

Abstract
Cell death has been reported in the CNS in models of neuropathic pain (Sugimoto et al., 1990; Whiteside and Munglani, 2001; Scholz et al., 2005; Fuccio et al., 2009). In our present study, we examined the effects of spinal nerve ligation (SNL) on the number of neurons in the rostral ventromedial medulla (RVM), a brain stem region involved in modulation of nociception. In rats receiving SNL, we found that the number of RVM neurons decreased by 23% in the side ipsilateral to the surgery. The loss of RVM neurons was also associated with a bilateral increase in the number of glia as well as bilateral activation of both astrocytes and microglia. Administration of tauroursodeoxycholic acid (TUDCA), which reportedly inhibits apoptosis, significantly reduced the loss of neurons, the increase in glia, and the mechanical hypersensitivity induced by SNL.
Among RVM neurons, we found that serotonergic (5-HT) neurons decreased by 35% ipsilateral to SNL. Consistent with these findings, the density of 5-HT-immunoreactive varicosities in the superficial dorsal horn of the spinal cord was 15–30% lower, ipsilateral to SNL. To test the function of the remaining 5-HT neurons, we administered the 5-HT neurotoxin, 5,7-dihydroxytryptamine (5,7-DHT). Interestingly, after 5,7-DHT, mechanical withdrawal thresholds increased significantly.
We conclude that nerve injury induces death of antinociceptive RVM neurons that can be reduced or abolished by TUDCA. We propose that the loss of RVM neurons shifts the balance of descending control from pain inhibition to pain facilitation.
Keywords: serotonin, neuropathic pain, apoptosis, spinal cord, raphe, hyperalgesia, tauroursodeoxycholic acid
INTRODUCTION
Trauma or disease affecting peripheral nerves frequently results in the development of chronic, sometimes intractable, neuropathic pain. Existing treatments for neuropathic pain have limited effectiveness and produce relatively frequent adverse effects (Wallace, 2007). Therefore, understanding the basic mechanisms contributing to the generation and/or maintenance of neuropathic pain in preclinical animal models is key to providing predictable and efficient therapies in patient populations.
Studies have shown that enhanced pain induced by peripheral nerve injury is associated with increased spontaneous and evoked discharges from injured and/or adjacent nerves (Koltzenburg et al., 1992; Koltzenburg et al., 1994; Amir and Devor, 2000; Michaelis et al., 2000; Liu et al., 2001). Although this increased afferent discharge is vital in establishing spinal sensitization in the period immediately following nerve injury, the time course of such abnormal afferent activity is inconsistent with the long duration of heightened pain (Chaplan et al., 1994; Burgess et al., 2002; Porreca et al., 2002). It has been suggested that descending facilitation from the rostral ventromedial medulla (RVM), including its serotonergic (5-HT) neurons, may play an essential role by maintaining neuropathic pain after its initiation (Burgess et al., 2002; Suzuki et al., 2004; Vera-Portocarrero et al., 2006).
In studies where neuropathic pain is modeled by spinal nerve ligation (SNL) (Kim and Chung, 1992), microinjections of lidocaine into the RVM reduced the hypersensitivity observed after SNL (Pertovaara et al., 1996), as did ablation of populations of RVM neurons (Porreca et al., 2001), and lesions of the dorsolateral funiculus (DLF), in which RVM axons descend (Ossipov et al., 2000). In these studies, reversal of hypersensitivity was not observed immediately after nerve injury, but only several days later, consistent with a role for the RVM in the maintenance (but not initiation) of neuropathic pain.
Previous work has also suggested that apoptosis may play a role in the pathology of neuropathic pain. Apoptosis has been reported in dorsal root ganglia, the spinal dorsal horn and in cerebral cortex in models of neuropathic pain (Coggeshall et al., 2001; Whiteside and Munglani, 2001; Siniscalco et al., 2007; Fuccio et al., 2009; Sekiguchi et al., 2009). However, the loss of neurons has been disputed and its importance in neuropathic pain remains unclear (Polgar et al., 2004; Polgar et al., 2005).
To help understand the importance of the RVM and its neurons in neuropathic pain, we examined RVM neurons and their spinal projections after SNL. We found that after SNL, the numbers of RVM neurons (including 5-HT neurons) were significantly reduced. Our findings suggest that the pathology of neuropathic pain may be due in part to death of antinociceptive RVM neurons, leading to decreased descending inhibition of nociception.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (150–250 g; Harlan, Madison, WI) were used for these studies; three to ten animals were used for each experimental group. All experiments and procedures were performed using protocols approved by the University of Minnesota Institutional Animal Care and Use Committee.
SNL surgery
Rats were divided into two experimental groups: a group in which the L5 spinal nerve was ligated and cut (Blenk et al., 1997; Ringkamp et al., 1999; Li et al., 2000), and a sham-operated control group. The left L5 spinal nerve was isolated, tightly ligated and cut distal to the ligation under isoflurane anesthesia (1.5%). The surgical procedure for the sham-operated group was identical to that of the SNL group, except that the L5 spinal nerve was not ligated or cut. General behavior of the rats was monitored before and after the surgery. Any rats showing difficulty elevating a hindpaw were discarded from the study.
Drug treatments
Rats were treated with 5,7-dihydroxytryptamine (5,7-DHT: 100 μg/10 μl, intracisternal injection) to induce loss of RVM 5-HT neurons. Rats were randomly assigned to one of four groups for these experiments: (1) L5 SNL with 5,7-DHT; (2) L5 SNL with vehicle injection; (3) sham with 5,7-DHT; and (4) sham with vehicle. Injections were given ten days after surgery; animals receiving vehicle injections received sterile saline (10 μl, intracisternal injection). To prevent damage to noradrenergic (NE) neurons, we pre-treated animals 30 minutes before 5,7-DHT (or saline) administration with desipramine (20 mg/kg i.p.), an inhibitor of neuronal NE reuptake (Bjorklund et al., 1975).
To test the effects of inhibiting apoptosis on SNL, we administered tauroursodeoxycholic acid (TUDCA: 300 mg/kg i.p.) or saline prior to and after surgery. TUDCA has been reported to inhibit apoptosis and to be active systemically (Rodrigues et al., 2003); systemic administration avoids the potential for damage associated with microinjections made directly into the brain parenchyma. For each group, injections were given every-other day, starting 3 days prior to surgery and continuing until sacrifice.
We tested the efficacy of TUDCA as an inhibitor of apoptosis by examining its effects on dexamethasone-induced apoptosis in thymus. Two TUDCA injections (300 mg/kg i.p.) were given: one two days prior to dexamethasone (1 mg/kg i.p.) administration and one the day of dexamethasone administration. Control animals were given saline injections instead of TUDCA solution. Rats were killed one day after dexamethasone administration.
Perfusion
Rats were deeply anesthetized with a mixture of ketamine (67.5 mg/kg), xylazine (22.5 mg/kg) and acepromazine (1 mg/kg) and perfused via the ascending aorta with 180 ml oxygenated Ca 2+-free Tyrode’s solution (pH 7.2) followed by 500 ml of 4% formaldehyde (freshly made from paraformaldehyde) in 0.16 M phosphate buffer (pH 6.9). Immediately after fixation, brains were removed and stored in a 5% sucrose solution prior to sectioning.
Histology and Immunocytochemistry
The RVM and spinal cord were sectioned using a freezing microtome (Leica, SM2400) at a nominal thickness of 50 μm. The free-floating sections were washed in phosphate-buffered saline (PBS: 0.8% (w/v) NaCl, 0.02% KCl, 0.144% Na2HPO4, 0.024% KH2PO4, pH = 7.4) for three 5-minute intervals. RVM sections were incubated overnight at 4° C in solutions containing one of the following antibodies: (1) mouse anti-TPH (T0678, Sigma, Saint Louis, MO, 1:1000), (2) mouse anti-NeuN (MAB377, Millipore, Temecula, CA, 1:500), (3) mouse anti-CD11b (MCA75GA, AbD Serotec, Oxford, UK, 1:1000), or (4) mouse anti-GFAP (GA5, #3670, Cell Signaling Technology, Danvers, MA, 1:1000). Spinal cord sections were incubated with goat anti-5-HT (#20079, ImmunoStar, Hudson, WI, 1:1000). Sections were then washed in PBS and incubated for 4 h with Cy2-conjugated donkey anti-mouse IgG or Cy3-donkey anti-goat IgG (1:500). Secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). The fluorescent Nissl stain ethidium bromide (30 nM, Sigma) was used to counterstain RVM tissue (Schmued et al., 1982).
To stain for apoptosis markers, ten micron-thick cryostat sections were incubated with antibodies raised against activated caspase-3 (AF835, R&D Systems, Minneapolis, MN), cleaved caspase-6 (Asp162, Cell Signaling Technology), or stained for terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL kit; Roche, Mannheim, Germany). For caspase staining, RVM sections were incubated overnight at 4° in a solution containing rabbit anti-cleaved caspase-6 (1:500) or mouse anti-active caspase-3 (1:500). Sections were then washed in PBS and incubated for 2 h with Cy3-conjugated donkey anti-rabbit (1:500) or Cy3-conjugated donkey anti-mouse (1:500) and the Nissl counterstain SyBr RNA II Green Gel Stain (S-7564, Invitrogen, Carlsbad, CA, 1:10,000). For TUNEL labeling, sections were rinsed in dH2O and then permeabilized with tris-buffered saline (TBS; 135 mM NaCl and 25 mM tris-HCl; pH = 7.4) containing 0.2% Triton X-100 and 0.2% Tween-20 for 1 h at room temperature. Following permeabilization, the sections were rinsed in TBS then incubated in TdT reaction buffer (Roche, 1600 U/ml terminal transferase, 40 nM biotin-16-dUTP, 200 mM potassium cacodylate, 5 mM CoCl2, 250 μg/ml bovine serum albumin, 25 mM tris-HCl; pH 6.6) for 2 h at 37°C. The reaction was terminated in TBS containing 20 mM EDTA; the sections were washed in several changes of TBS and incubated in streptavidin conjugated to Cy3 (Jackson Immunoresearch) and counterstained with SyBr RNA II Green Gel Stain (Invitrogen, 1:10,000) in TBS containing 0.2% Tween-20 and 0.2% casein for 2 h at room temperature.
Tissue was stained for the mu-opioid receptor (MOR) after heat-induced epitope retrieval. Ten micron-thick cryostat sections were exposed to a temperature of 101°C for 30 min in a 10 mM solution of citric acid. Tissue was then washed three times (five minutes each) in TBS and incubated overnight at 4°C in guinea pig anti-MOR and mouse anti-TPH. The guinea pig anti-MOR was the generous gift of Dr. Robert Elde (University of Minnesota, Minneapolis, MN) and was characterized as specific in MOR knockout mice (Liu et al., 2011). Tissue was washed in TBS and then incubated in Cy3-conjugated donkey anti-guinea pig IgG, Cy5-conjugated donkey anti-mouse IgG and counterstained with SYBR Green II (Invitrogen, 1:10,000) for 2 h.
All tissue was dehydrated in graded alcohols (50–100%) and cleared in xylene. The slides were mounted with coverslips using DPX (Fluka, Ronkonkoma, NY).
Microscopy and Quantification
Conventional microscopy was used to collect images of the RVM for cell counting. An Olympus BX50 fluorescence microscope (Tokyo, Japan) equipped with filter sets designed to allow selective visualization of Cy2 and Cy3 was used. Microscopic images were collected with a Scion 1346 digital camera. Confocal images were collected using an Olympus FluoView FV1000 microscope (Tokyo, Japan). ImageJ 1.29 (developed at NIH and available at http://rsb.info.nih.gov/ij/) or Photoshop were used to adjust contrast and brightness in images.
The RVM was defined as an isosceles triangle that lies at the level of the facial nucleus with a base having a width equal to that of the combined pyramidal tracts, and its height equal to half the width of the base (Figure 1) (Gu and Wessendorf, 2007). The RVM extended from the rostral end of inferior olive to the caudal end of the trapezoid body. It was divided into ipsilateral and contralateral sides by the midline.
Figure 1.

Definition of RVM and description of cell-counting methods. Line Diagram: The RVM was defined as an isosceles triangle, the base of which was the width of the pyramidal tracts. The height of the triangle on the midline was half the width of the base. VII: facial nucleus. Crosses mark examples of randomly selected points used for cell counting. A,B. Nissl-stained sections from sham-treated and SNL-treated rats, respectively. Fewer RVM neurons were observed in rats after SNL. Stars in A and B represent the locations of the counting frames in C and D, respectively. C, D: Examples of high-magnification sequences of images through the thickness of tissue sections used for cell counting. Color of counting frames corresponds to the color of the stars in A and B. Arrows indicate neurons that would be counted. A neuron might appear in multiple images through the thickness of a section but was only counted when its nucleolus was first observed. Neurons were counted if their nucleoli fell entirely within the counting frame, or if they were partially within the counting frame without being in contact with a rejection line (solid line). E1–E3. Comparison of Nissl and NeuN labeling. Arrows indicate Nissl-stained neurons that were also NeuN-ir. Arrowheads indicate Nissl-stained glia that were not NeuN-ir. Cells with dark cytoplasm, a vacuous nucleus and a dark nucleolus (the Nissl criteria we used for a neuron) were always labeled for NeuN. Scale bar in B applies to A and B. Scale bar in D4 applies to C1–4 and D1–4. Scale bar in E3 applies to E1–E3.
In Nissl-stained sections of the RVM, two distinct populations of cells were found: (1) cells with dark cytoplasm, a vacuous nucleus and a single distinct nucleolus (Figure 1E1, arrows), and (2) cells with very lightly-stained cytoplasm, a dark nucleus, and nuclear granulations rather than a nucleolus (Figure 1E1, arrowheads). The first group was classified as neurons and the second as glia. These distinct Nissl-stained populations were confirmed by immunohistochemistry (e.g., NeuN for neurons and GFAP and CD11B for astrocytes and microglia, respectively). Cell counting was based on cells’ Nissl staining (including counting of TPH-immunoreactive (-ir) neurons, which were Nissl counter-stained); the same sections were used for counting Nissl-stained neurons, glia and TPH-ir neurons. NeuN-ir neurons were counted in a separate population of rats and their counting was not based on Nissl-staining (see below).
The numbers of cells were estimated separately for each half of the RVM (i.e. both ipsilateral and contralateral to the surgery). Systematic random sampling and unbiased stereological methods were used for quantification as described previously (Gu and Wessendorf, 2007). Five RVM sections (each 50 μm thick) per animal were sampled; the first section sampled was selected randomly. The sampling interval thereafter was determined based on the number of sections within the RVM and was usually every sixth section. For each selected section, a low-magnification (2×) image of the RVM was captured. A grid was randomly cast over the image (Figure 1, line drawing) and systematic random sampling was again used when choosing the grid intersections to be evaluated (Howard and Reed, 1998; Reed and Howard, 1998). Once an intersection was chosen, a stack of images was made at 40× while focusing through the full thickness of the section (Figure 1, C–D). Cells were counted as follows. A counting frame was superimposed on the stack of images (Howard and Reed, 1998). The right boundary and the upper boundary of the counting frame were used as acceptance lines (Figure 1, C–D, dotted lines); the other two boundaries were forbidden lines (Figure 1, C–D, solid lines). Neurons were counted only if nuclei (for NeuN-labeled neurons) or nucleoli (for Nissl-stained neurons) (Popken and Farel, 1997) either fell entirely inside the counting frame or if they crossed an acceptance line without also crossing a forbidden line. NeuN-labeled neurons were counted when the top of the nucleus was found within the thickness of the tissue section. Nissl-stained neurons were counted when the nucleolus was present within the thickness of the tissue section and TPH-ir neurons were counted based on their Nissl counterstaining. To estimate the total number of neurons (N) in the RVM per animal, we used the following equation:
where D = the neuronal density of the RVM, V = the volume of the RVM. To estimate the neuronal density of the RVM in each rat, we used the following equation:
where N = the number of neurons counted per rat, a = 2,500 μm2 (the area of the counting frame sampled), t = 50 μm (the thickness of the section sampled), and g = 20 (the number of counting frames sampled per rat). To estimate the volume of the RVM from each rat, we used the following equation:
where A = the cross sectional area of RVM in which the sections were sampled, I = the interval between adjacent sampled sections, and t = 50 μm (the thickness of section sampled).
To determine the effects of SNL on spinal 5-HT innervation, the density of 5-HT labeling was examined in the superficial dorsal horn using a 40×/1.4 NA objective. The number of 5-HT varicosities in the lateral and medial superficial dorsal horn was determined using the “Finding Maxima” routine of ImageJ. The density was then calculated by dividing those numbers by the corresponding cross-sectional areas of the regions being examined. The same value for noise tolerance was used in all cases.
The intensity of MOR-ir in the RVM was also determined with ImageJ. A region of interest was outlined and the mean intensity determined using the measurement function.
Statistics
Differences among treatment groups were identified either by 1-way or 2-way analyses of variances (ANOVA); post-hoc comparisons were made after 2-way ANOVAs with a Bonferroni test. Chi-square tests were used to detect differences in proportions. P< 0.05 was considered significant. Statistical tests were performed using the GraphPad Prism software (La Jolla, CA) and the statistical tools available on the GraphPad website (http://www.graphpad.com).
Von Frey Testing for Tactile Hypersensitivity
Mechanical sensitivity was determined by measuring the paw withdrawal threshold in response to the application of von Frey filaments, using the up-down method of Chaplan (Chaplan et al., 1994). In brief, rats were placed on a wire mesh surface, covered by an inverted plastic cage and allowed to habituate for 15 min. Filaments were then applied to the plantar surface of the hindpaw. If application of the filament evoked no response, the next-larger filament was applied; if application elicited withdrawal, the next-smaller filament was applied. This sequence was repeated until the largest filament was used or until four filaments were applied after the first withdrawal was noted. Withdrawal thresholds were measured prior to surgery and at 2–3 day intervals after surgery. In all cases, thresholds were measured bilaterally.
RESULTS
SNL induced tactile hypersensitivity ipsilateral to the surgery
As previously reported (Kim and Chung, 1992; Blenk et al., 1997; Ringkamp et al., 1999; Li et al., 2000), SNL resulted in hypersensitivity to tactile stimuli ipsilateral, but not contralateral, to the lesion. Ten days after surgery the withdrawal threshold for the hindpaw ipsilateral to the surgery was 2.7±0.5 g (mean± standard error of the mean, SEM) in rats receiving SNL, which was significantly lower than that in sham-operated rats (14.0±2.7 g; p<0.05, n=8 animals for each group, total degrees of freedom [d.f] = 31). Thresholds in the ipsilateral paws of rats subjected to SNL were also significantly lower than those in their contralateral hindpaws (14.5±1.3 g; p<0.05, Figure 2A). Paw withdrawal thresholds determined before surgery were not significantly different between experimental groups.
Figure 2.
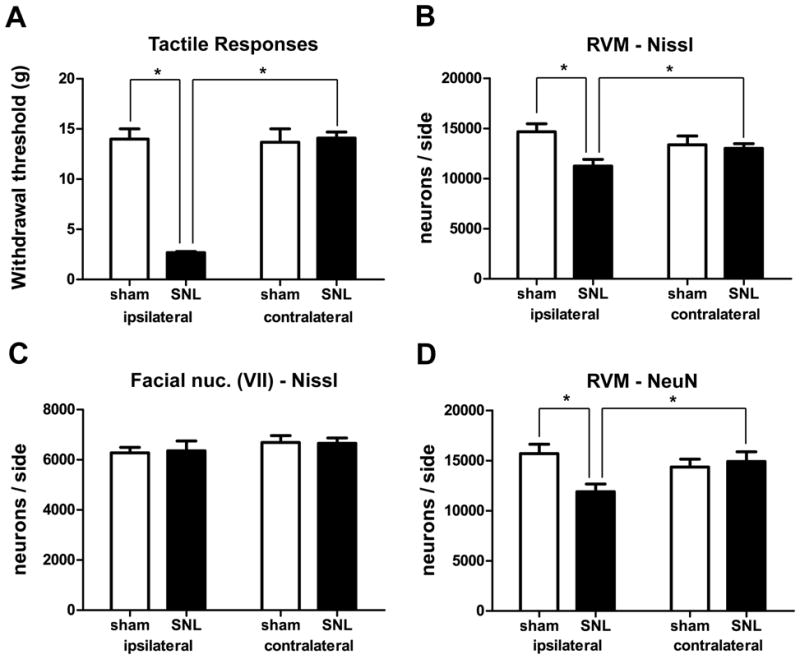
SNL induced tactile hypersensitivity and neuronal loss in RVM. A. Paw withdrawal thresholds, measured by von Frey filaments, ipsilateral to the ligation were significantly decreased compared to those in sham-operated rats or to those observed on the contralateral side in SNL rats (*, p<0.05; 2-way ANOVA). B. Significantly fewer Nissl-stained neurons were observed ipsilateral to SNL (*, p<0.05, 2-way ANOVA). C. SNL resulted in no significant differences in the number of neurons in the facial nucleus (p>0.05, 2-way ANOVA). D. SNL resulted in a significant decrease in the number of NeuN-labeled neurons ipsilateral to the surgery (*, p<0.05, 2-way ANOVA).
SNL reduced the number of RVM neurons
SNL resulted in a significant decrease in the number of RVM neurons ipsilateral to the lesion (Figure 1A,B; Figure 2B). In rats that had received SNL, the number of Nissl-stained neurons in the half of the RVM ipsilateral to the lesion was 11,248±668 (mean±SEM), which was significantly less (by 23%) than the number found in the ipsilateral side of the RVM in sham-operated rats (14,665±800, p<0.05, n=8 animals in each group, total d.f. = 31). The number of neurons ipsilateral to SNL was also significantly less (by 14%) than that on the contralateral side of the same animals (p<0.05, Figure 2B) but the number of RVM neurons on the contralateral side was not significantly different between rats receiving SNL and sham-operated rats (SNL: 13,003±471 vs. sham: 13,365±872, p>0.05, Figure 2B). No significant differences were observed between the volumes of the RVMs in sham-operated and experimental rats (sham ipsilateral=1.123±0.050 mm3; sham contralateral=1.131±0.057 mm3; SNL ipsilateral=1.075 ±0.079 mm3; SNL contralateral=1.077±0.083 mm3, p>0.05, n=8 animals, in each group, total d.f. = 31).
The loss of neurons after SNL was not non-specific. The number of neurons in the facial nucleus, which is adjacent to the RVM but not involved in pain modulation, was unchanged (ipsilateral sham: 6,267±219; ipsilateral SNL 6,204±422; contralateral sham: 6,693±270; contralateral SNL: 6,542±187: p>0.05, n = 8 animals in each group, total d.f. = 31, Figure 2C). This suggests that cell loss in the RVM was not due to any indiscriminant brain stem pathology resulting from SNL.
To confirm that SNL did not simply transform RVM neurons to cells with a glia-like morphology, we repeated the experiment in a second group of animals, staining sections for the neuronal marker NeuN (Mullen et al., 1992). SNL resulted in a significant decrease in the number of RVM NeuN-ir neurons ipsilateral to the lesion. In rats subjected to SNL, the number of NeuN-ir neurons in the half of the RVM ipsilateral to the lesion was 11,898±768, which was significantly less (24%) than the number found in the ipsilateral side of sham-operated rats (15,688±945, p<0.05, n=8 animals in each group, total d.f. = 31, Figure 2D). The number of NeuN-ir neurons ipsilateral to SNL was also significantly less (by 20%) than that on the contralateral sides of the same animals (14,913±964, p<0.05, Figure 2D). Thus, the NeuN results validated those obtained using Nissl staining and the latter method was used as the basis of neuronal counting for the remainder of these experiments.
RVM TPH-ir neurons were decreased in rats receiving SNL
TPH, the rate-limiting enzyme in 5-HT synthesis, was used as a marker for 5-HT neurons in the RVM. In both rats receiving SNL and sham-operated rats, TPH-ir neurons were found throughout the RVM. However, the number of RVM TPH-ir neurons ipsilateral to SNL was significantly less (by 35%) than the number of TPH-ir neurons ipsilateral to sham surgery (SNL ipsilateral: 773±134 vs. sham ipsilateral: 1,184±92, p<0.05, n=8 animals in each group, total d.f. = 31, Figure 3). Within SNL animals, the number of TPH-ir neurons ipsilateral to the lesion was also significantly less (27%) than the number contralateral to the lesion (SNL contralateral: 1,055±222 p<0.05, Figure 3C). The proportion of 5-HT cells lost (35%) was significantly larger than the percent decrease observed among all RVM neurons (23%), suggesting that 5-HT neurons are more sensitive to the effects of peripheral nerve damage than RVM neurons as a whole (p<0.0001; chi-square with Yates correction).
Figure 3.
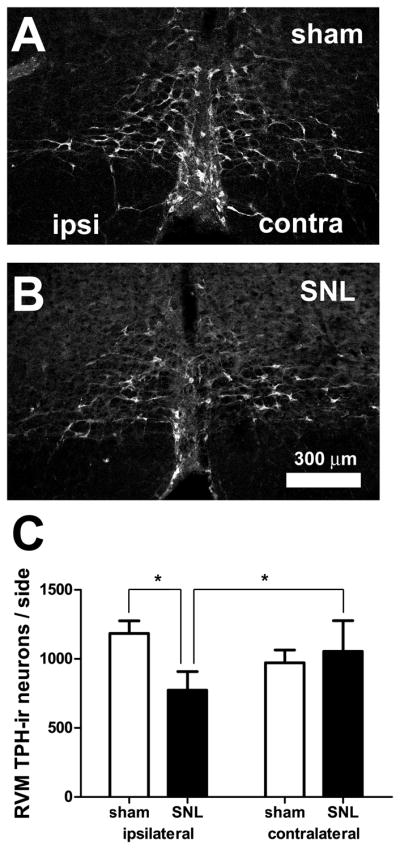
SNL induced loss of tryptophan hydroxylase-immunoreactive (TPH-ir) neurons in the RVM. A–B. Confocal images showing TPH-ir in the RVM in sham-operated (A) and SNL-treated (B) rats. Parameters for image acquisition and display (brightness, contrast, filtering) were identical. “Ipsilateral” and “contralateral” are relative to the nerve ligation. C. The number of RVM TPH-ir neurons ipsilateral to SNL was significantly less than contralateral to SNL. In addition, the number was significantly less than that found ipsilateral to sham-surgery (*, p<0.05; 2-way ANOVA). TPH-ir neurons were counted based on their Nissl counter-staining, as described in the Methods section. Scale bar in B applies to A and B.
Fewer 5-HT-ir varicosities found in substantia gelatinosa ipsilateral to SNL
Since RVM 5-HT neurons project to dorsal spinal cord (Bowker et al., 1981; Skagerberg and Bjorklund, 1985; Kwiat and Basbaum, 1992; Kalyuzhny et al., 1996), we examined whether spinal 5-HT-ir decreased after SNL (Figure 4). At spinal levels C2, T2 and L4, the density of 5-HT-ir varicosities in the ipsilateral superficial dorsal horn was significantly lower in rats receiving SNL than in sham-operated rats. At the cervical level, the density of 5-HT varicosities in rats treated with SNL was 0.033±0.002 varicosities / μm2 on the ipsilateral side (where 1 μm2 = 10−12 m2), which was significantly lower (by 30%) than in sham-operated rats (0.047±0.004 / μm2, p<0.05, n=6 animals receiving SNL, n=8 sham-operated animals, total d.f. = 27, Figure 4A). Densities in animals treated with SNL at the thoracic and lumbar levels (0.039±0.005 / μm2 and 0.043±0.004 / μm2, respectively) were also significantly lower on the ipsilateral side (decreases of 22% and 15%, respectively) than those in sham-operated rats (Figure 4B,C). There were no significant differences on the contralateral sides of the superficial dorsal horn at any spinal level.
Figure 4.

Effect of SNL on serotonin-immunoreactivity (5-HT-ir) in the superficial dorsal horn of the spinal cord. A–C. SNL significantly decreased the density of 5HT-ir varicosities at all spinal levels examined. (A) Segment C2, (B) Segment T2, (C) Segment L4 (*, p<0.05; 2-way ANOVA). D. Images of 5-HT-ir in superficial dorsal horn of rats receiving sham surgery or SNL. Note reduced 5-HT-ir in SNL image. Box in inset denotes area from which images were obtained.
Depletion of 5-HT reversed mechanical hypersensitivity after SNL
To determine whether loss of the RVM 5-HT neurons remaining after SNL would further exacerbate cutaneous hypersensitivity, we used intracisternal injection of the serotonin neurotoxin 5,7-DHT to deplete 5-HT in rats that had undergone SNL (Figure 5). Rather than being lower, mechanical withdrawal thresholds were significantly higher after 5,7-DHT (5.2 ±1.3 g prior to injection vs. 10.2± 1.7 g after injection; p<0.05, n=10 animals in each group, total d.f. = 39, Figure 5A). These thresholds were also significantly higher than those in rats that had received SNL and that were subsequently treated with saline rather than 5,7-DHT (10.2±1.7 g vs 4.5±1.3 g, respectively; p<0.05). Treatment with 5,7-DHT markedly reduced 5-HT-ir in the spinal cord (Figure 5B).
Figure 5.

Intracisternal injection of the serotonin neurotoxin 5,7 dihydroxytryptamine (5,7-DHT) partially reversed mechanical hypersensitivity after SNL. A. Withdrawal thresholds became significantly higher after rats receiving SNL were injected with 5,7-DHT. Also, thresholds were significantly higher in rats receiving SNL that were treated with 5,7-DHT than in rats receiving SNL that were given saline (*, p<0.05; 2-way ANOVA). B. Image of 5-HT-ir in superficial dorsal horn of a rat that had received SNL and that was treated with 5,7-DHT (compare to Figure 4D). Note lack of 5-HT-ir. Box in inset denotes area from which image was obtained.
Administration of tauroursodeoxycholic acid (TUDCA) reduced both the loss of RVM neurons and cutaneous hypersensitivity after SNL
Previous studies have reported that neuronal apoptosis occurs in the spinal cord and cerebral cortex after nerve injury and that inhibiting caspases reduces cell loss (Coggeshall et al., 2001; Whiteside and Munglani, 2001; Moore et al., 2002; Scholz et al., 2005; Siniscalco et al., 2007; Fuccio et al., 2009). To test whether apoptosis might mediate the cell loss we observed in the RVM, we administered an apoptosis inhibitor; to avoid the local damage that could result from a direct microinjection of a caspase inhibitor into the RVM, we gave rats a long-acting inhibitor of apoptosis that can be administered systemically. Ursodeoxycholic acid (UDCA) is a bile acid used for treating hepatic disease (Ward et al., 1984) that has been reported to inhibit apoptosis (for reviews, see Rodrigues and Steer, 2001; Solá et al., 2007) and that acts at least in part by inhibiting the translocation of Bax from the cytosol to the mitochondrion (Rodrigues et al., 1998). UDCA also activates cell survival pathways (Solá et al., 2007). The taurine conjugate of UDCA (TUDCA) has similarly been reported to inhibit apoptosis (Rodrigues et al., 2003; Ramalho et al., 2008) and promote cell survival (Schoemaker et al., 2004), but has greater solubility in water. For this reason, TUDCA was used in these experiments.
We first tested the actions of TUDCA by examining its effects on apoptosis in the thymus. We induced apoptosis in the thymus by treatment with 1 mg/kg dexamethasone, an immunosuppressant glucocorticoid (Compton et al., 1987; Quaglino and Ronchetti, 2001), which greatly enhanced staining for caspase-3. However, in rats treated with both TUDCA and dexamethasone, we found a marked decrease in caspase-3 labeling compared to treatment with dexamethasone alone (Figure 6).
Figure 6.

TUDCA inhibited dexamethasone-induced apoptosis in thymus. A. Dexamethasone (1 mg/kg) increased staining for activated caspase-3 in thymus. B. Treatment with TUDCA decreased caspase-3-ir in dexamethasone–treated rats. C. TUDCA administration alone did not cause increased caspase-3 staining. D. Thymus tissue from naïve rats showed little apoptosis.
In the brain stem, we found that administration of TUDCA prevented loss of RVM neurons after SNL (Figure 7). In rats receiving SNL and treated with TUDCA, the number of RVM neurons ipsilateral to the lesion was 17,976±1,202, which was significantly higher (by 43%) than the number found in the ipsilateral sides of vehicle-treated rats that had received SNL (12,540±930; p<0.05, n=8 animals in each group, total d.f. = 31, Figure 7A). As expected, in vehicle-treated rats, the number of RVM neurons in the ipsilateral side (12,540±929) was significantly less (by 26%) than the number found in the contralateral side (16,949±1,074, p<0.05, Figure 7A). There was no significant difference between the number of neurons in the ipsilateral and contralateral sides of TUDCA-treated rats receiving SNL (TUDCA ipsi: 17,976±1,202, TUDCA contra: 16,675±828, Figure 7A), nor between the numbers of neurons found in the contralateral sides of the two treatment groups (SNL with TUDCA: 16,675±828, SNL with saline: 16,949±1,074, p>0.05, Figure 7A).
Figure 7.

TUDCA prevented neuronal loss after SNL, including serotonergic cell loss, and partially reversed SNL-induced mechanical hypersensitivity. (A) Compared to vehicle-treated rats, TUDCA-treated rats had significantly more Nissl-stained neurons ipsilateral to SNL (*, p<0.05, 2-way ANOVA). (B) TUDCA-treated rats also had significantly more TPH-ir neurons after SNL (*, p<0.05, 2-way ANOVA). In contrast to saline-treated rats, there were no significant differences between the numbers of RVM neurons ipsilateral and contralateral to SNL in TUDCA-treated animals. C. Beginning four days post-SNL, the withdrawal thresholds of the ipsilateral hindpaws of TUDCA-treated rats were significantly higher than those in animals treated with saline (p<.05; 2-way ANOVA). There were no significant differences between the withdrawal thresholds of the contralateral hindpaws at any time point. Inverted triangles = contralateral saline; squares = contralateral TUDCA; circles = ipsilateral TUDCA; triangles = ipsilateral saline.
TUDCA also prevented the loss of 5-HT neurons in the RVM. The number of RVM 5-HT neurons ipsilateral to SNL in TUDCA-treated animals (1,353±96) was significantly higher (by 30%) than the number in vehicle-treated rats (1,043±118, p<0.05, n=8 animals in each group, total d.f. = 31, Figure 7B). Again, as expected, the number of 5-HT-ir RVM neurons in the ipsilateral side of vehicle-treated rats (1,043±118) was significantly less (by 27%) than that in the contralateral side (1,427±211, p<0.05, Figure 7B). In TUDCA-treated rats receiving SNL, there was no significant difference between the number of neurons in the ipsilateral and contralateral sides (TUDCA ipsi: 1,353±96, TUDCA contra: 1,251±99, p>0.05, Figure 7B). Despite the efficacy of TUDCA, though, we found neither caspase-3 labeling, caspase-6 labeling nor TUNEL in the RVM after SNL.
TUDCA administration also reduced SNL-induced mechanical hypersensitivity. Beginning at day 4 post-SNL, the withdrawal thresholds of the ipsilateral hindpaws of TUDCA-treated rats (Day 4: 3.5±0.5 g; Day 6: 4.0±0.5 g; Day 8: 4.6±1.1 g; Day 10: 5.2±1.3 g) were significantly higher than those animals treated with vehicle (Day 4: 1.1±0.3 g; Day 6: 1.3±0.2 g; Day 8: 1.2±0.3g; Day 10: 1.2±0.2 g; p<0.05, n=10 animals treated with TUDCA, n=9 animals treated with vehicle, d.f. interaction = 7, d.f. treatment = 1, d.f. time = 7, d.f. subjects = 17, d.f. residual = 119, total d.f. = 151, Figure 7C).
Glia: activation and increased numbers after SNL
RVM glial activation appears to have a role in descending facilitation of nociception in a model of persistent pain (Wei et al., 2008). To determine whether glial activation might be responsible for the neuronal loss, we stained for markers of astrocytes (GFAP) and microglia (CD11B) in RVM after SNL. At day three, we found a bilateral increase in activation of astrocytes in animals receiving SNL as compared to sham-operated animals (Figure 8A–D); GFAP staining was decreased by day 10. Conversely, although little labeling was observed at day three, we found markedly stronger labeling for CD11B at day 10 in rats receiving SNL. Again, the labeling increased both ipsilateral and contralateral to SNL (Figure 8E–H). Using Nissl-stained tissue, we also found a significant bilateral increase in numbers of glia in the RVM of SNL-treated animals as compared to sham-operated animals (SNL ipsi: 87,277± 10,354 vs. sham ipsi: 60,430± 7,551 or 44% higher, SNL contra: 85,903± 9,091 vs. sham contra: 56,104± 7,912 or 52% higher; p<0.05, n=5 animals in each group, total d.f. = 19, Figure 8I). To determine whether this increase in glial activation and number was non-selective and found across the brain stem, we examined the adjacent facial nucleus. In that nucleus we found no significant increase in the number Nissl-stained glia (Figure 8J) or in the activation of glia in SNL-treated rats, compared to sham-operated rats. Treatment with TUDCA significantly reduced the increase in RVM glia observed after SNL (TUDCA ipsi: 62,306±3,736 vs. vehicle ipsi: 85,541±2,972, or 27% fewer; TUDCA contra: 60,437±4,315 vs. vehicle contra: 80,086±2,899, or 25% fewer; p<0.05 in both cases, n=7 animals treated with TUDCA, n=6 animals treated with vehicle, total d.f. = 25), suggesting that the increase in glia was in response to cell death.
Figure 8.

SNL induced a bilateral activation of astrocytes and microglia and an increase in glial number. A–D. At day 3, astrocytes were activated (as shown by increased GFAP labeling) in the ipsilateral and contralateral RVM after SNL, compared to sham. E–H. At day 10, microglia were activated after SNL compared to sham, as shown by increased labeling for the microglial marker, CD11b. I. We observed significantly more Nissl-stained glia in the RVM after SNL and the increase was bilateral. J. No significant increases in glial number were found in Facial nucleus after SNL. Scale bar in H applies to A–H.
MOR expression in RVM after SNL
Previous studies have suggested that RVM neurons expressing the mu-opioid receptor (MOR) mediate descending facilitation of nociception after SNL (Porreca et al., 2001). Although RVM neurons were lost after SNL (see above), MOR-ir in the RVM appeared to be unchanged (Figure 9A,B). When the intensity of MOR staining was measured there was no significant difference between sham animals (55.5±2.6 arbitrary intensity units) and animals receiving SNL (53.7±3.2 arbitrary intensity units; n=3 animals in each group, t-test, d.f.=4, p>0.05). MOR-ir after SNL was observed in TPH-ir neurons as well as in neurons that were not immunoreactive for TPH (Figure 9B).
Figure 9.

MOR-immunoreactivity in the RVM appears to be unchanged after SNL. A. MOR-ir in RVM ipsilateral to sham surgery. B. MOR-ir in RVM ipsilateral to SNL. RVM neurons, including TPH-ir neurons expressed MOR-ir after SNL. Section was double-stained; insets show expression of MOR-ir by TPH-ir RVM neurons. Insets were taken from the region outlined by the square. C. Absorption control. Section of RVM serially adjacent to that in B; note marked reduction in labeling. Section was also double-stained and the same TPH-ir neurons visible in B can be seen in the insets. Again, the square indicates the region from which the insets were taken. Scale bar in insets = 30 μm. The 300 μm scale bar in C applies to A–C.
DISCUSSION
Nerve injury and disease can trigger a range of responses in both the peripheral and central nervous systems that contribute to chronic neuropathic pain (Devor, 2006). In the periphery, injured primary sensory neurons develop hyperexcitability and abnormal impulse generation (Blumberg and Janig, 1984; Devor, 2006). Other pathophysiological changes observed in injured primary sensory neurons include the altered regulation and expression of certain molecules (e.g. neuropeptides, ion channels, enzymes, etc.) (Hokfelt et al., 2006). Increased spinal cord excitability ipsilateral to the injury (i.e. central sensitization) also appears to contribute crucially to abnormal pain conditions after tissue injury (Ji et al., 2003). N-methyl-D-aspartate (NMDA) receptor activation is one of the principal mechanisms in central sensitization, and its role in neuropathic pain is implied by preclinical studies showing that NMDA receptor antagonists are effective in alleviating experimental neuropathic pain (Wei and Pertovaara, 1999; Woolf and Salter, 2000; Hokfelt et al., 2006).
Pathophysiological changes in the RVM also contribute to neuropathic pain. Microinjection of a NMDA receptor antagonist before SNL (Wei and Pertovaara, 1999), and microinjection of lidocaine in the RVM after SNL have both been shown to reduce allodynia (Pertovaara et al., 1996), suggesting that activation of (and activity in) the RVM contributes to cutaneous hypersensitivity. Consistent with this idea, lesioning the DLF, in which RVM axons descend to the spinal cord, has also been reported to reverse both thermal hyperalgesia and allodynia after SNL (Ossipov et al., 2000). In addition, selective ablation of MOR-expressing RVM neurons has been reported to prevent SNL-induced experimental hypersensitivity (Porreca et al., 2001). However, neither DLF lesions nor microinjection of lidocaine into the RVM reduced cutaneous hypersensitivity within three days after SNL--they did so only 4–6 or more days after SNL (Burgess et al., 2002). Based on these and similar findings, it has been proposed that physiological changes in the RVM contribute to the maintenance of neuropathic pain but not to its initiation.
Fewer RVM neurons after SNL
In our present studies, it was found that the number of RVM neurons ipsilateral to SNL was significantly decreased compared to sham-operated rats. Several prior studies have shown that neurons at the spinal level undergo apoptosis ipsilateral to peripheral nerve injury (Sugimoto et al., 1990; Whiteside and Munglani, 2001; Scholz et al., 2005) and apoptosis has been reported in cortex in response to nerve injury (Fuccio et al., 2009). Apoptosis would be expected to result in cell loss, although to date the loss of neurons in those regions has been controversial (Polgar et al., 2004; Polgar et al., 2005).
Our finding of neuronal loss in the RVM suggests that death of pain modulatory neurons contributes to the pathophysiology of neuropathic pain. Although it is possible that both pro- and anti-nociceptive neurons are lost after SNL, the most parsimonious interpretation of our findings is that SNL selectively kills antinociceptive RVM neurons, thereby facilitating cutaneous hypersensitivity (Figure 10).
Figure 10.

Summary Diagram. The loss of antinociceptive neurons underlies, in part, the hypersensitivity observed in neuropathic pain. Gray triangle indicates the borders of the RVM. We propose that after SNL, antinociceptive RVM neurons (filled circles) retrogradely degenerate after apoptosis of intrinsic spinal cells (squares) and primary afferent fibers. The resulting loss of descending inhibitory tone contributes to cutaneous hypersensitivity.
Treatment with TUDCA blocked RVM cell loss and significantly decreased tactile hypersensitivity four or more days after SNL but did not reduce hypersensitivity one-to-three days after SNL (Figure 7). Thus our findings suggest that cell loss underlies the RVM’s contribution to the maintenance phase of neuropathic pain (see above). Glial activation in the RVM may also contribute to cutaneous hypersensitivity (Wei et al., 2008), although since we found that it occurs bilaterally it would not by itself explain the unilateral decrease in withdrawal thresholds; instead, glial activation may be an important factor in the initiation of neuropathic pain (Mei et al., 2011). In our studies, we also found that the number of glia increased after SNL. This increase was blocked by TUDCA, suggesting it may represent gliosis in response to RVM neuronal death.
Although a significant number of 5-HT neurons were lost after SNL (see below), most cells that were lost (88%) did not express 5-HT. The identity of these neurons is not known but it has been proposed that the antinociceptive neurons of the RVM are physiologically characterized as OFF-cells (Fields et al., 1983b; Fields et al., 1983a; Heinricher et al., 1989; Fields et al., 1991). Many OFF-cells project to the dorsal horn (Fields et al., 1995) and express GABA (Winkler et al., 2006). Previous studies have suggested that changes in ON-cell and OFF-cell firing drive the hypersensitivity observed in SNL (Carlson et al., 2007; Goncalves et al., 2007), but it has not yet been determined whether or not the number of OFF-cells changes. It has also been reported that neuropathic pain is maintained by RVM neurons expressing MOR (Porreca et al., 2001; Burgess et al., 2002). Consistent with this finding, MOR-ir appeared not to decrease after SNL and RVM neurons (including 5-HT neurons) expressing MOR were still observed after SNL.
Mechanism of cell loss
The cause of RVM cell loss is unclear. It has been reported that hypersensitivity after SNL could be blocked by a single microinjection of an NMDA antagonist into the RVM (Wei and Pertovaara, 1999), suggesting that excessive activity among glutamatergic afferents immediately after injury might be sufficient to kill RVM neurons. Although NMDA receptor activation may contribute to cell loss, it is unclear how activation only of ipsilateral glutamatergic afferents could be evoked physiologically. Similarly, although RVM glial activation might contribute to cell loss, it appears unlikely to be the sole cause since activation was observed bilaterally. An alternative explanation is that loss of RVM neurons arises from their spinal projections. RVM neurons project predominantly ipsilaterally to the spinal cord dorsal horn (Skagerberg and Bjorklund, 1985) and nerve damage or primary afferent activity have been reported to result in cell loss or apoptosis in the dorsal horn and in dorsal root ganglia (Coggeshall et al., 2001; Whiteside and Munglani, 2001; Maione et al., 2002; de Novellis et al., 2004; Polgar et al., 2005; Scholz et al., 2005; Hassanzadeh and Ahmadiani, 2006; Jalalvand et al., 2008; Sekiguchi et al., 2009; Meisner et al., 2010). Thus we propose that apoptosis of spinal cells (either neurons, glia or primary afferent fibers) in the terminal fields of bulbospinal neurons triggers retrograde degeneration of RVM neurons.
TUDCA administration blocked RVM cell loss. TUDCA reportedly inhibits apoptosis and has been shown to have beneficial effects in in vivo and in vitro models of neurological damage that result in apoptosis, including hemorrhagic and ischemic stroke (Rodrigues et al., 2002; Rodrigues et al., 2003). However, to date we have been unable to demonstrate caspase-3, caspase-6 or TUNEL in the RVM after SNL, and in a study of another model of neuropathic pain, chronic constriction injury, apoptosis was not observed in the RVM (Wei et al., 2008). Thus, since TUDCA was given systemically, we conclude that it may have acted indirectly (i.e. on spinal cord or dorsal root ganglia: see above) rather than directly on the RVM. Nevertheless, apoptotic events are brief (Kerr et al., 1972; Bursch et al., 1990; Sanchez et al., 1992), which can result in few or no apoptotic cells being detected (Lawen, 2003). Moreover, TUDCA also promotes cell survival by other means than inhibiting apoptosis (Schoemaker et al., 2004) and it is possible that RVM neurons are lost via some mechanism other than the classical apoptotic pathway. Thus it remains possible that TUDCA blocks RVM cell loss by acting directly on RVM neurons.
Pain facilitation by 5-HT
In the present study, about 8% of RVM neurons were found to be serotonergic (i.e. they expressed TPH-ir). Of these, about one-third was lost ipsilateral to SNL. The remaining 5-HT neurons appeared to facilitate nociception: administration of 5,7-DHT, a serotonin neurotoxin, significantly improved (i.e. increased) withdrawal thresholds after SNL. This facilitatory effect might either be direct (e.g., via 5-HT3 receptors (Suzuki et al., 2005)) or indirect (e.g., via gating the facilitatory or inhibitory output of the RVM). Since 5,7-DHT was given intracisternally, this finding could be due at least in part to 5,7-DHT acting on 5-HT neurons outside the RVM. However, previous studies have suggested that bulbospinal 5-HT neurons do contribute to persistent pain (McCleane et al., 2003; Suzuki et al., 2004; Rahman et al., 2006; Dogrul et al., 2009; Wei et al., 2010) and our findings are consistent with there being a role for 5-HT in maintaining neuropathic pain.
In summary, our data indicate that almost one-fourth of RVM neurons are lost after SNL, and that preventing this loss reduces the hypersensitivity observed after SNL. We propose that death of antinociceptive neurons in the RVM is responsible, at least in part, for maintaining tactile hypersensitivity after nerve damage and that changes in the RVM and the spinal cord together contribute to the phenomenon of neuropathic pain. Reducing RVM neuronal death and the associated hypersensitivity with anti-apoptotic drugs may prove useful for preventing neuropathic pain in humans.
Acknowledgments
Supported by PHS grant DA017758 to MW, T32 DA 007234, the Minnesota Medical Foundation, and the Office of the Vice President for Research, University of Minnesota.
References
- Amir R, Devor M. Functional cross-excitation between afferent A- and C-neurons in dorsal root ganglia. Neuroscience. 2000;95:189–195. doi: 10.1016/s0306-4522(99)00388-7. [DOI] [PubMed] [Google Scholar]
- Bjorklund A, Baumgarten HG, Rensch A. 5,7-Dihydroxytryptamine: improvement of its selectivity for serotonin neurons in the CNS by pretreatment with desipramine. J Neurochem. 1975;24:833–835. [PubMed] [Google Scholar]
- Blenk KH, Habler HJ, Janig W. Neomycin and gadolinium applied to an L5 spinal nerve lesion prevent mechanical allodynia-like behaviour in rats. Pain. 1997;70:155–165. doi: 10.1016/s0304-3959(96)03314-3. [DOI] [PubMed] [Google Scholar]
- Blumberg H, Janig W. Discharge pattern of afferent fibers from a neuroma. Pain. 1984;20:335–353. doi: 10.1016/0304-3959(84)90111-8. [DOI] [PubMed] [Google Scholar]
- Bowker RM, Westlund KN, Coulter JD. Origins of serotonergic projections to the spinal cord in rat: an immunocytochemical-retrograde transport study. Brain Res. 1981;226:187–199. doi: 10.1016/0006-8993(81)91092-1. [DOI] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–5136. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursch W, Paffe S, Putz B, Barthel G, Schulte-Hermann R. Determination of the length of the histological stages of apoptosis in normal liver and in altered hepatic foci of rats. Carcinogenesis. 1990;11:847–853. doi: 10.1093/carcin/11.5.847. [DOI] [PubMed] [Google Scholar]
- Carlson JD, Maire JJ, Martenson ME, Heinricher MM. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. J Neurosci. 2007;27:13222–13231. doi: 10.1523/JNEUROSCI.3715-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Lekan HA, White FA, Woolf CJ. A-fiber sensory input induces neuronal cell death in the dorsal horn of the adult rat spinal cord. J Comp Neurol. 2001;435:276–282. doi: 10.1002/cne.1029. [DOI] [PubMed] [Google Scholar]
- Compton MM, Caron LA, Cidlowski JA. Glucocorticoid action on the immune system. J Steroid Biochem. 1987;27:201–208. doi: 10.1016/0022-4731(87)90311-6. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Siniscalco D, Galderisi U, Fuccio C, Nolano M, Santoro L, Cascino A, Roth KA, Rossi F, Maione S. Blockade of glutamate mGlu5 receptors in a rat model of neuropathic pain prevents early over-expression of pro-apoptotic genes and morphological changes in dorsal horn lamina II. Neuropharmacology. 2004;46:468–479. doi: 10.1016/j.neuropharm.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Devor M. Responses of nerves to injury in relation to neuropathic pain. In: McMahon S, Koltzenburg M, editors. Wall and Melzack’s Textbook of Pain. London: Elsevier Limited; 2006. pp. 905–927. [Google Scholar]
- Dogrul A, Ossipov MH, Porreca F. Differential mediation of descending pain facilitation and inhibition by spinal 5HT-3 and 5HT-7 receptors. Brain Res. 2009;1280:52–59. doi: 10.1016/j.brainres.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in Nociceptive Modulatory Circuits. Annual Review of Neuroscience. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Fields HL, Malick A, Burstein R. Dorsal horn projection targets of ON and OFF cells in the rostral ventromedial medulla. J Neurophysiol. 1995;74:1742–1759. doi: 10.1152/jn.1995.74.4.1742. [DOI] [PubMed] [Google Scholar]
- Fields HL, Vanegas H, Hentall ID, Zorman G. Evidence that disinhibition of brain stem neurones contributes to morphine analgesia. Nature. 1983a;306:684–686. doi: 10.1038/306684a0. [DOI] [PubMed] [Google Scholar]
- Fields HL, Bry J, Hentall I, Zorman G. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J Neurosci. 1983b;3:2545–2552. doi: 10.1523/JNEUROSCI.03-12-02545.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccio C, Luongo C, Capodanno P, Giordano C, Scafuro MA, Siniscalco D, Lettieri B, Rossi F, Maione S, Berrino L. A single subcutaneous injection of ozone prevents allodynia and decreases the over-expression of pro-inflammatory caspases in the orbito-frontal cortex of neuropathic mice. Eur J Pharmacol. 2009;603:42–49. doi: 10.1016/j.ejphar.2008.11.060. [DOI] [PubMed] [Google Scholar]
- Goncalves L, Almeida A, Pertovaara A. Pronociceptive changes in response properties of rostroventromedial medullary neurons in a rat model of peripheral neuropathy. Eur J Neurosci. 2007;26:2188–2195. doi: 10.1111/j.1460-9568.2007.05832.x. [DOI] [PubMed] [Google Scholar]
- Gu M, Wessendorf M. Endomorphin-2-immunoreactive fibers selectively appose serotonergic neuronal somata in the rostral ventral medial medulla. J Comp Neurol. 2007;502:701–713. doi: 10.1002/cne.21343. [DOI] [PubMed] [Google Scholar]
- Hassanzadeh P, Ahmadiani A. Nitric oxide and c-Jun N-terminal kinase are involved in the development of dark neurons induced by inflammatory pain. Synapse. 2006;59:101–106. doi: 10.1002/syn.20219. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Barbaro NM, Fields HL. Putative nociceptive modulating neurons in the rostral ventromedial medulla of the rat: firing of on- and off-cells is related to nociceptive responsiveness. Somatosens Mot Res. 1989;6:427–439. doi: 10.3109/08990228909144685. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Zhang X, Wiesenfeld-Hallin Z. Central consequences of peripheral nerve damage. In: McMahon S, Koltzenburg M, editors. Wall and Melzack’s Textbook of Pain. London: Elsevier; 2006. pp. 947–959. [Google Scholar]
- Howard CV, Reed MG. Unbiased Stereology: Three-Dimensional measurement in microscopy. Oxford: BIOS Scientific Publishers; 1998. [Google Scholar]
- Jalalvand E, Javan M, Haeri-Rohani A, Ahmadiani A. Stress- and non-stress-mediated mechanisms are involved in pain-induced apoptosis in hippocampus and dorsal lumbar spinal cord in rats. Neuroscience. 2008;157:446–452. doi: 10.1016/j.neuroscience.2008.08.052. [DOI] [PubMed] [Google Scholar]
- Ji R-R, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends in Neurosciences. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Kalyuzhny AE, Arvidsson U, Wu W, Wessendorf MW. mu-Opioid and delta-opioid receptors are expressed in brainstem antinociceptive circuits: studies using immunocytochemistry and retrograde tract-tracing. J Neurosci. 1996;16:6490–6503. doi: 10.1523/JNEUROSCI.16-20-06490.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Lundberg LE, Torebjork HE. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51:207–219. doi: 10.1016/0304-3959(92)90262-A. [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Torebjork HE, Wahren LK. Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117 (Pt 3):579–591. doi: 10.1093/brain/117.3.579. [DOI] [PubMed] [Google Scholar]
- Kwiat G, Basbaum A. The origin of brainstem noradrenergic and serotonergic projections to the spinal cord dorsal horn in the rat. Somatosens Mot Res. 1992;9:157–173. doi: 10.3109/08990229209144768. [DOI] [PubMed] [Google Scholar]
- Lawen A. Apoptosis-an introduction. Bioessays. 2003;25:888–896. doi: 10.1002/bies.10329. [DOI] [PubMed] [Google Scholar]
- Li Y, Dorsi MJ, Meyer RA, Belzberg AJ. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85:493–502. doi: 10.1016/S0304-3959(00)00250-5. [DOI] [PubMed] [Google Scholar]
- Liu CN, Raber P, Ziv-Sefer S, Devor M. Hyperexcitability in sensory neurons of rats selected for high versus low neuropathic pain phenotype. Neuroscience. 2001;105:265–275. doi: 10.1016/s0306-4522(01)00161-0. [DOI] [PubMed] [Google Scholar]
- Liu NJ, Chakrabarti S, Schnell S, Wessendorf M, Gintzler AR. Spinal synthesis of estrogen and concomitant membrane estrogen receptor signaling regulate spinal kappa- and mu-opioid receptor heterodimerization and female-specific spinal morphine antinociception. J Neurosci. 2011;31:11836–11845. doi: 10.1523/JNEUROSCI.1901-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Siniscalco D, Galderisi U, de Novellis V, Uliano R, Di Bernardo G, Berrino L, Cascino A, Rossi F. Apoptotic genes expression in the lumbar dorsal horn in a model neuropathic pain in rat. Neuroreport. 2002;13:101–106. doi: 10.1097/00001756-200201210-00024. [DOI] [PubMed] [Google Scholar]
- McCleane GJ, Suzuki R, Dickenson AH. Does a single intravenous injection of the 5HT3 receptor antagonist ondansetron have an analgesic effect in neuropathic pain? A double-blinded, placebo-controlled cross-over study. Anesth Analg. 2003;97:1474–1478. doi: 10.1213/01.ANE.0000085640.69855.51. [DOI] [PubMed] [Google Scholar]
- Mei XP, Xu H, Xie C, Ren J, Zhou Y, Zhang H, Xu LX. Post-injury administration of minocycline: An effective treatment for nerve-injury induced neuropathic pain. Neurosci Res. 2011;70:305–312. doi: 10.1016/j.neures.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Meisner JG, Marsh AD, Marsh DR. Loss of GABAergic interneurons in laminae I-III of the spinal cord dorsal horn contributes to reduced GABAergic tone and neuropathic pain after spinal cord injury. J Neurotrauma. 2010;27:729–737. doi: 10.1089/neu.2009.1166. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Liu X, Janig W. Axotomized and intact muscle afferents but no skin afferents develop ongoing discharges of dorsal root ganglion origin after peripheral nerve lesion. J Neurosci. 2000;20:2742–2748. doi: 10.1523/JNEUROSCI.20-07-02742.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Hong Sun T, Malan P, Jr, Lai J, Porreca F. Mediation of spinal nerve injury induced tactile allodynia by descending facilitatory pathways in the dorsolateral funiculus in rats. Neurosci Lett. 2000;290:129–132. doi: 10.1016/s0304-3940(00)01338-0. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Wei H, Hamalainen MM. Lidocaine in the rostroventromedial medulla and the periaqueductal gray attenuates allodynia in neuropathic rats. Neurosci Lett. 1996;218:127–130. doi: 10.1016/s0304-3940(96)13136-0. [DOI] [PubMed] [Google Scholar]
- Polgar E, Gray S, Riddell JS, Todd AJ. Lack of evidence for significant neuronal loss in laminae I–III of the spinal dorsal horn of the rat in the chronic constriction injury model. Pain. 2004;111:144–150. doi: 10.1016/j.pain.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Polgar E, Hughes DI, Arham AZ, Todd AJ. Loss of neurons from laminas I–III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. J Neurosci. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popken GJ, Farel PB. Sensory neuron number in neonatal and adult rats estimated by means of stereologic and profile-based methods. J Comp Neurol. 1997;386:8–15. [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J Neurosci. 2001;21:5281–5288. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaglino D, Ronchetti IP. Cell death in the rat thymus: a minireview. Apoptosis. 2001;6:389–401. doi: 10.1023/a:1011394304691. [DOI] [PubMed] [Google Scholar]
- Rahman W, Suzuki R, Webber M, Hunt SP, Dickenson AH. Depletion of endogenous spinal 5-HT attenuates the behavioural hypersensitivity to mechanical and cooling stimuli induced by spinal nerve ligation. Pain. 2006;123:264–274. doi: 10.1016/j.pain.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Ramalho RM, Viana RJ, Low WC, Steer CJ, Rodrigues CM. Bile acids and apoptosis modulation: an emerging role in experimental Alzheimer’s disease. Trends Mol Med. 2008;14:54–62. doi: 10.1016/j.molmed.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Reed MG, Howard CV. Surface-weighted star volume: concept and estimation. J Microsc. 1998;190:350–356. doi: 10.1046/j.1365-2818.1998.00331.x. [DOI] [PubMed] [Google Scholar]
- Ringkamp M, Eschenfelder S, Grethel EJ, Habler HJ, Meyer RA, Janig W, Raja SN. Lumbar sympathectomy failed to reverse mechanical allodynia- and hyperalgesia-like behavior in rats with L5 spinal nerve injury. Pain. 1999;79:143–153. doi: 10.1016/s0304-3959(98)00186-9. [DOI] [PubMed] [Google Scholar]
- Rodrigues CM, Steer CJ. The therapeutic effects of ursodeoxycholic acid as an anti-apoptotic agent. Expert Opin Investig Drugs. 2001;10:1243–1253. doi: 10.1517/13543784.10.7.1243. [DOI] [PubMed] [Google Scholar]
- Rodrigues CM, Fan G, Ma X, Kren BT, Steer CJ. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest. 1998;101:2790–2799. doi: 10.1172/JCI1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues CM, Spellman SR, Sola S, Grande AW, Linehan-Stieers C, Low WC, Steer CJ. Neuroprotection by a bile acid in an acute stroke model in the rat. J Cereb Blood Flow Metab. 2002;22:463–471. doi: 10.1097/00004647-200204000-00010. [DOI] [PubMed] [Google Scholar]
- Rodrigues CM, Sola S, Nan Z, Castro RE, Ribeiro PS, Low WC, Steer CJ. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci U S A. 2003;100:6087–6092. doi: 10.1073/pnas.1031632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Lucas M, Sanz A, Goberna R. Decreased protein kinase C activity is associated with programmed cell death (apoptosis) in freshly isolated rat hepatocytes. Biosci Rep. 1992;12:199–206. doi: 10.1007/BF01121789. [DOI] [PubMed] [Google Scholar]
- Schmued L, Swanson L, Sawchenko P. Some fluorescent counterstains for neuroanatomical studies. J Histochem Cytochem. 1982;30:123–128. doi: 10.1177/30.2.6174560. [DOI] [PubMed] [Google Scholar]
- Schoemaker MH, Conde de la Rosa L, Buist-Homan M, Vrenken TE, Havinga R, Poelstra K, Haisma HJ, Jansen PL, Moshage H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology. 2004;39:1563–1573. doi: 10.1002/hep.20246. [DOI] [PubMed] [Google Scholar]
- Scholz J, Broom DC, Youn D-H, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking Caspase Activity Prevents Transsynaptic Neuronal Apoptosis and the Loss of Inhibition in Lamina II of the Dorsal Horn after Peripheral Nerve. J Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi M, Sekiguchi Y, Konno S, Kobayashi H, Homma Y, Kikuchi S. Comparison of neuropathic pain and neuronal apoptosis following nerve root or spinal nerve compression. Eur Spine J. 2009;18:1978–1985. doi: 10.1007/s00586-009-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniscalco D, Fuccio C, Giordano C, Ferraraccio F, Palazzo E, Luongo L, Rossi F, Roth KA, Maione S, de Novellis V. Role of reactive oxygen species and spinal cord apoptotic genes in the development of neuropathic pain. Pharmacol Res. 2007;55:158–166. doi: 10.1016/j.phrs.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Skagerberg G, Bjorklund A. Topographic principles in the spinal projections of serotonergic and non-serotonergic brainstem neurons in the rat. Neuroscience. 1985;15:445–480. doi: 10.1016/0306-4522(85)90225-8. [DOI] [PubMed] [Google Scholar]
- Solá S, Aranha MM, Steer CJ, Rodrigues CM. Game and players: mitochondrial apoptosis and the therapeutic potential of ursodeoxycholic acid. Curr Issues Mol Biol. 2007;9:123–138. [PubMed] [Google Scholar]
- Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–213. doi: 10.1016/0304-3959(90)91164-E. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Rahman W, Hunt SP, Dickenson AH. Descending facilitatory control of mechanically evoked responses is enhanced in deep dorsal horn neurones following peripheral nerve injury. Brain Research. 2004;1019:68–76. doi: 10.1016/j.brainres.2004.05.108. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Rahman W, Rygh LJ, Webber M, Hunt SP, Dickenson AH. Spinal-supraspinal serotonergic circuits regulating neuropathic pain and its treatment with gabapentin. Pain. 2005;117:292–303. doi: 10.1016/j.pain.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, Ossipov MH, Xie JY, King T, Lai J, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains nerve injury-induced central sensitization. Neuroscience. 2006;140:1311–1320. doi: 10.1016/j.neuroscience.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Wallace J. Update on pharmacotherapy guidelines for treatment of neuropathic pain. Pain & Headache Reports. 2007;11:208–214. doi: 10.1007/s11916-007-0192-6. [DOI] [PubMed] [Google Scholar]
- Ward A, Brogden RN, Heel RC, Speight TM, Avery GS. Ursodeoxycholic acid: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1984;27:95–131. doi: 10.2165/00003495-198427020-00001. [DOI] [PubMed] [Google Scholar]
- Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28:10482–10495. doi: 10.1523/JNEUROSCI.3593-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Dubner R, Zou S, Ren K, Bai G, Wei D, Guo W. Molecular depletion of descending serotonin unmasks its novel facilitatory role in the development of persistent pain. J Neurosci. 2010;30:8624–8636. doi: 10.1523/JNEUROSCI.5389-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Pertovaara A. MK-801, an NMDA receptor antagonist, in the rostroventromedial medulla attenuates development of neuropathic symptoms in the rat. Neuroreport. 1999;10:2933–2937. doi: 10.1097/00001756-199909290-00011. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Munglani R. Cell death in the superficial dorsal horn in a model of neuropathic pain. J Neurosci Res. 2001;64:168–173. doi: 10.1002/jnr.1062. [DOI] [PubMed] [Google Scholar]
- Winkler CW, Hermes SM, Chavkin CI, Drake CT, Morrison SF, Aicher SA. Kappa opioid receptor (KOR) and GAD67 immunoreactivity are found in OFF and NEUTRAL cells in the rostral ventromedial medulla. J Neurophysiol. 2006;96:3465–3473. doi: 10.1152/jn.00676.2006. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]