

Abstract
Quantifying antigens in formalin-fixed tissue is challenging and limits investigation in population-based studies of brain aging. To address this major limitation, we have developed a new technique that we call “Histelide”: immunohistochemistry (HIST-) and ELISA (-EL-) performed on a glass slide (-IDE). We validated Histelide in sections of prefrontal cortex from 20 selected cases: 12 subjects with clinically and neuropathologically diagnosed Alzheimer’s disease (AD), either autosomal dominant or late-onset forms, and 8 clinical and neuropathologic Controls. AD cases had significantly increased amyloid beta (Aβ) peptide and paired helical filament– (PHF-) tau per area of neocortex that was proteinase K-sensitive, and significantly decreased amount of synaptophysin. We next investigated prefrontal cortex from 81 consecutive cases of high cognitive performers from the Adult Changes in Thought (ACT) study, a population-based study of brain aging and incident dementia. As expected, latent AD was common in this group; however, our results quantified widely individually-varying levels of Aβ peptides and PHF-tau among these high cognitive performers. This novel approach obtains quantitative data from population-based studies, and our initial studies with high cognitive performers provide important quantitative insights into latent AD that should help guide expectations from neuroimaging and prevention studies.
INTRODUCTION
First described by Dr. Alzheimer, the histologic hallmarks of the disease that bears his name, neuritic plaques (NPs) and neurofibrillary tangles (NFTs), are key to the neuropathologic diagnosis of this most common cause of dementia (15). Indeed, there has been an evolution of diagnostic criteria for the neuropathologic diagnosis of Alzheimer’s disease (AD) over the last thirty years that now relies on histologic estimates of the amount or anatomic distribution of these two abnormal structures (1, 6, 15). Although imperfect, these and other studies provide overwhelming observational data to support the use of these histologic assessments of NP and NFT accumulation in the diagnosis of AD as distinct from other causes of dementia. Further advantages of histologic assessments are obvious since they provide localizing information and build on over a century of experience in recognizing and classifying a broad range of diseases. However, these classic histologic features developed to diagnose AD also have limitations; notable, it is now clear that NPs represent only one form of amyloid beta (Aβ) peptides (11, 38), and there is more PHF-tau in tissue than is represented by NFTs (26).
Many, including us, have used these diagnostic criteria as tools for estimating the burden of NPs or NFTs in observational research attempting to correlate the level of these diagnostic markers with the extent of cognitive impairment, behavioral change, environmental factors, or drug exposures (28, 32, 36, 39).
Here the limitations are more severe. A theoretical limitation is that NPs are largely characterized by the presence of PHF-tau-immunoreactive processes and so the extent to which NP score provides disease information independent of NFTs is not clear. A practical limitation is that these diagnostic criteria are not quantitative measures but rather ordered rankings. Quantitation denotes measurement that has a demonstrated limit of detection, a dynamic range that can be captured in an equation, and an assay that can be standardized. The quantitative limitations of the diagnostic criteria for AD are in fact well recognized and have led many labs to develop assays for Aβ peptides or PHF-tau that use homogenates from fresh or frozen brain (12, 17, 22, 40). However, this approach has limitation of its own since not all of the pathologic forms of Aβ peptides (41) or PHF-tau (19) can be extracted from tissue for biochemical analyses, and since only a subset of cases will have tissue collected in a manner compatible with these assays.
This last point about a subset of tissue collected appropriately for biochemical extraction and quantitative analysis is especially germane to large research-, community-, or population-based studies. For example, the National Alzheimer’s Coordinating Center records that 72% of brain autopsies from research centers have at least some frozen tissue. In addition, the Adult Changes in Thought (ACT) study, a population-based study in Seattle, has one of the highest percentages of frozen brain samples among population-based studies, yet only about one-half of cases are collected with time between death and autopsy short enough to remove concern about post mortem biochemical changes in lipids and protein (35). Although there are multiple reasons for not obtaining frozen tissue, having only a subset of cases for quantitative analysis raises vexing statistical issues concerning potential bias and generalizability of results (13). Given the humbling commitment by subjects who participate in these longitudinal studies and the extensive resources dedicated to obtaining highly annotated clinical data, it seems to us a tragic loss not to be able to extract robustly quantitative data on Aβ peptides and PHF-tau from fixed and processed tissue.
Several groups have attempted to address this issue of quantifying Aβ peptides and PHF-tau in fixed and processed tissue. For example, some groups use computer-assisted assessment of NPs and NFTs revealed by histochemical stains (29) or of Aβ- and PHF-tau-immunoreactive structures (7, 26), although this is less common with human tissue than with mouse models. While this approach provides a more comprehensive assessment, it is limited by non-standardized observer-determined image threshold, the observer’s judgment as to whether an abnormal immunoreactive structure is a “real” plaque or tangle and should be included or not, and the well-known non-quantitative features of immunohistochemistry as commonly performed (9). Others have developed a technique called “PET blot” where tissue sections are adhered to membrane, probed, and then developed by techniques similar to a Western blot. This approach does have advantages for quantification but carries the serious sacrifice of greatly limited microscopic evaluation of the tissue section (33). Still others have developed an approach called the “Midwestern assay” or the “microtiter immunocytochemical ELISA” (MICE); however, neither were demonstrated to be robustly quantitative in human brain sections and in aggregate have been used to evaluate Aβ peptides and PHF-tau in an unspecified region of two cases of AD (10, 31). We have developed a technique that maintains the strengths of histologic assessment while also providing quantitative measure of Aβ peptides and PHF-tau in human brain samples that have been fixed and processed by standard protocols. Our method is a combination of histologic evaluation and ELISA performed on a standard glass slide (“histelide”). Here we describe our new technique and apply it to 101 cases: eight selected clinical and pathologic controls, six cases of autosomal dominant AD, six late-onset AD (LOAD), and the initial 81 consecutive ACT cases who had high cognitive function within two years of death.
METHODS
Subjects and Materials
This study was approved by the University of Washington Institutional Review Board. All cases were from the Neuropathology Core of the University of Washington that serves both the AD Research Center (ADRC), ACT study, and the National Prion Pathology Surveillance Center. In our initial studies we selected the 20 cases shown in Table 1. Eight cases were Controls from ACT who were individuals diagnosed as “not dementia” by DSM-IV criteria and who scored in the upper four quintiles on the Cognitive Assessment Screening Instrument (CASI) within two years of death, and who by neuropathologic examination had Braak stage II or below for NFTs (6) and absent or sparse NP score based on Consortium to Establish a Registry for Alzheimer's Disease (CERAD) (25). We also selected 12 cases of AD from our ADRC. Six of these were individuals with LOAD who did not have a family history of AD, who were diagnosed with dementia, and who by neuropathologic examination had a high probability that their dementia was caused by AD according to consensus criteria (15). Individuals in this group younger than 65 years of age were tested for known disease-causing autosomal dominant mutations; none were identified. Another 6 individuals had a family history of dementia, had inherited a known autosomal dominant AD-causing mutation in PSEN1 or PSEN2, had been diagnosed with dementia, and had intermediate or high probability that their dementia was caused by AD. The 12 AD cases had Braak stage IV, V or VI and moderate or frequent NP scores. All 20 selected cases had two or fewer cerebral microinfarcts (35) and no Lewy body disease (21). Post-mortem interval was less than six hours for all 20 selected cases. In addition to these selected cases, we also analyzed the initial 81 consecutive autopsies from ACT participants who were last evaluated clinically within two years of death and who at that time were “not dementia” by DSM-IV criteria and who also performed in the upper four quintiles on CASI; average PMI was 5 hr with no case exceeding 8 hr PMI. These 81 ACT high cognitive performers were not further selected by neuropathologic features. These 81 subjects (46 women) were 83 ± 7 years-old at time of death and had the following neuropathologic findings (mode, range): Braak stage (I, none to VI), CERAD neuritic plaque score (sparse, none to frequent), cerebral microinfarcts (1, 0 to 3); one case had neocortical Lewy body disease.
Table 1.
Demographic characteristics of subject population and corresponding Histelide measurements.
| CASE | GROUP | AGE (years) |
GENDER | BRAAK STAGE |
CERAD SCORE |
HISTELIDE MEASUREMENTS | ||
|---|---|---|---|---|---|---|---|---|
|
Amyloid β SIGM (Absorbance/cm2) |
PHF-tau SIGM (Absorbance/cm2) |
Synaptophysin SIGM (Absorbance/cm2) |
||||||
| 1 | Control | 83 | F | I | none | 0.08 | 0.00 | 0.34 |
| 2 | 95 | F | II | sparse | 0.14 | 0.01 | 0.78 | |
| 3 | 87 | F | None | sparse | 0.03 | 0.00 | 0.50 | |
| 4 | 80 | F | None | none | 0.29 | 0.01 | 0.66 | |
| 5 | 88 | F | I | none | 0.14 | 0.00 | 0.52 | |
| 6 | 88 | M | I | sparse | 0.19 | 0.02 | 0.54 | |
| 7 | 78 | M | None | none | 0.15 | 0.00 | 0.47 | |
| 8 | 81 | M | I | sparse | 0.14 | −0.01 | 0.44 | |
| 9* | Autosomal Dominant AD |
44 | F | VI | frequent | 0.73 | 0.41 | 0.74 |
| 10† | 51 | F | VI | frequent | 0.57 | 0.35 | 0.48 | |
| 11† | 64 | M | VI | moderate | 0.48 | 0.45 | 0.41 | |
| 12† | 55 | M | VI | frequent | 0.26 | 0.27 | 0.66 | |
| 13† | 52 | F | VI | frequent | 0.24 | 0.34 | 0.58 | |
| 14† | 66 | M | VI | frequent | 0.40 | 0.26 | 0.65 | |
| 15 | Late-onset AD |
78 | M | IV | frequent | 0.11 | 0.01 | 0.54 |
| 16 | 81 | F | VI | frequent | 0.38 | 0.72 | 0.59 | |
| 17 | 94 | M | V | frequent | 0.43 | 0.03 | 0.63 | |
| 18 | 61 | M | VI | frequent | 0.30 | 0.29 | 0.50 | |
| 19 | 63 | M | V | frequent | 0.35 | 0.40 | 0.74 | |
| 20 | 82 | M | VI | frequent | 0.38 | 0.29 | 0.70 | |
PSEN1 A260V,
PSEN2 N141I
ABBREVIATIONS. AD: Alzheimer’s disease, CERAD: Consortium to Establish a Registry for AD, GM: gray matter, PHF: paired helical filament, SI: signal intensity.
Middle frontal gyrus (MFG, Brodmann area 9) was evaluated in these 101 cases. In addition to these, we selected at random three cases of rapidly progressive dementia that were diagnosed as sporadic Creutzfeldt-Jakob disease (CJD) for evaluation of Proteinase K (PK) digestion. Because of limited tissue, some samples used for PK digestion used superior and middle temporal gyri (SMTG) rather than MFG.
Histelide method
All tissue except CJD cases was processed by usual methods of immersion fixation in formalin for two to three weeks. Cases of CJD were processed according to established protocols that minimize transmissibility (2). All tissue was dissected, embedding in paraffin, and cut at 5µm thick sections on a microtome. Tissue sections were placed in a water bath, collected on charged microscope slide or nitrocellulose membranes (5.0 µm pore size, Sterlitech Corporation, Kent, WA), and dried for at least 2 hours at 65°C. Duplicate sections from two blocks were processed for each antibody: two consecutive sections were from blocks that contained cortical gyri with some underlying white matter and two consecutive sections were from blocks of only white matter immediately adjacent to the cortical block.
Slides containing tissue sections were deparaffinized and rehydrated by standard means: xylene (4 times, 10 min each), followed by 1:1 mixture of xylene and 100% isopropanol (2 times, 5 min each), 100% isopropanol (2 times, 5 min each), 96% isopropanol in water (3 min), 70% isopropanol in water (3 min), 50% isopropanol in water (3 min), followed by two 5 min washes with TBST (10 mM Tris-HCl pH 7.8 + 100 mM NaCl + 0.05% Tween), and then pre-treated as described in Table 2. All of the following steps (Figure 1A) were carried out in 5-slide mailer plastic containers. Slides were washed with TBST 3 times for 10 min, and incubated in blocking solution (5% normal goat serum, 2% bovine serum albumin, 0.25% triton in TBST) overnight at 4°C. Slides were then incubated with primary antibody (see Table 2 for details on antibodies) for 8 hours at room temperature, washed three times for 10 min each in blocking solution, and then incubated with alkaline phosphatase–conjugated goat anti-mouse or anti-rabbit (as appropriate) IgG (Jackson Immunoresearch Laboratories, West Grove, PA) overnight at room temperature. Incubation with the secondary antibody was followed by three 15 min TBST washes. Omission of secondary antibody yielded no signal so efforts to block endogenous phosphatase activity were not pursued.
Table 2.
Antibodies and pretreatments.
| ANTIBODY | PRETREATMENT | ||
|---|---|---|---|
| PROTEIN | MANUFACTURER, PRODUCT NUMBER, TYPE, DILUTION |
SPECIFICITY | |
| Synaptophysin | Novus Biologicals NB300–653 Rabbit polyclonal 1:300 | Epitope: amino acids 41–62 of human synaptophysin. Does not cross-react with other synaptic vesicle proteins, such as synaptotagmin, GAP45, and SNAP25 proteins. | Boil in 0.1M citrate buffer (pH 6.5) in a water bath for 20 min |
| Amyloid beta (Aβ) | Covance Research Products, SIG-39320 (6E10), Mouse monoclonal 1:4000 | Eptitope: amino acids 3–8 of Aβ1–16 (18). Reacts with the abnormally processed isoforms, as well as precursor forms. | 88% formic acid for 10 min |
| Paired helical filament–tau | Thermo Scientific, MN1020 (Clone AT8), Mouse monoclonal 1:100 | Epitope: Serl99–Ser202 region; depends on the phosphorylation of one or both of these two serines (5). Binds to PHF-tau doubly phosphorylated at Ser202 and Thr205, and cross-reacts with tau doubly phosphorylated at Ser199/202 and Ser205/208. Does not bind the six human isoforms of non-phosphorylated tau. | 88% formic acid for 5 min |
| Drebrin | MBL International, D029-3, (Clone M2F6) Mouse monoclonal 1:100, 1:200, 1:500, 1:1000 | Immunogen: purified chicken drebrin E (34). Reacts with drebrin E and drebrin A. | Boil in 0.1M citrate buffer (pH 6.5) in a water bath for 20 min, or 88% formic acid for 5 min, or no pretreatment |
| Alpha-synuclein | Invitrogen, 18-0215, Mouse monoclonal 1:100, 1:300 | Immunogen: Lewy bodies purified from patients (3). Reacts with human alpha-synuclein and partially truncated forms of alph-synuclein. Does not react with beta-synuclein. | Boil in 0.1M citrate buffer (pH 6.5) in a water bath for 20 min |
Figure 1. Histelide method.

A. 1 – Formalin-fixed, paraffin-embedded tissue was cut, mounted on glass slides, and deparaffinized; 2 – Slides were washed with TBST and incubated overnight in the blocking solution; 3 – Incubated with primary antibody in blocking solution, followed by washes with blocking solution; 4 - Incubated with secondary antibody in blocking solution, then washed with TBST and DEA solution; 5 - Incubated in the PNPP solution; 100 µL of the solution collected at predetermined intervals, and absorbance read at 405 nm; 6 – Slides were washed with TBST and NTM, incubated in BCIP solution for 30 min, and then washed in PBS and water, dried, coverslipped, and examined by light microscopy. B. PNPP100 was determined by averaging absorbance values at 80, 100, and 120 min of incubation if all three values were ≥ 0.5. If absorbance values at these time points were < 0.5, then PNPP100 was calculated by averaging absorbance at 200, 220, and 240 min time points and extrapolating to 100 min. C. Signal Intensity for gray matter, SIGM, was calculated as the difference between the gray matter unit intensity (gray matter signal per cm2 of tissue) and background unit intensity; we assumed that any signal from pure white matter section for synaptophysin, PHF-tau, or Aβ was background immunoreactivity. Gray matter unit intensity was obtained by subtracting white matter signal from PNPP100 for the entire section that was composed of gray and white matter (Step 2).
Determining soluble alkaline-phosphatase reaction product using p-nitrophenyl phosphate (PNPP)
To measure PNPP absorbance, slides were first washed with diethanolamine (DEA) solution (10 mM diethanolamine and 0.5 mM MgCl2, pH 9.5) for 5 min. Afterwards, each slide was placed in a 2-slide mailer filled with 5 ml of PNPP solution (Sigma-Aldrich or Thermo Scientific, Waltham, MA), placed on a rocker, and allowed to rock for 2–4 hours, depending on the rate of PNPP development. One hundred microliters of the PNPP solution were drawn at 60, 80, 100, and 120 minutes and transferred into wells of a 96-well plate filled with 100µL of 1M NaOH. The plate was then read at 405 nm. At that point, if the absorbance reading of the well containing the 80-min draw reached or exceeded 0.5, the slides were taken out of the PNPP solution. Otherwise, the slides were allowed to rock for another 2 hours, and 100µL of the solution were drawn at 180, 200, 220, and 240 min. In our early experiments, replacing the withdrawn volumes did not alter the outcomes or the linearity of results, and so we did not include this step.
Visualizing precipitated alkaline-phosphatase reaction product 5-Bromo-4-chloro-3-indolyl phosphate (BCIP)
After the slides were removed from PNPP solution, they were again washed with TBST, for 10 min, and NTM solution (100 mM Tris-HCl pH 9.5 + 100 mM NaCl + 50 mM MgCl2), 2 times for 5 min. The slides were then placed into NBT/BCIP solution (Sigma-Aldrich) for 30 min at 4°C, after which they were washed with PBS (pH 7.4) and water, 15 min each, dried at room temperature, and cover slipped with Clear Mount mounting medium (Electron Microscopy Sciences, Hatfield, PA).
Area and length measurements
After the slides were cover slipped and dried overnight, gray and white matter were traced separately under a microscope, and respective areas were calculated. Average thickness of cerebral cortex was estimated in the same section by measuring depth of the cortical gray matter at 3 randomly selected points, and the results averaged. Nikon 90 microscope and Stereo Investigator software (MBF Bioscience, Williston, VT) were used for all measurements.
Data analysis
All absorbance data was between 0.5 and 2.5 absorbance units to maximize linearity of signal. The average signal of PNPP solution at the 100 min time point (PNPP100) was determined by averaging the absorbance measured at 80, 100, and 120 min time points, provided that the signal at all three time points was above 0.5. Otherwise, PNPP100 was calculated by averaging absorbance at 200, 220, and 240 min time points and dividing by 2.2 (220 min/100min = 2.2) (Figure 1B). Because tissue section size varied among cases, PNPP100 had to be normalized to the area of tissue on the slide. Also, because we were interested in measuring the signal coming from gray matter, but all of the tissue blocks used in the study contained some white matter as well, the signal coming from white matter had to be subtracted from the total signal. To achieve these goals, for each case, we first determined the PNPP100 signal per cm2 of the “white matter only” section (Figure 1C, Step 1), multiplied this value by the area of white matter on the “gray and white matter” slide, and then subtracted this product from the total signal to obtain PNPP100 for gray matter; this was then divided by the area of the gray matter on the slide to determine the gray matter signal unit intensity (Figure 1C, Step 2). For the three antibodies used in this study, we assumed that immunoreaction that we observed in the white matter was the result of non-specific binding of antibodies or non-specific chromogen product generation (“background”). Because background varied substantially among cases, our final result was calculated by subtracting the background unit signal from the gray matter unit signal, and expressed as Signal Intensity for Gray Matter, SIGM, in units of absorbance/per cm2 gray matter (Figure 1C, Step 3).
Determining the relationship between the amount of antigen and absorbance measurements
To determine the relationship between the amount of antigen present in a tissue section and the corresponding signal intensity, we determined absorbance values for ¼, ½, 1, 2, and 4 tissue sections, which reflected a 16-fold range of values, processed simultaneously as described above.
Tissue digestion with PK
These experiments used tissue blocks of superior and middle temporal gyri (SMTG) from a subset of the twenty cases in Table 1 because we were exhausting the MFG blocks. After deparaffinization, rehydration, and pretreatment (Table 2), slides were incubated with 1 mg/mL PK (Sigma) in buffer (10 mM Tris-HCl pH 7.8 + 100 mM NaCl + 0.1% Brij35 (Sigma), a nonionic polyoxyethylene surfactant) or buffer only at 60°C for 48 hours. The solution was renewed twice during the incubation time. For each case, we processed 4 pairs of slides: 2 pairs were digested with PK, while the other 2 were incubated with buffer only. After digestion, slides were washed three times with TBST, and 1 pair of slides from each group (digested and undigested) was processed as described above, while the remaining slides (digested and undigested) were processed without primary antibody. For each slide, PNPP100 signal per slide was averaged for the pair, and the signal obtained from those incubated without primary antibody was subtracted. Since accurate area measurements could not be performed on digested tissue, we compared PNPP100 values instead of calculating SIGM.
Statistical analysis
All data were expressed as mean ± SEM. Data were analyzed by using unpaired one-tailed Student’s t-test and one-way ANOVA. Correlations were evaluated by calculating Pearson’s correlation coefficient, and linearity was assessed by linear regression. Alpha was set at 0.05.
RESULTS
Our goals in developing this new technique were to devise a method that yields robustly quantitative data while still providing high quality histologic assessment of formalin-fixed paraffin-embedded tissue blocks. Our first step was to determine whether the relationship between the amount of antigen present on the slide or a membrane and PNPP signal was linear. To accomplish this, we determined absorbance values for different amounts of a given tissue section(s): ¼, ½, 1, 2, or 4, which were placed on a slide processed simultaneously. In parallel, we performed the same experiment using blotting membrane instead of glass slides to complement the PET blot technique (33). Figure 2 shows the 5-point curve between amount of tissue over a 16-fold range and PNPP100 for synaptophysin when tissue was adhered to glass slides or membranes (linear regression had R2 = 1.00 for both). The inset to Figure 2 presents these same data in an alternative format that does not require extrapolation to PNPP100 for some of the data points; it plots the time to achieve absorbance = 1.0. Again, these 5-point curves were nearly perfectly linear (R2 = 1.00) whether tissue was adhered to membranes or glass slides. Since glass slides and membranes yielded comparably robust quantitative data from our modified antibody capture ELISA and because glass slides were much more easily integrated into standard histologic protocols, all future studies focused on the use of our novel Histelide technique.
Figure 2. Quantification of synaptophysin signal.

Plot of PNPP100 absorbance values and best fit line from linear regression. PNPP100 absorbance values for ¼, ½, 1, 2, or 4 sections of a given tissue block that were adhered either on glass slides (●, Case 5 in Table 1,) or nitrocellulose membranes (■, Case 18 in Table 1,) and processed simultaneously. Linear regression showed a near perfect linear relationship (R2=1.00) for both glass slides and membranes over this 16-fold range. Insert: Incubation time in PNPP solution for ¼, ½, 1, 2, or 4 tissue sections to reach absorbance = 1.00 also was linear (R2=1.00) for tissue on both slides and membranes.
We compared levels of the presynaptic vesicle protein synaptophysin in the frontal cortex of patients with LOAD, autosomal dominant AD caused by mutations in PSEN1 (PSEN1-AD) or PSEN2 (PSEN2-AD), and Control subjects (Table 1). As expected, immunoreactivity as revealed by precipitated NBT was localized to gray matter (Figure 3A), and had a characteristic dot-like appearance when observed using standard light microscopy (Figure 3B). We did not observe a significant difference in SIGM between AD and Control groups (0.60 ± 0.03 vs. 0.53 ± 0.05, Figure 3C); however, it is critical to stress that SIGM is absorbance per area of gray matter. Next we measured average cortical thickness in each tissue section, and as expected it was significantly lower in AD patients than Control subjects (0.22 ± 0.01 cm vs. 0.32 ± 0.01 cm, p <0.0001, Figure 3D). We multiplied SIGM for each tissue section by average cortical thickness to yield a product that had units of absorbance per length of cortical gray matter for each section; these values were significantly lower in AD than Control subjects (0.13 ± 0.01 vs. 0.17 ± 0.02, p < 0.05, Figure 3E). Since we were exhausting tissue in frontal lobe blocks, data for Figure 3F came from a corresponding subset of SMTG sections. Our primary objective was to demonstrate that PNPP signal for synaptophysin in both AD and Controls could be ablated by digestion with PK; extensive digestion was required to accomplish this in formalin-fixed paraffin-embedded tissue sections (1mg/mL for 48 hours). Using lower concentrations of PK (0.5 mg/ml) or shorter incubation time (24 hours) yielded incomplete tissue digestion (not shown). PK digestion decreased PNPP100 to background levels for Controls (from 1.60 ± 0.12 to 0.10 ± 0.01 absorbance units) and AD (from 1.23 ± 0.55 to 0.12 ± 0.01 absorbance units). These data demonstrate that PNPP100 derived almost exclusively from molecules that were sensitive to PK digestion. Our results demonstrated the utility of Histelide in quantifying widely distribute antigens, like a presynaptic protein, and although in a relatively small number of cases, indicate that the amount of synaptophysin immunoreactivity in AD cerebral cortex varies widely and is related mostly to cortical atrophy.
Figure 3. Synaptophysin Histelide.

A. Section of middle frontal gyrus (MFG) from a Control case using Histelide technique. BCIP deposits are localized to gray matter. B. Gray matter (×400) from panel A showing the characteristic dot-like pattern of synaptophysin immunoreactivity with neuron soma and axons in relief. C–E. Scatterplots for sections of MFG from Control (●), late-onset AD (■), and autosomal dominant AD (▲) cases. Average (—) and SEM (⊤) are indicated. C. Synaptophysin SIGM was not statistically different between Control and AD groups. D. Average thickness of MFG cortical gray matter in MFG sections was significantly less in AD than Control groups (P < 0.0001). E. Synaptophysin SIGM multiplied by average MFG cortical thickness was significantly less in AD than Control groups (P < 0.05). F. Scatterplot of PNPP100 values from temporal cortex (SMTG) sections. Predigestion with proteinase K (open symbols) completely ablated the PNPP100 signal compared to no pre-digestion (closed symbols). Note that one AD case had very low SMTG synaptophysin PNPP100 and correspondingly very thin SMTG gray matter (not shown).
There are inherent limitations in quantifying a molecule that is decreasing in amount, like synaptophysin in MFG of AD. So, next we used Histelide to measure accumulation of PHF-tau, a component of NPs, neuropil threads, and neurofibrillary tangles (NFTs) in AD. Immunoreactivity for PHF-tau revealed the typical “ribbon” of superficial and deep deposits in MFG in AD subjects (Figure 4A), while no such staining was present in Control subjects (Figure 4D). Higher magnifications of NBT-stained sections revealed multiple PHF-tau immunoreactive structures (Figure 4B) including a classic NFT (Figure 4C). As expected, we observed that MFG from AD subjects had significantly higher average SIGM for PHF-tau compared to Controls (0.004 ± 0.003 vs. 0.318 ± 0.054, p <0.0001, Figure 4E). Interestingly, while all PSEN1-AD or PSEN2-AD cases had MFG PHF-tau SIGM greater than Controls, two LOAD cases had PHF-tau SIGM that was near Control values. Indeed, on inspection, these two cases did harbor a few isocortical NFTs but they were not in MFG. Using SMTG sections from the same subset of cases as above, we demonstrated that PHF-tau SIGM could be ablated by PK digestion, decreasing PNPP100 from 0.12 ± 0.05 to 0.05 ± 0.01 in Controls, and from 1.44 ± 0.20 to 0.04 ± 0.01 in AD; no PHF-tau immunoreactive structures remained on the slide following PK digestion (not shown). Next, we plotted MFG PHF-tau SIGM vs. Braak stage for all 81 ACT high cognitive performers, as well as the 12 selected AD cases for comparison (Figure 4F), and observed a significant increase in signal with increasing Braak stage among the 81 ACT high cognitive performers (P < 0.05), and between AD and ACT cases with Braak NFT stage III or IV (P < 0.001). Interesting, the two cases from among the ACT high cognitive performers who had high Braak stage showed low MFG PHF-tau SIGM signal similar to Controls.
Figure 4. Paired helical filament– (PHF-) tau Histelide.

A. Section of middle frontal gyrus (MFG) from a late-onset AD (LOAD) case using Histelide technique. BCIP deposits reveal the typical “ribbon” of superficial and deep PHF-tau accumulation in cortical gray matter. B. Light microscopy showed abundant neurofibrillary tangles (NFTs) and neuropil threads (×100). C. Higher magnification (×400) revealed a classic NFT. D. Section of MFG from a Control case; BCIP deposits are absent from cortical gray matter. E–F. Scatterplots for PHF-tau SIGM in sections of MFG from Control (●), late-onset AD (■), autosomal dominant AD (▲), and ACT high cognitive performer (+) cases. Average (—) and SEM (⊤) are indicated. E. Average PHF tau SIGM was significantly greater in both LOAD and autosomal dominant AD cases (n=12) compared to Controls (n=8, P < 0.0001). F. PHF-tau SIGM increased significantly with increasing Braak NFT stage among the ACT high performers (n=79, P < 0.05), and was significantly greater in AD cases (n=12) compared to ACT cases with Braak NFT stage III or IV (P < 0.0001).
We next used Histelide to quantify accumulation of Aβ peptides. Deposition of NBT in MFG sections from AD subjects revealed plaque-like deposits localized to the cortical gray matter as well as deposits in the subarachnoid space (Figure 5A). Minimal, if any, immunoreactivity was observed in Control subjects (Figure 5D). Low power microscopic examination of AD cases showed amyloid angiopathy as well as multiple plaques in gray matter (Figure 5B), some of which showed dense amyloid deposits (Figure 5C). SIGM for Aβ was significantly greater among patients with AD compared to Controls (0.39 ± 0.05 vs. 0.14 ± 0.03, p<0.0002, Figure 5E). As a result of digestion with proteinase K, PNPP100 decreased from 0.07 ± 0.02 to 0.01 ± 0.01 for Control SMTG sections, and from 0.30 ± 0.11 to 0.01 ± 0.01 for AD cases; no Aβ immunoreactive structures remained on the slide following PK digestion (not shown). We plotted MFG Aβ SIGM vs. Braak stage for all 81 ACT high cognitive performers, as well as the 12 selected AD cases for comparison (Figure 5F), and observed a no significant change in signal with increasing Braak NFT stage among the 81 ACT high cognitive performers; however, MFG Aβ SIGM was significantly greater in AD than ACT cases with Braak NFT stage III or IV (P < 0.001).
Figure 5. Amyloid (A) β Histelide.

A. Section of middle frontal gyrus (MFG) from a late-onset AD (LOAD) case processed using Histelide technique. BCIP deposits reveal plaque-like structures in cortical gray matter as well as deposits in the subarachnoid space. B. Light microscopy showed amyloid angiopathy and numerous senile plaques in gray matter (×100). C. Higher magnification (×400) showed a dense amyloid deposit. D. Section of MFG from a Control case; BCIP deposits are absent from cortical gray matter. E–F. Scatterplots for Aβ SIGM in sections of MFG from Control (●), late-onset AD (■), autosomal dominant AD (▲), and ACT high cognitive performer (+) cases. Average (—) and SEM (⊤) are indicated. E. Aβ SIGM was significantly greater in both LOAD and autosomal dominant AD cases (n=12) compared to Controls (n=8, P < 0.001). F. Aβ SIGM did not change significantly across Braak NFT stages among the ACT high performers (n=79). However, Aβ SIGM was significantly greater in AD cases (n=12) compared to ACT cases with Braak NFT stage III or IV (P < 0.0001).
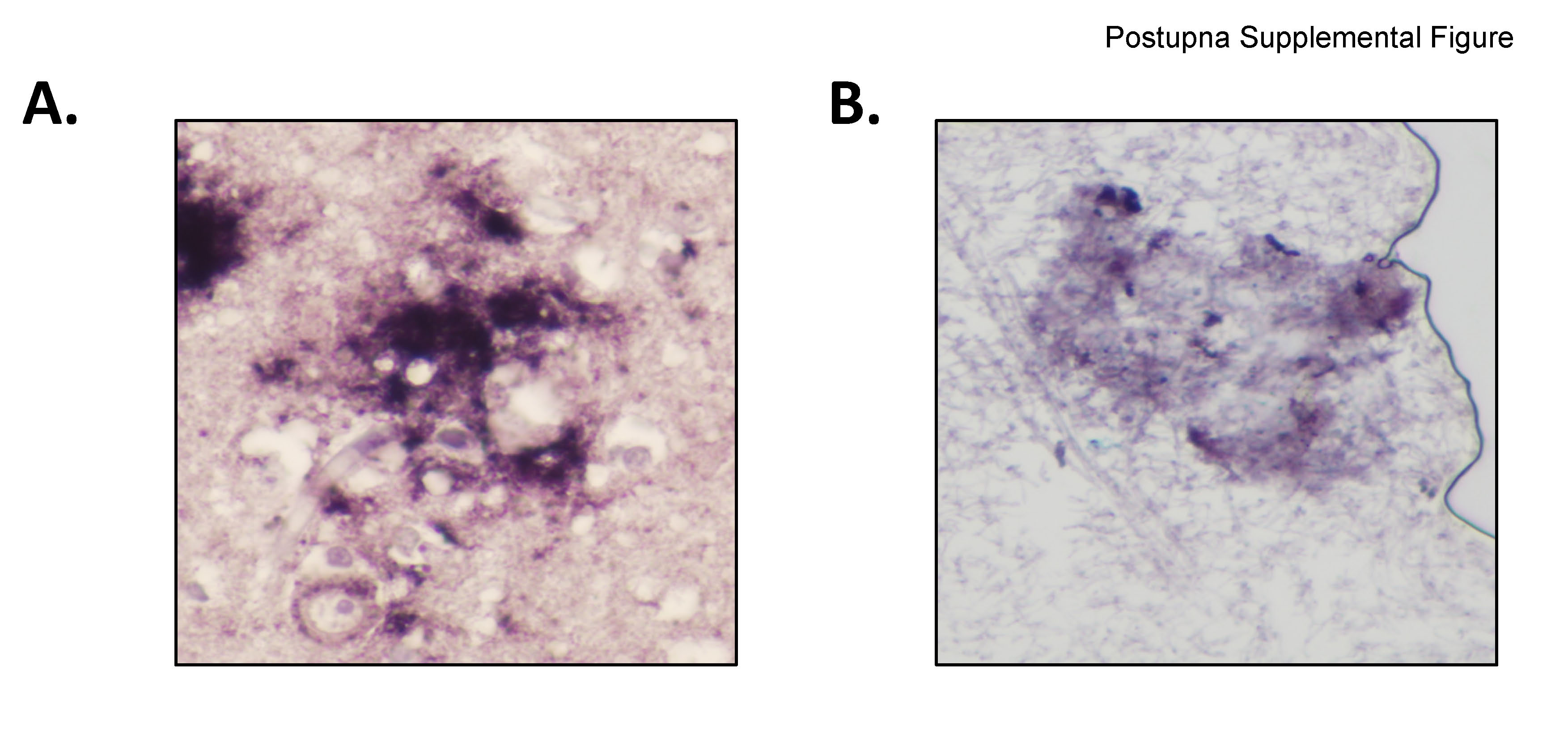
It is important to realize that our PK digestion protocol to ablate PNPP100 for Aβ and PHF-tau in formalin-fixed paraffin-embedded tissue is more intense than what is recommended for reducing transmissibility of CJD; however, the latter is meant for fresh tissue. To determine if prion protein would be relatively resistant to our PK digestion protocol for fixed and embedded tissue, we performed the exact same procedure on sections of MFG (Supplemental Figure) of three randomly selected cases of sporadic CJD. Our results showed that while most PrP-immunoreactive material was sensitive to digestion, a component of PrP-immunoreactivity in CJD plaques was resistant to our PK digestion protocol. Thus Aβ and PHF-tau in AD cases were completely ablated, while PrP was relatively resistant, to our PK digestion protocol.
As noted, our goal was to obtain robustly quantitative data from formalin-fixed paraffin-embedded tissue sections for evaluation of burden of AD pathologic changes. Therefore, we plotted MFG SIGM for PHF-tau vs. SIGM for Aβ for all 101 individuals in this study (Figure 6). Control cases formed a tight cluster with low SIGM for both PHF-tau and Aβ. In contrast, AD cases varied widely from partial overlap with selected Controls values to wide dispersion along both axes. Not unexpectedly, ACT high performers formed a continuum between selected Controls and AD cases. Interestingly, correlation between MFG SIGM PHF-tau and Aβ was significant for the 8 selected Controls (Pearson r =0.90, p<0.002), but not (P > 0.05) for the 81 ACT cases or the 12 AD cases, perhaps a reflection of the proposed maximum accumulation of Aβ early in the course of AD (16).
Figure 6. Scatterplot of SIGM for paired helical filament– (PHF-) tau vs. amyloid (A) β.

Scatterplot of all cases studied for PHF-tau SIGM vs. Aβ SIGM in MFG. Symbols represent Control (●), late-onset AD (■), autosomal dominant AD (▲), and ACT high cognitive performer (+) cases. Arrow points to Control case # 4, open arrowhead points to LOAD case # 17, filled arrowhead points to LOAD case # 15.
We attempted to adopt other commonly used antibodies to Histelide, specifically a different antibody to tau protein, which recognizes both phosphorylated and non-phosphorylated form; antibodies to a postsynaptic protein, drebrin, and alpha-synuclein, another presynaptic protein that accumulates in Lewy bodies and abnormal neurites (Table 2). Staining with the antibody to tau produced results very similar to those obtained when using the antibody to PHF-tau (data not shown). With other antibodies, although we were able to observe immunoreaction in the appropriate structures, it was difficult to obtain a reliable SIGM because of high and variable signal in white matter (drebrin) or very low signal overall (alpha-synuclein). While these results do not disqualify the use of other antibodies to quantify these proteins by Histelide, they do highlight the dependence of this approach on relatively abundant antigens with signal localized to gray matter.
DISCUSSION
Here we describe the development and initial application of Histelide, a method to obtain robustly quantitative data on antigens while maintaining high quality histologic assessment in the evaluation of neurodegenerative diseases. There are several advantages to our approach that we think will provide new opportunities for molecular analyses of human neurodegenerative diseases. These include improved analytical performance over current commonly used methods, optimization for use with formalin-fixed paraffin-embedded tissue, and seamless integration into conventional immunohistochemical approaches.
Histelide is designed to quantify antigens in paraffin-embedded tissue sections of cortical gray matter. By applying this technique to varying amounts of tissue, we demonstrated that the measurements were almost perfectly linear over a 16-fold dynamic range. Due only to physical limitations of how much tissue can be applied to a slide and dissecting very small pieces of tissue, we were not able to test for a broader range of linearity. Proteins from human brain commonly are quantified by extraction of fresh or frozen tissue followed by western blots. While this method allows quantification of different forms of the same antigen which have different physical properties, commonly used detection such as chemiluminescence is well-known not to produce a linear response and has a dynamic range of approximately one order of magnitude (14). More advanced detection systems are more reliable and allow a greater dynamic range (24).
In addition to superior analytical performance, an enormously helpful feature of Histelide is that it was designed to use formalin-fixed paraffin-embedded tissue sections. This key feature obviates the previous necessity for fresh or rapidly frozen tissue in order to perform quantitative molecular analyses. While dependence on fresh or frozen tissue has worked well in research centers, it has severely limited quantitative molecular analyses of brains collected in other settings, such as from individuals with rare genetic variants who reside at sites remote from academic medical centers, e.g., the PSEN2-AD cases presented here, or from individuals enrolled in large population-based studies with autopsy endpoint that are precluded logistically from routine collection of frozen tissue (32, 36, 39). We initially developed our method using nitrocellulose membranes, similar to the PET blot technique (33), but discovered that using glass slides instead of membranes was superior in three aspects. First, in contrast to glass slides, nitrocellulose membranes produced a relatively strong background signal (data not shown), which was likely due to nonspecific binding of antibodies to the membrane itself, making it more challenging to interpret the results. Also, membranes could not be examined under a traditional non-dissecting microscope, which made area measurements difficult to obtain. Finally, use of glass slides allows ready incorporation into the traditional workflow of an anatomic pathology laboratory.
There are potential limitations to Histelide that mostly revolve around fixation and processing. Formalin-fixation and paraffin-embedding alters antigens and can influence binding by primary antibody. Pathologists who perform immunohistochemistry as part of their clinical duties are very familiar with this issue and a variety of methods exist to mitigate these effects or “retrieve” antigens. Indeed, we observed that older cases (≥ 8 years or older) generally showed higher background white matter signal compared to more recent cases (< 3 years). To determine if not correcting for variation in background would alter results, we compared our Control and AD group using gray matter unit signal instead of SIGM. For synaptophysin, PHF-tau, and Aβ, omitting background increased the variance of each (data not shown). While extraction of fresh or rapidly frozen tissue circumvents these potential problems that derive from fixation, it has unique problems of its own, such as proteolysis following tissue homogenization, liberation of competing substances following extraction, and incomplete extraction. The last is an especially difficult problem when trying to assess PHF-tau and Aβ accumulation in human brain samples.
One of the goals of developing Histelide was to obtain quantitative data on antigens that are widely dispersed and so challenging to other quantitative methods, like stereological counting. For this goal we selected synaptophysin, a presynaptic protein expressed throughout the cerebral cortex, because it has been used extensively as a marker of synapse loss in AD, although many other measures also have been used including postsynaptic proteins and ultrastructural analysis. Literature reports concerning levels of synaptophysin in cerebral cortex of patients with AD are conflicting with virtually every possible outcome observed, likely due to methodological differences. For example, Mukaetova-Ladinska and colleagues used ELISA on extracted tissue to assess the levels of synaptic proteins in frontal cortex, and reported a transitional increase in synaptophysin in the frontal cortex at Braak stage III, but no significant difference at later stages (27). Others using immunoblotting with chemiluminescence of extracted tissue, observed a significant decrease in synaptophysin in frontal cortex in AD (30), while still others, who used a similar technique, observed no significant difference between controls and AD (8). Rather than using extracted tissue, others have used immunohistochemistry with densitometry. Some have reported that synaptophysin immunoreaction in frontal cortex is not significantly different between control and AD subjects (20), while others have reported that synaptophysin staining is decreased (23). When we examined levels of synaptophysin in MFG, we did not observe a significant difference in SIGM between the Controls and AD patients. However, SIGM is normalized to area of tissue. When we multiplied synaptophysin SIGM by the MFG thickness to account for cortical atrophy in AD, the difference between groups was statistically significant. Although we cannot resolve the conflicts in the literature, our Histelide results suggest that patients with AD have reduced MFG synaptophysin that parallels cortical atrophy, and that the amount of synaptophysin per remaining unit area of MFG in AD is not significantly different from Controls. We hasten to add that cortical atrophy may be due to synapse loss, that our results are from a relatively small group of patients and focused on MFG, and that our limited analysis of SMTG with our PK digestion experiments suggested that synaptophysin loss may vary substantially among regions of cerebral cortex.
We also examined neocortical Aβ and PHF-tau burden in AD and Control cases. As mentioned previously, Aβ and PHF-tau accumulate in tissue in a variety of forms or structures, and it is important to realize that these are transparent to the approach developed here, e.g., signal generated from Histelide does not distinguish between Aβ deposited in plaques and cerebral amyloid angiopathy. On average, AD patients showed significantly higher levels of both proteins than Controls. When Aβ and PHF-tau values were plotted together (see Figure 6), Controls formed a cluster in the lower left corner of the graph (low Aβ and low PHF-tau SIGM), with the exception of case # 4 (indicated by an arrow). Interestingly, case #4 also had CASI score of 93; while still interpreted as cognitively normal, it was, interestingly, the lowest CASI score is our Control group. Moreover, even though staining for Aβ did not reveal any plaque-like deposits in MFG of case #4, we did observe a distinct diffuse immunoreaction throughout gray matter. Unlike the Control group, AD cases demonstrated a wide dispersion of MFG PHF-tau and Aβ accumulation. Cases #15 (Braak stage IV) and #17 (Braak stage V), designated by a filled or open arrowhead, respectively, both showed low MFG PHF-tau SIGM. This is expected in MFG for a Braak stage IV case. PHF-tau-stained sections from case #17 revealed two NFTs in parietal cortex, consistent with both Braak stage V and low MFG PHF-tau SIGM. Interestingly, PSEN1-AD and PSEN2-AD cases formed a “horizontal” cluster with relatively uniform PHF-tau SIGM but varying levels of Aβ SIGM; indeed, the three highest SIGM measurements for Aβ were from inherited forms of AD. In contrast, LOAD cases formed a more “vertical” cluster with relatively constant Aβ SIGM but varying PHF-tau SIGM; the greatest PHF-tau SIGM was a LOAD case. Although these potential clusterings are speculative, our results do at least suggest a non-uniform relationship between MFG PHF-tau and Aβ accumulation among individuals with LOAD vs. PSEN1-AD or PSEN2-AD.
We chose to focus on ACT cases from cognitive high performers because of intense interest in latent AD (37). Our results showed wide variance among Aβ and PHF-tau burden in MFG among these cases that spanned from selected Controls to AD. Moreover, MFG PHF-tau burden progressively increased with Braak stage, even at stages where neocortex is not involved by neurofibrillary tangles, likely a reflection of accumulating PHF-tau in dystrophic neurites prior to widespread appearance of neurofibrillary tangles. In contrast, MFG Aβ burden did not increase with increasing Braak stage among the ACT high performers, perhaps reflecting maximum accumulation of Aβ in MFG early in the course of AD (16). While fitting these quantitative data into a broader image of AD awaits results on ongoing Histelide of other brain regions, these outcomes in MFG underscore the highly individually-varying levels of Aβ and PHF-tau in cognitively normal individuals. Although there were only two individuals in among the 81 ACT high cognitive performers with Braak stage V or VI disease, each had MFG PHF-tau burden that was similar to selected Controls and much lower than the average for AD cases with comparable Braak stage. Re-evaluation of these cases confirmed their Braak stage, suggesting that while they had neurofibrillary tangles somewhere in neocortex, the burden of PHF-tau in MFG was like other Controls and not like AD. While we are limited by a small number of observations and evaluation of MFG only, these unusual cases suggest that Braak stage may be an imperfect surrogate for PHF-tau burden in specific neocortical regions, similar to AD case #17 discussed above. These results might be helpful when interpreting pending neuroimaging studies that may soon probe for PHF-tau in older cognitively normal individuals (4).
Histelide is a novel approach to obtain rigorous quantification of antigens in formalin-fixed paraffin-embedded tissue while maintaining high quality immunohistochemical results within the traditional workflow of an anatomic pathology laboratory. Because our interests lie in the field of neurodegenerative diseases, we concentrated on antigens of high importance to this area of research. However, we think Histelide may be successfully applied to other areas of histopathology, for example, surgical pathology of neoplasia, where quantification of an antigen has a clinical significance, such as Ki-67 antigen in a variety of neoplasms and HER2 in breast carcinoma. We have shown that Histelide is a new tool to obtain quantitative data about molecular pathologic changes of AD in formalin-fixed paraffin-embedded tissue. Our method will permit robust quantitative investigation of full tissue archives from community- and population-based studies that up to now has been limited to fresh or frozen samples only. Moreover, our results quantify the highly individual nature of neocortical Aβ and PHF-tau burden in cognitively normal older adults, and these should guide planning of and expectations for prevention trials and neuroimaging studies.
Supplementary Material
A. High power magnification (400X) of a PrPSc plaque immunoreactive with 3F4 antibody utilizing Histelide without Proteinase K digestion. B. High power magnification (400X) of a PrPSc plaque immunoreactive with 3F4 antibody utilizing Histelide following Proteinase K digestion as described in Methods. Note background tissue has been digested from glass slide while the PrPSc plaque was partially resistant to digestion. Due to extensive tissue digestion an air bubble is present in the right side of the image.
{kind=link}
ACKNOWLEDGEMENTS
This work was supported by grants from the NIH (AG05136, AG23801, AG006781, NS62684, T32AG000258), and the Nancy and Buster Alvord Endowment. We thank Dr. Kathy Montine for editorial assistance.
Footnotes
The authors confirm that we have no disclosures or conflicts of interest for this submission.
References
- 1.Alafuzoff I, Thal DR, Arzberger T, Bogdanovic N, Al-Sarraj S, Bodi I, Boluda S, Bugiani O, Duyckaerts C, Gelpi E, Gentleman S, Giaccone G, Graeber M, Hortobagyi T, Hoftberger R, Ince P, Ironside JW, Kavantzas N, King A, Korkolopoulou P, Kovacs GG, Meyronet D, Monoranu C, Nilsson T, Parchi P, Patsouris E, Pikkarainen M, Revesz T, Rozemuller A, Seilhean D, Schulz-Schaeffer W, Streichenberger N, Wharton SB, Kretzschmar H. Assessment of beta-amyloid deposits in human brain: a study of the BrainNet Europe Consortium. Acta Neuropathol. 2009;117:309–320. doi: 10.1007/s00401-009-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen CT, Sonnen J, Leslie MJ, Kidoguchi L, Harris C, Gambetti P, Montine TJ. Washington statewide pathology surveillance for prion disease. Ann Neurol. 2007;61:371–372. doi: 10.1002/ana.21096. [DOI] [PubMed] [Google Scholar]
- 3.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 4.Berti V, Pupi A, Mosconi L. PET/CT in diagnosis of dementia. Ann N Y Acad Sci. 2011;1228:81–92. doi: 10.1111/j.1749-6632.2011.06015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. Embo J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 7.Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F, Carpenter AP, Flitter ML, Krautkramer MJ, Kung HF, Coleman RE, Doraiswamy PM, Fleisher AS, Sabbagh MN, Sadowsky CH, Reiman EP, Zehntner SP, Skovronsky DM. Use of florbetapir-PET for imaging beta-amyloid pathology. Jama. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Dabbs DJ. Diagnostic Immunohistochemistry. 3rd Edition. Philadelphia: Saunders; 2010. [Google Scholar]
- 10.de la Monte SM, Ganju N, Wands JR. Microtiter immunocytochemical ELISA assay. Biotechniques. 1999;26:1073–1076. doi: 10.2144/99266bm15. 8. [DOI] [PubMed] [Google Scholar]
- 11.Esiri MM, Hyman BT, Beyreuther K, Masters C. Aging and dementia. In: Graham DI, Lantos PL, editors. Greenfield's Neuropathology. Sixth Edition. London: Arnold Publishing; 1997. [Google Scholar]
- 12.Forman MS, Mufson EJ, Leurgans S, Pratico D, Joyce S, Leight S, Lee VM, Trojanowski JQ. Cortical biochemistry in MCI and Alzheimer disease: lack of correlation with clinical diagnosis. Neurology. 2007;68:757–763. doi: 10.1212/01.wnl.0000256373.39415.b1. [DOI] [PubMed] [Google Scholar]
- 13.Haneuse S, Schildcrout J, Crane P, Sonnen J, Breitner J, Larson E. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology. 2009;32:229–239. doi: 10.1159/000197389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harlow E, Lane D. Antibodies: A Laboratory Manual. NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 15.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khatoon S, Grundke-Iqbal I, Iqbal K. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 1994;351:80–84. doi: 10.1016/0014-5793(94)00829-9. [DOI] [PubMed] [Google Scholar]
- 18.Kim KS, Miller DL, Sapienza VJ, Chen CMJ, Bai C, Grundke-Iqbal I, Currie JR, Wisniewski HM. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun. 1988;2:121–130. [Google Scholar]
- 19.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 20.Leuba G, Savioz A, Vernay A, Carnal B, Kraftsik R, Tardif E, Riederer I, Riederer BM. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis. 2008;15:139–151. doi: 10.3233/jad-2008-15112. [DOI] [PubMed] [Google Scholar]
- 21.Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB, Kukull WA, Lopez O, Galasko D, Masliah E, Kaye J, Woltjer R, Clark C, Trojanowski JQ, Montine TJ. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol. 2008;18:220–224. doi: 10.1111/j.1750-3639.2007.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maarouf CL, Daugs ID, Kokjohn TA, Kalback WM, Patton RL, Luehrs DC, Masliah E, Nicoll JA, Sabbagh MN, Beach TG, Castano EM, Roher AE. The biochemical aftermath of anti-amyloid immunotherapy. Mol Neurodegener. 2010;5:39. doi: 10.1186/1750-1326-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol. 1991;138:235–246. [PMC free article] [PubMed] [Google Scholar]
- 24.Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell TW, Mufson EJ, Schneider JA, Cochran EJ, Nissanov J, Han LY, Bienias JL, Lee VM, Trojanowski JQ, Bennett DA, Arnold SE. Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Ann Neurol. 2002;51:182–189. doi: 10.1002/ana.10086. [DOI] [PubMed] [Google Scholar]
- 27.Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, Gertz HJ, Xuereb JH, Hills R, Brayne C, Huppert FA, Paykel ES, McGee M, Jakes R, Honer WG, Harrington CR, Wischik CM. Staging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer's disease. Am J Pathol. 2000;157:623–636. doi: 10.1016/s0002-9440(10)64573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2010;20:66–79. doi: 10.1111/j.1750-3639.2008.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, Niinisto L, Halonen P, Kontula K. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333:1242–1247. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 30.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–117. doi: 10.3233/jad-2005-7203. discussion 73–80. [DOI] [PubMed] [Google Scholar]
- 31.Roth KA, Brenner JW, Selznick LA, Gokden M, Lorenz RG. Enzyme-based antigen localization and quantitation in cell and tissue samples (Midwestern assay) J Histochem Cytochem. 1997;45:1629–1641. doi: 10.1177/002215549704501206. [DOI] [PubMed] [Google Scholar]
- 32.Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, Groschup MH, Kretzschmar HA. The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am J Pathol. 2000;156:51–56. doi: 10.1016/S0002-9440(10)64705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shirao T, Obata K. Immunochemical homology of 3 developmentally regulated brain proteins and their developmental change in neuronal distribution. Brain Res. 1986;394:233–244. doi: 10.1016/0165-3806(86)90099-4. [DOI] [PubMed] [Google Scholar]
- 35.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 36.Sonnen JA, Larson EB, Haneuse S, Woltjer R, Li G, Crane PK, Craft S, Montine TJ. Neuropathology in the adult changes in thought study: a review. J Alzheimers Dis. 2009;18:703–711. doi: 10.3233/JAD-2009-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 39.White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia aging study. J Alzheimers Dis. 2009;18:713–725. doi: 10.3233/JAD-2009-1178. [DOI] [PubMed] [Google Scholar]
- 40.Woltjer RL, Sonnen JA, Sokal I, Rung LG, Yang W, Kjerulf JD, Klingert D, Johnson C, Rhew I, Tsuang D, Crane PK, Larson EB, Montine TJ. Quantitation and mapping of cerebral detergent-insoluble proteins in the elderly. Brain Pathol. 2009;19:365–374. doi: 10.1111/j.1750-3639.2008.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong CW, Quaranta V, Glenner GG. Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc Natl Acad Sci U S A. 1985;82:8729–8732. doi: 10.1073/pnas.82.24.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. High power magnification (400X) of a PrPSc plaque immunoreactive with 3F4 antibody utilizing Histelide without Proteinase K digestion. B. High power magnification (400X) of a PrPSc plaque immunoreactive with 3F4 antibody utilizing Histelide following Proteinase K digestion as described in Methods. Note background tissue has been digested from glass slide while the PrPSc plaque was partially resistant to digestion. Due to extensive tissue digestion an air bubble is present in the right side of the image.