation and regulation of stem/progenitor cell self- renewal remain to be further investigated. Our mouse model demonstrates the genetic and histological changes during the initiation of gastric carcinogenesis [47,48], serving as a useful tool to study the early molecular events during the process of tumorigenesis.
Acknowledgments
We thank Ben Stanger for providing us with K-rasG12D/+ mice, Eric Brown for providing the Ubc9 Cre-ER mice, Hai Sheng for help with breeding of the mice, and the NIH/NIDDK P30-DK050306 Center for Molecular Studies in Digestive and Liver Diseases Morphology Core (Gary Swain), Molecular Biology Core, Mouse Core and Histology core. We also thank Shivani Sethi for critical reading of the manuscript and Yuquing Yang for useful suggestions.
This work was supported in part by grants from the National institute of health [R01-CA-113962, X.H. and R-01-DK-085121-01A1, X.H.]
Footnotes
Conflict of interest: Authors have no conflict of interest.
References
- [1].Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- [2].Lauren P. The Two Histological Main Types Of Gastric Carcinoma: Diffuse And So-Called Intestinal-Type Carcinoma. An Attempt At A Histo-Clinical Classification. Acta Pathol Microbiol Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- [3].Correa P. Human gastric carcinogenesis: a multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–40. [PubMed] [Google Scholar]
- [4].Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–8. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- [5].Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9. [PubMed] [Google Scholar]
- [6].Yashiro M, Nishioka N, Hirakawa K. K-ras mutation influences macroscopic features of gastric carcinoma. J Surg Res. 2005;124:74–8. doi: 10.1016/j.jss.2004.09.020. [DOI] [PubMed] [Google Scholar]
- [7].Watari J, Tanaka A, Tanabe H, Sato R, Moriichi K, Zaky A, Okamoto K, Maemoto A, Fujiya M, Ashida T, Das KM, Kohgo Y. K-ras mutations and cell kinetics in Helicobacter pylori associated gastric intestinal metaplasia: a comparison before and after eradication in patients with chronic gastritis and gastric cancer. J Clin Pathol. 2007;60:921–6. doi: 10.1136/jcp.2006.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Das N, Majumder J, DasGupta UB. ras gene mutations in oral cancer in eastern India. Oral Oncol. 2000;36:76–80. doi: 10.1016/s1368-8375(99)00058-5. [DOI] [PubMed] [Google Scholar]
- [9].Lord RV, O’Grady R, Sheehan C, Field AF, Ward RL. K-ras codon 12 mutations in Barrett’s oesophagus and adenocarcinomas of the oesophagus and oesophagogastric junction. J Gastroenterol Hepatol. 2000;15:730–6. doi: 10.1046/j.1440-1746.2000.02163.x. [DOI] [PubMed] [Google Scholar]
- [10].Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, Campuzano V, Barbacid M. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–20. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- [12].Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004;64:8804–7. doi: 10.1158/0008-5472.CAN-04-2623. [DOI] [PubMed] [Google Scholar]
- [13].Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- [14].Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev. 2008;22:1856–64. doi: 10.1101/gad.1674008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- [16].Giannakis M, Stappenbeck TS, Mills JC, Leip DG, Lovett M, Clifton SW, Ippolito JE, Glasscock JI, Arumugam M, Brent MR, Gordon JI. Molecular properties of adult mouse gastric and intestinal epithelial progenitors in their niches. J Biol Chem. 2006;281:11292–300. doi: 10.1074/jbc.M512118200. [DOI] [PubMed] [Google Scholar]
- [17].Bjerknes M, Cheng H. Multipotential stem cells in adult mouse gastric epithelium. Am J Physiol Gastrointest Liver Physiol. 2002;283:G767–77. doi: 10.1152/ajpgi.00415.2001. [DOI] [PubMed] [Google Scholar]
- [18].Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- [19].Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, Gordon SA, Shimada Y, Wang TC. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27:1006–20. doi: 10.1002/stem.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci U S A. 1996;93:10887–90. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Karam SM, Leblond CP. Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. Anat Rec. 1993;236:259–79. doi: 10.1002/ar.1092360202. [DOI] [PubMed] [Google Scholar]
- [22].Canfield VA, Levenson R. Structural organization and transcription of the mouse gastric H+, K(+)-ATPase beta subunit gene. Proc Natl Acad Sci U S A. 1991;88:8247–51. doi: 10.1073/pnas.88.18.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Busuttil RA, Boussioutas A. Intestinal metaplasia: a premalignant lesion involved in gastric carcinogenesis. J Gastroenterol Hepatol. 2009;24:193–201. doi: 10.1111/j.1440-1746.2008.05774.x. [DOI] [PubMed] [Google Scholar]
- [24].Silberg DG, Sullivan J, Kang E, Swain GP, Moffett J, Sund NJ, Sackett SD, Kaestner KH. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology. 2002;122:689–96. doi: 10.1053/gast.2002.31902. [DOI] [PubMed] [Google Scholar]
- [25].Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003;3:592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- [26].Atsumi T, Kato K, Uno K, Iijima K, Koike T, Imatani A, Ohara S, Shimosegawa T. Pathophysiological role of the activation of p38 mitogen-activated protein kinases in poorly differentiated gastric cancer. Pathol Int. 2007;57:635–44. doi: 10.1111/j.1440-1827.2007.02152.x. [DOI] [PubMed] [Google Scholar]
- [27].Zavros Y, Eaton KA, Kang W, Rathinavelu S, Katukuri V, Kao JY, Samuelson LC, Merchant JL. Chronic gastritis in the hypochlorhydric gastrindeficient mouse progresses to adenocarcinoma. Oncogene. 2005;24:2354–66. doi: 10.1038/sj.onc.1208407. [DOI] [PubMed] [Google Scholar]
- [28].Leung WK, To KF, Go MY, Chan KK, Chan FK, Ng EK, Chung SC, Sung JJ. Cyclooxygenase-2 upregulates vascular endothelial growth factor expression and angiogenesis in human gastric carcinoma. Int J Oncol. 2003;23:1317–22. [PubMed] [Google Scholar]
- [29].Uefuji K, Ichikura T, Mochizuki H, Shinomiya N. Expression of cyclooxygenase-2 protein in gastric adenocarcinoma. J Surg Oncol. 1998;69:168–72. doi: 10.1002/(sici)1096-9098(199811)69:3<168::aid-jso9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- [30].Lim HY, Joo HJ, Choi JH, Yi JW, Yang MS, Cho DY, Kim HS, Nam DK, Lee KB, Kim HC. Increased expression of cyclooxygenase-2 protein in human gastric carcinoma. Clin Cancer Res. 2000;6:519–25. [PubMed] [Google Scholar]
- [31].Sheng H, Shao J, Dubois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61:2670–5. [PubMed] [Google Scholar]
- [32].Taylor MT, Lawson KR, Ignatenko NA, Marek SE, Stringer DE, Skovan BA, Gerner EW. Sulindac sulfone inhibits K-ras-dependent cyclooxygenase-2 expression in human colon cancer cells. Cancer Res. 2000;60:6607–10. [PubMed] [Google Scholar]
- [33].Zeilstra J, Joosten SP, Dokter M, Verwiel E, Spaargaren M, Pals ST. Deletion of the WNT target and cancer stem cell marker CD44 in Apc(Min/+) mice attenuates intestinal tumorigenesis. Cancer Res. 2008;68:3655–61. doi: 10.1158/0008-5472.CAN-07-2940. [DOI] [PubMed] [Google Scholar]
- [34].Heider KH, Dammrich J, Skroch-Angel P, Muller-Hermelink HK, Vollmers HP, Herrlich P, Ponta H. Differential expression of CD44 splice variants in intestinal- and diffuse-type human gastric carcinomas and normal gastric mucosa. Cancer Res. 1993;53:4197–203. [PubMed] [Google Scholar]
- [35].Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG, Rustgi AK. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–9. [PubMed] [Google Scholar]
- [36].Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, Johnson L, Akashi K, Tuveson DA, Jacks T, Gilliland DG. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–38. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dragovich T, McCoy S, Fenoglio-Preiser CM, Wang J, Benedetti JK, Baker AF, Hackett CB, Urba SG, Zaner KS, Blanke CD, Abbruzzese JL. Phase II trial of erlotinib in gastroesophageal junction and gastric adenocarcinomas: SWOG 0127. J Clin Oncol. 2006;24:4922–7. doi: 10.1200/JCO.2006.07.1316. [DOI] [PubMed] [Google Scholar]
- [38].Gong C, Mera R, Bravo JC, Ruiz B, Diaz-Escamilla R, Fontham ET, Correa P, Hunt JD. KRAS mutations predict progression of preneoplastic gastric lesions. Cancer Epidemiol Biomarkers Prev. 1999;8:167–71. [PubMed] [Google Scholar]
- [39].Chandrasoma P, Wickramasinghe K, Ma Y, DeMeester T. Is intestinal metaplasia a necessary precursor lesion for adenocarcinomas of the distal esophagus, gastroesophageal junction and gastric cardia? Dis Esophagus. 2007;20:36–41. doi: 10.1111/j.1442-2050.2007.00638.x. [DOI] [PubMed] [Google Scholar]
- [40].Konturek PC, Kania J, Konturek JW, Nikiforuk A, Konturek SJ, Hahn EG. H. pylori infection, atrophic gastritis, cytokines, gastrin, COX-2, PPAR gamma and impaired apoptosis in gastric carcinogenesis. Med Sci Monit. 2003;9:SR53–66. [PubMed] [Google Scholar]
- [41].Naghshvar F, Torabizadeh Z, Emadian O, Enami K, Ghahremani M. Correlation of cyclooxygenase 2 expression and inflammatory cells infiltration in colorectal cancer. Pak J Biol Sci. 2009;12:98–100. doi: 10.3923/pjbs.2009.98.100. [DOI] [PubMed] [Google Scholar]
- [42].Lee CW, Rickman B, Rogers AB, Muthupalani S, Takaishi S, Yang P, Wang TC, Fox JG. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009;69:8166–74. doi: 10.1158/0008-5472.CAN-08-3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- [44].Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- [45].Zhang J, Wang J, Liu Y, Sidik H, Young KH, Lodish HF, Fleming MD. Oncogenic Kras-induced leukemogeneis: hematopoietic stem cells as the initial target and lineage-specific progenitors as the potential targets for final leukemic transformation. Blood. 2009;113:1304–14. doi: 10.1182/blood-2008-01-134262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–11. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- [47].Aqeilan RI, Hagan JP, Aqeilan HA, Pichiorri F, Fong LY, Croce CM. Inactivation of the Wwox gene accelerates forestomach tumor progression in vivo. Cancer Res. 2007;67:5606–10. doi: 10.1158/0008-5472.CAN-07-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Engel LS, Chow WH, Vaughan TL, Gammon MD, Risch HA, Stanford JL, Schoenberg JB, Mayne ST, Dubrow R, Rotterdam H, West AB, Blaser M, Blot WJ, Gail MH, Fraumeni JF., Jr. Population attributable risks of esophageal and gastric cancers. J Natl Cancer Inst. 2003;95:1404–13. doi: 10.1093/jnci/djg047. [DOI] [PubMed] [Google Scholar]