

Abstract
The activity of voltage-gated ion channels is critical for the maintenance of cellular membrane potential and generation of action potentials. In turn, membrane potential regulates cellular ion homeostasis, triggering the opening and closing of ion channels in the plasma membrane and, thus, enabling ion transport across the membrane. Such transmembrane ion fluxes are important for excitation-contraction coupling in pulmonary artery smooth muscle cells (PASMC). Families of voltage-dependent cation channels known to be present in PASMC include voltage-gated K+ (Kv) channels, voltage-dependent Ca2+-activated K+ (Kca) channels, L- and T- type voltage-dependent Ca2+ channels, voltage-gated Na+ channels and voltage-gated proton channels. When cells are dialyzed with Ca2+-free K+- solutions, depolarization elicits four components of 4-aminopyridine (4-AP)-sensitive Kvcurrents based on the kinetics of current activation and inactivation. In cell-attached membrane patches, depolarization elicits a wide range of single-channel K+ currents, with conductances ranging between 6 and 290 pS. Macroscopic 4-AP-sensitive Kv currents and iberiotoxin-sensitive Kca currents are also observed. Transcripts of (a) two Na+ channel α–subunit genes (SCN5A and SCN6A), (b) six Ca2+ channel α–subunit genes (α1A, α1B, α1x, α1D, α1E and α1G) and many regulatory subunits (α2δ1, β1-4, and γ6), (c) 22 Kv channel α–subunit genes (Kv1.1 - Kv1.7, Kv1.10, Kv2.1, Kv3.1, Kv3.3, Kv3.4, Kv4.1, Kv4.2, Kv5.1, Kv 6.1-Kv6.3, Kv9.1, Kv9.3, Kv10.1 and Kv11.1) and three Kv channel β-subunit genes (Kv(β1-3) and (d) four Kca channel α–subunit genes (Sloα1 and SK2-SK4) and four Kca channel (β-subunit genes (Kca(β1-4) have been detected in PASMC. Tetrodotoxin-sensitive and rapidly inactivating Na+ currents have been recorded with properties similar to those in cardiac myocytes. In the presence of 20 mM external Ca2+, membrane depolarization from a holding potential of -100 mV elicits a rapidly inactivating T-type Ca2+ current, while depolarization from a holding potential of -70 mV elicits a slowly inactivating dihydropyridine-sensitive L-type Ca2+ current. This review will focus on describing the electrophysiological properties and molecular identities of these voltage-dependent cation channels in PASMC and their contribution to the regulation of pulmonary vascular function and its potential role in the pathogenesis of pulmonary vascular disease.
Keywords: Ca2+ channel, K+ channel, membrane potential, Na+ channel, pulmonary hypertension
Introduction
Intracellular ion homeostasis, cell volume and membrane excitability are all important mechanisms regulated by the membrane permeability to cations and anions. It is this transmembrane ion flux that is the predominant factor in controlling excitation–contraction (EC) coupling mechanisms in pulmonary artery smooth muscle cells (PASMC). Electromechanical and pharmacomechanical coupling processes are the two major EC coupling mechanisms. Of these, it is the electric excitability that plays an important role in EC coupling in the pulmonary vasculature,[1,2] predominantly controlled by the transmembrane ion flux in PASMC. Indeed, many vasoactive substances also alter the membrane potential (Em) in these cells.[3,4] Expression and functionality of ion channels in the plasma membrane is also important in modulation of cell motility, migration and proliferation by governing the cytoplasmic free Ca2+ concentration ([Ca2+]cyt).
A rise in [Ca2+ ]cyt in PASMC triggers pulmonary vasoconstriction[5] and stimulates cell proliferation[6] and migration,[7] leading to pulmonary vascular remodeling.[8] The mechanisms involved in the regulation of [Ca2+ ]cyt directly control vasomotor tone and vascular wall thickness; two major determinants of pulmonary vascular resistance (PVR). Because PVR is inversely proportional to the fourth power of the radius (r) of the pulmonary arterial lumen (PVR=8Lη/πr4), a very small change in r would thus cause a large change in PVR. As a consequence, pulmonary vasoconstriction will also increase PVR by reducing the arterial radius. Pulmonary arterial pressure (PAP), a diagnostic criterion for PAH, is a product of PVR and cardiac output. Pulmonary vasoconstriction and vascular medial hypertrophy caused by excessive PASMC proliferation and migration contribute considerably to the elevated PVR in patients with pulmonary hypertension. Indeed, dysfunction of a number of ion channels has been implicated in a variety of cardiopulmonary diseases, such as pulmonary arterial hypertension,[8,9] spontaneous genetic systemic arterial hypertension[10-13] and heart failure.[14] Therefore, defining the molecular identities and electrophysiological properties of plasmalemmal ion channels in human PASMC will help to enhance our understanding of normal EC coupling mechanisms, to define the pathogenic roles of ion channels in pulmonary vascular disease and to develop new therapeutic approaches for patients with pulmonary hypertension.
As mentioned above, EC coupling requires a change in membrane potential to alter vascular tone. Ion channels are sarcolemmal pores selectively permeable to either cations (Na+, Ca2+, K+) or anions (Cl-). Both anions and cations are distributed on either side of the cell membrane, and their transmembrane movement is based on their electrochemical gradient, a potential- and concentration-based driving force for the ions, i.e. flowing from more-concentrated to less-concentrated zones and, for cations, from positive or less-negative sites to those with a more negative membrane potential. In human cells, Na+ (∼140 mM) and Ca2+ (∼2 mM) are the dominant cations in the external fluid (concentrations similar to those found in blood plasma), whereas K+ (∼140 mM) is the dominant cation in the cell cytoplasm. Cl-, the most dominant anion in vascular smooth muscle cells,[15] is unevenly distributed between the cytosol and the extracellular fluids, and plays an important role in controlling osmolarity, cell volume, excitability and ion homeostasis [Table 1]. Additionally, ion channels expressed in the plasma membrane also play important roles in the regulation of secretion, migration, proliferation, differentiation and apoptosis. In vascular smooth muscle cells, the resting Em is predominantly regulated by the permeability and the concentration gradients of K+ across the plasma membrane. The reason that the resting Em (-40 to -55 mV) in vascular smooth muscle cells is not equal to the K+ equilibrium potential (approximately -85 mV) indicates that other cation (e.g., Na+ and Ca2+) and anion (e.g., Cl-) channels also contribute to regulating the Em. This review will provide an in-depth summary of the molecular identities and electrophysiological properties of voltage-dependent cation channels in PASMC, focusing on Na+ and Ca2+ channels, which are opened by membrane depolarization and responsible for cell excitation, and voltage-gated Kv and Kca channels, which are responsible for controlling resting Em and repolarization when the cells are stimulated.
Table 1. Ionic composition of extracellular and intracellular solutions used for measurement of various ion channel currents.
| Current Type | Na+ mM | Ca2+ mM | K+ mM | Mg2+ mM | Cl- mM | Cs+ mM | ATP mM | EGTA mM | Glu mM | HEPES mM | pH |
|---|---|---|---|---|---|---|---|---|---|---|---|
| INa (whole cell) | |||||||||||
| Bath | 141 | - | 4.7 | 3 | 151.7 | - | - | 1 | 10 | 10 | 7.4 |
| Pipette | 10 | - | - | 4 | 143 | 135 | 5 | 10 | - | 10 | 7.2 |
| ICa (whole cell) | |||||||||||
| Bath | 110 | 20 | 4.7 | 1.2 | 157.1 | - | - | - | 10 | 10 | 7.4 |
| Pipette | 10 | - | - | 4 | 143 | 135 | 5 | 10 | - | 10 | 7.2 |
| IK(V) (whole cell) | |||||||||||
| Bath | 141 | - | 4.7 | 3 | 151.7 | - | - | 1 | 10 | 10 | 7.4 |
| Pipette | 10 | 0 | 135 | 4 | 143 | - | 5 | 10 | - | 10 | 7.2 |
| I K(Ca) (whole cell) | |||||||||||
| Bath | 141 | 1.8 | 4.7 | 1.2 | 151.7 | - | - | - | 10 | 10 | 7.4 |
| Pipette | 10 | 0 (8.8) | 135 | 4 | 143 | - | 5 | - (10) | - | 10 | 7.2 |
| IK(Ca) (cell attached) | |||||||||||
| Bath | 141 | 1.7 | 4.7 | 1.2 | 151.7 | - | - | - | 10 | 10 | 7.4 |
| Pipette | 10 | - | 125 | 4 | 129 | - | 5 | 0.1 | - | 10 | 7.2 |
| IK(V) (cell attached) | |||||||||||
| Bath | 141 | 1.8 | 4.7 | 1.2 | 151.7 | - | - | - | 10 | 10 | 7.4 |
| Pipette | 5 | 0 | 137 | 1.2 | 143.2 | - | 5 | 0.1 | 0 | 10 | 7.2 |
Passive cell membrane properties of human PASMC
The whole-cell patch clamp configuration[16] may be likened to an electrical circuit [Figures 1 and 2a]. A capacitor is made with two charged surfaces separated by a dielectric substance. The pipette itself is such a dielectric substance with two charged surfaces and, therefore, is represented by a capacitor in the circuit. Pipette capacitance (Cp, measured in Farads, F) is complicated in character, but its contribution to the overall circuit is usually minimized electronically by injecting a current transient designed to pre-charge the glass surface to the new desired potential. The pore of the pipette presents a resistance to current flow that may be easily measured before seal formation (Re, measured in Ohms, Ω). During whole-cell access, however, this resistance is increased by further resistance to current flow due to the contents or geometry of the cell itself (“series resistance” or “access resistance,” Ra), i.e. resistance to filling the entire cytosolic space with the desired amount of charge or potential due to interaction of charges with proteins or due to limited flux through long cell processes or narrow cell geometry.
Figure 1.
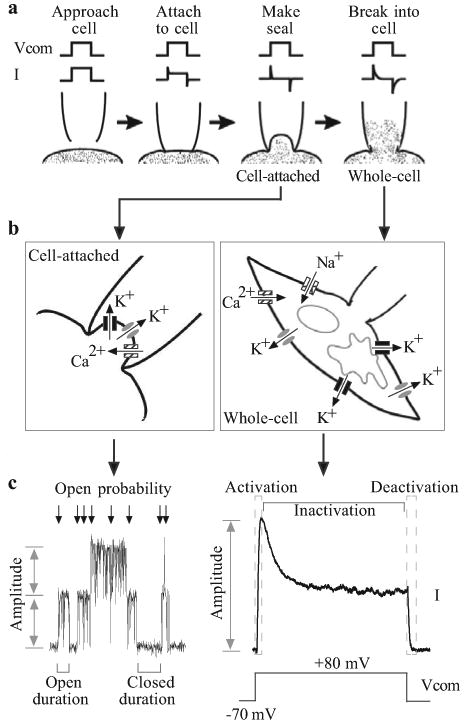
Patch-clamp electrophysiology. (a) Formation of the gigaΩ seal and the subsequent cell-attached and whole-cell configurations. (b) Enhanced view of the membrane–pipette arrangements in the cell-attached and whole-cell configurations. In a cell-attached patch, unitary currents are produced by ion flux through single channels. Three different channel types are shown. In the whole-cell mode, the macroscopic current recorded is the summation of all the currents generated by similar channels throughout the cell. (c) Measured parameters. Single-channel recordings can provide information relating to the amplitude the unitary currents, the open probability of the channels and the amount of time the channel(s) spend in open (open duration) or closed (closed duration) configurations. Macroscopic currents are characterized by the current amplitude, activation, inactivation and deactivation during a pulse protocol
Figure 2.
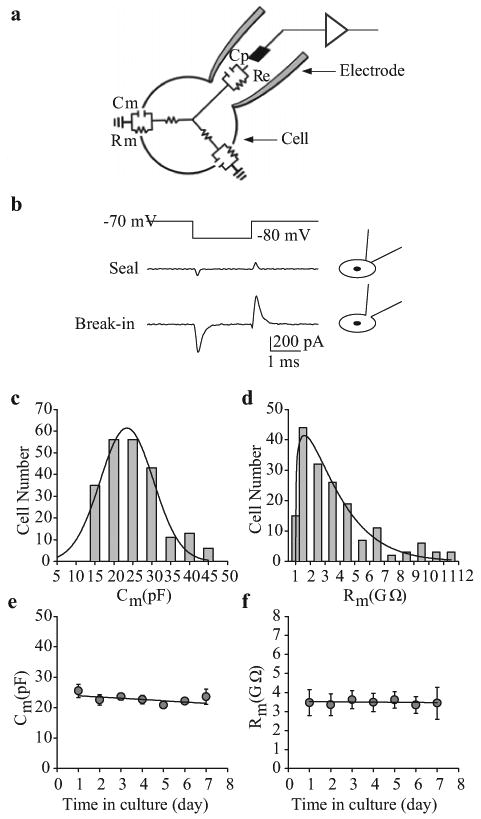
Passive membrane properties of human pulmonary artery smooth muscle cells (PASMC). (a) The cell and pipette form a circuit in the whole-cell patch-clamp configuration. Membrane capacitance (Cm) and resistance (Rm) are indicators of cell size and transmembrane ion flux, respectively. (b) Cm is often used to indicate that the membrane is ruptured. In the cell-attached configuration (“Seal”), Cm, measured as the surface area under the transient spikes, is small. Upon whole-cell access (“Break-in”), Cm is greatly increased. (c and d) Frequency distribution of Cm and Rm within a cell population. (e and f) Cm (n=220) and Rm (n=171) of human PASMC do not vary over time in culture
Once the cytosolic space of the cell is voltage clamped, the cell membrane also presents its own capacitance. Unlike the pipette, the cell membrane is of relatively uniform thickness and uniform dielectric content (lipids); therefore, in most cells, the specific membrane capacitance (Cm), which is normalized by the area of the plasma membrane, is ∼1 μF/cm2,[17] and a measure of the cell capacitance is a good indicator of cell size. The cell membrane itself is a very good dielectric, presenting a resistance of several gigaOhms (gigaΩ) when the membrane channels are closed at rest, in effect stopping the flow of charge across the membrane. However, the membrane resistance (Rm) is strongly influenced by the presence of ion conductances through the membrane ion channels.
Ion channels are selectively permeable to specific cations, and have a gating mechanism that may be controlled by voltage or other methods. Ion channels produce a conductance (g, measured in Siemens, S) that is dependent on the transmembrane electrical potential energy (ΔE, or Em, measured in Volts, V), and defined by Ohm's law, Ix=gE, where Ix is the conductance or current through that particular type of channel. Because Ohm's law defines resistance as the inverse of conductance, the overall membrane resistance is the inverse of the sum of all the conductances present on the membrane. The simple measurement of overall membrane resistance is therefore a good indicator of the amount of current carried through all the open channels on the membrane. As many membrane channels are voltage dependent, the membrane resistance likewise varies with membrane potential.
By employing a small hyperpolarizing command voltage step (Vcomm), for example from -70 mV to -85 mV (close to the equilibrium potential for K+), current transient (Itran) is induced [Figure 2b]. The cell membrane capacitance (Cm) can then be determined by pClamp software based on the equation: Cm=(integral of Itran)/)Vcomm). The membrane input resistance (Rm) is then calculated from the equation: Rm=(Rtotal×Rseal)/(Rseal -Rtotal), where Rseal and Rtotal are the resistance determined, respectively, from the steady currents of Itran in response to Vcomm (-5 mV) before and after break-in. As shown in Figure 2c, Cm can range from 15 pF to 45 pF, with an average Cm of 34±5 pF measured in 220 PASMC. The specific membrane capacitance can be calculated from the mean values of Cm and cell surface (capacitative) area; for PASMC, this is in the region of 1.25 μF/cm2, similar to the 1.3 μF/cm2 reported in rat caudal artery smooth muscle cells.[18] Rm under resting conditions is usually very high in the vascular smooth muscle cells.[19] Indeed, the calculated Rm in PASMC ranges from 1 GΩ to 12 GΩ, with an average Rm of 5±1 GΩ (n=171) [Figure 2d]. Importantly, the duration of PASMC in cell culture conditions does not significantly alter the values for Cm and Rm [Figure 2e and f].
Membrane potential can be measured in the current-clamp (I=0) mode. The resting Em in cultured human PASMC is approximately -45±5 mV [Figure 3a], and is slightly less negative than that observed in freshly dissociated PASMC from animals [20,21] As previously mentioned, Em is less negative than the EK (approximately -85 mV), which suggests that Em in these cells is also controlled by the permeability of other ions (e.g., Na+, Ca2+ and Cl-). The equilibrium potentials for Na+, Ca2+ and Cl- are believed to be +66, +122 and -26 mV, respectively, in native vascular smooth muscle cells.[15] In some PASMC, spontaneous electrical activity has been observed under resting conditions [Figure 3b], suggesting that these cells are electrically excitable.[22] This spontaneous electrical activity in PASMC is dependent upon the presence of extracellular Ca2+ [Figure 3b].[2,23-25]
Figure 3.

Electrically active human pulmonary artery smooth muscle cells (PASMC). (a) Histogram showing the wide distribution of resting Em in human PASMC. Em was measured in the current clamp (I=0) mode. (b) Spontaneous action potentials recorded in human PASMC are abolished when external Ca2+ is removed. Electrical activity is restored upon return to normal physiological Ca2+ (1.8 mM)
Evolution and diversity of the pore-forming voltage-gated cation channels
Ion-selective voltage-gated cation channels generate electrical activity in cells by undergoing rapid conformational changes from an impermeable structure to a highly permeable pore in the membrane through which ions can pass. Based on inherent similarities in the transmembrane domain structure of Na+, Ca2+ and K+ channel pore-forming α subunits, it is widely agreed upon that voltage-gated cation channels share a common ancestor. The basic building block of all these channels is a one-domain (1D) two-transmembrane segment (2TM) protein with an ion-selective pore/loop region between the transmembrane segments,[26] reminiscent of prokaryotic and eukaryotic K+-selective inward rectifier channels [Figure 4]. Indeed, K+ channels are the oldest of the voltage-gated cation channels as examples of these have been found in both prokaryotic and eukaryotic organisms.[27] Over time, multiple gene duplications and modifications elaborated this channel by the addition of four transmembrane segments, forming 1D six-transmembrane segment (6TM) protein that constitutes the basic pore-forming α–unit of mammalian voltage-gated cation channels. From this point, the evolution of voltage-gated ion channels diverged, with ion selectivity being a key element to channel diversification. Figure 4 provides a simple phylogenetic tree of voltage-gated cation channels based on sequence identity and domain arrangement.
Figure 4.
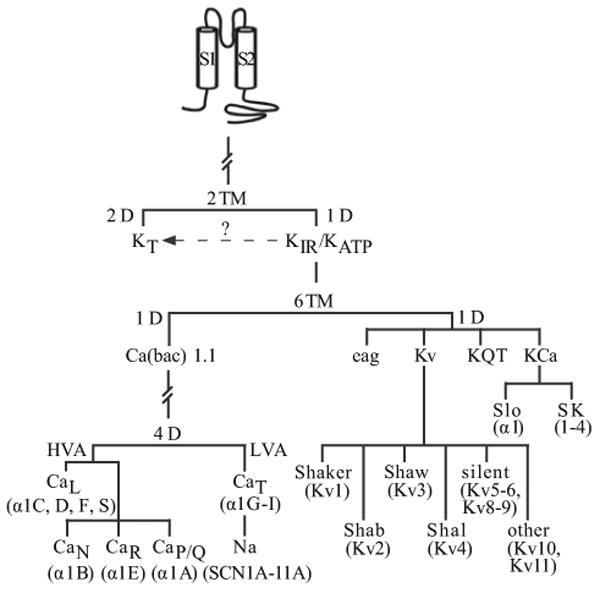
Proposed phylogenetic tree depicting the evolution of voltage-dependent cation channels. Pore-forming unit isoforms representing each channel are shown in parentheses. (TM – Transmembrane domain; D – Domain; KT – Two-pore domain K+ channel; KIR – Inward rectifier K+ channel; KATP – ATP-sensitive K+ channel; HVA – High-voltage activated; LVA – Low-voltage activated; Kv – Voltage-gated K+ channel; KQT – Long-QT K+ channel; Kca – Ca2+-activated K+ channel; SK – small-conductance Ca2+-activated K+ channel)
K+ channels evolved into the most diverse family of channels, mainly due to the sheer number of α–subunits and possible α4β4 subunit combinations. Four superfamilies of human K+ channels have maintained the 1D-6TM motif, and multiple α–subunits have surfaced for each: ether-a-go-go (eag, erk, elk; 3 isoforms in human), KQT (5 isoforms in humans), Kca (maxi-Kca and SKca; 6 isoforms in humans) and Kv (11 subfamilies and ≥30 isoforms) (see Coetzee et al. for review[28]). The sequence identity varies greatly within (e.g., 35–88% identity between Kv1 and Kv9) and between (e.g., 8–17% identity between Kv and Kca) the families.[28] Chromosomal site analysis of the known human isoforms also suggests that K+ channels have existed for a long time. Genes encoding 1D-6TM K+ channels are found on at least 13 human chromosomes, with little evidence of clustering of genes except in the case of a few Kv channels (Figure 5 depicts the chromosomal location of channel pore-forming and regulatory subunits identified in human PASMC). Kv channel α–subunits alone can be found on 10 chromosomes within the human genome.
Figure 5.

Chromosomal location of ion channel genes expressed in human pulmonary artery smooth muscle cells (PASMC). All isoforms of pore-forming and regulatory subunits of Na+, voltage-dependent Ca2+ channels, Kv, and Kca channels identified in human PASMC are shown. Chromosomal location is based on the primer sequences described in Table 2
Na+ and Ca2+ channels evolved after K+ channels were well established.[27] A few theories have been put forth to explain the development of four-domain (4D) -6TM channels from 1D-6TM channels:[26] (a) two rounds of gene duplication of 1D-6TM K+ channels (1D÷2D, 2D÷4D) and mutations within the pore region to alter ion selectivity created the 4D-6TM Ca2+ and Na+ channels.[26] Coupled with mutations within the pore to alter ion selectivity, this evolutionary cascade would have produced the 4D-6TM Ca2+ and Na+ channels. (b) 1D- and 4D-6TM channels have a common 1D-6TM cyclic-nucleotide gated (CNG) channel ancestor, with S4 and pore regions similar to voltage-gated K+, Ca2+ and Na+ channels. In addition to structural similarities, CNG channels also exhibit some voltage sensitivity and are permeable to both monovalent and divalent cations, making it an ideal common precursor.[26] (c) More recently, Durell and Guy[29] showed that a Ca2+ channel with a 1D-6TM motif could be detected in the akylaphilic bacterium Bacillus halodurans\pard plain. This suggests that more mutations conferring Ca2+ selectivity in bacterial 1D-6TM channels occurred before any gene duplication occurred and prior to the development of lower eukaryotes (protozoans), where 4D-6TM Ca2+ channels have been identified.[26,29]
The first 4D-6TM proteins were voltage-dependent Ca2+ channels (VDCC), with early gene identification revealing multiple channels within the same tissue or cell. Eventually, Ca2+ channels were classified as low-voltage activated (LVA) or high-voltage activated (HVA) VDCC (see below and Catterall[30] for review). Ca2+ influx via VDCC has already been established as an effector or trigger in numerous cellular processes, with the different channel subtypes sometimes playing different roles. The variety of functional roles for VDCC correlates well with the significant structural diversity between the 10 VDCC α subunits currently identified (α1A-1I, α1S).[30] Although the six isoforms identified in human PASMC represent each of the five Ca2+ channel subtypes, there is only electrophysiological evidence for the L- and T-type channels in PASMC[31,32] (also see below). As for voltage-gated K+ channels, the associated genes are encoded on at least five human chromosomes, with no grouping of α–subunits encoding for similar currents on the same chromosome. For example, α1C and α1D, both encoding for L-type VDCC in human PASMC, are located on chromosomes 12 and 3, respectively, while those encoding for T-, N-, R- and P/Q-type channels are scattered on chromosomes 17, 9, 1 and 19, respectively [Figure 5]. Furthermore, while the structure of T-type VDCC is very similar to that of LVA channels, the sequence identity between them is <25%, implying that the HVA and LVA subfamilies represent radically different evolutionary branches.[26]
Like T-type Ca2+ currents (ICa(T)), Na+ currents are rapidly activating transient currents activated at more negative membrane potentials. In lower eukaryotes, Ca2+ was the primary charge carrier;[26] purely Na+-dependent action potentials were not common until the advent of the early metazoans. This has led to speculation that low-voltage activated and rapidly activating Na+ channels evolved from Ca2+ channels in parallel with the evolution of the first nervous systems. Sequence analysis has shown that ligand-binding sites (e.g., carboxy-terminal calmodulin-binding site) may be conserved within the 4D-6TM voltage-gated Ca2+ and Na+ channels, suggesting a similar evolutionary precursor.[33,34] Of particular interest is a putative calmodulin (CaM)-binding site located in the carboxy-terminal regions of both Na+ and Ca2+ channels.[33,34] Cloning of the first LVA channels verified that voltage-dependent Na+ channels did evolve from T-type VDCC.[35] Eleven known Na+ channel α–subunit genes (SCN1A-11A) bearing strong biophysical and sequence (∼75% sequence identity) similarities have been identified in the skeletal, cardiac and uterine muscles and in the human brain,[26,36,37] and bear approximately 75% sequence identity to each other.[26] Because the isoforms bear strong similarities, even when expressed in heterologous systems, voltage-gated Na+ channels are not generally grouped into families like their Ca2+ and K+ counterparts. The SCN5A and 6A isoforms expressed in PASMC are typically found in cardiac and uterine muscle. Unlike K+ and Ca2+ channel α–subunit genes, all Na+ channel α–subunit genes map within four chromosomes (2, 3, 12 and 17) containing homeobox (HOX) gene clusters.[37] HOX genes have been predicted to have existed in ancestral chordates, suggesting that the initial expansion of Na+ channels is associated with multiple chromosome duplications occurring after the divergence from invertebrate to pre-vertebrate chordates. The fact that many of the SCN genes are clustered on two chromosomes also suggests that intrachromosomal duplications also occurred over time.
Voltage-gated Na+ Channels
In a variety of excitable cells, including smooth muscle cells, voltage-gated Na+ channels are responsible for generating action potentials. Activation of the channels induces membrane depolarization and thus increases [Ca2+ ]cyt by promoting Ca2+ influx through the sarcolemmal VDCC and the reverse mode Na+/Ca2+ exchanger.[38,39] While the activation of Na+ channels may underlie the spontaneous action potentials observed in cardiac and skeletal muscle myocytes,[40] removal of extracellular Ca2+ abolished spontaneous action potentials in PASMC [Figure 3b], suggesting that the electrical excitability of PASMC is induced by multiple ion channel functions. Furthermore, voltage-gated Na+ channels may serve as a pathway for Ca2+ entry under physiological and pathophysiological conditions.[41,42]
Biophysical properties of voltage-gated Na+ currents (INa)
The inward INa(V) observed in PASMC possesses similar biophysical and pharmacological characteristics to those previously identified in other human vascular smooth muscle cells:[39,43-46] sensitivity to tetrodotoxin (≤1 μM for total inhibition), -60 to -50 mV activation threshold, -15 to 10 mV peak amplitude potential, -70 to -65 mV half-inactivation voltage (τinact ≤4 ms) and -25 to -15 mV half-activation voltage (τact ∼1 ms). In cultured human PASMC dialyzed with Cs+-containing solution, a rapidly inactivating inward Na+ current is observed in the absence of extracellular Ca2+ [Figure 6a]. As mentioned above, the current activates at potentials close to -60 mV and peaks at approximately +10 mV [Figure 6a]. These currents also inactivate rapidly, with the half-inactivation (V0.5) occurring at approximately -65 mV and complete inactivation occurs at -20 mV [Figure 6b]. Equimolar replacement of external Na+ with N-methyl-D-glucamine (NMDG) or extracellular application of 1 μM tetrodotoxin (TTX) is sufficient to abolish the currents [Figure 6c], suggesting that the currents in PASMC are carried by Na+ influx through the TTX-sensitive, voltage-gated Na+ channels similar to those described in neurons and cardiomyocytes.[40] The window currents determined by the overlap between the activation and inactivation curves are in the voltage range of -60 to -20 mV in human PASMC cultured in growth medium [Figure 6b, right panel]; these values are similar to those in other vascular smooth muscle cells, suggesting the participation of Na+ currents in the regulation of resting Em in human PASMC.
Figure 6.

Electrophysiological and pharmacological properties of voltage-gated Na+ currents (INa(V)) in human pulmonary artery smooth muscle cells (PASMC). Cells are dialyzed with a Cs+-containing pipette solution [Table 1]. (a) Representative currents were elicited by depolarizing the cell to from a holding potential of −70 mV to test potentials between −80 mV and +80 mV (protocol at bottom). Upper left inset: Steady-state activation and inactivation of currents occurred within <5 ms and <16 ms, respectively. Lower right inset: Summarized INa(V) I-V relationship. (b) Currents were evoked by a step depolarization to 0 mV from different conditioning potentials (-120 mV and -20 mV) applied for 10 s prior to the test depolarization (left). Voltage-dependent steady-state availability (I/Imax) and normalized conductance-voltage relationship (gNa/gNa, max) of the peak INa(V) amplitude. The I/Imax and gNa/gNa, max curves were best fitted using exponential and Boltzman equations, respectively. (c) INa(V) is completely suppressed by equimolar replacement of extracellular Na+ with N-methyl-D-glucamine (NMG) or extracellular application of 1 μM tetrodotoxin. Currents were elicited by step depolarizations from −70 mV to 0 mV
Voltage-gated Na+ channel genes expressed in human PASMC
A complex of three glycoprotein subunits form functional Na+ channels: a pore-forming α–subunit and two β-subunits that modulate channel gating and membrane expression [Figure 7a].[47] The α–subunit alone can form a functional channel and is composed of four domains, each containing six transmembrane segments (S1–S6) and a pore loop (P region). Each S4 segment is believed to act as a voltage sensor, while the S5-pore loop-S6 segments form the transmembrane pore itself. Using reverse transcriptase-polymerase chain reaction (RT-PCR), seven Na+ channel-related gene transcripts (SCN1B, 2A, 2B, 4A, 8A, 9A and 11A) have been detected in PASMC [Figure 7b]. Transcripts for SCN5A and SCN6A have not yet been detected in PASMC. All of these isoforms are expressed in the brain [Figure 7b]. The combined functional and molecular identification of Na+ currents and channels in human PASMC suggest that voltage-gated Na+ channel activity and expression may relate to PASMC excitability, contractility, proliferation and differentiation.
Figure 7.
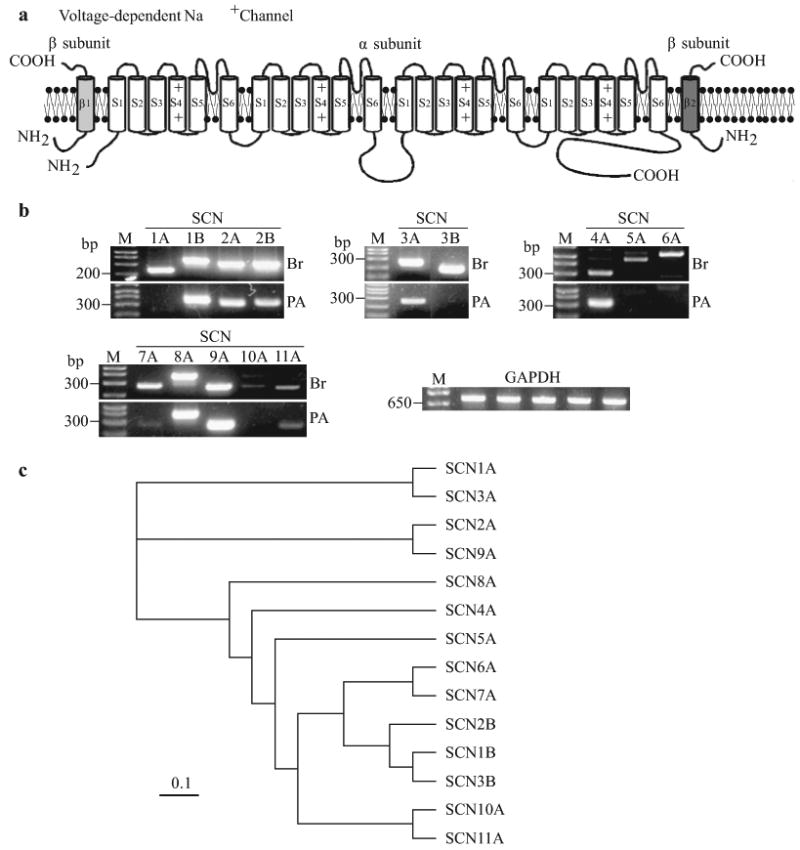
Molecular identity of voltage-gated Na+ channels in human pulmonary artery smooth muscle cells (PASMC). (a) Structural arrangement of Na+ channel α-, β1- and β2-subunits. (b) The mRNA expression of cloned Na+ channels in human brain (Br) and PASMC (PA). Polymerase chain reaction (PCR)-amplified products displayed for the transcripts of SCN2A, SCN4A, SCN5A, SCN6A and β-actin. “-RT”, PCR performed with no reverse transcriptase (RT). “M”, 100 bp DNA ladder. (c) A phylogenetic tree showing the inferred evolutionary relationships among different Na+ channel genes
Phenotypical change of voltage-gated Na+ channel expression in freshly dissociated and cultured VSMC
Voltage-gated TTX-sensitive Na+ currents (INa(V)) have been described in several types of human vascular smooth muscle cells cultured from the aorta[43,44] and coronary[39,46] and pulmonary[43,45] arteries. On only rare occasions have INa(V) been recorded in freshly dissociated human vascular smooth muscle cells, although they are readily detected when the same cells are cultured.[46] Although it may be due to a technical problem (e.g., the rapid inactivation of the currents and the large size of most freshly dissociated smooth muscle cells to record INa(V)), the relative inability to detect INa(V) in freshly dissociated, but not cultured, cells from the same vascular bed may bear some relation to cell dedifferentiation and proliferation.[46] More specifically, voltage-gated Na+ channel expression and activity may be required to facilitate the transition from a “contractile” to “synthetic” or “proliferative” phenotype.[48,49] However, INa(V) have been recorded in both freshly dispersed rabbit[24] and cultured human[45] PASMC. This raises the possibility that the development and expression of functional voltage-gated Na+ channels in cultured cells acts as a trigger for cell differentiation and proliferation, possibly via enhanced [Ca2+ ]cyt, as discussed below.
Functional properties of voltage-gated Na+ channels in human PASMC
Na+ channels appear to play an important role in the regulation of [Ca2+ ]cyt and sarcolemmal Ca2+ influx by different mechanisms. Firstly, in cardiac myocytes, enhanced TTX-sensitive INa(V) causes a localized transient increase in [Na+ ]cyt, thereby activating reverse-mode Na+/Ca2+ exchange and increasing [Ca2+ ]cyt with the subsarcolemmal space between the plasma membrane and SR.[41] The Ca2+ newly introduced into the cytoplasm can then trigger further Ca2+ release from the SR (which ultimately will cause contraction and stimulate proliferation and migration) or replenish SR Ca2+ pools by Ca2+-ATPase-mediated re-uptake.[42,50] Secondly, TTX-sensitive Na+ channels are promiscuous, i.e. they can allow permeation of other cations (such as Ca2+) under certain conditions (e.g., absence of extracellular Na+, presence of tracing doses of steroids such as ouabain and digoxin).[42,51] Ca2+ influx through promiscuous Na+ channels can contribute to local and global cardiac Ca2+ signaling, especially in heart failure patients treated with digoxin.[42] In addition to its modulating [Ca2+ ]cyt, the permeability of Na+ channels to Ca2+ may also play a role in the contractile-to-proliferative cellular transition. Thirdly, voltage-gated Na+ channels are essential in the generation of action potentials in many excitable cells, thereby regulating [Ca2+ ]cyt based on evidence from expressed SCN5A channels,[33,53] we can speculate that Ca2+/CaM-mediated regulation of voltage-gated Na+ channels may play an important role in the coupling of human PASMC excitation and contraction.
VDCC
In excitable cells, the opening of VDCC is a critical mechanism responsible for muscle contraction induced by neuronal and humoral stimulation. There are at least five types of VDCC described in neurons and cardiomyocytes: L-type, T-type, P/Q-type, R-type and N-type.[54,55] These Ca2+ channels have been sorted based on their electrophysiological, pharmacological, kinetic and molecular properties. VDCC have also been separated into two groups based on their activation voltage. HVA VDCC include all but T-type channels, with the latter classified as LVA VDCC. HVA channels activate at membrane potentials between -50 mV and -20 mV, while LVA channels activate at more negative potentials approximating -70 mV. Typically, only currents generated by L- and T-type channels have been measured in cardiovascular tissues, while all current types have been recorded in neuronal tissues.[54]
Whole-cell VDCC currents (ICa)
In human PASMC, a large, slowly inactivating inward Ca2+ current is observed when cells are held at -70 mV and depolarized to 0 mV [Figure 8a]. The current activates close to -20 mV, with a maximal activation of approximately +15 mV. Removal of extracellular Ca2+ abolishes the currents, confirming that the currents are due to Ca2+ influx. Nifedipine, a dihydropyridine blocker of VDCC, is also able to significantly inhibit the currents. The currents present in PASMC are, therefore, mainly due to Ca2+ influx through dihydropyridine-sensitive L-type Ca2+ channels. Less frequently, and while being held at a very negative potential (-100 mV), depolarization to a test potential to -20 mV can elicit a rapidly activating transient inward Ca2+ current [Figure 8b and c]. This transient current activates and inactivates rapidly in comparison with the L-type current [Figure 8a], with a threshold potential for activation of approximately -36 mV at a holding potential of -90 mV. The biophysical properties of these currents are very similar to the T-type Ca2+ current observed in aortic[56] and renal artery[57] smooth muscle cells, rat PASMC[58] and cardiomyocytes.[59]
Figure 8.

Electrophysiological and pharmacological properties of L- and T-type voltage-dependent Ca2+ currents (ICa) in human pulmonary artery smooth muscle cells (PASMC). Cells were dialyzed with a Cs+-containing pipette solution [Table 1]. (a) A representative current (L-type ICa), elicited by depolarizing a cell from a holding potential of -70 mV to 0 mV. (b) A representative current (L-type ICa), elicited by depolarizing a cell from a holding potential of -100 mV to -20 m V. (c) Activation (left) and inactivation (right) kinetics of L-type and T-type ICa
Endogenously expressed genes that encode VDCC in human PASMC
As for voltage-gated Na+ channels, the pore-forming VDCC α1-subunits (10 identified isoforms) are composed of four, six transmembrane segment domains [Figure 9a] that, when expressed alone, can create functional channels.[47] The pore-forming S5-loop-S6 segments and the voltage-sensing S4 segments are integral to the function of α1-subunits. Three different regulatory subunits are also part of the greater Ca2+ channel complex.[54,60-62] β–subunits (four isoforms with their associated subtypes) play multiple roles in regulating channel membrane expression of α1-subunits, current kinetics and biophysical properties. Extracellular α2δ-subunits (two isoforms plus their subtypes) that are attached to the plasma membrane via a disulfide linkage can influence current amplitude and inactivation rates, and likely play a major role in stabilizing the incorporation of the Ca2+ channel complex into the plasma membrane. Finally, γ-subunits (six known isoforms) may modulate channel assembly and channel subtype-specific current kinetics, both effects being highly dependent on the nature of the co-expressed β– and α2δ-subunits [Figure 9a]. At the RNA level, transcripts for six pore-forming α1-subunits have been detected in human PASMC, encoding for all five VDCC types: α1A (P/Q-type), α1B (N-type), α1C and α1D (L-type), α1E (R-type) and α1G (T-type) [Figure 9b]. Additionally, a variety of regulatory subunit isoforms are also present, including α2δ1 [Figure 9c], β1-4 [Figure 9d] and γ6 [Figure 9e] in human PASMC. From the current molecular and electrophysiological evidence, it may be speculated that the α1C-subunit may encode the L-type VDCC while the α1G encodes for the T-type VDCC in human PASMC.
Figure 9.
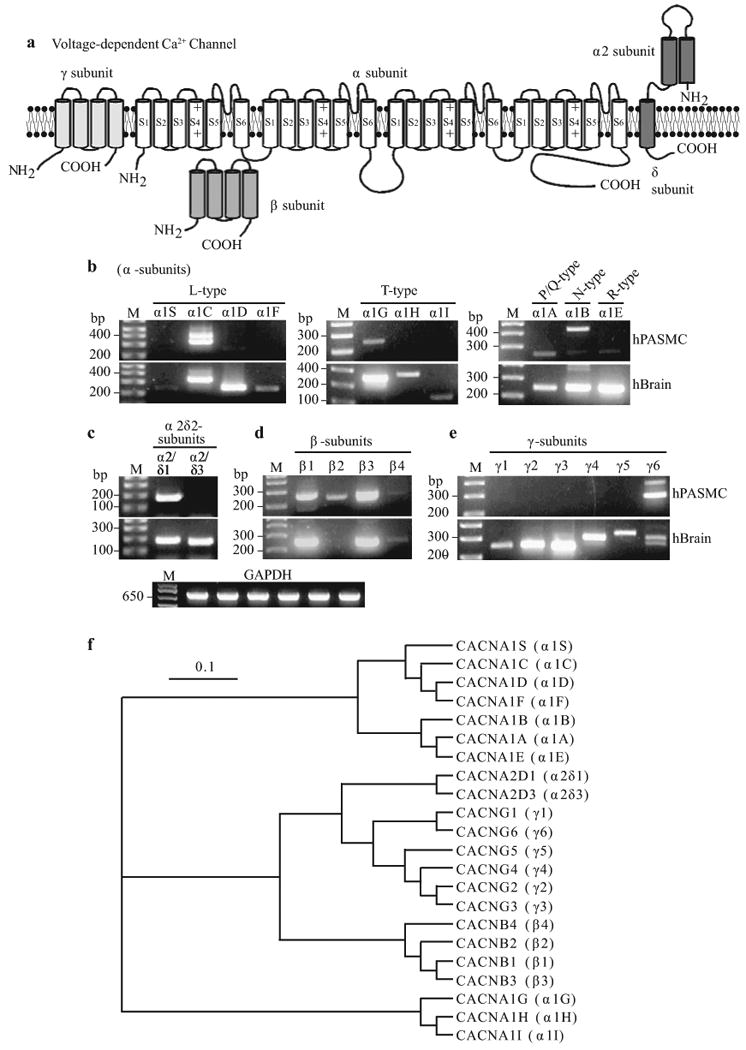
Molecular identity of voltage-dependent Ca2+ channels (VDCC) in pulmonary artery smooth muscle cells (PASMC). (a) Structural arrangement of Ca2+ channel α-, β-, α2β- and γ-subunits. (b–e) The mRNA expression of α (1A-1F, 1S), α2δ (δ1 and δ3), β (1-4) and γ (1-6) subunits for L-, T-, P/Q-, N- and R-type VDCC in human PASMC (hPASMC) and brain tissues (hBrain). “M,” 100 bp DNA ladder. (f) A phylogenetic tree showing the inferred evolutionary relationships among different Ca2+ channel genes
Voltage-gated K+ Channels
Functionally, both voltage-gated (Kv) channels and Ca2+-activated K+ (Kca) channels (see below) are sensitive to voltage changes. In other words, these channels are activated by membrane depolarization and are deactivated by membrane hyperpolarization. A fundamental difference between Kv and Kca channels is their response to Ca2+: in vascular smooth muscle cells, Kv channels are inhibited by cytoplasmic Ca2+[21,63] and Kca channels are activated by cytosolic Ca2+.[19,64] The existence of other types of K+ channels, such as inward rectifier (KIR), ATP-sensitive (KATP) and tandem-pore (KT) channels, has also been demonstrated in vascular smooth muscle cells.[19,65,66] This review focuses only on the voltage-dependent channels; Kv and Kca channels.
Classification based on unitary conductance
Macroscopic currents of Kv (IK(V)) and Kca (IK(Ca)) channels can be readily dissociated based on their pharmacological properties and Ca2+-dependence. Additionally, the single-channel conductance for each of these channels can also serve to distinguish them from each other. The traces shown in Figure 10 are representative cell-attached recordings from PASMC, where multiple channel subtype openings can be recorded from the same patch using identical Ca2+-containing perfusion solutions. As shown in Figure 10a, large amplitude K+ currents (a) and several small amplitude currents (b–f) can be recorded in a cell-attached membrane patch. In addition to the various amplitudes of the recorded K+ currents, the duration of the channel openings varies in human PASMC. Examples of long-lasting channel and “flickery” openings are shown in Figures 10b and c. In cell-attached patches of PASMC, multiple amplitudes of outward K+ currents can be elicited by steadily holding the patch at different potentials. Representative openings for channels with seven different conductance levels are shown in Figure 11. The large amplitude current (225 pS and 189 pS) openings are likely generated by the activation of large-conductance Kca channels,[67] while the 33 pS, 81 pS and 6 pS channels may represent unitary currents through different Kv channels or small to intermediate conductance Kca channels.
Figure 10.

Single-channel K+ currents in cell-attached patches of human pulmonary artery smooth muscle cells (PASMC). (a) Recordings from a human PASMC showing the variability of current amplitudes (a-f) within the same patch. The horizontal broken line indicates the level of currents when the channels are closed. Unitary Kv (b) and Kca (c) openings can be sustained (a) or flickery (b). (c). View of flickery and sustained iK(Ca) on expanded time scales
Figure 11.

Range of single-channel conductances of K+ channels observed in human pulmonary artery smooth muscle cells (PASMC). Floating bar graph showing modes, medians and ranges of the seven conductance classes identified in human PASMC. The number of cells exhibiting particular channel conductances is indicated by the gray-shaded bars
In addition to its regulation of current amplitude, membrane potential can also affect the gating properties of these channels, e.g. the open probability (Popen). For the 189 pS channel shown in Figure 10, Popen increased with membrane depolarization from 0.0005 at 0 mV to 0.014 at +50 mV and 0.27 at +90 mV. Similarly Popen for the 33 pS channel increased from 0.04 at +60 mV to 0.43 at +90 mV, from 0.009 at +40 mV to 0.01 at +90 mV for the 141 pS channel and from 0.007 at +40 mV to 0.02 at +90 mV for the 6 pS channel. Therefore, both the single channel amplitude and the open probability of Kca and Kv channels are influenced by membrane potential in human PASMC.
Whole-cell voltage-gated K+ (Kv) currents
In order to record optimal whole-cell (macroscopic) Kv currents (IK(V)), cells are commonly perfused with Ca2+-free bath solution (plus 1 mM EGTA) and dialyzed with Ca2+-free pipette solution (plus 10 mM EGTA). Depolarizing the cells from a holding potential of -70 mV to a series of test potentials ranging from -60 mV to +80 mV elicits outward K+ currents, with a threshold potential of activation at approximately -45 mV. Four families of whole-cell IK(V) currents can be distinguished based on their activation and inactivation kinetics: (i) rapidly activating and slowly inactivating IK(V) [Figure 12a], (ii) rapidly activating and non-inactivating IK(V) [Figure 12b], (iii) slowly activating and non-inactivating IK(V) [Figure 12c] and (iv) rapidly activating and rapidly inactivating IK(V) [Figure 12d]. Activation time constants (τact) can be separated into two components corresponding to the rapidly and slowly activating currents (<3 ms and >3 ms, respectively) [Figure 12e, top panel]. Inactivation constants (τinact) are much more variable, as shown in Figure 12e, bottom panel, with the midpoint between rapid and slow inactivation being approximately 100 ms. The half-activation occurs at +25 mV for each type of current. In PASMC, the family of Kv channels can thus be grossly divided into two categories: (a) delayed rectifier Kv channels generating slowly activating and non- or slowly inactivating currents and (b) rapidly activating and rapidly inactivating currents originating from the activation of transient “A”-type currents similar to those observed in phasic smooth muscle, cardiomyocytes and neurons.[68-71]
Figure 12.

Whole-cell voltage-gated K+ (Kv) currents (IK(V)) in human pulmonary artery smooth muscle cells (PASMC). (a–d) Four different types of Kv currents were elicited by step depolarizations from a holding potential of −70 mV to test potentials between -80 mV and +80 mV in 20 mV increments. Representative families of currents (left panels), enlarged trace segments showing steady-state activation (middle, top panels) and inactivation (middle, bottom panels) and I-V curves are presented for each type of current. (e) Activation (top) and inactivation (bottom) time constants are plotted as a function of cell number. The majority of currents activated rapidly (within 1-4 ms). The range of inactivation constants is more varied, reflecting the different current types
Extracellular application of 5 mM 4-AP, a common Kv channel inhibitor, reversibly decreases Kv currents [Figure 13]. While the slow inactivation kinetics of three of the different currents are typical of most native delayed-rectifier K+ currents recorded in vascular SMCs,[19,25] the 4-AP-sensitive rapidly activating and inactivating current may represent a different class of K+ current less commonly observed in vascular SMC. Based on its rapid inactivation (<100 ms) kinetics, this component closely resembles the transient IA-type current that has been observed in phasic smooth muscle cells,[71,72] cardiac cells[68] and neurons.[70] Heteromeric assembly of K+ channel α–subunits may account for the notable diversity of K+ currents within the same cell system. When the electrophysiological properties of PASMC Kv currents are compared with those generated by cloned Kv channel α–subunits,[28,73-75] it is clear that the native channels' properties are intermediaries of those different clones forming the functional channels.
Figure 13.

Inhibitory effect of 4-amynopyridine (4-AP) on macroscopic voltage-gated K+ (Kv) currents (IK(V)) in pulmonary artery smooth muscle cells (PASMC). Rapidly activating and slowly inactivating (a), rapidly activating and non-inactivating (b) and rapidly activating and rapidly inactivating (c) were elicited by step depolarizations between -80 mV and +80 mV from a holding potential of -70 m V. Representative traces are shown before (Cont), during (4-AP) and after (washout) the application of 5 mM 4-AP. I-V curves of 4-AP-sensitive currents (subtraction of the currents recorded during 4-AP from the control currents) are presented in the middle panels for each current type. The 4-AP-sensitive current components depicted to the right were obtained by digital subtraction (Cont − 4-AP)
The behavior of single channels within a patch provides some evidence for the heteromeric assembly of the pore-forming units [Figure 10]. Cloned Kv channels have a wide range of single-channel conductances that do not always match with the conductance of native Kv channels. For example, the single-channel conductances for Kv1.1, Kv1.2 and Kv1.5 channels expressed in heterologous expression systems are reported to be 10 pS, 9–17 pS and 8 pS, respectively.[28,76,77] The conductance of native Kv channels in vascular SMC at physiological K+ concentrations (5 mM internal, 140 mM external) ranges between 5 pS and 11 pS,[72,78] and between 15 pS and 70 pS in symmetrical (140 mM) K+ conditions.[79-81] While the differences between native and cloned Kv conductances may relate to differences in the expression systems (e.g., pulmonary artery vs. HEK 293 cells), splicing or post-translational modifications, it is quite likely that native Kv channels are heterotetramers.
The association of multiple β–subunits with the functional α–tetramer may further influence the biophysical properties of native currents,[73] including those in human PASMC. Cytoplasmic Kv channel β–subunits associate with the S6 segment and carboxy-terminal region of Kvα–subunits via their own highly conserved carboxy terminus.[82] The most dramatic functional effect of Kv channel β-subunit association with Kv channel α is to confer inactivation onto the non-inactivating channels [Figure 14] and to confer redox and O2 sensitivity onto the Kv channels.[83,84] In extreme cases, they convert non-inactivating Kv currents into rapidly inactivating transient currents.[85] In the case of the Kv channel β1-subunit, this occurs via the pore-blocking effect of an amino-terminal inactivation ball domain similar to that found on the “subunit.[85] Other β-subunits modulate current kinetics by shifting the activation curve, slowly deactivating the current, enhancing slow inactivation or altering peak current amplitude by acting as an open-channel blocker.[82,86] Finally, Kv channel β–subunits may participate in α–subunit assembly and transport to the plasma membrane, and enhance the interaction of α–subunits with protein kinases.[82,87] Given the diversity of roles and properties of Kv α– and β-subunits, it is not altogether surprising that Kv channel activity is central to numerous processes that rely on membrane potential regulation, such as hypoxic pulmonary vasoconstriction,[21,88-90] cell proliferation[6,91] and myogenic reactivity.[92]
Figure 14.

Co-transfection of Kvβ-subunits affects KCNA5 channel kinetics. HEK-293 cells were transfected with wild-type KCNA5 alone (KCNA5) or in the presence of Kvβ1.3-HA (KCNA5/Kvβ1.3). Representative current recordings (a) and I-V curves (b) are shown (pulse protocol, lower panel). (c) Averaged currents (left) and normalized currents (right) at +60 mV in cells transiently transfected with WT KCNA5 alone (KCNA5) or WT KCNA5 + Kvβ1.3-HA (KCNA5/Kvβ1.3)
Kv channel genes expressed in human PASMC
As mentioned above, native Kv channels are believed to be heteromeric tetramers composed of the pore-forming α–subunits and regulatory cytoplasmic β-subunits (α4/β4)[28,93] [Figure 15a]. Transcripts of Kv channel genes detected by RT-PCR on mRNA isolated from human PASMC and brain tissues are shown for each Kv channel subunit in Table 2. Brain tissue is commonly used as a positive control for the mRNA expression of ion channels due to its high expression of the majority of known ion channels. In human PASMC, at least 22 Kvα–subunits and 3 Kvβ-subunits [Figure 15b] have been identified. It is currently unknown as to how many of these transcripts are transcribed leading to expression of a functional channel/protein in PASMC. Figure 15c shows a phylogenetic diagram of Kv channels.
Figure 15.
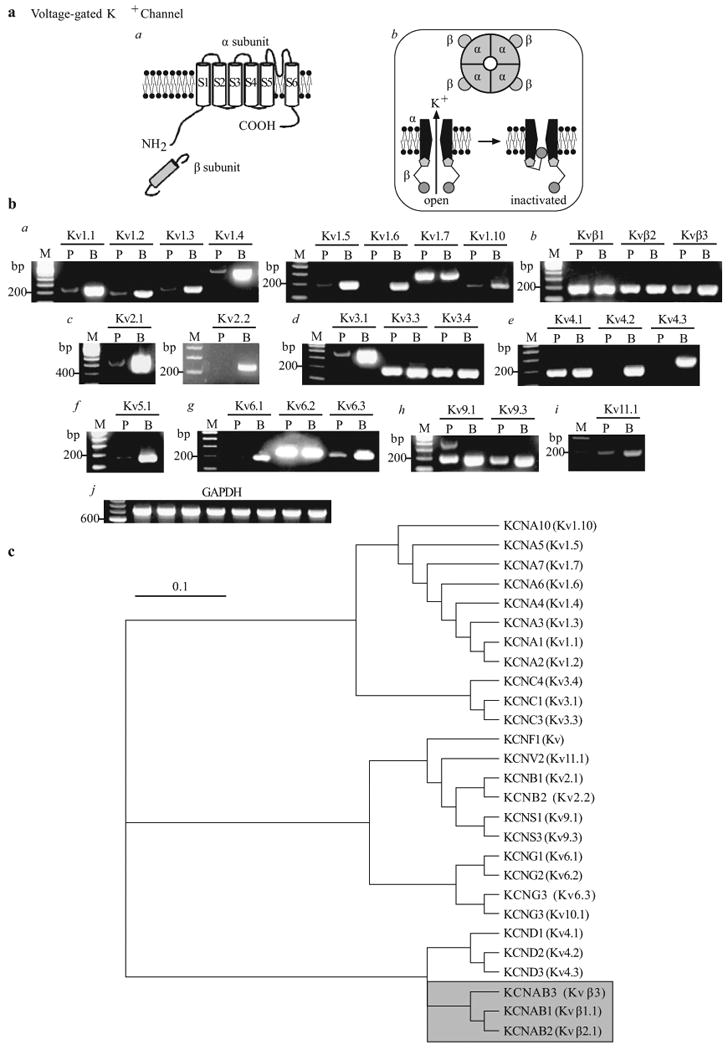
Molecular identity of voltage-gated K+ (Kv) channels in pulmonary artery smooth muscle cells (PASMC). (a) Structural arrangement of Kv channel α- and β-subunits (a), the tetrameric association of α–subunits (b) and the ball-and-chain inactivation mechanism for IK(V) (b). (b) The mRNA expression of reverse transcriptase-polymerase chain reaction products using Kv1 (a), Kvβ (b), Kv2 (c), Kv3 (d), Kv4 (e), Kv5 (f), Kv6 (g), Kv9 (h) and Kv11 (i) primers in human PASMC (P) and brain (b) tissues. “M,” 100 bp DNA ladder. (c) A phylogenetic tree showing the inferred evolutionary relationships among different Kv channel genes
Table 2. Oligonucleotide sequences of the RT-PCR primers.
| Standard names (Accession no.)* | Size (bp) | Predicted sense/antisense | Location (nt) | Gene (chromosome) |
|---|---|---|---|---|
| Voltage-gated Na+ channels | ||||
| SCN2A | 629 | 5′-ACATCTGTGTGAAGGCTGGTAG-3′/ | 1157-1179 | 2q23-q24 |
| (X65361) | 5′-CAGTAAGGACTGGTGTGGAGAA-3′ | 1763-1785 | ||
| SCN4A | 684 | 5′-GCCGTTCAACGACACCAACACC-3′/ | 932-954 | 17q23.1-q25.3 |
| (M81758) | 5′-GATGTGTCCAGGCTGCCATTGC-3′ | 1593-1615 | ||
| SCN5A | 466 | 5′ -AGAAGATGGTCCCAGAGCAATG-3′/ | 1647-1669 | 3p21 |
| (M77235.1) | 5′-CTCGAAGCCATCTACACACGGA-3′ | 2090-2112 | ||
| SCN6A | 507 | 5′-CAGATGAGGCCAAGACCATACA-3′/ | 1422-1444 | 2q21-q23 |
| (M91556) | 5′-ATCGAAGAAGAGCCATTCCTGC-3′ | 1906-1928 | ||
| β-actin | 661 | 5′-GACGGGGTCACCCACACTGTGCCCATCTA-3′/ | 2134-2162 | 7p22-p12 |
| (M10277) | 5′-CTAGAAGCATTTGCGGTGGACGATGGAGG-3′ | 2971-3000 | ||
| β-actin | 314 | 5′-CACTCTTCCAGCCTTCCTTC-3′/ | 820-840 | 7p22-p12 |
| (X00351) | 5′-CTCGTCATACTCCTGCTTGC-3′ | 1113-1133 | ||
| Voltage-gated Ca2+ channels | ||||
| L-type | ||||
| α1S | 246 | 5′-GGGAGCGAGGAGAGTAATCC-3′/ | 46-65 | 1q32 |
| (NM_000069) | 5′-CTCAGGAACTGGCTTCTTGG-3′ | 272-291 | ||
| α1C | 357 | 5′-TCTTTCACCCCAATGCCTAC-3′/ | 1875-1894 | 12p13.3 |
| (NM_000719 | 5′-CCTCCTGGTTGTAGCAGGTC-3′ | 2212-2231 | ||
| α1D | 258 | 5′-GCCAGATTGGTTGACACAGA-3′/ | 1819-1838 | 3p14.3 |
| (XM_003238) | 5′-CAGTGCCTGGTCACTTTGAA-3′ | 2057-2076 | ||
| α1F | 251 | 5′-CTACTTCCTGGGATCCGACA-3′/ | 863-882 | Xp11.23 |
| (NM_005183 | 5′-ACACCCAGGGCAGTTCATA-3′ | 1095-1113 | ||
| T-type | ||||
| α1G | 250 | 5′-TTCCCAAAGATGCACCTCAT-3′/ | 494-513 | 17q22 |
| (AF029229) | 5′-TGTGCCTGGGTACTTGACTG-3′ | 724-743 | ||
| α1H | 5′-ACCGTGTTCCAGATCCTGAC-3′/ | 3144-3163 | 22q13.1 | |
| (NM_021096) | 5′-TGAAGAGCACATAGTTGCCG-3′ | 3251-3270 | ||
| α1I | 270 | 5′-CCGTGGTTTGAATGTGTCAG-3′/ | 235-254 | 22q13.1 |
| (XM_010005) | 5′-GTCCAGGGAGTACTCGACCA-3′ | 485-504 | ||
| N-type | ||||
| α1B | 254 | 5′-CCCAGGCAGTGGAAGAAATA-3′/ | 2041-2060 | 9q34 |
| (XM_015804) | 5′-TGCTCCTGGAAGGTGATGAT-3′ | 2275-2294 | ||
| P/Q-type | ||||
| α1A | 250 | 5′-TTGCCCTACAGAAAGCCAAG-3′/ | 2458-2477 | 19p13.2-p13.1 |
| (XM_051369) | 5′-TCCAAGTGCGTCTTCATGTC-3′ | 2688-2707 | ||
| R-type | ||||
| α1E | 253 | 5′-GTGGCAAGTTACATCGAGCA-3′/ | 988-1007 | 1q25-q31 |
| (XM_001815) | 5′-CAATTCCAGGTGGCTCCTAA-3′ | 1221-1240 | ||
| α2δ1 | 178 | 5′-AGTGGATGGCCTGTGAAAAC-3′/ | 1231-1250 | 7q21-q22 |
| (XM_167505) | 5′-ACAAGTCCCAGTTCCAATGC-3′ | 1389-1408 | ||
| α2δ3 | 167 | 5′-CCTGTGCCAACAAAGGATTT-3′/ | 555-574 | 3p21.1 |
| (XM_035446) | 5′-CTTTTGTGCCTGAGGGAGAG-3′ | 702-721 | ||
| β1 | 253 | 5′-CTGGCTAAGCGCTCAGTTCT-3′/ | 976-995 | 17q21-q22 |
| (XM_054993) | 5′-GGGACTTGATGAGCCTTTGA-3′ | 1209-1228 | ||
| β2 | 241 | 5′-CCACAACCACAGAGACGAGA-3′/ | 2097-2116 | 10p12 |
| (NM_000724) | 5′-AACACAAAAGGGCAAAACTC-3′ | 2318-2337 | ||
| β3 | 248 | 5′-AGGAGATCTGGGAACCCTTC-3′/ | 406-425 | 12q13 |
| (XM_028766) | 5′-GTGACTCGGGTGATGGAGAT-3′ | 634-653 | ||
| β4 | 262 | 5′-CGCACCCTGAGAGTCTTTGT-3′/ | 342-361 | 2q22-q23 |
| (AF216867) | 5′-CCACCTGGACTCGACACAC3′ | 585-603 | ||
| γ1 | 250 | 5′-GATTTGTACCAAGCGCATCC-3′/ | 236-255 | 17q24 |
| (XM_008262) | 5′-TCGCAGCAGATAGTCCCTCT-3′ | 466-485 | ||
| γ2 | 249 | 5′-GGCTGTTTGATCGAGGTGTT-3′/ | 5-24 | 22q13.1 |
| (NM_006078) | 5′-TGCATCCTCTGGGAAGTGAT-3′ | 242-261 | ||
| γ3 | 259 | 5′-CCTTGGGGGAAGTGTACAGA-3′/ | 1327-1346 | 1612-p13.1 |
| (NM_006539) | 5′-CAAGGAGGGAAGAATGTGGA-3′ | 1566-1585 | ||
| γ4 | 299 | 5′-CTACAGCCGCAAGAACAACA-3′/ | 407-426 | 17q24 |
| (NM_014408) | 5′-AAGGAGAAGAGGAAGACGCC-3′ | 686-705 | ||
| γ5 | 337 | 5′-CGATGAGATGCTCAACAGGA-3′/ | 471-490 | 17q24 |
| (AF361351) | 5′-CTGATCATAGTCGGGGCACT-3′ | 788-807 | ||
| γ6 | 271 | 5′-TTGGGGTGCCTCTGTATCAT-3′/ | 460-479 | 19q13.4 |
| (AF361352) | 5′-GCAGTGTGAGCAGCAGAAAG-3′ | 711-730 | ||
| Voltage-gated K+ channels | ||||
| Kv1.1 (KCNA1) | 258 | 5′-CGGGGTCATCCTGTTTTCTA-3′/ | 1005-1024 | 12p13 |
| (NM_000217) | 5′-CCCTCAGTTTCTCGGTGGTA-3′ | 1243-1262 | ||
| Kv1.2 (KCNA2) | 200 | 5′-ATGAGAGAATTGGGCCTCCT-3′/ | 986-1005 | 1p13 |
| (NM_004974) | 5′-CCCACTATCTTTCCCCCAAT-3′ | 1166-1185 | ||
| Kv1.3 (KCNA3) | 259 | 5′-TCTGCCTATGCCCTTGTTTT-3′/ | 1711-1730 | 1p21-p13.3 |
| (NM_002232) | 5′-TTCCTCCCAGGATGTACTGC-3′ | 1950-1969 | ||
| Kv1.4 (KCNA4) | 571 | 5′-TGGCGGCTACAGTTCAGTC-3′/ | 1640-1658 | 11q13.4-q14.1 |
| (NM_002233) | 5′-ATCATTCAACAACCCACCAT-3′ | 2191-2210 | ||
| Kv1.5 (KCNA5) | 201 | 5′-ACTTGCGGAGGTCCCTTTAT-3′/ | 2013-2032 | 12p13 |
| (NM_002234) | 5′-GGAGGGAGGAAAGGAGTGAA-3′ | 2194-2213 | ||
| Kv1.6 (KCNA6) | 197 | 5′-CAGAGGAATCGTGTTGCAGA-3′/ | 3380-3399 | 12p13 |
| (NM_002235) | 5′-TGCCCATAAAATGGGAAGAA-3′ | 3557-3576 | ||
| Kv1.7 (KCNA7) | 300 | 5′-CTTCCAGGGGCATGTTATTTTA-3′/ | 2254-2275 | 19q13.3 |
| (NM_031886) | 5′-CTCAATGGAACTCAATTCAGCA-3′ | 2532-2553 | ||
| Kv1.10 (KCNA10) | 201 | 5′-TGGGGTTGCTCATCTTCTTT-3′/ | 1124-1143 | 1p13.1 |
| (NM_005549) | 5′-CACACAGAGTGCCCACAATC-3′ | 1305-1324 | ||
| Kvβ1 (KCNAB1) | 197 | 5′-AGGCTGCAGCTCGAGTATGT-3′/ | 701-720 | 3q26.1 |
| (NM_003471) | 5′-ACCGGTGGGATCATATTGAA-3′ | 878-897 | ||
| Kvβ2 (KCNAB2) | 195 | 5′-TGGGCAATAAACCCTACAGC-3′/ | 1242-1261 | 1p36.3 |
| (NM_003636) | 5′-CAGCGACTTGGGAGATCATT-3′ | 1417-1436 | ||
| Kvβ3 (KCNAB3) | 200 | 5′-GTGGTGTTCGGGTATCCTGT-3′/ | 257-276 | 17p13.1 |
| (NM_004732) | 5′-TGATCTCCTCCAACCTTTGC-3′ | 437-456 | ||
| Kv2.1 (KCNB1) | 383 | 5′-ACAGAGCAAACCAAAGGAAGAAC-3′/ | 1656-1678 | 20q13.2 |
| (NM_004975) | 5′-CACCCTCCATGAAGTTGACTTTA-3′ | 2016-2038 | ||
| Kv3.1 (KCNC1) | 387 | 5′-GGAGGCCTTCCTTACCTACATC-3′/ | 791-812 | 11p15 |
| (NM_004976) | 5′-CCTATCCTCTCGGCGTAGTAGA-3′ | 1156-1177 | ||
| Kv3.3 (KCNC3) | 199 | 5′-TGCTGCTCATCATCTTCCTG-3′/ | 1641-1660 | 19q13.3-q13.4 |
| (NM_004977) | 5′-GACCACGTCTTGGGGTACAT-3′ | 1820-1839 | ||
| Kv3.4 (KCNC4) | 196 | 5′-CTACCTGGAGGTGGGACTGA-3′/ | 1131-1150 | 1p21 |
| (NM_004978) | 5′-CAGGGCCAGGAAGATGATAA-3′ | 1307-1326 | ||
| Kv4.1 (KCND1) | 199 | 5′-GAGAAGACAACGTGCCATGA-3′/ | 1456-1475 | Xp11.23-p11.3 |
| (NM_004979) | 5′-TGACTGAGGCAGTGGAGTTG-3′ | 1635-1654 | ||
| Kv4.2 (KCND2) | 201 | 5′-GCCAATGTGTCAGGAAGTCA-3′/ | 1857-1876 | 7q31-q32 |
| (NM_012281) | 5′-TTCTGGGGTGGTTACTGGAG-3′ | 2038-2057 | ||
| Kv4.3 (KCND3) | 275 | 5′-CATGGCCATCATCATCTTTG-3′/ | 1465-1484 | 1p13.3 |
| (NM_004980) | 5′-CCCTGCGTTTATCAGCTCTC-3′ | 1720-1739 | ||
| Kv5.1 (KCNF1) | 195 | 5′-GCCAGCGACGACATAGAGAT-3′/ | 303-322 | 2p25 |
| (NM_002236) | 5′-CGGGTCCCTGTCAAAGTAGA-3′ | 478-497 | ||
| Kv6.1 (KCNG1) | 191 | 5′-CTACTGGTGGGCTGTCATCA-3′/ | 1242-1261 | 20q13 |
| (NM_002237) | 5′-TCACCCTCTCTTGCTCCTGT-3′ | 1413-1432 | ||
| Kv6.2 (KCNG2) | 254 | 5′-CTGCTGCTGCTGTTCCTCTG-3′/ | 347-366 | 18q22-q23 |
| (NM_012283) | 5′-AAAGGTGTGGAAGATGGAGGT-3′ | 581-601 | ||
| Kv6.3 (KCNG3) | 227 | 5′-TGCATAGGTTGGTTCACTGC-3′/ | 682-701 | 16q24 |
| (NM_133329) | 5′-GGCAAGCTTAATCACCCAAA-3′ | 890-909 | ||
| (Identical gene to Kv10.1) | ||||
| Kv9.1 (KCNS1) | 202 | 5′-ATACCAGCCCTTCTGCACAC-3′/ | 4234-4253 | 20q12 |
| (NM_002251) | 5′-AGGCCAGATGATTCCCTCTT-3′ | 4416-4435 | ||
| Kv9.3 (KCNS3) | 200 | 5′-CAGTGAGGATGCACCAGAGA-3′/ | 1652-1671 | 2p24 |
| (NM_002252) | 5′-TTGCTGTGCAATTCTCCAAG-3′ | 1832-1851 | ||
| Kv10.1 (KCNG3) | 227 | 5′-TGCATAGGTTGGTTCACTGC-3′/ | 682-701 | 2p21 |
| (AF-348982) | 5′-GGCAAGCTTAATCACCCAAA-3′ | 890-909 | ||
| (Identical gene to Kv6.3) | ||||
| Kv11.1 | 210 | 5′-CCTTCCTCTGCATTGCTTTT-3′/ | 1425-1444 | 9p24.2 |
| (NM_133497.1) | 5′-CTTGTCTTGGGGTGAGCTGT-3′ | 1615-1634 | ||
| Voltage-dependent Ca2+-activated K+ channels | ||||
| MaxiKca-α1 | 442 | 5′-CTACTGGGATGTTTCACTGGTGT-3′/ | 2210-2232 | 10q22 |
| (NM_002247) | 5′-TGCTGTCATCAAACTGCATA-3′ | 2634-2653 | ||
| MaxiKca-β1 | 363 | 5′-TCTACTGCTTCTCCGCAC-3′/ | 557-574 | 5q34 |
| (NM_004137) | 5′-GAGCAGGCAATGACTTCA-3′ | 902-919 | ||
| MaxiKca-β2 | 449 | 5′-GGGACTGGCTATGATGGT-3′/ | 502-519 | 3q26.2-q27.1 |
| (NM_005832) | 5′-GTGAATGGAACAGCACGTTG-3′ | 931-950 | ||
| MaxiKca-β3 | 351 | 5′-GCTCAACAGTGCTCTGGACA-3′/ | 1013-1032 | 3q26.3-q27.1 |
| (NM_014407) | 5′-TGGCCACCGTCTTAAGATTT-3′ | 1344-1363 | ||
| MaxiKca-β4 | 300 | 5′-CTGAGTCCAACTCTAGGGCG-3′/ | 612-631 | 12q14.1-q15 |
| (NM_014505) | 5′-TGGTCAGGACCACAATGAGA-3′ | 892-911 | ||
| Kca-SK1 | 357 | 5′-CTTCCTCTCCATTGGCTACG-3′/ | 1301-1320 | 19p13.1 |
| (NM_002248) | 5′-TTCCCTTGCTCGATCTTCAC-3′ | 1638-1657 | ||
| Kca-SK2 | 451 | 5′-CAAGCAAACACTTTGGTGGA-3′ | 1880-1899 | 5q22.3 |
| (NM_021614) | 5′-TGTTCAGGTTCCCAGGATTC-3′ | 2311-2330 | ||
| Kca-SK3 | 349 | 5′-CTTGATCATCGCCTACCACA-3′/ | 1344-1363 | 1q21.3 |
| (NM_002249) | 5′-GCGGGTGTTGAAGTTGATCT-3′ | 1673-1692 | ||
| Kca-SK4 | 399 | 5′-GCCCTGGAGAAACAGATTGA-3′/ | 1567-1586 | 19q13.2 |
| (NM_002250) | 5′-AGAGCTGGAGGTCGTCCATA-3′ | 1946-1965 |
The accession numbers in GenBank for the sequence used in designing the primers
Macroscopic Ca2+-activated K+ currents
To record Kca currents, cells need to be superfused with 1.8 mM Ca2+-containing bath solution and dialysed with an EGTA-free pipette solution. Depolarization from a holding potential of -70 mV to a series of test potentials ranging from -60 mV to +80 mV will elicit both Kca and Kv currents. The noisy currents dominant at positive potentials are representative of whole-cell Kca currents (IK(Ca)) observed in freshly dissociated animal vascular smooth muscle cells.[21,69] In direct comparison with IK(V), IK(Ca) activate slowly with relatively little inactivation. Extracellular application of known inhibitors of Kca channels such as 1 mM TEA, 50 nM iberiotoxin or 50 nM charybdotoxin can significantly block the noisy IK(Ca) while having a negligible effect on IK(V). These findings in PASMC are consistent with observations in systemic vascular smooth muscle cells.[19,78,80] Dialysis of PASMC with a high (500 μM) Ca2+ pipette solution (containing 8.8 mM EGTA and 10 mM CaCl2) yields slowly activating outward currents that are significantly inhibited by extracellular application of the Kca channel blockers iberiotoxin and charybdotoxin. The slow activation kinetics of the IK(Ca) is consistent with the kinetics of the currents measured in cells transfected with the maxi-K+ channel gene, hSlo-α1.[94,95]
In cell-attached patches of PASMC, increased [Ca2+]cyt by FCCP (which depolarizes and releases Ca2+ from mitochondria[96]) causes a significant increase in the steady-state open probability of the large-conductance IK(Ca) [Figure 16a]. Extracellular application of dihydroepiandrosterone, an agent that opens Kca channels via cAMP/cGMP-independent pathways,[97] also increases the Popen of large-conductance IK(Ca) [Figure 16b]. Cyclopiazonic acid causes Ca2+ mobilization from intracellular stores and, when applied in the cell-attached configuration, can increase Popen of a smaller-conductance IK(Ca) (47 pS) [Figure 16c] at +70 mV. It can therefore be inferred that two types of Kca channels, a large- and a small- or intermediate-conductance channel, are functionally expressed in human PASMC, and that they are synergistically regulated by membrane potential and [Ca2+]cyt.
Figure 16.

Single-channel Ca2+-activated K+ (Kca) currents (iK(Ca)) in cell-attached membrane patches of human pulmonary artery smooth muscle cells (PASMC). (a) FCCP (5 μM) enhances large-amplitude iK(Ca) open probability (Popen) (a) by causing a transient [Ca+]cyt (b) increase. (b) dihydroepiandrosterone (0.1 mM) also enhances the large-amplitude iK(Ca) Popen. (c) Cyclopiazonic acid (5 μM), an inhibitor of sarcoplasmic reticulum (SR) Ca2+ pump, increases the activity of a smaller amplitude iK(Ca) (a) by causing SR Ca2+ release from the SR to the cytosol (b)
Molecular identities of Kca channels in human PASMC
Unlike the mainly heterotetrameric Kv channels, Kca channels are predominantly homomeric tetramers composed of the pore-forming α–subunits and the auxiliary β-subunits [Figure 17a].[95] Several human Kca channel α–subunits that encode the large (maxi-Kca)- and small (SKca)-conductance Kca channels have been cloned and characterized in vascular SMC.[98,99] In addition to the pore-forming α–subunit, several β-subunits have also been identified.[99] Maxi-Kca α1 (hSlo-α1) is highly expressed in human PASMC [Figure 17a]. Four β-subunits (Maxi-Kca β1-4) are also detected by RT-PCR in PASMC [Figure 17b]. Three (SK2-4) pore-forming subunits are observed at the mRNA level for SKca channels [Figure 17c].
Figure 17.
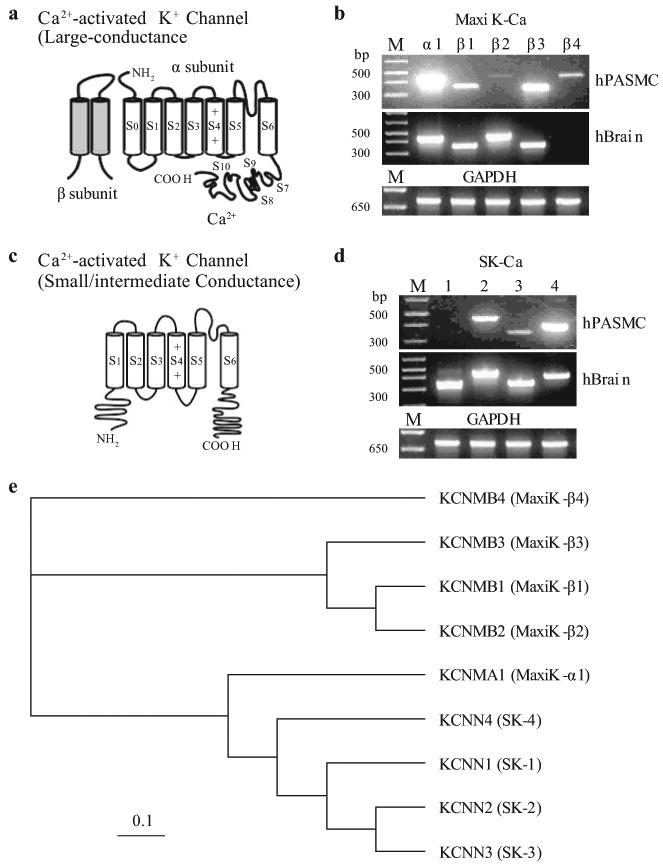
Molecular identity of Ca2+-activated K+ (Kca) channels in human pulmonary artery smooth muscle cells (PASMC). (a) Structural arrangement of Kca channel α- and β-subunits. The putative binding site for Ca2+ is shown on the C-terminal region of the α-subunit. (b–d) The mRNA expression of reverse transcriptase-polymerase chain reaction products for maxi Kca channel α1 and β1-4 subunits (b) and small- (SK1-3) and intermediate- (SK4) channels (c and d) are shown in human PASMC and brain tissues. “M,” 100 bp DNA ladder. (e) A phylogenetic tree showing the inferred evolutionary relationships among different Kca channel genes
Table 3 shows the biophysical and pharmacological properties, along with the molecular identifies of, voltage-dependent cation channels in human PASMC. The information shown in Table 3 is certainly incomplete and it is important to conduct more studies to reveal all voltage-dependent cation channels that are functionally expressed in animal and human PASMC.
Table 3. Biophysical properties and molecular identities of voltage-dependent cation channels expressed in human PASMC.
| Types of currents | Activation threshold | Blocked by (EC50) | Gene transcripts identified |
|---|---|---|---|
| INa | -34 mV | Tetrodotoxin | SCN5A |
| SCN6A | |||
| ICa | -24 mV (HVA) | Nickel | L-type (α1C, α1D) |
| -50 mV (LVA) | Nifedipine | T-type (α1G) | |
| P/Q-type (α1A) | |||
| N-type (α1B) | |||
| R-type (α1E) | |||
| α2δ1 | |||
| β1, β2, β3, β4 | |||
| γ6 | |||
| IK(V) | -55 mV | 4-AP | Kv1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, |
| 1-10 | |||
| Kv2.1 | |||
| Kv3.1, 3.3, 3.4 | |||
| Kv4.1, 4.2 | |||
| Kv5.1 | |||
| Kv6.1, 6.2, 6.3 | |||
| Kv9.1, 9.3 | |||
| Kv10.1 | |||
| Kv11.1 | |||
| Kvβ1, β2, β3 | |||
| IK(Ca) | -25 mV | Iberiotoxin | Maxi-Kca α1 |
| Charybdotoxin | Maxi-Kca β1, β2, β3, β4 | ||
| SK2, 3, 4 |
Contribution of Cation Channels to the Regulation of Em and [Ca2+]cyt in Human PASMC
As discussed, membrane potential is primarily determined by the concentration gradients across the plasma membrane of electrically charged ions, mainly Na+, Ca2+, K+ and Cl+, and their relative permeability. At rest, the concentration of intracellular K+ (∼140 mM) is much greater than that of the extracellular space (∼5 mM) because of Na+-K+ ATPase pump activity, and the K+ permeability across the plasma membrane is far greater than that of Na+, Ca2+ and Cl-.[15,106] Therefore, the resting Em is mainly determined by the permeability of K+ (EK ≈ -85 mV) and the activity of Na+-K+ ATPase. Extracellular application of 5 mM 4-AP, which is known to reduce IK(V) [Figure 13], can reversibly cause membrane depolarization in human PASMC, whereas iberiotoxin, an inhibitor of Kca channels, has little effect on Em. Increasing extracellular K+ concentration (e.g., from 4.7 mM to 60 mM) also shifts the K+ equilibrium potential and depolarizes Em. It can, therefore, be proposed that 4-AP-sensitive Kv channels are active and contribute to the regulation of the resting Em in PASMC.[25] Em is an important determinant of [Ca2+]cyt in SMC because of the voltage dependence of Ca2+ influx via voltage-dependent L-type VDCC[106,109] and the reverse mode of Na+/Ca2+ exchanger.[110,111] Consistent with its inhibitory effect on Kv channels and depolarizing effect on PASMC membrane, extracellular application of 4-AP also reversibly increases [Ca2+]cyt in PASMC and causes pulmonary vasoconstriction [Figure 18]. Membrane depolarization generated by raising the extracellular K+ concentration from 4.7 mM to 60 mM results from an ∼20 mV shift of EK toward less-negative potentials. As a result of this rightward shift in EK and the subsequent depolarization of Em, [Ca2+]cyt is elevated [Figure 18b], an effect significantly attenuated by the removal of extracellular Ca2+. Membrane depolarization-mediated elevation of [Ca2+]cyt is therefore mainly due to Ca2+ influx through nifedipine-sensitive VDCC in human PASMC.
Figure 18.

Inhibition of K+ channels causes membrane depolarization and causes pulmonary vasoconstriction. (a) Closure of K+ channels in pulmonary artery smooth muscle cells (PASMC) causes membrane depolarization, which subsequently opens voltage-dependent Ca2+ channels (VDCC), enhances Ca2+ influx, increases [Ca2+]cyt and induces pulmonary vasoconstriction. Opening of K+ channels, on the other hand, causes membrane hyperpolarization (close to the K+ equilibrium potential), decreases VDCC activity and causes pulmonary vasodilation. (b) Representative records of whole-cell K+ currents (a), membrane potential (Em, b) and [Ca2+]cyt (c) in PASMC before (control), during (4-AP) and after (wash) extracellular application of 5 mM 4-amynopyridine (4-AP), an inhibitor of Kv channels. (d) A representative record of tension measurement in an isolated mouse pulmonary arterial ring before, during and after 4-AP treatment is shown
Both excitable and quiescent cells possess a negative resting Em. Em is known to control electrical excitability (e.g., generation and propagation of action potentials), muscle contraction, apoptosis[112-114] and gene expression.[115,116] From the latter functions, it is apparent that the mechanisms controlling Em and [Ca2+]cyt are interrelated. Membrane depolarization elevates [Ca2+]cyt mainly by activating VDCC[19,25,109,117,118] and the reverse-mode Na+/Ca2+ exchanger[110,111,119] in the plasma membrane. In smooth muscle cells, the voltage window of sarcolemmal L-type voltage-gated Ca2+ channels for sustained elevation of [Ca2+]cyt ranges from -40 mV to -20 mV, and peaks at -30 mV,[109] similar to what has been observed in human PASMC. The Na+/Ca2+ exchanger has a reversal potential (ENa-Ca) of approximately -47 mV at rest, based on the equation: ENa-Ca=3ENa-2ECa,[111] where ENa is the Na+ equilibrium potential (approximately +66 mV) and ECa is the Ca2+ equilibrium potential (approximately +122 mV). Membrane depolarization to potentials less negative than ENa-Ca would activate the reverse-mode Na+/Ca2+ exchanger and promote Ca2+ influx.[110,111,119] Thus, the sustained membrane depolarization in PASMC may produce a constant Ca2+ influx through voltage-gated Ca2+ channels[106,109] and an inward Ca2+ transportation via the reverse mode of Na+/Ca2+ exchange, and contribute to maintain the elevated [Ca2+]cyt that is crucial for PASMC contraction and proliferation. As discussed earlier, Na+ channel activation by membrane depolarization can also modulate [Ca2+]cyt by (i) controlling [Na+]cyt, thereby modulating Na+/Ca2+ exchange activity, (ii) non-selective permeation of Ca2+ ions through Na+ channels and (iii) rapid Na+-induced depolarization and subsequent VDCC activation.
While Na+ and Ca2+ channel activation has a tendency to depolarize cells and enhance [Ca2+]cyt, K+ channel activation hyperpolarizes the membrane and decreases sarcolemmal Ca2+ influx. Because of their voltage- and/or Ca2+-dependence, K+ channels are key elements in the maintenance of Em to the “near-resting” level. This review reflects on data that an inhibition of Kv channels with 4-AP induces membrane depolarization and increases [Ca2+]cyt by opening the nifedipine-sensitive L-type VDCC in human PASMC. An increase in [Ca2+]cyt is believed to play an important role in stimulating cell growth by activating protein kinases and transcription factors that are essential for the progression of cell cycle.[115,116,120-122] Kv channels in PASMC may play an important role in modulating pulmonary vascular contractility and remodeling via regulating Em and [Ca2+]cyt. Indeed, the roles of both Kca and Kv channels as feedback modulators of myogenic tone and agonist-induced vascular tone in systemic[123-125] and pulmonary arteries[63,126-132] are well documented.
Conclusions
In PASMC, EC-coupling is mainly achieved by a rise in [Ca2+]cyt, which is controlled by two related mechanisms, voltage-sensitive Ca2+ influx (electromechanical coupling) and ligand-mediated Ca2+ influx and mobilization (pharmacomechanical coupling). Membrane potential (Em) and ion diffusion across the plasma membrane are dominantly regulated by the function and expression of ion-selective channels embedded in the plasma membrane. In addition, the activity of plasmalemmal ion channels and homeostasis of intracellular ions play important roles in the regulation of cell excitability, contraction, gene expression, proliferation, differentiation and apoptosis.[22,102,133] Electromechanical coupling mechanisms cause tonic and phasic vasomotor tone in blood vessels[106,134] and participate in regulating cell proliferation[6] and protein/gene expression.[135] An understanding of the electrophysiological properties and molecular composition of voltage-dependent ion channels in human PASMC may provide important information for the development of effective therapeutic approaches for patients with pulmonary vascular diseases.
Pulmonary vasoconstriction and vascular remodeling (i.e., intimal and medial hypertrophy due to smooth muscle cell proliferation and migration) greatly contribute to the elevated pulmonary vascular resistance in patients with pulmonary hypertension.[136,137] Because both Em and Ca2+ are recognized as important modulators of both vascular tone and cell growth, it is plausible that ion channels also play a role in these processes, particularly those ion channels that regulate and can be regulated by Em and Ca2+. Dysfunctional K+ channels have been demonstrated to be involved in the pathogenesis of idiopathic pulmonary arterial hypertension.[8,9] There is no direct evidence to suggest alterations in Na+ channel gene expression or function in pulmonary hypertension-induced vascular remodeling. Nonetheless, Na+ channel-mediated regulation of [Ca2+]cyt may be important in the modulation of cell proliferation. Similarly, VDCC upregulation or “gain-in-function” has not been directly involved in the pathogenesis of pulmonary arterial hypertension, although any abnormalities in its expression or function may alter the remodeling process. However, recent observations have reported an increase in store-operated Ca2+ channel activity during human PASMC proliferation,[138] suggesting that alternative Ca2+ influx pathways may be involved in the pulmonary vascular remodeling process. Targeting Ca2+ - (and Na+-) permeable channels in the plasma membrane of pulmonary vascular smooth muscle cells and myofibroblasts is an efficient approach to develop a novel therapy for patients with pulmonary arterial hypertension.
Acknowledgments
This work was supported by grants from the National Heart, Lung and Blood Institute of the National Institutes of Health (HL 066012 and HL 098053). We would like to thank Mehran Mandegar, Jian Wang, Tiffany Sison and Elyssa D. Burg for their excellent work for generating the unpublished data shown in this review.
Footnotes
Conflict of Interest: None declared.
References
- 1.Casteels R, Kitamura K, Kuriyama H, Suzuki H. Excitation-contraction coupling in the smooth muscle cells of the rabbit main pulmonary artery. J Physiol. 1977;271:63–79. doi: 10.1113/jphysiol.1977.sp011990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madden JA, Dawson CA, Harder DR. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol. 1985;59:113–9. doi: 10.1152/jappl.1985.59.1.113. [DOI] [PubMed] [Google Scholar]
- 3.Beech DJ. Actions of neurotransmit ers and other messengers on Ca2+ channels and K+ channels in smooth muscle cells. Pharmacol Ther. 1997;73:91–119. doi: 10.1016/s0163-7258(97)87271-3. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Large WA. Action of histamine on single smooth muscle cells dispersed from the rabbit pulmonary artery. J Physiol. 1993;468:125–39. doi: 10.1113/jphysiol.1993.sp019763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devine CE, Somlyo AV, Somlyo AP. Sarcoplasmic reticulum and excitation-contraction coupling in mammalian smooth muscles. J Cell Biol. 1972;52:690–718. doi: 10.1083/jcb.52.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, et al. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2000;279:C1540–9. doi: 10.1152/ajpcell.2000.279.5.C1540. [DOI] [PubMed] [Google Scholar]
- 7.Pauly RR, Bilato C, Sollot SJ, Monticone R, Kelly PT, Lakat a EG, et al. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation. 1995;91:1107–15. doi: 10.1161/01.cir.91.4.1107. [DOI] [PubMed] [Google Scholar]
- 8.Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr, Gaine SP, et al. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–6. doi: 10.1161/01.cir.98.14.1400. [DOI] [PubMed] [Google Scholar]
- 9.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351:726–7. doi: 10.1016/S0140-6736(05)78495-6. [DOI] [PubMed] [Google Scholar]
- 10.Cox RH, Folander K, Swanson R. Differential expression of voltage-gated K+ channel genes in arteries from spontaneously hypertensive and Wistar-Kyoto rats. Hypertension. 2001;37:1315–22. doi: 10.1161/01.hyp.37.5.1315. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Hudetz AG, Knaus HG, Rusch NJ. Increased expression of Ca2+-sensitive K+ channels in the cerebral microcirculation of genetically hypertensive rats. Evidence for their protection against cerebral vasospasm. Circ Res. 1998;82:729–37. doi: 10.1161/01.res.82.6.729. [DOI] [PubMed] [Google Scholar]
- 12.Martens JR, Gelband CH. Alterations in rat interlobar artery membrane potential and K+ channels in genetic and nongenetic hypertension. Circ Res. 1996;79:295–301. doi: 10.1161/01.res.79.2.295. [DOI] [PubMed] [Google Scholar]
- 13.Martens JR, Gelband CH. Ion channels in vascular smooth muscle: Alterations in essential hypertension. Proc Soc Exp Biol Med. 1998;218:192–203. doi: 10.3181/00379727-218-44286. [DOI] [PubMed] [Google Scholar]
- 14.Beuckelmann DJ, Näbauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–85. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- 15.Jones AW. Content and fluxes of electrolytes. In: Bohr DF, Somlyo AP, Sparks HV, editors. Handbook of Physiology, The Cardiovascular System Vascular Smooth Muscle. Baltimore, MD: Williams and Wilkins; 1980. pp. 253–99. [Google Scholar]
- 16.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 17.Hille B. Ionic channels of excitable membranes. 2nd. Sunderland, Massachusetts: Sinauer Associates Inc.; 1992. [Google Scholar]
- 18.Toro L, Gonzalez-Robles A, Stefani E. Electrical properties and morphology of single vascular smooth muscle cells in culture. Am J Physiol. 1986;251:C763–73. doi: 10.1152/ajpcell.1986.251.5.C763. [DOI] [PubMed] [Google Scholar]
- 19.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 20.Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol. 1992;262:C882–90. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- 21.Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery. Novel mechanism for hypoxia-induced membrane depolarization. Circ Res. 1995;77:131–9. doi: 10.1161/01.res.77.1.131. [DOI] [PubMed] [Google Scholar]
- 22.Somlyo AP, Somlyo AV. Smooth muscle: Excitation-contraction coupling, contractile regulation, and the cross-bridge cycle. Alcohol Clin Exp Res. 1994;18:138–43. doi: 10.1111/j.1530-0277.1994.tb00893.x. [DOI] [PubMed] [Google Scholar]
- 23.Kuriyama H, Suzuki H. Electrical property and chemical sensitivity of vascular smooth muscles in normotensive and spontaneously hypertensive rats. J Physiol. 1978;285:409–24. doi: 10.1113/jphysiol.1978.sp012579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okabe K, Kitamura K, Kuriyama H. The existence of a highly tetrodotoxin sensitive Na channel in freshly dispersed smooth muscle cells of the rabbit main pulmonary artery. Pflugers Arch. 1988;411:423–8. doi: 10.1007/BF00587722. [DOI] [PubMed] [Google Scholar]
- 25.Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res. 1995;77:370–8. doi: 10.1161/01.res.77.2.370. [DOI] [PubMed] [Google Scholar]
- 26.Anderson PA, Greenberg RM. Phylogeny of ion channels: Clues to structure and function. Comp Biochem Physiol B Biochem Mol Biol. 2001;129:17–28. doi: 10.1016/s1096-4959(01)00376-1. [DOI] [PubMed] [Google Scholar]
- 27.Strong M, Chandy KG, Gutman GA. Molecular evolution of voltage-sensitive ion channel genes: On the origns of electrical excitability. Mol Biol Evol. 1993;10:221–42. doi: 10.1093/oxfordjournals.molbev.a039986. [DOI] [PubMed] [Google Scholar]
- 28.Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–85. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- 29.Durell SR, Guy HR. A putative prokaryote voltage-gated Ca2+ channel with only one 6TM motif per subunit. Biochem Biophys Res Comm. 2001;281:741–6. doi: 10.1006/bbrc.2001.4408. [DOI] [PubMed] [Google Scholar]
- 30.Catterall WA. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 31.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol. 2000;279:L884–94. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- 32.Zhou C, Wu S. T-type calcium channels in pulmonary vascular endothelium. Microcirculation. 2006;13:645–56. doi: 10.1080/10739680600930289. [DOI] [PubMed] [Google Scholar]
- 33.Mori M, Konno T, Ozawa T, Murata M, Imoto K, Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: Does VDSC acquire calmoduln-mediated Ca2+ sensitivity? Biochemistry. 2000;39:1316–23. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- 34.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–62. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 35.Spafford JD, Spencer AN, Gallin WJ. Genomic organization of a voltage-gated Na+ channel in a hydrozoan jellyfish: Insights into the evolution of voltage-gated Na+ channel genes. Receptors Channels. 1999;6:493–506. [PubMed] [Google Scholar]
- 36.Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–94. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 37.Plummer NW, Meisler MH. Evolution and diversity of mammalian sodium channel genes. Genomics. 1999;57:323–31. doi: 10.1006/geno.1998.5735. [DOI] [PubMed] [Google Scholar]
- 38.Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am J Physiol Cell Physiol. 2000;278:C163–73. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- 39.Boccara G, Choby C, Frapier JM, Quignard JF, Nargeot J, Dayanithi G, et al. Regulation of Ca2+ homeostasis by atypical Na+ currents in cultured human coronary myocytes. Circ Res. 1999;85:606–13. doi: 10.1161/01.res.85.7.606. [DOI] [PubMed] [Google Scholar]
- 40.Fozzard HA, January CT, Makielski JC. New studies of the excitatory sodium currents in heart muscle. Circ Res. 1985;56:475–85. doi: 10.1161/01.res.56.4.475. [DOI] [PubMed] [Google Scholar]
- 41.Leblanc N, Hume JR. Sodium current induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–6. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- 42.Santana LF, Gómez AM, Lederer WJ. Ca2+ flux through promiscuous cardiac Na+ channels: Slip-mode conductance. Science. 1998;279:1027–33. doi: 10.1126/science.279.5353.1027. [DOI] [PubMed] [Google Scholar]
- 43.Choby C, Mangoni ME, Boccara G, Nargeot J, Richard S. Evidence for tetrodotoxin-sensitive sodium currents in primary cultured myocytes from human, pig and rabbit arteries. Pflugers Arch. 2000;440:149–52. doi: 10.1007/s004240000268. [DOI] [PubMed] [Google Scholar]
- 44.Cox RH, Zhou Z, Tulenko TN. Voltage-gated sodium channels in human aortic smooth muscle cells. J Vasc Res. 1998;35:310–7. doi: 10.1159/000025600. [DOI] [PubMed] [Google Scholar]
- 45.James AF, Okada T, Horie M. A fast transient outward current in cultured cells from human pulmonary artery smooth muscle. Am J Physiol. 1995;268:H2358–65. doi: 10.1152/ajpheart.1995.268.6.H2358. [DOI] [PubMed] [Google Scholar]
- 46.Quignard JF, Ryckwaert F, Albat B, Nargeot J, Richard S. A novel tetrodotoxin-sensitive Na+ current in cultured human coronary myocytes. Circ Res. 1997;80:377–82. [PubMed] [Google Scholar]
- 47.Felix R. Channelopathies: Ion channel defects linked to heritable clinical disorders. J Med Genet. 2000;37:729–40. doi: 10.1136/jmg.37.10.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li S, Sims S, Jiao Y, Chow LH, Pickering JG. Evidence from a novel human cell clone that adult vascular smooth muscle cells can convert reversibly between noncontractile and contractile phenotypes. Circ Res. 1999;85:338–48. doi: 10.1161/01.res.85.4.338. [DOI] [PubMed] [Google Scholar]
- 49.Sashihara S, Tshuji S, Matsui T. Oncogenes and signal transduction pathways involved in the regulation of Na+ channel expression. Crit Rev Oncog. 1998;9:19–34. doi: 10.1615/critrevoncog.v9.i1.20. [DOI] [PubMed] [Google Scholar]
- 50.Aggarwal R, Shorofsky SR, Goldman L, Balke CW. Tetrodotoxin-blockable calcium currents in rat ventricular myocytes: A third type of cardiac cell sodium current. J Physiol. 1997;505:353–69. doi: 10.1111/j.1469-7793.1997.353bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cole WC, Chartier D, Martin M, Leblanc N. Ca2+ permeation through Na+ channels in guinea pig ventricular myocytes. Am J Physiol. 1997;273:H128–37. doi: 10.1152/ajpheart.1997.273.1.H128. [DOI] [PubMed] [Google Scholar]
- 52.Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform-specifi c modulation of voltage-gated Na(+) channels by calmodulin. Circ Res. 2002;90:E49–57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- 53.Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–7. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- 54.Cat erall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 55.Randall AD. The molecular basis of voltage-gated Ca2+ channel diversity: Is it time for T? J Membr Biol. 1998;161:207–13. doi: 10.1007/s002329900327. [DOI] [PubMed] [Google Scholar]
- 56.Sturek M, Hermsmeyer K. Calcium and sodium channels in spontaneously contracting vascular muscle cells. Science. 1986;233:475–8. doi: 10.1126/science.2425434. [DOI] [PubMed] [Google Scholar]
- 57.Hansen PB, Jensen BL, Andreasen D, Skøt O. Diff erential expression of T- and L-type voltage dependent calcium channels in renal resistance vessels. Circ Res. 2001;89:630–8. doi: 10.1161/hh1901.097126. [DOI] [PubMed] [Google Scholar]
- 58.Muramatsu M, Tyler RC, Rodman DM, McMurtry IF. Possible role of T-type Ca2+ channels in L-NNA vasoconstriction of hypertensive rat lungs. Am J Physiol. 1997;272:H2612–21. doi: 10.1152/ajpheart.1997.272.6.H2616. [DOI] [PubMed] [Google Scholar]
- 59.Balke CW, Rose WC, Marban E, Wier WG. Macroscopic and unitary properties of physiological ion flux through T-type Ca2+ channels in guinea-pig heart cells. J Physiol. 1992;456:247–65. doi: 10.1113/jphysiol.1992.sp019335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Restituito S, Cens T, Rousset M, Charnet P. Ca2+ channel inactivation heterogeneity reveals physiological unbinding of auxiliary b subunits. Biophys J. 2001;81:89–96. doi: 10.1016/S0006-3495(01)75682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Varadi G, Mori Y, Mikala G, Schwartz A. Molecular determinants of Ca2+ channel function and drug action. Trends Pharmacol Sci. 1995;16:43–9. doi: 10.1016/s0165-6147(00)88977-4. [DOI] [PubMed] [Google Scholar]
- 62.Walker D, De Waard M. Subunit interaction sites in voltage-dependent Ca2+ channels: Role in channel function. Trends Neurosci. 1998;21:148–54. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 63.Gelband CH, Gelband H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation. 1997;96:3647–54. doi: 10.1161/01.cir.96.10.3647. [DOI] [PubMed] [Google Scholar]
- 64.Peng W, Hoidal JR, Karwande SV, Farrukh IS. Effect of chronic hypoxia on K+ channels: Regulation in human pulmonary vascular smooth muscle cells. Am J Physiol. 1997;272:C1271–8. doi: 10.1152/ajpcell.1997.272.4.C1271. [DOI] [PubMed] [Google Scholar]
- 65.Koh SD, Monaghan K, Sergeant GP, Ro S, Walker RL, Sanders KM, et al. TREK-1 regulation by nitric oxide and cGMP-dependent protein kinase. An essential role in smooth muscle inhibitory neurotransmission. J Biol Chem. 2001;276:44338–46. doi: 10.1074/jbc.M108125200. [DOI] [PubMed] [Google Scholar]
- 66.Mandegar M, Yu Y, Platoshyn O, Lapp BR, Rubin LJ, Yuan JX. Expression and function of tandem-pore K+ channels in human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. 2002;195:B53. [Google Scholar]
- 67.Peng W, Karwande SV, Hoidal JR, Farrukh IS. Potassium currents in cultured human pulmonary arterial smooth muscle cells. J Appl Physiol. 1996;80:1187–96. doi: 10.1152/jappl.1996.80.4.1187. [DOI] [PubMed] [Google Scholar]
- 68.Barry DM, Nerbonne JM. Myocardial potassium channels: Electrophysiological and molecular diversity. Annu Rev Physiol. 1994;58:363–94. doi: 10.1146/annurev.ph.58.030196.002051. [DOI] [PubMed] [Google Scholar]
- 69.Beech DJ, Bolton TB. Two components of potassium current activated by depolarization of single smooth muscle cells from the rabbit portal vein. J Physiol. 1989;418:293–309. doi: 10.1113/jphysiol.1989.sp017841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogawski MA. The A-current: How ubiquitous a feature of excitable cells is it? Trends Neurosci. 1985;8:214–9. [Google Scholar]
- 71.Vogalis F, Lang RJ. Identification of single transiently opening (‘A-type’) K channels in guinea-pig colonic myocytes. Pflugers Arch. 1994;429:160–4. doi: 10.1007/BF00374307. [DOI] [PubMed] [Google Scholar]
- 72.Beech DJ, Bolton TB. A voltage-dependent outward current with fast kinetics in single smooth muscle cells isolated from rabbit portal vein. J Physiol. 1989;412:397–414. doi: 10.1113/jphysiol.1989.sp017623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coppock EA, Tamkun MM. Differential expression of Kv channel a- and b-subunits in the bovine pulmonary arterial circulation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1350–60. doi: 10.1152/ajplung.2001.281.6.L1350. [DOI] [PubMed] [Google Scholar]
- 74.Hulme JT, Coppock EA, Felipe A, Martens JR, Tamkun MM. Oxygen sensitivity of cloned voltage-gated K+ channels expressed in the pulmonary vasculature. Circ Res. 1999;85:489–97. doi: 10.1161/01.res.85.6.489. [DOI] [PubMed] [Google Scholar]
- 75.Patel AJ, Honoré E. Molecular physiology of oxygen-sensitive potassium channels. Eur Respir J. 2001;18:221–7. doi: 10.1183/09031936.01.00204001. [DOI] [PubMed] [Google Scholar]
- 76.Clément-Chomienne O, Ishii K, Walsh MP, Cole WC. Identification, cloning and expression of rabbit vascular smooth muscle Kv1.5 and comparison with native delayed rectifier K+ current. J Physiol. 1999;515:653–67. doi: 10.1111/j.1469-7793.1999.653ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hart PJ, Overturf KE, Russell SN, Carl A, Hume JR, Sanders KM, et al. Cloning and expression of a Kv1.2 class delayed rectifier K+ channel from canine colonic smooth muscle. Proc Natl Acad Sci U S A. 1993;90:9659–63. doi: 10.1073/pnas.90.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Volk KA, Matsuda JJ, Shibata EF. A voltage-dependent potassium current in rabbit coronary artery smooth muscle cells. J Physiol. 1991;439:751–68. doi: 10.1113/jphysiol.1991.sp018691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aiello EA, Malcolm AT, Walsh MP, Cole WC. b-Adrenoceptor activation and PKA regulate delayed rectifier K+ channels of vascular smooth muscle cells. Am J Physiol. 1998;275:H448–59. doi: 10.1152/ajpheart.1998.275.2.H448. [DOI] [PubMed] [Google Scholar]
- 80.Gelband CH, Hume JR. Ionic currents in single smooth muscle cells of the canine renal artery. Circ Res. 1992;71:745–58. doi: 10.1161/01.res.71.4.745. [DOI] [PubMed] [Google Scholar]
- 81.Ishikawa T, Hume JR, Keef KD. Modulation of K+ and Ca2+ channels by histamine H1-receptor stimulation in rabbit coronary artery cells. J Physiol. 1993;468:379–400. doi: 10.1113/jphysiol.1993.sp019777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martens JR, Kwak YG, Tamkun MM. Modulation of Kv channel a/b subunit interactions. Trends Cardiovasc Med. 1999;9:253–8. doi: 10.1016/s1050-1738(00)00037-2. [DOI] [PubMed] [Google Scholar]
- 83.Gulbis JM, Mann S, MacKinnon R. Structure of a voltage-dependent K+ channel beta subunit. Cell. 1999;97:943–52. doi: 10.1016/s0092-8674(00)80805-3. [DOI] [PubMed] [Google Scholar]
- 84.Pérez-García MT, López-López JR, González C. Kvb1.2 subunit coexpression in HEK293 cells confers O2 sensitivity to Kv4.2 but not to Shaker channels. J Gen Physiol. 1999;113:897–907. doi: 10.1085/jgp.113.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, et al. Inactivation properties of voltage-gated K+ channels altered by presence of b-subunit. Nature. 1994;369:289–94. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 86.Rasmusson RL, Wang S, Castellino RC, Morales MJ, Strauss HC. The b subunit, Kvb1.2, acts as a rapid open channel blocker of NH2 terminal deleted Kv1.4 a-subunits. Adv Exp Med Biol. 1997;430:29–37. doi: 10.1007/978-1-4615-5959-7_3. [DOI] [PubMed] [Google Scholar]
- 87.Gong J, Xu J, Bezanilla M, van Huizen R, Derin R, Li M. Diff erential stimulation of PKC phosphorylation of potassium channels by ZIP1 and ZIP2. Science. 1999;285:1565–9. doi: 10.1126/science.285.5433.1565. [DOI] [PubMed] [Google Scholar]
- 88.Park MK, Bae YM, Lee SH, Ho WK, Earm YE. Modulation of voltage-dependent K+ channel by redox potential in pulmonary and ear arterial smooth muscle cells of the rabbit. Pflugers Arch. 1997;434:764–71. doi: 10.1007/s004240050463. [DOI] [PubMed] [Google Scholar]
- 89.Smirnov SV, Robertson TP, Ward JP, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. Am J Physiol. 1994;266:H365–70. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- 90.Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol. 1993;264:L116–23. doi: 10.1152/ajplung.1993.264.2.L116. [DOI] [PubMed] [Google Scholar]
- 91.Yu Y, Platoshyn O, Zhang J, Krick S, Zhao Y, Rubin LJ, et al. c-Jun decreases voltage-gated K+ channel activity in pulmonary artery smooth muscle cells. Circulation. 2001;104:1557–63. doi: 10.1161/hc3801.095662. [DOI] [PubMed] [Google Scholar]
- 92.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol. 1995;269:H348–55. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 93.Chandy KG, Gutman GA. Voltage-gated K+ channels. In: North RA, editor. Ligand- and Voltage-Gated Ion Channels. Boca Raton, FL: CRC; 1995. pp. 1–71. [Google Scholar]
- 94.Alioua A, Tanaka Y, Wallner M, Hofmann F, Ruth P, Meera P, et al. The large-conductance, voltage-dependent, and calcium-sensitive K+ channel, Hslo, is a target of cGMP-dependent protein kinase phosphorylation in vivo. J Biol Chem. 1998;273:32950–6. doi: 10.1074/jbc.273.49.32950. [DOI] [PubMed] [Google Scholar]
- 95.Toro L, Wallner M, Meera P, Tanaka Y. Maxi-Kca, a unique member of the voltage-gated K channel superfamily. News Physiol Sci. 1998;13:112–7. doi: 10.1152/physiologyonline.1998.13.3.112. [DOI] [PubMed] [Google Scholar]
- 96.Krick S, Platoshyn O, McDaniel SS, Rubin LJ, Yuan JX. Augmented K+ currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am J Physiol Lung Cell Mol Physiol. 2001;281:L887–94. doi: 10.1152/ajplung.2001.281.4.L887. [DOI] [PubMed] [Google Scholar]
- 97.Farrukh IS, Peng W, Orlinska U, Hoidal JR. Effect of dehydroepiandrosterone on hypoxic pulmonary vasoconstriction: A Ca2+-activated K+-channel opener. Am J Physiol. 1998;274:L186–95. doi: 10.1152/ajplung.1998.274.2.L186. [DOI] [PubMed] [Google Scholar]
- 98.Neylon CB, Lang RJ, Fu Y, Bobik A, Reinhart PH. Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: Relationship between Kca channel diversity and smooth muscle cell function. Circ Res. 1999;85:e33–43. doi: 10.1161/01.res.85.9.e33. [DOI] [PubMed] [Google Scholar]
- 99.Tanaka Y, Meera P, Song M, Knaus HG, Toro L. Molecular constituents of maxi Kca channels in human coronary smooth muscle: Predominant a + b subunit complexes. J Physiol. 1997;502:545–57. doi: 10.1111/j.1469-7793.1997.545bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stull JT, Kamm KE, Taylor DA. Calcium control of smooth muscle contractility. Am J Med Sci. 1988;296:241–5. doi: 10.1016/s0002-9629(15)40865-1. [DOI] [PubMed] [Google Scholar]
- 101.Berridge MJ. Calcium signalling and cell proliferation. Bioessays. 1995;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- 102.Means AR. Calcium calmodulin and cell cycle regulation. FEBS Lett. 1994;347:1–4. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- 103.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–8. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 104.Ginty DD. Calcium regulation of gene expression: Isn't that spatial? Neuron. 1997;18:183–6. doi: 10.1016/s0896-6273(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 105.Johnson CM, Hill CS, Chawla S, Treisman R, Bading H. Calcium controls gene expression via three distinct pathways that can function independently of the Ras/mitogen-activated protein kinases (ERKs) signaling cascade. J Neurosci. 1997;17:6189–202. doi: 10.1523/JNEUROSCI.17-16-06189.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 107.Short AD, Bian J, Ghosh TK, Waldron RT, Rybak SL, Gill DL. Intracellular Ca2+ pool content is linked to control of cell growth. Proc Natl Acad Sci U S A. 1993;90:4986–90. doi: 10.1073/pnas.90.11.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.He H, Lam M, McCormick TS, Distelhorst CW. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J Cell Biol. 1997;138:1219–28. doi: 10.1083/jcb.138.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fleischmann BK, Murray RK, Kotlikoff MI. Voltage window for sustained elevation of cytosolic calcium in smooth muscle cells. Proc Natl Acad Sci U S A. 1994;91:11914–8. doi: 10.1073/pnas.91.25.11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kohomoto O, Levi AJ, Bridge JH. Relation between reverse sodium-calcium exchange and sarcoplasmic reticulum calcium release in guinea pig ventricular cells. Circ Res. 1994;74:550–4. doi: 10.1161/01.res.74.3.550. [DOI] [PubMed] [Google Scholar]
- 111.Blaustein MP, Lederer WJ. Sodium/calcium exchange: Its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 112.Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: Requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–64. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koike T, Martin DP, Johnson EM., Jr Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: Evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci U S A. 1989;86:6421–5. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–7. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 115.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium infl ux stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–21. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 116.Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–82. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- 117.Tsien RW, Tsien RY. Calcium channels, stores, and oscillations. Annu Rev Cell Biol. 1990;6:715–60. doi: 10.1146/annurev.cb.06.110190.003435. [DOI] [PubMed] [Google Scholar]
- 118.Van Breemen C, Saida K. Cellular mechanisms regulating [Ca2+]i smooth muscle. Annu Rev Physiol. 1989;51:315–29. doi: 10.1146/annurev.ph.51.030189.001531. [DOI] [PubMed] [Google Scholar]
- 119.Sham JS, Cleemann L, Morad M. Gating of the cardiac Ca2+ release channel: The role of Na+ current and Na+-Ca2+ exchange. Science. 1992;255:850–3. doi: 10.1126/science.1311127. [DOI] [PubMed] [Google Scholar]
- 120.Chao TS, Byron KL, Lee KM, Villereal M, Rosner MR. Activation of MAP kinases by calcium-dependent and calcium-independent pathways.Stimulation by thapsigargin and epidermal growth factor. J Biol Chem. 1992;267:19876–83. [PubMed] [Google Scholar]
- 121.Dynlacht BD. Regulation of transcription by proteins that control the cell cycle. Nature. 1997;389:149–52. doi: 10.1038/38225. [DOI] [PubMed] [Google Scholar]
- 122.Husain M, Jiang L, See V, Bein K, Simons M, Alper SL, et al. Regulation of vascular smooth muscle cell proliferation by plasma membrane Ca(2+)-ATPase. Am J Physiol. 1997;272:C1947–59. doi: 10.1152/ajpcell.1997.272.6.C1947. [DOI] [PubMed] [Google Scholar]
- 123.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–5. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 124.Jaggar JH, Mawe GM, Nelson MT. Voltage-dependent K+ currents in smooth muscle cells from mouse gallbladder. Am J Physiol. 1998;274:G687–93. doi: 10.1152/ajpgi.1998.274.4.G687. [DOI] [PubMed] [Google Scholar]
- 125.Khan SA, Mathews WR, Meisheri KD. Role of calcium-activated K+ channels in vasodilation induced by nitroglycerine, acetylcholine and nitric oxide. J Pharmacol Exp Ther. 1993;267:1327–35. [PubMed] [Google Scholar]
- 126.Barman SA. Pulmonary vasoreactivity to endothelin-1 at elevated vascular tone is modulated by potassium channels. J Appl Physiol. 1996;80:91–8. doi: 10.1152/jappl.1996.80.1.91. [DOI] [PubMed] [Google Scholar]
- 127.Barman SA. Potassium channels modulate canine pulmonary vasoreactivity to protein kinase C activation. Am J Physiol. 1999;277:L558–65. doi: 10.1152/ajplung.1999.277.3.L558. [DOI] [PubMed] [Google Scholar]
- 128.Hasunuma K, Rodman DM, McMurtry IF. Effects of K+ channel blockers on vascular tone in the perfused rat lung. Am Rev Respir Dis. 1991;144:884–7. doi: 10.1164/ajrccm/144.4.884. [DOI] [PubMed] [Google Scholar]
- 129.Peng W, Hoidal JR, Farrukh IS. Regulation of Ca2+-activated K+ channels in pulmonary vascular smooth muscle cells: Role of nitric oxide. J Appl Physiol. 1996;81:1264–72. doi: 10.1152/jappl.1996.81.3.1264. [DOI] [PubMed] [Google Scholar]
- 130.Shimoda LA, Sylvester JT, Sham JS. Inhibition of voltage-gated K+ current in rat intrapulmonary arterial myocytes by endothelin-1. Am J Physiol. 1998;274:L842–53. doi: 10.1152/ajplung.1998.274.5.L842. [DOI] [PubMed] [Google Scholar]
- 131.Yuan XJ, Sugiyama T, Goldman WF, Rubin LJ, Blaustein MP. A mitochondrial uncoupler increases Kca currents but decreases Kv currents in pulmonary artery myocytes. Am J Physiol. 1996;270:C321–31. doi: 10.1152/ajpcell.1996.270.1.C321. [DOI] [PubMed] [Google Scholar]
- 132.Zhao YJ, Wang J, Rubin LJ, Yuan XJ. Inhibition of Kv and Kca channels antagonizes NO-induced relaxation in pulmonary artery. Am J Physiol. 1997;272:H904–12. doi: 10.1152/ajpheart.1997.272.2.H904. [DOI] [PubMed] [Google Scholar]
- 133.Yu SP, Choi DW. Ions, cell volume, and apoptosis. Proc Natl Acad Sci U S A. 2000;97:9360–2. doi: 10.1073/pnas.97.17.9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Somlyo AV, Somlyo AP. Electromechanical and pharmacomechanical coupling in vascular smooth muscle. J Pharmacol Exp Ther. 1968;159:129–45. [PubMed] [Google Scholar]
- 135.Morgan KG. Calcium and vascular smooth muscle tone. Am J Med. 1987;82:9–15. doi: 10.1016/0002-9343(87)90205-1. [DOI] [PubMed] [Google Scholar]
- 136.Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol. 1997;59:89–144. doi: 10.1146/annurev.physiol.59.1.89. [DOI] [PubMed] [Google Scholar]
- 137.Rubin LJ, Rich S. Primary Pulmonary Hypertension. New York, NY: Marcel Dekker Inc.; 1997. [Google Scholar]
- 138.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, et al. Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol. 2001;280:H746–55. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]