

Abstract
Since over-activation of the hypothalamic-pituitary-adrenal (HPA) axis occurs in Alzheimer’s disease (AD), dysregulation of stress neuromediators may play a mechanistic role in the pathophysiology of AD. However, the effects of stress on tau phosphorylation are poorly understood and the relationship between corticosterone and corticotropin-releasing factor (CRF) on both Aβ and tau pathology remain unclear. Therefore, we first established a model of chronic stress which exacerbates Aβ accumulation in Tg2576 mice and then extended this stress paradigm to a tau transgenic mouse model with the P301S mutation (PS19) which displays tau hyperphosphorylation, insoluble tau inclusions and neurodegeneration. We show for the first time that both Tg2576 and PS19 mice demonstrate a heightened HPA stress profile in the unstressed state. In Tg2576 mice, one month of restraint/isolation (RI) stress increased Aβ levels, suppressed microglial activation, and worsened spatial and fear memory compared to non-stressed mice. In PS19 mice, RI stress promoted tau hyperphosphorylation, insoluble tau aggregation, neurodegeneration and fear-memory impairments. These effects were not mimicked by chronic corticosterone administration but were prevented by pre-stress administration of a CRF receptor type 1 (CRF1) antagonist. The role for a CRF1-dependent mechanism was further supported by the finding that mice over-expressing CRF had increased hyperphosphorylated tau compared to wildtype littermates. Together, these results implicate HPA dysregulation in AD neuropathogenesis and suggest that prolonged stress may increase Aβ and tau hyperphosphorylation. These studies also implicate CRF in AD pathophysiology and suggest that pharmacological manipulation of this neuropeptide may be a potential therapeutic strategy for AD.
Keywords: stress, corticosterone, corticotropin-releasing factor, hypothalamic-pituitary-adrenal axis, Alzheimer’s disease, β-amyloid, tau
Introduction
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder characterized by memory deficits, neuron loss, Aβ deposited in amyloid plaques and neurofibrillary tangles composed of hyperphosphorylated tau fibrils. Convergent evidence implicates stress in AD neuropathology. For example, AD and stress disorders are characterized by hypothalamic-pituitary-adrenal (HPA) dysfunction (Swaab et al., 2005; Holsboer and Ising, 2008; Pervanidou and Chrousos, 2010), while psychological stress increases AD risk (Wilson et al., 2006) and AD is associated with stress-related disorders (Aznar and Knudsen, 2011).
This notwithstanding, the molecular mechanisms linking stress to AD pathogenesis remain unknown. With stress, hippocampal glucocorticoid receptor activation increases neuronal metabolism, decreases cell survival and neurogenesis, promotes dendritic atrophy, and causes long-term potentiation and cognition deficits (Sapolsky et al., 1985; McEwen, 1999; Kim and Diamond, 2002; Rothman and Mattson, 2010). Elevated glucocorticoid levels, characteristic of chronic stress, are associated with cognitive symptoms of dementia in elderly and AD subjects (Csernansky et al., 2006; Lee et al., 2008). From these observations the glucocorticoid hypothesis of brain aging emerged, proposing that chronic stress and/or glucocorticoid exposure promotes hippocampal aging and even AD pathogenesis (Landfield et al., 2007). Although it has been well established that chronic stress exacerbates Aβ accumulation and memory deficits in AD transgenic (Tg) mice (Green et al., 2006; Jeong et al., 2006; Kang et al., 2007; Lee et al., 2009; Huang et al., 2011), the involvement of a glucocorticoid receptor-dependent pathway remains controversial. Recent studies have implicated corticotropin-releasing factor (CRF) as a more likely causal factor in stress-induced Aβ increases (Kang et al., 2007) although it may also offer protective effects (Bayatti and Behl, 2005). CRF acts through G-protein-coupled receptor type 1 (CRF1) with widespread brain expression to orchestrate the stress response (Bale and Vale, 2004). CRF overproduction is implicated in stress-related psychopathology, such as depression, anxiety, and post-traumatic stress disorder (Hillhouse and Grammatopoulos, 2006; Hauger et al., 2009; Pervanidou and Chrousos, 2010; Stengel and Tache, 2010).
The majority of studies examining stress contributions to AD pathology have focused on Aβ plaques, while less has been reported about stress-related effects on disease-relevant tau pathology. A substantial body of literature demonstrates that tau phosphorylation is induced by various environmental insults such as cold (Korneyev et al., 1995; Feng et al., 2005), heat (Papasozomenos, 1996), hypoglycemia (Yanagisawa et al., 1999), starvation (Planel et al., 2001), hibernation (Arendt et al., 2003), social isolation (Zhang et al., 2011) and restraint (Yan et al., 2010), which may be CRF1-mediated (Rissman et al., 2007). However, these data were generated almost exclusively in non-Tg rodents. Thus, no studies have addressed CRF1-mediated mechanisms of tau hyperphosphorylation in a disease-relevant Tg mouse model. Therefore, we sought to establish and validate a chronic stress paradigm using the Tg2576 mouse model in which mutated Aβ precursor protein found in familial AD is over-expressed, and then investigate the mechanisms behind stress-induced tau pathology using established Tg mouse models which either over-express mutant tau (PS19) found in familial frontotemporal degeneration or CRF. Our data implicate CRF1-dependent mechanisms in the pathophysiology of AD.
Materials and Methods
Mouse cohorts
Three separate cohorts of different Tg mice were used in this study; a) female hemizygous Tg2576 harboring the APPswe mutation and wildtype (WT) mice of the same genetic background (Bl6SJL/J) in Experiment 1, b) male hemizygous PS19 harboring P301S mutant human tau (Yoshiyama et al., 2007) and WT mice of the same genetic background (Bl6/C3H) in Experiment 2–3, and c) female CRF-over-expressing (CRF-OE) (Stenzel-Poore et al., 1992) and WT mice of the same genetic background (C57BL/6J) in Experiment 4. Tg2576 and PS19 mice were housed with same sex littermates at The University of Pennsylvania School of Medicine and CRF-OE mice were purchased from Jackson Labs and housed at The Children’s Hospital of Philadelphia. All procedures were approved by the University of Pennsylvania IACUC. Separate groups of Tg2576 and WT mice (n=10/group) were evaluated at 4 or 14 months (mo) of age in Experiment 1. PS19 and WT mice (n=8/group) were evaluated at 6 mo of age in Experiment 2, or 8 mo of age in Experiment 3 and 4. Finally, the third cohort of female WT and CRF-OE mice (n=6/group) were evaluated at 10 mo of age.
In general, mice were evaluated on behavioral tests, and then exposed to either drug treatment or chronic stress, or no stress for 1 mo. Following 1 mo of chronic stress and/or treatment, mice were again administered behavioral tests, followed by corticosterone analysis after an acute stress and then sacrificed. At time of sacrifice, mice were deeply anesthetized with ketamine/xylazine (100mg ket/10mg xyl/kg) and were perfused with 10 mM phosphate buffered serum with heparin. Brains were bisected and one hemibrain was fixed for 24 hrs in 10% neutral buffered formalin for immunohistochemical analysis while the other hemibrain was dissected, weighed and frozen for biochemical analysis. The adrenal cortex was weighed as bioassay of stress.
Stress Paradigms
Mice were exposed to 1 mo of no stress (NS), variable stress (VS), or restraint/isolation (RI) stress as summarized in Figures 1A, 3A and 6A in order to compare the effects of different stress types. To achieve chronic VS resistant to habituation, mice experienced 1 stressor per day for 6 days (d) per week for 4 weeks. Six stressors were used and the order, time of day (morning or afternoon), and location (testing room) of which were randomized (adapted as previously described, (Alfarez et al., 2003; Teegarden and Bale, 2008). The 6 stressors included: 1) long swim for 20 minute (min) in a 30° water bath, 2) cold swim for 2.5 min in a 15° water bath, 3) restraint for 15 min in a 50 ml conical tube, 4) housed in isolation for 24 hrs, 5) housed with soiled bedding for 24 hrs, 6) housed under lights-on conditions for 24 hrs. In contrast, to achieve RI, mice were housed individually for 1 mo and were restrained in a conical tube for 6 hrs per day for 6 d per week in Experiments 1–2, or 3 hrs per day in Experiment 4. Non-stressed control mice were housed with same sex littermates (n=3–5/cage) and were transported to/from testing rooms but were not otherwise handled. Body weight of all mice was monitored at 0, 2, and 4 weeks during all the 1 mo treatment periods.
Figure 1. Chronic RI stress exacerbates Aβ levels in stress-sensitive Tg2576 mice.
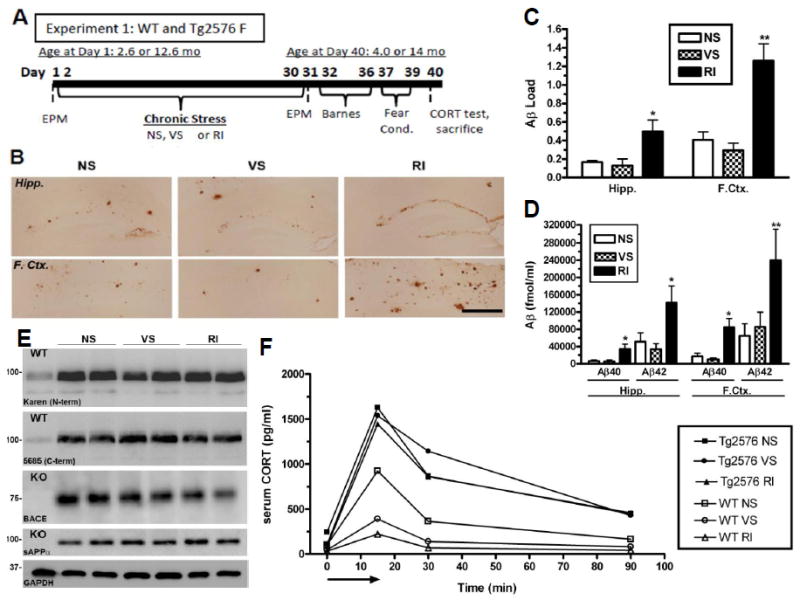
(A) Experimental design of Experiment 1; WT and Tg2576 female mice (n=10/group) were exposed to NS, VS or RI stress for 1 mo. (B) Representative images in the hippocampus and frontal cortex show increased Aβ-IR in RI mice, quantified in (C) (F(3,18)=5.61, p=0.014) and (F(3,24)=15.64, p<0.0001). Data show mean Aβ load ± SEM. (D) By ELISA, insoluble Aβ40 was significantly increased by RI in the hippocampus (F(5,28)=4.47, p=0.005) and frontal cortex (F(5,28)=2.08, p=0.11), as was Aβ42 in the hippocampus (F(2,21)=6.51, p=0.007) and frontal cortex (F(2,15)=12.4, p=0.001). Data show mean Aβ levels ± SEM (n=8/group). (E) Hippocampal protein levels of APP, sAPPα and BACE were measured with either WT or BACE knockout (KO) mouse hippocampal homogenate (n=2/group) as a negative control. In 14 mo old Tg2576 mice, neither stress altered hippocampal levels of 5685, Karen, BACE or sAPP. (F) CORT levels were plotted over time after an acute 15 min restraint stress (arrow) in 4 mo WT and Tg2576 mice. In WT mice, both VS (F(5,28)=4.9, p=0.05) and RI stress (F(2,20)=1.3, p=0.31) decreased the area under the curve compared to NS. However, CORT levels were elevated significantly in all conditions of Tg2576 mice, regardless of stress treatment. Data show mean ± SEM (n=10/group). p*<0.05, **<0.01 from NS. Scale bar = 0.5mm.
Figure 3. Chronic RI stress exacerbates hyperphosphorylated tau, aggregated tau inclusions and fear-related memory in PS19 mice.
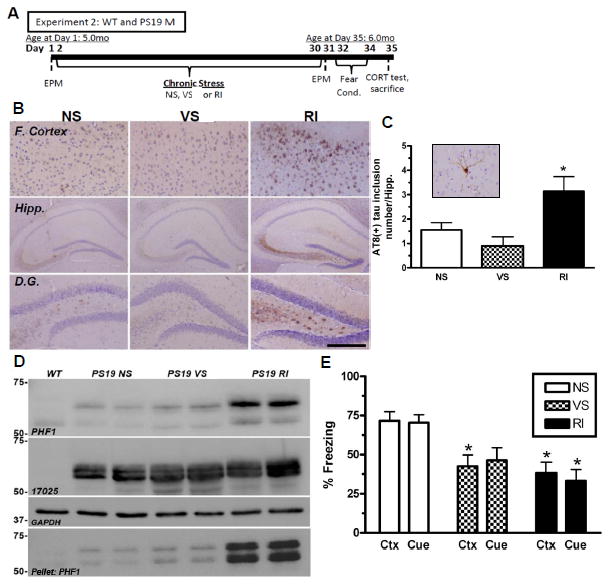
(A) Experimental design of Experiment 2; WT and PS19 male mice (n=8/group). (B) Representative images demonstrate that in the frontal cortex and the hippocampus, specifically the dentate gyrus, chronic RI increased AT8 (+)-IR compared to NS. (C) The number of AT8 (+) tau inclusions (inset) in the hippocampus was counted and showed a significant increase in RI compared to NS mice (F(2,23)=14.42, p<0.0001). Data show mean number of tau inclusions ± SEM. (D) Tau was also evaluated by western blot using both PHF1 and 17025 antibodies and probed for GAPDH as a loading control. PS19 RI stressed mice displayed a significant increase in soluble PHF1 (F(2,5)=8.21, p=0.0024) and insoluble (F(2,5)=1.96, p=0.009) but not 17025 expressions in the hippocampus. (E) PS19 RI mice showed significant impairment compared to NS mice in both context (F(5,54)=3.88, p=0.0048) and cued fear conditioning (F(5,54)=4.19, p=0.003). p*<0.05, **<0.01 and ***<0.001 from NS of same genotype. Scale bar = 0.5mm for (B), Cortex and Hipp., and 200mm for D.G.
Figure 6. CRF1 mediates stress-induced pathological tau inclusions, neurodegeneration and fear-associated learning.
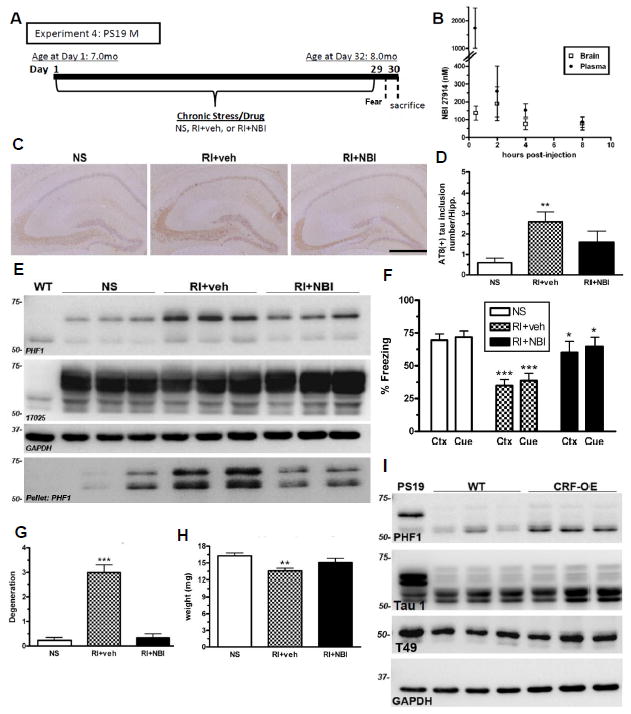
(A) Experimental design of Experiment 4; male PS19 mice were exposed to NS or RI stress with pre-stress CRF1 antagonist (NBI) or vehicle subcutaneous injections (n=8/group). (B) A small pilot utilizing adult WT female mice (n=3/group) was conducted to measure brain and plasma levels of NBI after a 10mg/kg sc injection. (C) Representative pictomicrographs demonstrate that vehicle-treated RI mice displayed significantly higher AT8 (+) tau inclusions in the hippocampus, quantified in (D) (F(2,25)=7.15, p=0.05), as well as soluble and insoluble PHF1, but not 17025 levels by western blot (E) that was blocked by a pre-stress injection of NBI. (F) Vehicle-treated RI mice also displayed fear-related learning impairments in both context (F(2,28)=12.4, p<0.05) and cued (F(2,28)=9.3, p<0.05) fear conditioning which was significantly prevented by NBI. Data show mean % freezing ± SEM. (G) Compared to NS, vehicle-treated mice had significantly lower hippocampal weight (F(2,44)=6.44, p=0.004) and significantly increased NeuN-IR (+) degeneration scores (F(2,28)=6.19, p<0.001) (H) while NBI treatment significantly prevented both effects. (I) Lastly, phosphorylated tau was quantified in the hippocampus of 10 mo old CRF-OE mice and WT littermate controls. A substantial elevation was observed in PHF1 levels in CRF-OE compared to WT littermates without a change in Tau 1 and T49 level. Data show mean PHF1 score ± SEM. Scale bar = 0.5mm. p***<0.001, p**<0.01, from NS. p*<0.05, from RI+veh.
Drug Treatments
To achieve chronic hormone administration of corticosterone (CORT) in PS19 mice in Experiment 3, subjects were implanted with slow-release, subcutaneous CORT pellets (10mg, 21-day release) purchased from Innovative Research of America (Sarasota, Fl). This dose corresponds to an approximate dose of 12mg/kg/day and has previously been shown to induce physiological plasma CORT levels (Herrmann et al., 2009). PS19 mice were briefly anesthetized with isoflourane, shaved at the nape of their neck, implanted with a hormone pellet, and the site was closed with surgical staples. Pellets were implanted on day 1, 10 and 20 and animals were sacrificed on day 30, as summarized in Figure 6B. Control mice were also anesthetized but received sham surgery where no pellet was implanted. This pellet administration schedule was based on results of a small pilot study in 6 mo old male WT mice (n=3/group). These pilot mice were implanted with corticosterone pellets and 10 μl of serum was collected from the tail 0.5, 2, 6, 18, 24 hrs post-injection and then on each subsequent day for 9 d. Serum was then analyzed for CORT concentration.
The CRF1 antagonist, NBI 27914 (NBI; Sigma), was chronically administered to a subset of PS19 mice in Experiment 4. These mice were administered a subcutaneous (sc) injection of 10 mg/kg NBI dissolved in a solution of DMSO:PEG400:dH20. Injections occurred 15 min before onset of restraint stress. Vehicle-treated mice were also handled and underwent RI stress but were injected with DMSO:PEG400:dH20. All mice underwent treatment 6 d/week for 4 weeks. The selectivity of NBI 27914 for the CRF1 receptor has been demonstrated as NBI has a much higher receptor affinity for CRF1 (Ki = 3.5 nM) over CRF2 (Ki = > 1000 nM) (Hoare et al., 2004) and this NBI dose previously has been shown to be effective (Lee et al., 2009). NBI brain penetration was confirmed in a small pilot study. In this pilot, 7 mo old female WT mice (n=3/group) were injected sc with 10 mg/kg NBI and sacrificed 30 min, 2 hrs, 4 hrs, or 8 hrs post-injection. NBI concentrations in both plasma and brain were assessed to determine blood-brain-barrier penetration, drug half-life in brain, and a standard brain/plasma pharmacokinetic profile.
Corticosterone Assay
To evaluate the corticosterone response to acute stress, blood was collected before and after a 15 min restraint stress on the day of sacrifice for all mice in all treatment groups in Experiments 1–3. After normal lights-on (7:00 AM) and habituation from 7:00–8:00 AM, mice were restrained in 50 ml conical tubes and blood was drawn at time 0, 15, 30 and 90 min after 15 min restraint stress, as adapted by (Teegarden and Bale, 2008). Serum was frozen and later analyzed using a corticosterone (CORT) EIA kit (Assay Designs, cat # 901–097) according to the manufacturer’s instructions.
Determination of Plasma and Brain NBI27914 Concentrations
Sample preparation and LC/MS/MS was conducted as previously described by our lab (Brunden et al., 2011). To prepare samples, n=3 brains/group were homogenized in 10 mM ammonium acetate, pH 5.7 (1:2; w/v) using a handheld sonic homogenizer. NBI 27914 from 50 μL of brain homogenate or plasma was extracted with 200 μL acetonitrile, centrifuged, and the supernatant removed for LC/MS/MS analysis. The LC/MS/MS system was comprised of an Aquity UPLC, a TQ MS, and controlled using MassLynx software (Waters Corporation, Milford, MA, USA). Samples were separated on an Aquity BEH C18 column (1.7 μm, 2.1 × 50 mm) at 50°C. The mobile phase A was 0.1% (v/v) formic acid, and B was 0.1% (v/v) formic acid in acetonitrile. Injections of 5 μL were separated at a flow rate of 0.6 mL/min using a gradient from 70% to 95% B over two min followed by wash and re-equilibration steps. NBI 27914 was detected in positive ionization mode using multiple reactions monitoring of specific collision induced ion transitions and quantified by peak area. The MS was operated with a desolvation temperature of 450°C and a source temperature of 150°C. Desolvation and source nitrogen gas flows were 900 L/hr and 50 L/hr, respectively. Collision cell argon gas flow was 0.1 mL/min. Source and analyzer voltages were optimized using the MassLynx auto tune utility. NBI 27914 standards at 1000 ng/mL in 50% acetonitrile were infused at 30 μL/min and combined with flow from the UPLC at 0.6 mL/min and 50% B. Standard curves were generated from spiked brain homogenate and plasma prepared at 0.01, 0.1, 1 and 10 μM and extracted as above. Peak area was plotted versus concentration and a 1/x weighed linear regression curve was used to quantify the unknowns.
Behavioral Analysis
To evaluate anxiety-related behavior and cognitive learning and memory function, either before and/or after stress exposure, mice were tested in the elevated plus maze (EPM), Barnes maze test, and/or context and cued fear conditioning as summarized in Figures 1A, 3A and 6A. In Experiment 1, all mice in all treatment groups were tested on all behavioral tasks. In Experiment 2, all mice in all treatment groups were tested on EPM and fear conditioning. In Experiment 3, mice were only tested on EPM and in Experiment 4, mice were tested on fear conditioning. In all tests, mice were habituated to the testing room 1 hr before tests and the apparatus was cleaned with 70% ethanol in between animals to minimize odor cues. To assess anxiety-related behavior, the EPM was conducted as previously described (Carroll et al., 2010) where mice were allowed to explore the maze for 5 min and scored for % time spent in open arms. Mice were tested for hippocampal-dependent spatial memory on the Barnes Maze (San Diego Instruments). On all testing days (Day 1–6), mice were exposed to the Barnes maze for three 2.5 min trials, 15 min apart and mice were tested on their ability to learn and remember the fixed position of this escape compartment and their performance was scored for success rate and latency to find the escape compartment, as adapted from (Patil et al., 2009) and previously described in our lab (Brunden et al., 2010). Also, to assess fear-related learning, mice were tested on the context and cued fear conditioning test (Med Associates). Mice were exposed to 3 repeats of an 80 db tone paired with a 0.7 mA footshock. After 24 hrs, mice were scored for % time freezing during a 5 min test in the same context, and 48 hrs later mice were scored for % time freezing during a 3 min tone exposure in a novel context (altered odor, flooring, and chamber shape), as adapted from (Wellman et al., 2007).
Aβ Sandwich ELISA
Hippocampal and frontal cortex samples (n=8/group) were homogenized in RIPA buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P-40, 5 mM EDTA in TBS, pH 8.0) containing protease inhibitors (1 μg/mL each of pepstatin A, leupeptin, L-1-tosylamido-2-phenylethyl chloromethyl ketone, 1-chloro-3-tosylamido-7-amino-2-heptanone, soybean trypsin inhibitor, and 0.5 mM phenylmethylsulfonyl fluoride). Homogenates were sonicated and spun at 40,000 × g for 30 min at 4°C. The supernatant was collected and the pellet was rehomogenized in 70% formic acid to a final concentration of 2 μL/mg. Samples were sonicated and spun at 40,000×g for 30 min at 4°C. Aβ 1–40 and Aβ1–42 were detected using a Sandwich ELISA protocol described previously (Lee et al., 2003). Briefly, Ban50 (anti-Aβ1–10) was used as a capturing antibody coated in 384 well Nunc Maxisorp plates. After application of brain lysate samples overnight at 4°C, horseradish peroxidase-conjugated BA-27 and BC-05 were incubated for 4 hrs to report Aβ species ending at position 40 and 42, respectively. Plates were washed, blocked and exposed to reagents with a Thermo Multidrop 384 and a Bio Tek Elx450 Select CW plate washer. For quantification of Aβ levels, synthetic Aβ1–40 and Aβ1–42 purchased from Bachem Bioscience Inc. (King of Prussia, PA) were serially diluted to generate standard curves and read on a Spectra Max M5 plate reader.
Immunohistochemistry (IHC)
Neutral buffered formalin was leeched from fixed hemibrains (n=6–10/group) after 24 hrs with Tris/NaCl then brains were paraffin embedded and 6 μm sections were cut and slide mounted. Slides were processed as previously described (Nakagawa et al., 1999; Yoshiyama et al., 2007). Briefly, sections were deparaffinized in xylene, hydrated in a series of ethanol and deionized water, quenched using a freshly prepared mixture of methanol (150 mL) and hydrogen peroxide (33%,30 ml), washed in deionized water followed by 0.1M Tris, blocked with 0.1M Tris/2% fetal bovine serum and incubated overnight in primary antibody at 4°C. The following antibodies were used in this study: Aβ1–11 (nab228), 1:30,000; Ser202/Thr205 phosphorylated tau (ptau; AT8), 1:7,500; activated microglia (Iba1), 1:10,000 (Wako, 019-19741); neuronal nuclei (NeuN), 1:1000 (Millipore, MAB377) and anti-glucocorticoid receptor (GR), 1:300, (Santa Cruz Biotechnology, H-300: sc-8992). To complete IHC, the avidin-biotin complex method was used according to the instructions of the vendor (Vector Laboratories, Burlingame, CA) and processed using Biogenex Reagents and the Biogenex i6000 automated staining system.
Triple Immunoflourescence
Tissue was deparaffinized and washed as described above and subjected to antigen retrieval using boiling citrate treatment. Next, sections were stained with Thioflavin-S using a freshly made 0.0125% Thioflavin-S/40%EtOH/60%PBS solution, washed and co-incubated overnight at 4°C with primary antibodies Nab228 (monoclonal) and Iba1 (polyclonal). To complete immunoflourescent staining, sections were then co-incubated with fluorescent secondary antibodies (FITC red anti-rabbit and AMCA blue anti-mouse, 1:200) and coverslipped using VectaShield antifade medium.
Immunoreactivity (IR) Quantification
Aβ, Iba1 and GR-IR in the frontal cortex and/or hippocampus were quantified by a researcher blinded to experimental condition using the IHC load technique as adapted from a previously described protocol (Carroll et al., 2007; Carroll and Pike, 2008; Carroll et al., 2010). Load values were determined from selected 420 μm × 330 μm fields of immunolabeled sections that were captured and digitized using a video capture system (Nikon camera coupled to an Olympus DP71 upright microscope). Using Image J software, digital grayscale images were converted into binary positive/negative data using a constant threshold limit. The percentage of positive pixels (i.e., immunoreactive area) was quantified for each image to generate immunoreactive load values (i.e., % area occupied by the immunoreactivity). For GR-IR in the CA1 of hippocampus, 3 sequential sections of hippocampus were analyzed/brain. The capture frame was centered over the pyramidal layer with the captured field corresponding from between the narrow zone that marks the division between the end of the regio inferior (CA2/3) and the start of regio superior (CA1) and the beginning of the subiculum, as previously defined (West et al., 1991). Each of the 3 images/brain were analyzed and load values were averaged. For Aβ-IR in the hippocampus, 1 field of classic hippocampus was captured from each of 3 sequential sections per brain. The capture field was drawn free-hand around the entire hippocampus corresponding to the region from the posterior CA2 to the anterior subiculum and from the ventral tip of dentate gryus to the dorsal arch of the CA1. Each free-hand drawn hippocampus was analyzed and load values were averaged. The same method was applied for Aβ and Iba1-IR in the frontal cortex, however the quantification field was held constant (50 μm × 75 μm). Here, fields were centered over layers 4–5, beginning in cingulate cortex area 1 and progressing laterally to somatosensory cortex as previously described (Carroll et al., 2007). To achieve the ratio of Iba1-IR:Nab228-IR, identical quantification fields were overlaid on adjacent sections stained with each non-flourescent antibody. Thus, quantification of each antibody was achieved on the exact same quantification field.
To quantify neurofibrillary tangle-like inclusions, AT8-IR, and degeneration in the hippocampus, digital pictures were taken of the hippocampus (every 5th longitudinal section was analyzed across the entire hippocampus, a total of approximately 10 sections per brain), beginning with a randomly selected section of dorsal hippocampus. AT8-IR aggregates were counted within the hippocampal formation as those tau inclusions stained thoroughly with AT8(+)-IR and dystrophic neuronal processes. Subregional AT8(+)-IR was quantified in 7 subregions of hippocampus and 6 cortical layers using a semi-quantitative scale (Stage 0–5 of increasing IR presence and intensity) as modified from a previously described paradigm in our lab (Hurtado et al., 2010). Degeneration was scored using the same semi-quantitative scale (Stage 0–5 of increasing cell loss).
Western Blots
To measure soluble proteins, hippocampal and frontal cortex samples (n=2–6/group) were homogenized in RIPA buffer in the presence of protease inhibitors and briefly sonicated. Samples were centrifuged at 40,000×g for 30 min at 4°C, and protein was measured by bicinchoninic acid (BCA) assay. To measure insoluble tau, the supernatant was collected and the pellet was rehomogenized in 70% formic acid to a final concentration of 2 μL/mg. Samples were sonicated and spun at 40,000×g for 30 min at 4°C. Later, 15 μg protein was electrophoresed on 10% Tris-glycine acrylamide gels, and transferred to a nitrocellulose membrane. Immunoblots were probed with the following antibodies: N-terminal APP (Karen), 1:1000; C-terminal APP (5685), 1:1000; β-secretase cleavage enzyme (BACE), 1:1000; soluble APP (sAPPα), 1:1000; Ser396/Ser404 ptau (PHF1), 1:1000; total human tau (17025), 1:1000; non-phosphorylated tau (Tau 1), 1:1000; total mouse tau (T49), 1:1000; and a loading control (GAPDH), 1:2000. As a negative control to demonstrate BACE antibody specificity, hippocampal homogenate from a BACE knockout (KO) mouse (Luo et al., 2001) was also probed when appropriate. Immunoblots were then exposed to species-specific horseradish peroxidase-conjugated anti-IgG antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) and visualized by enhanced chemiluminescence (PerkinElmer Life Sciences) using a Fuji Imager. Images were quantified using Multi Gauge v3.2 FujiFilm software.
Statistical Analysis
To statistically evaluate treatment effects, raw data were first analyzed by ANOVA and then subjected to between group comparisons using the Bonferonni post-hoc test. Effects achieving 95% probability (i.e., p < 0.05) were interpreted as statistically significant. Barnes maze was analyzed using Repeated Measures ANOVA.
Results
Experiment 1: Establishment of a chronic stress model capable of exacerbating pathology in Tg2576 mice
Two cohorts of Tg2576 and WT littermate control mice were exposed to either NS, VS or RI stress for 1 mo and were subsequently sacrificed at either 4 or 14 mo of age (Figure 1A). To ensure that stress did not affect general health, mice were monitored daily and weighed at 0, 2 and 4 weeks of stress. No body weight differences were observed between the NS and stress groups in either 4 mo (F(5,39)=2.97, p=0.23) or 14 mo old (F(5,42)=4.41, p=0.46) WT cohorts. To ensure that both VS and RI stress paradigms induced sufficient stress, adrenal gland weight was measured and found to be significantly greater in the VS and RI WT cohorts than in the NS WT mice at both 4 mo (F(5,44)=4.93, p=0.0014) and 14 mo of age (F(5,48)=6.25, p<0.0001).
After 1 mo of stress, Tg2576 mice were sacrificed and assessed for levels of Aβ both by IHC and sandwich ELISA. Figure 1B shows representative examples of Aβ-IR load, as assessed by quantification of Aβ IHC, in both hippocampus and frontal cortex of 14 mo old Tg2576 mice exposed to NS, VS and RI. Quantification of the Aβ-IR load values revealed that mice exposed to RI displayed significantly greater Aβ-IR compared to NS, while VS mice displayed no change (Figure 1C). Further, insoluble levels of Aβ42 were also measured by ELISA in both frontal cortex and hippocampus. In 14 mo old Tg2576 mice, RI treatment significantly increased Aβ40 and Aβ42 levels compared to NS in both brain regions (Figure 1D). Again, VS mice displayed no change in Aβ levels. The same trend was also demonstrated in 4 mo old Tg2576 mice. Although these mice have not yet developed appreciable Aβ load levels, soluble Aβ40 levels in both the frontal cortex (F(2,27)=8.78, p=0.0001) and hippocampus (F(5,27)=5.22, p=0.0026) were strongly increased in RI (p<0.01), but not VS mice, relative to NS mice. These data demonstrate that chronic RI stress, but not VS, exacerbates levels of Aβ in Tg2576 mice.
To investigate potential mechanisms by which RI stress exacerbates Aβ accumulation, we evaluated indices of APP production and proteolytic processing by western blot (Figure 1E). In 14 mo old Tg2576 mice, neither VS nor RI stress altered hippocampal levels of amyloid precursor protein (APP) detected with a C-terminal (5685) antibody (F(2,5)=8.01, p=0.53), an N-terminal (Karen) antibody (F(2,5)=2.05, p=0.83), or an antibody that detects secreted APP cleaved at the α-secretase site (sAPPα) (F(2,5)=1.88, p=0.30), suggesting that stress does not lead to higher APP production or non-amyloidogenic cleavage products. Further, neither VS nor RI stress altered hippocampal levels of beta-site APP-Cleaving enzyme (BACE) cleavage products of APP (F(2,5)=3.19, p=0.75) compared to a BACE knockout (KO) mouse as a negative control to demonstrate antibody specificity. This result suggests that stress does not induce altered APP processing at this site. Further, stress did not alter the levels of neprilysin, an Aβ degrading enzyme (data not shown).
Finally, to investigate the relationship between Aβ accumulation and HPA function, we measured serum CORT levels in response to an acute restraint after 1 mo of chronic stress. The 4 mo old, WT, naïve, NS mice displayed a striking but transient increase in CORT after a 15 min acute restraint stress which returns to baseline within 90 min (Figure 1F). However, young WT mice previously exposed to 1 mo of chronic VS or RI stress display a significantly blunted CORT response, characterized by smaller area under the curve (data not shown) as well as a smaller CORT peak and rise rate. The blunted CORT response to an acute stressor in rats with a history of chronic stress is consistent with previous reports (Watanabe et al., 1992; Magarinos and McEwen, 1995). In contrast, young Tg2576 mice displayed a significantly higher CORT peak than their WT littermates and the CORT responses were not blunted by previous exposure to VS or RI stress (Figure 1F). This effect was masked in 14 mo old mice, as all mice displayed a maximal CORT response regardless of treatment group or genotype (data not shown). This is consistent with previous studies demonstrating altered HPA responsiveness in aging rodents (Bao et al., 2008). Together, these data suggest that even a relatively low level of Aβ accumulation is associated with a heightened HPA sensitivity and a weakened ability to habituate to chronic stress. While the hypothalamus, the central controller of the CORT response, is generally spared from Aβ accumulation in AD, these data suggest that the regulation of hypothalamic function by the hippocampus and cortical regions may be disrupted in this mouse model.
To address the mechanism of increased Aβ in the absence of increased APP levels, we examined microglia activation to determine if chronic stress might suppress Aβ clearance through an alteration of microglia phagocytosis. We analyzed activated microglia levels by Iba1 IHC in the frontal cortex of 14 mo old Tg2576 and WT mice (Figure 2A–B). In WT mice, no change in Iba1 load in the frontal cortex was observed between different stress conditions (F(2,22)=6.47, p=0.81). In Tg2576 mice, while activated microglia were strongly associated with Thioflavin-S (+) Aβ plaques in NS mice and VS mice, RI mice demonstrated a strong trend towards a lower ratio of Thioflavin-S (+) to total Aβ plaque load (p=0.18). Interestingly, a determination of load values for Iba1 and nab228 in adjacent sections revealed that the ratio of activated microglia to Aβ deposits was significantly lower in RI mice, an effect driven by the increase in Aβ despite unchanged Iba1-IR levels in the same area (Figure 2B). Further, this effect was specific to Tg2576 mice with Aβ deposits as neither VS nor RI significantly altered Iba1 load values in PS19 mice (F(5,37)=9.44, p=0.47). These observations suggest a failure in RI mice to recruit and/or activate microglia at Aβ deposits.
Figure 2. Chronic stress impairs spatial and fear-related learning, but does not correlate with activated microglia-associated Aβ plaques in Tg2576 mice.
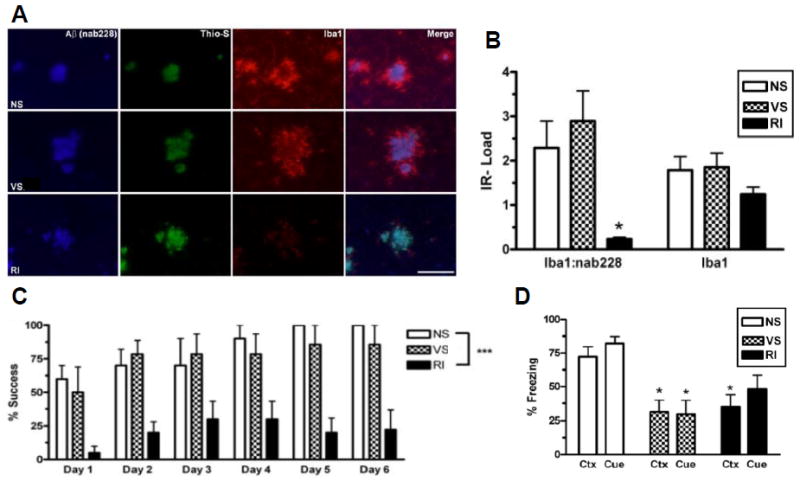
Tg2576 mice were assessed for activated microglia associated with Aβ plaques. (A) Representative images show that regardless of stress condition, all mice display Nab228-IR (+) Aβ plaques (blue) that are Thioflavin-S (+) (green) in the frontal cortex. However, RI mice showed significantly less Iba1-IR (+) activated microglia (red) around Aβ plaques without a decrease in overall Iba1-IR. (B) This effect is quantified by non-fluorescent immunoreactivity load ratios ± SEM of adjacent sections (F(2,21)=6.51, p=0.007) and (F(2,21)=6.51, p=0.007) (n=8/group). Fourteen mo old Tg2576 mice (n=10/group) were also tested on the Barnes maze (C) and fear conditioning (D). RI but not VS mice demonstrated significantly worse % success (F(3,35)=12.8, p<0.0001) compared to NS mice. In both context (F(2,20)=6.22, p=0.009) and cued (F(2,21)=5.98, p=0.01) fear conditioning, both VS and RI stressed Tg2576 mice froze significantly less than NS mice. Data show mean ± SEM. p*<0.05, **<0.01, ***<0.001 from NS. Scale bar = 100 μm.
To evaluate whether the stress models affect spatial memory and fear-related memory, WT and Tg2576 mice were tested on the Barnes maze and for both context and cued fear conditioning (Figure 2C–D). We observed a significant overall RI stress effect in the Barnes maze, as fourteen mo old RI stressed Tg2576 mice had a lower success rate (Figure 2C) and took significantly longer to find the target (increased latency) (F(5,35)=5.99, p=0.006) compared to NS mice, while VS mice displayed no spatial memory impairment compared to NS mice. Both VS and RI Tg2576 mice displayed a significant impairment (lower % freezing) on both context and cued fear conditioning compared to NS mice (Figure 2D). Importantly, these stress-induced differences were not observed in 14 mo old WT mice, as there was no effect of either stress on the Barnes maze success (F(2,17)=1.18, p=0.33) and latency measures (F(2,17)=1.84, p=0.19). Further, WT mice under different stress conditions were indistinguishable on both context (F(2,24)=5.86, p=0.48) and cued (F(2,24)=1.54, p=0.24) fear conditioning. These data suggest that both the stress-induced spatial and fear-associated memory deficits observed in RI Tg2576 mice are related to increased brain Aβ levels. Our data also suggest the possibility that VS differentially impairs fear memory through a pathway which occurs before overt Aβ deposition such as synaptic plasticity disruption.
Experiment 2: Effects of stress on hyperphosphorylated tau, aggregated tau inclusions, neurodegeneration and fear-related memory in PS19 mice
Having established a model of chronic stress capable of exacerbating AD-related neuropathology in Tg2576 mice, we sought to apply this paradigm to a previously characterized Tg mouse model in which tau hyperphosphorylation, pathological aggregated tau inclusions and neurodegeneration develop with age. A cohort of PS19 and WT littermate control mice were exposed to either NS, VS or RI stress (6hrs/day) for 1 mo and sacrificed at 6 mo of age (Figure 3A), at which point the PS19 normally begin to show modest tau pathology. No body weight differences were observed between treatment groups (F(5,56)=2.01, p=0.09) in WT mice, indicating that stress did not affect general health. To ensure that both VS and RI stress paradigms induced sufficient stress, adrenal gland weight was measured in WT mice and found to be stress-regulated (F(5,56)=4.15, p=0.003) and significantly greater after VS and RI relative to NS. Further, EPM performance in WT mice was measured as a reflection of anxiety and found to be stress-regulated (F(5,54)=6.11, p=0.0002) as both WT VS and RI mice demonstrated higher anxiety compared to WT NS mice (data not shown).
After 1 mo of stress, PS19 mice were assessed for levels of tau hyperphosphorylation both by IHC and western blot (Figure 3B–3D) as well as neurofibrillary tangle-like inclusions (Figure 3C). Figure 3B shows representative examples of AT8 (+)-IR in both the hippocampus (specifically in the mossy fibers, hilus and dentate gyrus) and frontal cortex. Quantification of AT8(+)-IR neurofibrillary tangle-like inclusions (inset) revealed that RI stressed mice displayed significantly more hyperphosphorylated tau (ptau) in the hippocampus by IHC, while VS stressed mice displayed no change (Figures 3B–3C). In addition, subregional quantification of AT8(+)-IR using a semi-quantitative rating scale (0–5) revealed that the most vulnerable hippocampal areas with the highest ptau scores were the mossy fibers (1.07 ± 0.2), hilus (0.93 ± 0.1) and dentate gyrus (0.43 ± 0.1), and the most vulnerable cortical area was layer 1 (1.17 ± 0.2). The subiculum (0.28 ± 0.1), CA1 (0.35 ± 0.1), CA2 0.26 ± 0.1 and CA3 (0.39 ± 0.1) and cortical layer 6 (0.07 ± 0.0) were rarely affected. However, this pattern persevered regardless of stress condition, suggesting that while subregions of the hippocampus and cortex are differentially vulnerable to tau pathology, chronic stress does not induce additional subregional vulnerability.
Further, levels of soluble and insoluble tau phosphorylated at Ser396 and Ser404 were assessed by PHF1 western blots. PHF1 levels were normalized to total tau, as determined with the 17025 antibody, and showed significantly higher levels in RI, but not VS, stressed mice compared to NS controls. However no change was observed in levels of a total tau when normalized to GAPDH (F(2,5)=1.42, p=0.37) (Figure 3D). These data demonstrate that chronic RI, but not VS stress, exacerbates both soluble and insoluble levels of pathologic ptau in PS19 mice, similar to the enhanced pathology induced by RI stress in Tg2576 mice.
To evaluate whether stress induced deficits in fear-related memory, both WT and PS19 mice were tested for both context and cued fear conditioning (Figure 3E). In both fear conditioning tests, significant overall group differences were observed. PS19 mice showed a statistically significant impairment in both context and cued fear conditioning after chronic RI, and the VS mice demonstrated a significant deficit in contextual fear conditioning, as well as a trend toward impaired performance in cue conditioning that did not reach statistical significance. In contrast, WT mice displayed only a non-significant trend towards impaired freezing behavior after either VS or RI stress in both context (F(2,28)=2.72, p=0.25) and cued (F(2,26)=1.52, p=0.09) fear conditioning. These data suggest that the decreased cognitive performance in the RI stressed PS19 mice is associated with the exacerbated tau pathology observed in these animals.
To evaluate the effects of chronic stress on neurodegeneration, we completed a semi-quantitative analysis of hippocampal degeneration by NeuN immunohistochemistry in the PS19 mice and also measured hemi-hippocampal weight in WT and PS19 mice. We observed that RI, but not VS chronic stress induced cell loss in the hippocampus of PS19 mice compared to NS mice (Figure 4A), and these were statistically significant results when analyzed using a semi-quantitative rating scale of degeneration (Figure 4B). Further, hemi-hippocampal weights were measured and found to be significantly lower in PS19 RI mice compared to NS or VS PS19 or WT mice (Figure 4C), suggesting that PS19 RI mice are susceptible to neurodegeneration.
Figure 4. Chronic RI stress induces neurodegeneration, altered glucocorticoid receptor (GR) expression and altered corticosterone response in PS19 mice.
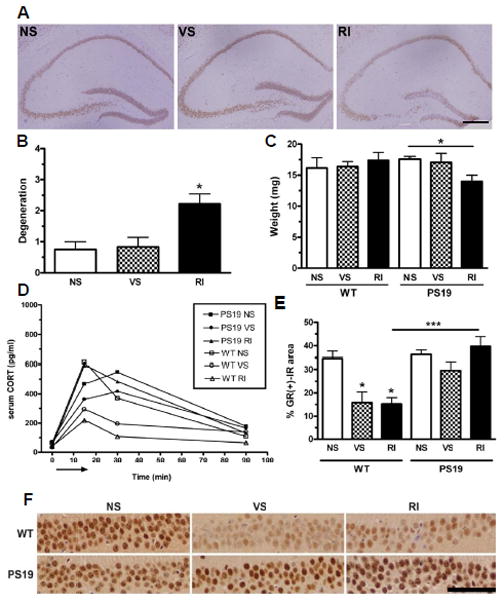
PS19 mice were evaluated for hippocampal neurodegeneration. (A) Representative pictomicrographs of NeuN-IR (+) show that PS19 RI mice displayed significantly higher degeneration as quantified in (B) (F(2,18)=6.89, p=0.007). (C) Hemi-hippocampal weight assessment also revealed that PS19 (but not WT) RI mice displayed lower hippocampal weight (F(2,21)=3.62, p=0.04) compared to VS or NS mice. Data show mean ± SEM (n=8/group). p*<0.05 from NS. Scale bar= 0.5mm. (D) CORT levels were plotted over time after an acute 15 min restraint stress (arrow). In WT mice, both VS (F(2,18)=24.7, p<0.001) and RI (F(2,18)=24.7, p<0.001) stress decreased the area under the curve compared to NS. However, CORT levels were elevated significantly in all conditions of PS19 mice, regardless of stress treatment. Data show mean ± SEM (n=10/group). (F) These mice were also assessed for GR-IR in the CA1. Representative pictomicrographs of both WT and PS19 mice under different stress conditions show that VS and RI significantly reduced GR levels in WT but not PS19 mice. (E) This effect was quantified to demonstrate a significant group effect (F(5,25)=11.6, p<0.0001). Data show mean GR-IR load ± SEM. p*<0.05, **<0.01 and ***<0.001. Scale bar = 50 μm.
To investigate the relationship between tau hyperphosphorylation and HPA function, we measured serum CORT levels in response to an acute stress after 1 mo of chronic stress. Consistent with our findings in Experiment 1, WT NS mice again displayed a striking but transient increase in CORT after a 15 min acute restraint stress which returns to baseline within 90 min (Figure 4D). However, WT mice previously exposed to 1 mo of chronic VS or RI stress display a significantly blunted CORT response, characterized by smaller area under the curve (Figure 4D). In contrast, NS and VS PS19 mice displayed a somewhat delayed CORT peak relative to their WT littermates, and the CORT response in the PS19 mice appeared to be independent of their previous stress exposure (Figure 4D). These data suggest that tau hyperphosphorylation alters HPA sensitivity and weakens the ability for PS19 mice to habituate to chronic stress.
To further investigate the effect of stress in the brain, we analyzed the level of glucocorticoid receptor (GR), the most prominent corticosterone receptor, in the hippocampus of WT and PS19 mice by IHC (Figure 4E–F). This brain region is particularly vulnerable to tau pathology and we observed interesting differences in GR expression. In WT mice, both chronic VS and RI stress cause a significant decrease in GR (+)-IR in the CA1 compared to NS naïve WT mice (Figure 4F), which is consistent with the implication that WT mice are able to habituate to chronic stress and display a blunted HPA response when challenged with an acute stress. In contrast, in PS19 mice both VS and RI mice maintained a high level of GR (+)-IR in the hippocampus. This suggests that GR receptors are unable to habituate to chronic stress in the PS19 mice and thus raises the possibility that excessive signaling through the GR may be involved in stress-induced enhancement of tau hyperphosphorylation.
Experiment 3: CORT involvement in stress-induced tau hyperphosphorylation in PS19 mice
Based on the glucocorticoid hypothesis of brain aging, which proposes that chronic exposure to stress and/or glucocorticoids promotes hippocampal aging and even AD pathogenesis (Landfield et al., 2007), we sought to mimic RI stress-induced changes on tau phosphorylation using direct CORT administration. Non-stressed, male PS19 mice at 8 mo of age were evaluated for changes in ptau after 1 mo of chronic CORT administration (Figure 5A). A separate group of control mice received subcutaneous pellets containing vehicle only. In a small pilot study, CORT treatment induced a significant spike in serum CORT levels, suggesting that the pellets released an initial bolus of corticosterone into the bloodstream. However, 72 hrs post-implantation, mice still maintained a CORT level which was higher than a pre-stress, baseline CORT measurement in PS19 mice (~50pg/ml) (Figure 5B). This treatment did not cause any significant changes in body weight (F(5,5)=2.6, p=0.93), suggesting that it was well-tolerated. However, PS19 mice treated with chronic CORT were indistinguishable from vehicle-treated mice with regard to the level of AT8 (+)-IR in the hippocampus (Figure 5C–D). Further, as measured by immunoblotting, neither PHF1 levels (F(5,8)=1.78, p=0.47) nor total tau levels (F(2,8)=4.31, p=0.17) were altered by CORT administration (Figure 5E). In addition, chronic direct administration of the synthetic glucocorticoid, dexamethasone (5mg/kg), also had no effect on tau hyperphosphorylation, as measured by PHF1 western blot analysis (data not shown). Taken together, these data suggest that CORT is not the crucial stress hormone mediating the stress-induced changes in tau hyperphosphorylation observed in this study. While this finding is surprising in light of prior literature supporting the glucocorticoid hypothesis of brain aging, it is consistent with emerging evidence that CORT does not mediate stress-induced changes in either Aβ (Kang et al., 2007) or ptau (Rissman et al., 2007).
Figure 5. CORT treatment does not mimic chronic stress-induced tau hyperphosphorylation in PS19 mice.
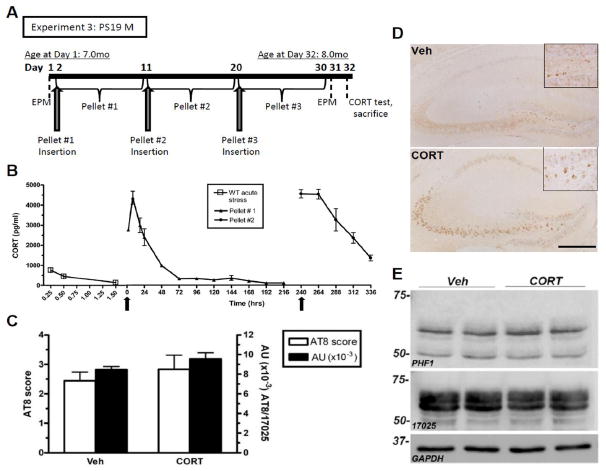
(A) Experimental design of Experiment 3; PS19 male mice were not stressed but were administered CORT pellets (n=8/group). (B) First, a small pilot (n=3/group) of adult WT male mice were implanted with CORT pellets (arrows) to assess the plasma hormone levels achieved by these pellets. A high spike in CORT within the first 72 hrs was significantly higher (F(2,7)=2.25, p<0.0001) than after an acute stress from Experiment 1 (data also shown in Figure 4A). Further, neither representative images of AT8-IR in the hippocampus (C) nor IR quantification using a semi-quantitative rating scale (Stage I–VI) (D) showed a treatment difference. Data shows mean AT8 score ± SEM. (E) A representative western blot probed for both PHF1, 17025 and GAPDH as a loading control also demonstrate no difference after CORT treatment in the hippocampus. Scale bar = 0.5mm.
Experiment 4: CRF1 mediates stress-induced tau pathological inclusions, neurodegeneration and fear-associated learning
Based on our results suggesting that the mechanism behind stress-induced increases in ptau may be CORT-independent and previous studies suggesting involvement of CRF (Kang et al., 2007; Rissman et al., 2007), we sought to determine the role of CRF1 in stress-induced tau phosphorylation. Therefore, the selective CRF1 antagonist, NBI 27914 (NBI), was administered before stress in an effort to block tau phosphorylation. PS19 mice were evaluated after 1 mo treatment with either NS, 3 hrs/day of RI stress, or RI stress after treatment with NBI, as depicted in Figure 6A. Examination of brain and plasma drug concentrations following sc injections of 10 mg/kg NBI revealed that effective concentrations of the drug reached the brain at levels that exceeded the reported binding constant to the CRF1 receptor for several hrs (Figure 6B). This dose demonstrated high drug levels in brain and a high but transient plasma drug level which was cleared within 2–4 hrs. After chronic administration, the NBI and vehicle treatment groups did not display significant differences in body weight (F(2,23)=2.78, p=0.08), suggesting that the drug was well-tolerated. Further, the experimental animals receiving 1 mo of chronic NBI treatment displayed high brain NBI levels (66.2 ± 9.5 nM) at the time of sacrifice, which was 3 d after the last injection. This suggests that relatively high brain concentrations of NBI were maintained throughout the study period. Consistent with our result in Figure 3, vehicle-treated RI mice displayed significantly more neurofibrillary tangle-like inclusions and higher AT8 (+)-IR levels in the hippocampus compared to NS mice (Figure 6C), as quantified in Figure 6D. Further, vehicle-treated RI mice displayed both significantly higher soluble and insoluble PHF1 levels when normalized to total tau (17025 antibody), although total tau levels were unchanged when normalized to GAPDH (F(3,10)=12.9, p=0.3) (Figure 6E). Lastly, consistent with our results in Figure 3, RI stress significantly impaired both contextual and cued fear-associated memory (Figure 6F). Importantly, the pre-stress injection of a CRF1 antagonist NBI significantly prevented the stress-induced impairment in fear-associated memory, AT8 (+) tau inclusions and ptau (p<0.05). These effects of NBI suggest that CRF acting at CRF1 is a crucial mediator of the stress-induced exacerbation of AD-related ptau that leads to impairment of fear conditioning.
To evaluate the involvement of CRF1 in mediating neurodegeneration, we completed a semi-quantitative analysis of hippocampal degeneration by NeuN immunohistochemistry and also measured hemi-hippocampal weight. Similar to our results in Figure 3, we observed that RI induced cell loss in the hippocampus (Figure 6G) and hemi-hippocampal weight (Figure 6H) compared to NS mice, while NBI treatment significantly prevented this loss. Together, these results suggest that CRF1 is the primary mediator of stress-induced neurodegeneration in PS19 mice.
Lastly, to further confirm our hypothesis that CRF is integrally involved in stress-mediated elevation of tau phosphorylation, we utilized CRF-OE mice that have a chronic overproduction of CRF driven by the murine metallothionine 1 promoter and which display Cushing’s disease-like features, as well as increased anxiety and stress sensitivity (Stenzel-Poore et al., 1992). The levels of phosphorylated PHF1 tau were quantified in the hippocampus of 10 mo old CRF-OE mice and littermate controls, and we observed a substantial elevation in PHF1 levels in CRF-OE mice compared to WT littermates without a significant change in total tau levels (assessed with Tau 1 and T49 antibodies) when normalized to GAPDH (F(2,4)=4.39, p=0.5) (Figure 6I). While these mice do show altered CORT levels (Stenzel-Poore et al., 1992), our data above suggest that stress-induced change in tau phosphorylation is CORT-independent, but CRF-dependent. Together, these results suggest that enhanced CRF signaling is likely responsible for stress-induced exacerbation of tau phosphorylation.
Discussion
We sought to apply a paradigm of chronic stress, capable of exacerbating Aβ accumulation in the Tg2576 mouse model of Aβ plaque pathology, to a tau Tg mouse model in order to investigate the interaction between stress and disease-relevant tau pathology. Our data demonstrate that chronic RI stress worsens memory in both Tg2576 and PS19 mice, exacerbates Aβ accumulation and suppresses microglial activation in the Tg2576 mice, and increases tau phosphorylation, neurodegeneration and AT8 (+) tau inclusions in the PS19 mice. Further, both Aβ and tau pathology alter the HPA profile. Importantly, CORT administration did not mimic the stress-induced promotion of tau pathology. However, CRF1 antagonism blocked tau pathology development, neurodegeneration and learning impairments, while CRF-OE enhanced this effect, thereby implicating CRF signaling in mediating stress-induced tau pathology. These results represent a very comprehensive description of stress-induced changes in both Aβ and tau pathology in vivo, and also implicate microglia for the first time in stress-induced augmentation of Aβ levels. This is the first report of chronic stress-induced changes in pathological tau hyperphosphorylation and tau inclusions in a mouse that over-expresses a disease-relevant tau mutation.
That chronic stress induces Aβ accumulation has been well-established in a large body of research. Our results are consistent with these analogous findings after similar paradigms of restraint and/or isolation in APPV717I-CT100 (Jeong et al., 2006), Tg2576 (Lee et al., 2009), APPxPS1 (Huang et al., 2011) and 5XFAD mice (Devi et al., 2010). Also, stress increased Aβ in brain interstitial fluid, possibly through a CRF1 but not CORT-dependent mechanism (Kang et al., 2007). However, our report and that by others of CORT-independence are inconsistent with GR-dependent effects in 3xTg-AD mice (Green et al., 2006). This discrepancy may be dependent on the different mouse models (Tg2576 and PS19 vs. 3xTg-AD), different methods of ptau evaluation, different hormones (natural vs. synthetic) and modes of delivery (i.p. injections vs. hormone pellets). However, it is possible that glucocorticoids still play an important role in this process. Corticosterone has been shown to exacerbate kainate-induced hippocampal ptau, neuron loss and cytoskeletal pathology in vivo (Elliott et. al., 1993; Stein-Behrens et al., 1994). It would be informative to investigate the AD-related pathological consequences of excess glucocorticoid production in patients with endocrine disorders such as Cushing’s disease (Guldiken and Guldiken, 2008) to further understand the relationship between glucocorticoids and AD.
Most importantly, our data strengthen the emerging hypothesis that stress induces tau pathology through a CRF1-dependent but CORT-independent mechanism (Dong and Csernansky, 2009). Our results suggest that stress-induced pathologic tau inclusion development, insoluble tau levels, neurodegeneration and learning impairments are mediated by CRF1. Our findings also build on those from Sawchenko’s group that stress increases endogenous mouse tau phosphorylation in WT mice, possibly through CRF1 (Rissman et al., 2007). However, others have demonstrated that CRF offers neuroprotective effects which are relevant to AD (reviewed in Bayatti and Behl, 2005). In particular, known CRFR agonists were shown to be protective against cell death induced by Aβ, lipid peroxidation or excitotoxic glutamate in primary neuronal culture (Pederson et al., 2001; Pederson et al., 2002). However, these seemingly inconsistent results were obtained in vitro and represent a very different environment than a long-term, chronic stress paradigm in vivo. Acute and chronic stress and/or CRF exposure may have different effects on tau phosphorylation. Interestingly, we found that a single exposure to acute stress (15 minute restraint) decreased tau phosphorylation in PS19 mice as assessed by AT8 (+) immunohistochemistry and PHF1 biochemistry (Carroll and Trojanowski, unpublished observations), supporting the differential effects of acute vs. chronic stress on tau phosphorylation.
Based on the known downstream signaling cascades associated with CRF1, several downstream targets may potentially link CRF to tau hyperphosphorylation. Agonist binding to CRF1 initiates the adenylyl cyclase-cAMP signaling pathway, PKA-dependent protein phosphorylation and CREB-dependent transcription regulation, as well as other signaling pathways such as PKC, PKB/Akt, ERK and p38 MAPK (Hauger et al., 2009). One important downstream target that has been directly linked to tau hyperphosphorylation is glycogen synthase kinase (GSK)-3β, a major protein kinase thought to be responsible for phosphorylating tau on various disease-specific sites (Avila et al., 2010). In cultured neurons, CRF1 activation initiates CREB signaling which leads to phosphorylation of GSK3β (Bayatti et al., 2003; Facci et al., 2003). Recently, this link has also been reported in vivo, as stress induces GSK3β activation through phosphorylation at Tyr216 and increases ptau in wild type mice (Rissman et al., 2007). Nonetheless, other tau kinases, such as Fyn and Lck of the Src-kinase family (Scales et al., 2011), as well as various phosphatases such as protein phosphatase 2B (Qian et al., 2010), are also interesting candidates that may be linked to CRF1 signaling and are planned targets of investigation by our laboratory. These studies will guide future efforts by which the kinases or signaling molecules can be pharmacologically manipulated in an effort to attenuate stress-induced tau phosphorylation.
Our data also suggest the possibility that stress alters microglial activation and perhaps the ability of these cells to clear Aβ accumulation by phagocytosis. The link between AD, Aβ and activation of the brain immune response, with the release of pro-inflammatory cytokines and chemokines, is well documented (Farfara et al., 2008; Benzing et al., 1999). Our findings are also consistent with known stress-induced disruption of neuron-microglia crosstalk (Biber et al., 2007), which can lead to an aberrant brain immune response (Jurgens and Johnson, 2010). Both aging and stress can induce deleterious microglia morphology and physiology alterations (Neumann, 2001; Pocock and Kettenmann, 2007). Further, aged and diseased brains are more sensitive to stress effects and seem unable to adapt to the continued presence of a stressor (Buchanan et al., 2008). While these conditions are usually associated with excessive microglia activation, it is also possible that aging, disease and/or stress may suppress microglia activation depending on concentration, timing and duration (acute vs. chronic) of the stressor (Glezer and Rivest, 2004; Sorrells and Sapolsky, 2007; Dhabhar, 2009), a phenomenon that has been described in human brains (De la Fuente and Gimenez-Llort, 2010). Future studies designed to measure levels of cytokines such as interleukin IL-1β, IL-6 and tumor necrosis factor-α, as well as M1 and M2 macrophage markers, would help clarify these issues.
One very interesting observation from this study was the differential effects of various stressors. While RI had detrimental effects on spatial and fear memory, Aβ and tau, VS only impaired fear memory, presumably through an Aβ-independent mechanism or a process which occurs before overt Aβ deposition such as synaptic plasticity dysfunction. While both paradigms succeeded in invoking stress, they differed greatly in the amount of required physical activity. VS stress induced higher physical activity, either directly or indirectly, in the majority of its six stressors, while RI stress by definition reduced physical activity. It is possible that the VS induced an increase in exercise or environmental enrichment, both of which have consistently been shown to reduce indices of neuropathology in Tg mice (Radak et al., 2010; Nithianantharajah and Hannan, 2006). The differential effects of both stressors are also consistent with the hypothesis that the brain categorizes stressors and signals category-specific responses (Li et al., 1996; Herman and Cullinan, 1997; Sawchenko et al., 2000; Dayas et al., 2001). While the differential pathways are not completely understood, this observation raises interesting questions regarding the different molecular mechanisms that link stress-induced AD risk in patients with psychological, emotional or physical stress.
One limitation of this study is gender variation. Female Tg2576 and male PS19 mice were chosen for their higher and more consistent Aβ (Callahan et al., 2001) and tau pathology burden, respectively (Yoshiyama and Lee, unpublished observations). However, it has been well documented that gender differences exist in several important stress-sensitive indices of hippocampal plasticity such as dendrite remodeling, neurogenesis and synapse formation (McEwen, 2002). It is possible that the two stress paradigms used in this study could have different effects on AD-related neuropathology in different genders. While such investigation is beyond the scope of this study, appreciating gender differences in stress-induced AD-related pathology is important to understanding the link between stress, gender and risk for neurodegenerative disorders.
Finally, these data raise an interesting link between AD, stress-related psychiatric disorders, endocrine disorders and gender. Both AD and several stress disorders are characterized by HPA dysfunction (Holsboer and Ising, 2008; Pervanidou and Chrousos, 2010) (Swaab et al., 2005). Increased CRF release or function has been implicated in many stress-related psychiatric disorders, such as depression (Gold and Chrousos, 2002). Further, neither risk for stress-related psychiatric disorders nor neurodegenerative diseases is gender-neutral (Bachman et al., 1992; Ruitenberg et al., 2001; Cahill, 2006; Figueira and Ouakinin, 2010). Therefore, it is possible that stress differentially influences disease risk depending on gender. In this light, given the emerging potential role of CRF in Aβ aggregation and tau pathology, increased CRF in stress-related psychiatric disorders would be predicted to predispose to AD. Understanding this emerging link between CRF, AD-related pathology and stress is of crucial importance in future treatments of not only neurodegenerative diseases but also psychiatric disorders.
Acknowledgments
NIH grant PO1 AG17586 (V.M.-Y.L.), MH40008 (R.J.V.) and UPenn Comprehensive Neuroscience Center grant (R.J.V. and V.M.-Y.L.). Also, the authors would like to thank R. Vassar for the generous BACE antibody gift, T. Bale for expert advice on CORT measurements and K. Siniakowicz for technical assistance.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
References
- Alfarez DN, Joels M, Krugers HJ. Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci. 2003;17:1928–1934. doi: 10.1046/j.1460-9568.2003.02622.x. [DOI] [PubMed] [Google Scholar]
- Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila J, Wandosell F, Hernandez F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother. 2010;10:703–710. doi: 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- Aznar S, Knudsen GM. Depression and Alzheimer’s disease: is stress the initiating factor in a common neuropathological cascade? J Alzheimers Dis. 2011;23:177–193. doi: 10.3233/JAD-2010-100390. [DOI] [PubMed] [Google Scholar]
- Bachman DL, Wolf PA, Linn R, Knoefel JE, Cobb J, Belanger A, D’Agostino RB, White LR. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology. 1992;42:115–119. doi: 10.1212/wnl.42.1.115. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Bao AM, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev. 2008;57:531–553. doi: 10.1016/j.brainresrev.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Bayatti N, Behl C. The neuroprotective actions of CRH. Ageing Res Rev. 2005;4(2):258–70. doi: 10.1016/j.arr.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Bayatti N, Zschocke J, Behl C. Brain region-specific neuroprotective action and signaling of corticotropin-releasing hormone in primary neurons. Endocrinology. 2003;144:4051–4060. doi: 10.1210/en.2003-0168. [DOI] [PubMed] [Google Scholar]
- Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20(6):581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Briski K, Gillen E. Differential distribution of Fos expression within the male rat preoptic area and hypothalamus in response to physical vs. psychological stress. Brain Res Bull. 2001;55:401–408. doi: 10.1016/s0361-9230(01)00532-9. [DOI] [PubMed] [Google Scholar]
- Brunden KR, Yao Y, Potuzak JS, Ferrer NI, Ballatore C, James MJ, Hogan AM, Trojanowski JQ, Smith AB, 3rd, Lee VM. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol Res. 2011;63:341–351. doi: 10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB, 3rd, Lee VMY, Trojanowski JQ. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan JB, Sparkman NL, Chen J, Johnson RW. Cognitive and neuroinflammatory consequences of mild repeated stress are exacerbated in aged mice. Psychoneuroendocrinology. 2008;33:755–765. doi: 10.1016/j.psyneuen.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill L. Why sex matters for neuroscience. Nat Rev Neurosci. 2006;7:477–484. doi: 10.1038/nrn1909. [DOI] [PubMed] [Google Scholar]
- Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol. 2001;158:1173–1177. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Carroll JC, Pike CJ. Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice. Endocrinology. 2008;149:2607–2611. doi: 10.1210/en.2007-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JC, Rosario ER, Villamagna A, Pike CJ. Continuous and cyclic progesterone differentially interact with estradiol in the regulation of Alzheimer-like pathology in female 3xTransgenic-Alzheimer’s disease mice. Endocrinology. 2010;151:2713–2722. doi: 10.1210/en.2009-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007;27:13357–13365. doi: 10.1523/JNEUROSCI.2718-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 2001;14:1143–1152. doi: 10.1046/j.0953-816x.2001.01733.x. [DOI] [PubMed] [Google Scholar]
- De la Fuente M, Gimenez-Llort L. Models of aging of neuroimmunomodulation: strategies for its improvement. Neuroimmunomodulation. 2010;17:213–216. doi: 10.1159/000258727. [DOI] [PubMed] [Google Scholar]
- Devi L, Alldred MJ, Ginsberg SD, Ohno M. Sex- and brain region-specific acceleration of beta-amyloidogenesis following behavioral stress in a mouse model of Alzheimer’s disease. Mol Brain. 2010;3:34. doi: 10.1186/1756-6606-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16:300–317. doi: 10.1159/000216188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Csernansky JG. Effects of stress and stress hormones on amyloid-beta protein and plaque deposition. J Alzheimers Dis. 2009;18:459–469. doi: 10.3233/JAD-2009-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott EM, Mattson MP, Vanderklish P, Lynch G, Chang I, Sapolsky RM. Corticosterone exacerbates kainate-induced alterations in hippocampal tau immunoreactivity and spectrin proteolysis in vivo. J Neurochem. 1993;61(1):57–67. doi: 10.1111/j.1471-4159.1993.tb03537.x. [DOI] [PubMed] [Google Scholar]
- Facci L, Stevens DA, Pangallo M, Franceschini D, Skaper SD, Strijbos PJ. Corticotropin-releasing factor (CRF) and related peptides confer neuroprotection via type 1 CRF receptors. Neuropharmacology. 2003;45:623–636. doi: 10.1016/s0028-3908(03)00211-9. [DOI] [PubMed] [Google Scholar]
- Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease. J Cell Mol Med. 2008;12:762–780. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Cheng B, Yang R, Sun FY, Zhu CQ. Dynamic changes of phosphorylated tau in mouse hippocampus after cold water stress. Neurosci Lett. 2005;388:13–16. doi: 10.1016/j.neulet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Figueira ML, Ouakinin S. Gender-related endocrinological dysfunction and mental disorders. Curr Opin Psychiatry. 2010;23:369–372. doi: 10.1097/YCO.0b013e3283399b86. [DOI] [PubMed] [Google Scholar]
- Glezer I, Rivest S. Glucocorticoids: protectors of the brain during innate immune responses. Neuroscientist. 2004;10:538–552. doi: 10.1177/1073858404263494. [DOI] [PubMed] [Google Scholar]
- Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psych. 2002;7:254–275. doi: 10.1038/sj.mp.4001032. [DOI] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldiken S, Guldiken B. Subclinical Cushing’s syndrome is a potential cause of metabolic dementia and rapidly progressive Alzheimer-type dementia. Med Hypotheses. 2008;71:703–5. doi: 10.1016/j.mehy.2008.05.036. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM. Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci. 2009;1179:120–143. doi: 10.1111/j.1749-6632.2009.05011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Henneicke H, Street J, Modzelewski J, Kalak R, Buttgereit F, Dunstan CR, Zhou H, Seibel MJ. The challenge of continuous exogenous glucocorticoid administration in mice. Steroids. 2009;74:245–249. doi: 10.1016/j.steroids.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- Hoare SR, Sullivan SK, Schwarz DA, Ling N, Vale WW, Crowe PD, Grigoriadis DE. Ligand affinity for amino-terminal and juxtamembrane domains of the corticotropin releasing factor type I receptor: regulation by G-protein and nonpeptide antagonists. Biochemistry. 2004;43:3996–4011. doi: 10.1021/bi036110a. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Ising M. Central CRH system in depression and anxiety--evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol. 2008;583:350–357. doi: 10.1016/j.ejphar.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Huang HJ, Liang KC, Ke HC, Chang YY, Hsieh-Li HM. Long-term social isolation exacerbates the impairment of spatial working memory in APP/PS1 transgenic mice. Brain Res. 2011;1371:150–160. doi: 10.1016/j.brainres.2010.11.043. [DOI] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Iba M, Aboagye AK, Paul SM, Trojanowski JQ, Lee VMY. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol. 2010;177:1977–1988. doi: 10.2353/ajpath.2010.100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Tsuchiya K, Koyama T. Regional changes in dopamine and serotonin activation with various intensity of physical and psychological stress in the rat brain. Pharmacol Biochem Behav. 1994;49:911–920. doi: 10.1016/0091-3057(94)90243-7. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Park CH, Yoo J, Shin KY, Ahn SM, Kim HS, Lee SH, Emson PC, Suh YH. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer’s disease model. Faseb J. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Jurgens HA, Johnson RW. Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp Neurol. 2010 doi: 10.1016/j.expneurol.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci U S A. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Korneyev A, Binder L, Bernardis J. Rapid reversible phosphorylation of rat brain tau proteins in response to cold water stress. Neurosci Lett. 1995;191:19–22. doi: 10.1016/0304-3940(95)11546-3. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Blalock EM, Chen KC, Porter NM. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer’s disease. Curr Alzheimer Res. 2007;4:205–212. doi: 10.2174/156720507780362083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BK, Glass TA, Wand GS, McAtee MJ, Bandeen-Roche K, Bolla KI, Schwartz BS. Apolipoprotein e genotype, cortisol, and cognitive function in community-dwelling older adults. Am J Psychiatry. 2008;165:1456–1464. doi: 10.1176/appi.ajp.2008.07091532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VMY. Secretion and intracellular generation of truncated Abeta in beta-site amyloid-beta precursor protein-cleaving enzyme expressing human neurons. J Biol Chem. 2003;278:4458–4466. doi: 10.1074/jbc.M210105200. [DOI] [PubMed] [Google Scholar]
- Lee KW, Kim JB, Seo JS, Kim TK, Im JY, Baek IS, Kim KS, Lee JK, Han PL. Behavioral stress accelerates plaque pathogenesis in the brain of Tg2576 mice via generation of metabolic oxidative stress. J Neurochem. 2009;108:165–175. doi: 10.1111/j.1471-4159.2008.05769.x. [DOI] [PubMed] [Google Scholar]
- Li HY, Ericsson A, Sawchenko PE. Distinct mechanisms underlie activation of hypothalamic neurosecretory neurons and their medullary catecholaminergic afferents in categorically different stress paradigms. Proc Natl Acad Sci U S A. 1996;93:2359–2364. doi: 10.1073/pnas.93.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995;69:83–88. doi: 10.1016/0306-4522(95)00256-i. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging. 2002;23:921–939. doi: 10.1016/s0197-4580(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Nakamura M, McIntosh TK, Rodriguez A, Berlin JA, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Schmidt ML, Lee VM, Trojanowski JQ. Traumatic brain injury in young, amyloid-beta peptide overexpressing transgenic mice induces marked ipsilateral hippocampal atrophy and diminished Abeta deposition during aging. J Comp Neurol. 1999;411:390–398. [PubMed] [Google Scholar]
- Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7(9):697–709. doi: 10.1038/nrn1970. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC. Heat shock induces rapid dephosphorylation of tau in both female and male rats followed by hyperphosphorylation only in female rats: implications for Alzheimer’s disease. J Neurochem. 1996;66:1140–1149. doi: 10.1046/j.1471-4159.1996.66031140.x. [DOI] [PubMed] [Google Scholar]
- Patil SS, Sunyer B, Hoger H, Lubec G. Evaluation of spatial memory of C57BL/6J and CD1 mice in the Barnes maze, the Multiple T-maze and in the Morris water maze. Behav Brain Res. 2009;198:58–68. doi: 10.1016/j.bbr.2008.10.029. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McCullers D, Culmsee C, Haughey NJ, Herman JP, Mattson MP. Corticotropin-releasing hormone protects neurons against insults relevant to the pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2001;8(3):492–503. doi: 10.1006/nbdi.2001.0395. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Wan R, Zhang P, Mattson MP. Urocortin, but not urocortin II, protects cultured hippocampal neurons from oxidative and excitotoxic cell death via corticotropin-releasing hormone receptor type I. J Neurosci. 2002;15;22(2):404–12. doi: 10.1523/JNEUROSCI.22-02-00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervanidou P, Chrousos GP. Neuroendocrinology of post-traumatic stress disorder. Prog Brain Res. 2010;182:149–160. doi: 10.1016/S0079-6123(10)82005-9. [DOI] [PubMed] [Google Scholar]
- Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Qian W, Yin X, Hu W, Shi J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX, Liu F. Activation ofProtein Phosphatase 2B and Hyperphosphorylation of Tau in Alzheimer’s Disease. J Alzheimers Dis. 2010 doi: 10.3233/JAD-2010-100987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak Z, Hart N, Sarga L, Koltai E, Atalay M, Ohno H, Boldogh I. Exercise plays a preventive role against Alzheimer’s disease. J Alzheimers Dis. 2010;20:777–783. doi: 10.3233/JAD-2010-091531. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Lee KF, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27:6552–6562. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Mattson MP. Adverse stress, hippocampal networks, and Alzheimer’s disease. Neuromolecular Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruitenberg A, Ott A, van Swieten JC, Hofman A, Breteler MM. Incidence of dementia: does gender make a difference? Neurobiol Aging. 2001;22:575–580. doi: 10.1016/s0197-4580(01)00231-7. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. 1985;5:1222–1227. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE, Li HY, Ericsson A. Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog Brain Res. 2000;122:61–78. doi: 10.1016/s0079-6123(08)62131-7. [DOI] [PubMed] [Google Scholar]
- Scales TM, Derkinderen P, Leung KY, Byers HL, Ward MA, Price C, Bird IN, Perera T, Kellie S, Williamson R, Anderton BH, Reynolds CH. Tyrosine phosphorylation of tau by the SRC family kinases lck and fyn. Mol Neurodegener. 2011;6:12. doi: 10.1186/1750-1326-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21:259–272. doi: 10.1016/j.bbi.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanojevic S, Mitic K, Vujic V, Kovacevic-Jovanovic V, Dimitrijevic M. Exposure to acute physical and psychological stress alters the response of rat macrophages to corticosterone, neuropeptide Y and beta-endorphin. Stress. 2007;10:65–73. doi: 10.1080/10253890601181289. [DOI] [PubMed] [Google Scholar]
- Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 1994;14(9):5373–80. doi: 10.1523/JNEUROSCI.14-09-05373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel A, Tache Y. Corticotropin-releasing factor signaling and visceral response to stress. Exp Biol Med (Maywood) 2010;235:1168–1178. doi: 10.1258/ebm.2010.009347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Cameron VA, Vaughan J, Sawchenko PE, Vale W. Development of Cushing’s syndrome in corticotropin-releasing factor transgenic mice. Endocrinology. 1992;130:3378–3386. doi: 10.1210/endo.130.6.1597149. [DOI] [PubMed] [Google Scholar]
- Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. 2005;4:141–194. doi: 10.1016/j.arr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Teegarden SL, Bale TL. Effects of stress on dietary preference and intake are dependent on access and stress sensitivity. Physiol Behav. 2008;93:713–723. doi: 10.1016/j.physbeh.2007.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Gould E, McEwen BS. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992;588:341–345. doi: 10.1016/0006-8993(92)91597-8. [DOI] [PubMed] [Google Scholar]
- Wellman CL, Izquierdo A, Garrett JE, Martin KP, Carroll J, Millstein R, Lesch KP, Murphy DL, Holmes A. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci. 2007;27:684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27:143–153. doi: 10.1159/000095761. [DOI] [PubMed] [Google Scholar]
- Yan J, Sun XB, Wang HQ, Zhao H, Zhao XY, Xu YX, Guo JC, Zhu CQ. Chronic restraint stress alters the expression and distribution of phosphorylated tau and MAP2 in cortex and hippocampus of rat brain. Brain Res. 2010;6:1347–41. doi: 10.1016/j.brainres.2010.05.074. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Planel E, Ishiguro K, Fujita SC. Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer’s disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VMY. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang LF, Shi L, Liu H, Meng FT, Liu YJ, Wu HM, Du X, Zhou JN. Increased hippocampal tau phosphorylation and axonal mitochondrial transport in a mouse model of chronic stress. Int J Neuropsychopharm. 2011;22:1–12. doi: 10.1017/S1461145711000411. [DOI] [PubMed] [Google Scholar]