

Abstract
Therapeutic angiogenesis represents an attempt to relieve inadequate blood flow by the directed growth and proliferation of blood vessels. Neovascularization is a complex process involving multiple growth factors, receptors, extracellular matrix glycoproteins, intracellular and extracellular signaling pathways, and local and bone-marrow-derived constituent cells, all responding to a symphonic arrangement of temporal and spatial cues. In cardiovascular disease, patients with refractory angina and lower extremity intermittent claudication seem most amenable to early tests of therapeutic angiogenesis. Monotherapy with the recombinant protein basic fibroblast growth factor (FGF-2) has been tested in six human trials. These have shown provisional safety, and two have provided ‘proof of concept’ for the strategy of therapeutic angiogenesis. One large randomized phase II trial failed to show significant efficacy in coronary artery disease. Another showed significant efficacy in peripheral artery disease, although the magnitude of benefit was disappointing at the dose tested. This overview details the suitable clinical trial design and further steps toward the clinical development of FGF-2.
Keywords: Angiogenesis, fibroblast growth factor, clinical trials
Abbreviations: ABI, Ankle-brachial index; ANOVA, Analysis of variance; CAD, Coronary artery disease; CCS, Canadian cardiovascular society; CLI, Critical limb ischemia; ETT, Exercise treadmill test; FGF, Fibroblast growth factor; FGFR, Fibroblast growth factor receptor; G-CSF, Granulocyte colony-stimulating factor; HIF, Hypoxia-induced factor; HSPG, Heparan sulfate proteoglycan; IC, Intermittent claudication; Ig, Immunoglobulin; JNK, c-Jun N-terminal kinase; MAPK, Mitogen-activated protein kinase; NO, Nitric oxide; PAD, Peripheral artery disease; PDGF, Platelet-derived growth factor; PET, Positron emission tomography; PI3, Phosphatidylinositol 3; SAQ, Seattle angina questionnaire; SPECT, Single photon emission computed tomography; TGF, Transforming growth factor; VEGF, Vascular endothelial growth factor
Introduction
Chronic stable angina and peripheral artery disease are widely prevalent, affecting 16.5 million and 10 million patients in the United States, respectively (Criqui, 2001; Gibbons et al., 2003), and are associated with significant morbidity and mortality. Despite recent advances in medical therapy, catheter-based intervention, and surgical revascularization, a significant number of patients still suffer from unrelieved myocardial and peripheral ischemia.
Spontaneous neovascularization is a normal adaptive response to chronic myocardial or limb ischemia. This process involves a complex cascade of trophic factors, extracellular matrix components, cell surface receptors, and signaling pathways. Preclinical and clinical experience indicates that delivery of a single angiogenic growth factor, such as basic fibroblast growth factor (or FGF-2) or vascular endothelial growth factor (VEGF), to ischemic regions is feasible, safe, and may enhance or recapitulate this natural process and relieve ischemia by enhancing the collateral blood flow around arterial occlusion. The purpose of this review is to examine FGF-2 in the light of a larger family of signaling peptides, and to discuss the key aspects of study design relevant to testing therapeutic angiogenesis.
Angiogenesis and basic fibroblast growth factor
Blood vessels, vasculogenesis, angiogenesis, and arteriogenesis
The basic constituents of blood vessels are endothelial cells, smooth muscle cells, fibroblasts and pericytes, basement membrane, and extracellular matrix. The relative composition varies based on the mechanical or metabolic demands of a specific vascular bed. The endothelium, in addition to forming a single-cell semipermeable monolayer that lines the entire circulatory system, is a homeostatic organ that modulates angiogenesis, thrombosis–fibrinolysis, and inflammation.
In antenatal vasculogenesis, a vascular network is assembled from undifferentiated precursor cells or angioblasts. It is important to note that the resulting vessels are immature and incompletely functional. In contrast, postnatal angiogenesis refers to new small blood vessels sprouting off the pre-existing ones. This may require division into pillars of periendothelial cells (intussusception), or the formation of trans-endothelial cell bridges which subsequently split into single capillaries. The new capillary network may then mature into blood vessels with a distinct muscular media, a process referred to as arteriogenesis. Alternatively, the new capillary network may involute if the local milieu is no longer appropriate (Cao et al., 2003). Postnatal angiogenesis occurs to a greater extent than arteriogenesis (Isner & Asahara, 1999) and, in the setting of ischemia, is often insufficient to reconstitute baseline blood flow in response to vascular occlusion even in juveniles.
Fibroblast growth factor-2
FGF-2 belongs to the family of over 20 proteins (Ornitz & Itoh, 2001). Recombinant FGF-2 protein tested clinically is a 16.5-kDa peptide with 144 amino acids (Okada-Ban et al., 2000). Alternative splicing of the FGF-2 mRNA encoded by a gene on chromosome 4 generates four variant isoforms. Although all FGFs share ~30–70% homology in their amino-acid sequences, FGF-1 and FGF-2 are different from other FGF proteins in lacking a signal sequence for extracellular transport. Both FGFs are secreted into the extracellular space, where they exert their biologic activity. Plasma FGF-2 is generally nearly undetectable among healthy volunteers. Levels of 2.4 and 12.1 μg l−1, respectively (Heeschen et al., 2003), are reported in patients with stable coronary disease and in patients with acute coronary syndromes.
FGF receptors and signaling
The biologic activity of FGF-2 is mediated by four related high-affinity (10−11 M) transmembrane tyrosine kinase receptors: FGFR-1, -2, -3, and -4, a syndecan-4 core protein, and perhaps other transmembrane receptors (Powers et al., 2000; Khurana & Simons, 2003). FGFRs belong to a family of immunoglobulin (Ig)-like receptors, which also include platelet-derived growth factor-α, -β, and interleukin-1 receptors. Multiple splice variants of multiple genes generate a wide diversity of FGFRs. The structure of FGFRs consists of three extracellular Ig-like domains, an acidic region between IgI and IgII, a transmembrane domain, and an intracellular tyrosine kinase domain. The IgIII domain is responsible for three additional splice variants designated IgIIIa, IgIIIb, and IgIIIc for FGFR-1, -2, and -3. In contrast, FGFR-4 expresses only one variant of the IgIII domain. The IgIII domain is an important mechanism for specificity of FGF binding.
The diversity in FGFs and FGFRs results in a great deal of apparent redundancy within the FGF/FGFR family. Except for disruption of the gene encoding for FGFR-1 or -2 (Deng et al., 1994; Xu et al., 1998), which results in embryo death before gastrulation, modification of a single gene encoding for any FGF or FGFR results only in minor phenotypic alteration (Cross & Claesson-Welsh, 2001). For example, disruption of the genes for FGF-1 or -2, two angiogens with therapeutic potential, resulted in only minor phenotypic alterations and decreased vascular tone/low blood pressure in mice, respectively (Dono et al., 1998; Miller et al., 2000), and intact neovascular response to hindlimb ischemia (Sullivan et al., 2002).
FGFR binding results in receptor dimerization and autophosphorylation. Tyrosine kinase then contributes to endothelial cell proliferation via activation of the Ras/Raf-MAPK pathway by enabling adaptor proteins Grb2, shc, and Nck (Klein et al., 1997; Chin et al., 1999). After entering the nucleus, FGF-2 and -1 are also able to promote endothelial cell proliferation through other pathways. FGF-1 supports transition of the G1 stage to the S stage of the cell cycle, and FGF-2 is associated with phosphorylation of nucleolin, resulting in increased transcription of rDNA (Bouche et al., 1994). FGF-2 has also been shown to promote endothelial cell proliferation and angiogenesis in vivo via an αvβ3-dependent pathway (Friedlander et al., 1995).
FGF-2 and the extracellular matrix
All FGFs bind heparin, despite numerous structural differences. FGF-2 binds with high affinity to heparan sulfate proteoglycans (HSPGs) by two heparin-binding domains (Powers et al., 2000; Cross & Claesson-Welsh, 2001). HSPGs are associated with most cell surface receptors in the extracellular matrix. The functional importance of the FGF-2–HSPG interaction is manifold. The HSPG–FGF-2 complex serves as a reservoir for FGF-2, thus modulating FGF-2 availability in vivo, and it protects FGF-2 from enzymatic or acidic degradation. Hypoxia increases heparin sulfate-binding sites and facilitates FGF-2–endothelial cell interaction. FGF-2 is released from the FGF–HSPG complex by enzymatic cleavage (i.e. heparanase or protease activity), or by binding to a carrier protein (Li et al., 2002). Even circulating heparin alters the pharmacokinetics of FGF-2 in patients, by reducing the clearance of FGF-2–heparin complexes (Bush et al., 2001).
FGF-2 regulation
Hypoxia is a potent stimulus known to mediate increases in angiogenic factor expression. The precise signaling mechanism that leads to upregulation of FGF receptors and an increment in FGF-2 expression is not clear. Endothelial cell responsiveness to FGF-2 is enhanced by a HIF-1α-dependent process involving the upregulation of heparan sulfate FGF-2-binding sites. However, the increase of FGF-2 expression in response to hypoxia seems to be mediated by JNK-1 signaling, and not by HIF (Le & Corry, 1999; Fang et al., 2001; Khurana & Simons, 2003).
FGF-2 and apoptosis
Angiogenesis involves both growth and involution of blood vessels. FGF-2 seems to promote angiogenesis not only by stimulating the growth of new blood vessels, but also by abrogating their apoptotic potential. In fact, when considering the low-grade proliferative (two- to three-fold) effect of endothelial cells promoted by angiogenic factors (e.g., VEGF), increasing cell survival by inhibiting apoptosis may be particularly important during ischemia and hypoxia (Dimmeler & Zeiher, 2000). Activation of PI3/Akt by FGF-2 and VEGF confers both angiogenic factors antiapoptotic properties on endothelial cells (Gerber et al., 1998; Ong et al., 2001). The PI3/Akt pathway seems to enhance expression of the antiapoptotic protein survivin (Ong et al., 2001), and activation of NO synthase by Akt (which results in increased NO production) promotes endothelial cell survival by inhibition of the caspases’ cysteine protease activity (Dimmeler et al., 1997).
FGF-2 and other factors in angiogenesis
In addition to activating signaling pathways that result in endothelial cell activation, proliferation, increased survival, migration, and differentiation, fibroblast growth factors also activate other cell lines, such as smooth muscle cells and fibroblasts, which participate in arteriogenesis. FGF receptor-1 is expressed robustly on the surface of endothelial progenitor cells (Burger et al., 2002), suggesting that FGF-2 may be involved in the recruitment or homing of bone-marrow-derived cells into expanding neoarteries. In this way, it is possible that FGF-2 induces the growth not only of capillaries, but also of more mature arterioles. Moreover, variable local expression of the receptors for FGF receptors may modulate the action of paracrine FGF-2 in arteriogenesis (Deindl et al., 2003).
Other molecules also contribute to the formation, remodeling, and maturation of new blood vessels in response to injury or ischemia. The list of factors with important angiogenic activity includes the hypoxia-inducible factors, TFG-α, -β, G-CSF, epidermal growth factor, hepatocyte growth factor, placental growth factor, monocyte chemoattractant protein-1, angiopoietin-1, interleukin-1, -8, platelet-activating factor, etc. (Carmeliet, 2003).
Moreover, the temporal and spatial coordination of angiogen exposure probably impacts their biologic activity. Transient monotherapy may not be adequate to induce sustained neovascularization. For example, platelet-derived growth factor-BB (PDGF-BB) and FGF-2 act synergistically and durably to improve animal lower extremity blood flow and neovascularization. Transient treatment with only one agent leads to transient improvement followed by neovessel regression (Cao et al., 2003). Similarly, Ang-1 appears to stabilize neocapillary endothelial cells after their formation is mobilized by VEGF or FGF. In vitro and in vivo experiments have also demonstrated that FGF-2 and VEGF act synergistically to promote angiogenesis (Asahara et al., 1995; Pepper et al., 1998).
Testing clinical therapeutic angiogenesis with FGF-2
Target syndromes
‘Biological revascularization’ has long been sought to exaggerate the spontaneous formation of collateral arteries in response to chronic myocardial or lower extremity ischemia. Clinical targets for therapeutic angiogenesis include the cardiac and skeletal muscles.
Myocardial ischemia
Chronic myocardial ischemia, usually related to atherosclerotic coronary artery disease (CAD), may manifest as angina pectoris, or exertional chest discomfort that is relieved by rest. More severe myocardial ischemia may manifest as angina with minimal exercise or even at rest. Other manifestations of myocardial ischemia may be life-threatening ischemic dysrhythmia and chronic ischemic myocardial dysfunction. The latter includes a metabolic adaptation called myocardial hibernation, which may be reversible, or which may progress to irreversible ‘ischemic cardiomyopathy’ (Wijns et al., 1998). Chronic myocardial ischemia is usually attributed to large-vessel (epicardial coronary) atherosclerotic obstruction, but comparable syndromes may derive from small-vessel or microvascular dysfunction. While improved blood flow would be expected to attenuate the impact of acute myocardial infarction, the biological time scale for angiogenesis probably exceeds the time required to salvage the infarcting myocardium.
Conventional treatment of chronic myocardial ischemia is not successful in all patients with chronic myocardial ischemia. Pharmacologic agents to relieve the angina, including nitro-vasodilators, beta-adrenergic antagonists, and calcium-channel antagonists, are limited by cumulative blood-pressure lowering or other side effects that may be intolerable. Catheter-based therapeutic options (angioplasty or stenting) may be limited in patients who have diffuse coronary artery obstruction or chronic total occlusions. This remains true even in the emerging era of drug-eluting stents to reduce stent restenosis. Surgical options may be unattractive, especially in patients with advanced age, extensive comorbidity, severe left ventricular dysfunction, or after multiple prior revascularization procedures. An estimated 12% or more of patients with refractory myocardial ischemia may be ill suited for conventional revascularization and, thereby, as candidates for therapeutic angiogenesis (Mukherjee et al., 2001).
Skeletal muscle ischemia
Just as coronary artery obstruction induces myocardial ischemia, peripheral artery (usually atherosclerotic) obstruction is associated with inducible or constant skeletal muscle ischemia. An estimated 15% of North American adults over the age of 55 have detectable hemodynamic impairments attributed to such peripheral artery disease (PAD) (Criqui, 2001). While only a third are thought to have classic symptoms of lower extremity ischemia, an unknown but significant remainder are thought to have atypical symptoms. Many patients with PAD do not even report the symptoms unless specifically queried, attributing them instead to the many ‘indignities of aging.’
Intermittent claudication (IC), muscular leg discomfort provoked by exercise and promptly relieved by rest, is the most common manifestation of PAD. Both ‘atypical’ leg discomfort and marked functional impairment without claudication are common in patients even with otherwise ‘subclinical’ intermittent claudication (McDermott et al., 2001). Patients with IC are limited in walking distance, speed, functional status, and other measures of life quality. The annual incidence of progression to critical limb ischemia is low in nonsmokers and nondiabetics. While the site of vascular obstruction may be anywhere from the aorta, iliac, femoropopliteal, or tibial artery levels, the symptoms are typically first experienced in the antigravity muscles of the calf. When the vascular obstruction is more proximal, the symptoms may occur at a lower exercise threshold, or may extend to more proximal (thigh, buttock) muscles with progressively greater effort. There is enormous variability in the severity of clinical symptoms associated with a given lesion, probably related to baseline functional status, skeletal muscle metabolic ‘economy,’ adaptive collateral artery formation, etc. IC is an attractive clinical target for the study of therapeutic angiogenesis, because the functional impairment (walking) is directly attributable to the pathophysiology (impaired lower extremity blood flow) and can easily be measured in the majority of patients. Moreover, the affected organ is available for direct observation and measurement. In contrast, functional measures of myocardial ischemia are potentially confounded by nonmyocardial phenomena including pulmonary and skeletal muscle abnormalities.
Critical limb ischemia (CLI) is a less common but more profound manifestation of PAD. It includes a constellation of syndromes: intermittent or unremitting metatarsalgia even at rest (probably representing ischemic neuritis), ulcers that fail to heal, or frank gangrene. CLI represents blood flow impairment, so profound that limb loss is likely without revascularization. Patients with CLI tend to be older with significantly more comorbidity, significantly more diffuse obstructive atherosclerosis, and a significantly higher incidence of age-related malignancy that may preclude participation in early clinical trials of growth factors. In the course of a clinical trial, patients with CLI are substantially more likely to suffer from major adverse cardiovascular events or death. Therapy aims to improve blood flow, if only transiently, to permit wound healing. There is a significant incidence of transient or even durable improvement after conservative measures, a fact that complicates uncontrolled trials in CLI treatment.
Conventional PAD treatment includes risk-factor reduction (especially smoking cessation), pharmacotherapy of the underlying atherosclerosis (with antiplatelet, lipid-lowering, angiotensin-cascade antagonists and other antihypertensives), and nonpharmacologic therapy including limb hygiene and structured exercise therapy. Therapeutic strategies designed to improve perfusion, the primary pathophysiologic process in PAD, are not available. Pentoxifylline has little or no clinical benefit. Cilostazol, a phosphodiesterase inhibitor, confers a small but significant improvement in symptoms and function, and this drug remains contraindicated in the setting of heart failure. Medical treatments are instead directed at reducing the mortality and morbidity from systemic atherosclerosis, primarily with antiplatelet, angiotensin-converting enzyme, and HMG-coA-reductase inhibitors. Mechanical revascularization (angioplasty or bypass surgery) can be gratifying to patients with focal or ‘single-segment’ atherosclerotic obstruction confined to aortoiliac or femoropopliteal levels. Unfortunately, most patients with debilitating symptoms have multilevel disease, and more profoundly affected patients have more diffuse and difficult-to-revascularize target anatomy. The latter may be amenable to surgical revascularization, but the risk–benefit ratio of lower extremity bypass may not justify the perioperative morbidity and mortality, loss of conduit, and graft attrition that make this option less desirable except in cases of limb-threatening ischemia. Overall, surgical bypass using autologous or prosthetic conduits is generally offered to patients with CLI or most severe IC. Transcatheter treatment is feasible in most patients with PAD; however, intermediate-term (6–12 months) recurrent obstruction is common, especially in the large proportion of patients with diffuse or distal disease. For these reasons, and because of poor understanding of diagnosis and treatment by the majority of general medical practitioners, revascularization is currently not offered to most patients even with symptomatic PAD. Thus, a far greater proportion of patients with symptomatic PAD is probably eligible for putative therapeutic angiogenesis.
Route of administration and drug formulation
Angiogenic protein formulations such as FGF-2 can be administered systemically or locally. Systemic administration is attractive because of ease of access (i.e. intravenous bolus or infusion), but unattractive because of the greater risk of nonspecific or toxic responses to a pluripotent biological agent. Local administration has the potential to enhance target exposure to FGF-2 while reducing nonspecific exposure. Obvious options for local delivery include direct intra-arterial (Lazarous et al., 1997) or intramuscular (Unger et al., 1994) injection. More elaborate schemes include intrapericardial instillation (Baek et al., 2002; Laham et al., 2003) to create an epicardial depot in patients with intact pericardium, perivascular depot (Lopez et al., 1997; Arras et al., 1998) by surgical or transcatheter implantation, and retrograde venous infusion to overcome arterial inflow obstruction (Herity et al., 2000).
Potential toxicity
Preclinical studies suggest an important potential clinical toxicity (Mazue et al., 1991; Ornitz & Itoh, 2001; Post et al., 2001). These include nontarget organ neoangiogenesis, acceleration of atherosclerosis, and spread of malignancy. Non-target organ neoangiogenesis may be relevant in the retina, in the potential acceleration of macular degeneration or of diabetic proliferative retinopathy. Neoangiogenesis may contribute to diabetic or other nephropathy. FGF and other angiogens may promote favorable or deleterious neointimal formation after therapeutic vascular injury (angioplasty and stenting), or may accelerate the underlying atherosclerosis progression (Moulton et al., 1999). Angiogens were first characterized by their ability to promote tumor blood supply, and growth or metastasis of subclinical malignancy or angioma is a theoretical concern. Drug-related toxicities have included proteinuria, presumably caused by a membranous nephropathy, and transient nitric-oxide-mediated hypotension (Okada-Ban et al., 2000; Ornitz & Itoh, 2001; Post et al., 2001). However, six human trials of FGF-2 have not demonstrated any evidence of nontarget organ neoangiogenesis, and only low rates of proteinuria and spontaneously resolving episodes of hypotension.
End points for clinical investigation
Clinical and surrogate end points
Regulatory approval of FGF-2 for therapeutic angiogenesis requires evidence of efficacy and an acceptable safety profile in concordant well-designed clinical experiments. The demonstration of efficacy may be more difficult than it appears at first, because angiogenesis per se is not readily measurable nor necessarily clinically meaningful.
Ideal clinical end points include reduction in mortality or in demonstrable events such as myocardial infarction. Unfortunately, such ‘hard’ end points may require clinical studies that are inaccessibly prolonged and expensive. Moreover, the general mechanism of benefit (i.e. reduction in ischemia) may provide other meaningful benefits without necessarily reducing death or other hard end points. Early-phase clinical studies, intended to test safety and feasibility, should include clinical surrogates to corroborate the proposed mechanisms of biological action. Meaningful surrogate end points for therapeutic angiogenesis include measurements of arterial size and extent, measures of blood flow, and biomarkers of ischemia.
Since leg pain induced by exertion is the chief manifestation of PAD, appropriate clinical end points for IC might include validated pain or disease-specific quality of life questionnaires (e.g. Walking Impairment Questionnaire) (Hiatt et al., 1995). The functional measurements include walking impairment (peak walking time or claudication onset time on a standardized treadmill exam). Among patients with CLI, appropriate measurements might include not only pain or analgesic medication use, but when appropriate, ulcer size, ulcer healing, and frank tissue loss. The same concepts apply to CAD. Relevant measurements of pain perception include the angina class (i.e. CCS angina class), nitrovasodilator pill count, and self-reported angina (i.e. the Seattle Angina Questionnaire). Treadmill exercise testing is a less straightforward clinical end point for patients with coronary atherosclerosis, simply because exercise performance tests pulmonary, skeletal, and vascular function in addition to myocardial function, in contrast to patients with PAD. Imperfect as it is, treadmill exercise testing, with or without direct measurement of oxygen consumption, may be the best available clinical end point.
Measuring angiogenesis
No clinical study of therapeutic angiogenesis to date has measured de novo vessel formation. The most direct technique would be biopsy for histologic examination of target tissue before and after treatment. Unfortunately, suitable biopsy specimens of myocardium are difficult to obtain. Biopsy specimens in CLI may exacerbate impaired wound healing and should not be obtained. Even with tissue in hand, microscopic vascularity may be subject to sampling error.
Invasive or noninvasive angiography is not well suited for angiogenesis assessment. Conventional radiocontrast (X-ray) angiography has a spatial resolution of approximately 200 μm, whereas the caliber of useful new arterioles and small arteries may be in the range of 20–200 μm. Moreover, technical variables render radiocontrast angiography ill suited for serial comparative examinations. Acquired images may vary in contrast volume, injection rate, selected phase of imaging sequence (early or late after contrast arrival or microvascular tissue ‘blush’), and baseline vascular tone. Even assuming reproducible image acquisition, variables in image segmentation render these complex data sets difficult to compare. An example in Figure 1 demonstrates four different segmented renditions of the same angiographic image. X-ray computed tomography and available clinical magnetic resonance angiography may provide three-dimensional data sets, but the spatial resolution is slightly to dramatically worse than for projection radiocontrast angiography. Satisfactory spatial resolution may be achievable with synchrotron radiation microangiography, which is unfortunately not available for clinical applications.
Figure 1.
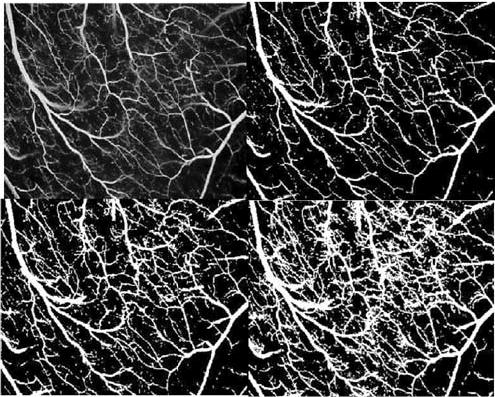
Four different renditions of the same angiographic view. The images are segmented for analysis using different gray-scale thresholds for vessel edge detection. This is a good example of the difficulty in quantitative analysis of complex angiographic trees in the measurement of therapeutic angiogenesis. Courtesy of Richard B. Thompson.
Pressure, perfusion, and related measurements
Perfusion and hemodynamics measures reflect the function of native and neoarterial circulation. In limbs, the most accessible measurement is the normalized ankle systolic blood pressure, called the ankle-brachial index (ABI). It is simple, objective, and reproducible, and can be combined with provocative measures such as exercise to extend its sensitivity or to produce a measure of ischemic burden (Feinberg et al., 1992). The disadvantage of ABI is that it is not especially sensitive, and is not useful in ‘noncompressible’ arteries, affecting approximately 10–25% of elderly and diabetic adults. Moreover, concurrent alterations in blood flow and vascular resistance can confound ABI findings.
Spectrophotometric and related techniques can measure superficial (skin or subcutaneous) perfusion noninvasively. These measures, such as transcutaneous pulse oximetry (TCPO2), near-infrared spectroscopy, and laser-Doppler flow, are simple, precise, and have a wide dynamic range. Skin perfusion impacts wound healing, with established prognostic utility in patients with CLI. Disadvantages include a nonlinear relation to perfusion, and a high biological and regional variability within patients. Resting perfusion and skin perfusion are usually normal in patients with IC, for which such measures are probably not useful.
Venous occlusion plethysmography is the classic ‘direct’ measure of limb perfusion. It works by detecting changes in limb volume (usually using a strain gauge) immediately after blocking the exit of venous blood using low-pressure occlusive cuffs. It is inexpensive and noninvasive. However, limbs consist of bone and fat in addition to skeletal muscle, so the denominator of the Δvolume/volume measure is misleading, and calf swelling is affected by tissue capacitance and compliance. Moreover, the technique is highly operator-dependent and ill suited for large multicenter trials.
Radionuclide scintigraphy is widely available, noninvasive, and fairly sensitive (Ritchie et al., 1995). Single-photon nuclear perfusion techniques (SPECT) measure tracer uptake, activity over the region of interest, rest and stress ratios to estimate flow reserve, and first-pass tracer arrival time. It is a noninvasive technique with a linear relation between tracer activity and blood flow, and the appropriate tracers can overcome temporal resolution limitations. Spatial resolution is limited, however, especially in the assessment of subendocardial defects of the myocardium. Unfortunately, measurements of ‘input function,’ which relate measured signal to absolute blood flow, are difficult, rendering most techniques difficult, nonquantitative, and ill suited to serial studies. With this in mind, ‘ischemia’ represents a relative defect compared with surrounding structures. The most common comparator, the adjacent myocardium or contralateral limb, is also often affected by the underlying atherosclerosis, thereby confounding interpretation. Dual-photon positron-emission tomography (PET) perfusion measures the tracer content of muscle in time and space with high precision but poor resolution. PET is quantitative and can be applied to myocardium or skeletal muscle, but capital and radiopharmaceutical costs are high. Importantly, there is a linear correlation between tracer activity and blood flow, and the extraction fraction can be calculated using two isotopes. There is limited experience with scintigraphy in PAD.
Myocardial function can be tested as a marker of myocardial blood flow, using echocardiography (Christian, 1999). Resting wall motion abnormalities may reflect reduced blood flow at rest, but they also may represent ischemic ‘stunning’ in response to a transient flow alteration, adaptive ‘hibernation’ in response to chronic flow impairment, or permanent myocardial necrosis. Provoked ischemia or shunting with catecholamines or vasodilators may be useful to distinguish among the latter. Endocardial border detection may be difficult unless sonographic contrast agents are added. Overall, as for the other tests described above, paired comparative ultrasound assessment of resting or provoked wall motion can be difficult to implement in a multicenter trial of angiogenesis.
Magnetic resonance imaging (MRI) may prove useful for myocardial and skeletal muscle perfusion measurement. MRI is especially good at distinguishing necrosis from hibernation or stunning (Kim et al., 2000). Qualitative techniques (such as contrast arrival time or upslope) are widely available, but quantitative techniques suitable for paired follow-up analysis remain investigational.
Half-life, formulation, dosing, and delivery strategies
For all putative angiogenic agents, the biological half-life should probably exceed the pharmacokinetic half-life. The target tissue should be exposed to the therapeutic agent long enough to have a biological effect, yet short enough to preclude toxic effects (such as hemangioma or other tumor growth). Endogenous peptide growth factors or other small molecules tend to have a short-circulating half-life, so they may require modification or repeat or prolonged administration for efficacy (Isner & Asahara, 1999). Gene transfer is attractive, particularly because it permits prolonged tissue exposure to the angiogenic growth factor by recruiting the patient’s tissues for the manufacture and delivery of the desired agent. Different gene transfer formulations (i.e., adenoviral, naked DNA) or tropisms (i.e., endothelial-specific) may be attractive because of longer but self-limited gene expression. Cellular agents (i.e., embryonic, umbilical, bone-marrow-derived progenitor or stem cells) with or without genetic modification may also achieve therapeutic angiogenesis (Isner & Asahara, 1999).
Angiogenic growth factors may be delivered systemically (i.e. orally or via a peripheral intravenous line), topically (e.g., topical becaplermin/platelet-derived growth factor for neuropathic ulcers), or locally by intra-arterial infusion, elution from an implanted prosthesis, or even via cell-mediated gene transfer of ex vivo transfected autologous cellular agents. Local residence after local intra-arterial administration is difficult, and may require first-pass extraction, local over-expression of conjugate receptors induced by ischemia, or extravasation. In any case, only a small fraction of radiolabeled but otherwise unmodified FGF-2 remains in the myocardium after systemic, intracoronary, or intramyocardial delivery (Laham et al., 1999a; Post et al., 2001).
Bilateral PAD usually underlies even predominantly unilateral symptoms of intermittent claudication or critical limb ischemia. Unilateral therapy can therefore be perilous in investigational trials for PAD: functional end points such as treadmill testing may show no improvement even after successful therapy when the contralateral limb remains untreated. For this reason, in the TRAFFIC study, all patients were treated in both limbs irrespective of clinical or angiographic pattern of obstruction (Lederman et al., 2001).
Clinical trials testing the safety and efficacy of FGF-2 (Table 1)
Table 1.
Summary of clinical trials of basic fibroblast growth factor*
| Reference | N | Study design | Control group | Formulation | Route | Dose | Primary end point | Secondary end point | Toxicity | Results |
|---|---|---|---|---|---|---|---|---|---|---|
| Trials of peripheral artery disease | ||||||||||
| Lazarous et al. (2000) | 19 | Phase I | Yes | FGF-2 protein | Intra-arterial | 0/10/30 μg kg−1 | Calf blood flow | Calf blood flow | None | ↑Blood flow |
| Lederman et al. (2002) | 190 | RCT, Phase II | Yes | FGF-2 protein | Intra-arterial | 0/30 μg kg−1 | WIQ, SF-36, ETT | ABI | Brief; ↓BP proteinuria | ↑PWT at 90 days; ↑ABI |
| Trials of coronary artery disease | ||||||||||
| Unger et al. (2000) | 25 | Phase I | Yes | FGF-2 protein | Intracoronary | 0/3/10/30/ 100 μg kg−1 | ETT, ECG | ↓BP; ↓HR proteinuria; ↓platelets | Null | |
| Laham et al. (2000)/Udelson et al. (2000) | 52/59 | Open-label, Phase I | No | FGF-2 protein | Intracoronary and intravenous | 0.33–48 μg kg−1 | SAQ | SPECT, MRI, ETT | ↓BP if >36 μg kg−1 | ↓Stress-induced ischemia and resting perfusion; ↑ETT time; ↑perfusion/function; ↓angina |
| Simons et al. (2002) | 337 | RCT, Phase I, II | Yes | FGF-2 protein | Intracoronary | 0/0.3/3/30 μg kg−1 | CCS, SAQ, ETT, SF-36 | SPECT | ↓BP (30 μg kg−1) | Null post hoc analysis; ↓angina |
| Laham et al. (1999b)/(Ruel et al. (2002) | 24 | RCT, Phase I, II | Yes | FGF-2, sustained- release heparin- alginate capsules | Intramuscular, surgical | 0/10/100 μg kg−1 | Death, MI, angina, revasc. | SPECT, MRI | None | ↑Perfusion; ↓angina |
Studies are presented in hierarchal order according to the reliability of results based on the robustness of study design. FGF-2 = recombinant fibroblast growth factor 2; ETT = exercise treadmill test; ABI = ankle-brachial index; WIQ = walking impairment questionnaire; SF-36 = short form 36; BP = blood pressure; PWT = peak walking time; CCS = Canadian Cardiovascular Society (angina class); SAQ = Seattle Angina Questionnaire; SPECT = single-photon emission computed tomography; MRI = magnetic resonance imaging; revasc = revascularization; MI = myocardial infarction.
The first human studies of parenteral FGF-2 were conducted at the US National Institutes of Health (Lazarous et al., 2000; Unger et al., 2000). Both used intra-arterial administration, and both used randomized concurrent controls. Unger et al. (2000) treated patients with mild baseline angina and observed sustained hypotension bolus therapy of 30 or 100 μg kg−1, yet no clinical signs of efficacy as determined by exercise tolerance or electrocardiographic indices of ischemia. Lazarous et al. (2000) treated patients with mild IC, and found no important toxicity, and found plethysmographic evidence of improved lower extremity blood flow. To avoid infusional hypotension, this study used 30 μg kg−1 on two consecutive days as the highest dose. Subsequent studies prevented hypotension with slower drug infusion and attention to prehydration.
Laham et al. (2000) performed a dose-escalating, open-labeled, uncontrolled, phase I trial of FGF-2. They studied 52 patients with CAD not amenable to revascularization therapy after intracoronary administration of FGF-2 between 0.33 and 48 μg kg−1 ideal body weight. While safety was the primary end point, an open-label efficacy substudy incorporated additional patients treated with intravenous FGF-2, and used myocardial scintigraphy for endpoint assessment. They found improved segmental perfusion in patients treated with FGF-2 having baseline resting perfusion abnormalities.
The FIRST trial
Based on these findings, a larger randomized, double-blind, placebo-controlled, dose-ranging study was conducted, the FGF-Initiating RevaScularization Trial (FIRST) (Simons et al., 2002). The population consisted of 337 patients with CAD not candidates for revascularization, most of whom had curiously few symptoms or exercise impairment. FGF-2 was administered via 10 min intracoronary infusions, and patients were randomly assigned in a 1 : 1 : 1 : 1 ratio to placebo, 0.3, 3, and 30 μg kg−1. The primary efficacy end point was change in exercise treadmill test (ETT) time from baseline to 90 days, and secondary end points included change in ETT time from baseline to 180 days, changes in the magnitude of ischemic segments using SPECT imaging, change in CCS angina class, and SAQ or SF-36 scorers from baseline to 90 and 180 days. There was no significant difference in exercise treadmill time or scintigraphy findings in patients treated with FGF-2 compared with placebo (Figure 2). A post hoc analysis compared all treated groups with placebo at 90 days, and suggested that symptomatic patients (SAQ score ≤40 or CCS angina class II/III) treated with FGF-2 had fewer episodes of angina.
Figure 2.
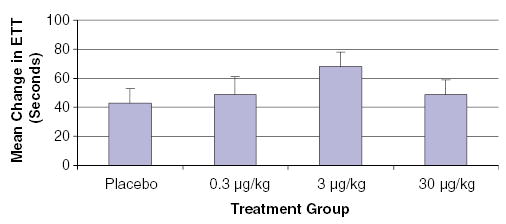
Mean change in ETT time among all treated groups in the FIRST study of patients with coronary artery disease (Simons et al., 2002). Although there was no significant difference between treatment groups, it is interesting to note that the effect of FGF-2 was highest among patients treated with 3 μg kg−1 and not among patients treated with the highest dose of FGF-2.
It is important to consider why this trial failed to show efficacy. Unfortunately, patients in FIRST were not very symptomatic, which limits any achievable treatment effect. Indeed, almost half the patients stopped their treadmill tests because of noncardiovascular symptoms. Moreover, by design, the study recruited patients with no or poor options for conventional revascularization, who may have inadequate potential to respond to the biological therapy. As noted above, ETT may be the best available functional end point, but ETT performance relies on more than myocardial blood flow alone. Also noteworthy was a trend to benefit in patients with the second-highest tested dose (3 μg kg−1), raising the possibility of dose-related confounding toxicity such as accelerated intimal hypertrophy or atherogenesis, and the possibility that dose optimization might be useful. Nevertheless, the conclusion of FIRST was that intracoronary FGF-2 protein at the doses tested is not efficacious in the treatment of myocardial ischemia.
Surgical delivery of sustained-release FGF-2
The Beth Israel group performed a double-blind, placebo-controlled, randomized clinical trial testing the safety and efficacy of a sustained-release FGF-2 formulation delivered locally to viable nongraftable ischemic regions of the myocardium at the time of coronary artery bypass surgery (Laham et al., 1999b). They used heparin alginate microspheres containing FGF-2 to prolong local epicardial drug administration. Patients were randomly assigned in a 1 : 1 : 1 ratio to 10 μg of placebo or 100 μg of FGF-2. The primary end point was nuclear perfusion imaging (SPECT). The protocol was modified following enrollment of 10 patients to perform baseline nuclear scans after and not before cardiac surgery. The purpose of this change was to avoid confounding by the effect of conventional surgical revascularization. In contrast to patients assigned to placebo or 10 μg of FGF-2, patients treated with 100 μg of FGF-2 were free of angina and had significant reduction in stress perfusion defects at 3 months (19 vs 9.1%, P<0.01). The long-term effects of FGF-2 persisted after 32.2±6.8 months of follow-up (Ruel et al., 2002). However, promising the findings, concurrent effective revascularization using conventional surgical techniques is widely considered to be a ‘lethal’ confounder of studies using this design.
FGF-2 for PAD: the TRAFFIC study
The Therapeutic Angiogenesis with FGF-2 for Intermittent Claudication Trial (TRAFFIC) was a randomized, double-blind, placebo-controlled, phase II clinical trial testing the efficacy and safety of FGF-2 in 190 patients with moderate-to-severe intermittent claudication with an ABI <0.8. Patients were randomly assigned to receive a bilateral intra-arterial infusion of placebo, single bolus, or double bolus (1 : 1 : 1) of 30 μg kg−1 of FGF-2 on days 1 and 30. The primary end point in TRAFFIC was 90-day change in peak walking time, and secondary end points included safety, ABI, claudication onset time at 90 and 180 days, and quality of life measures. In an intention-to-treat analysis, patients in the placebo, single bolus, and double bolus groups had 0.60, 1.77, and 1.54 min increases in peak walking time (P = 0.034), as shown in Figure 3. An alternative treadmill test (time to onset of claudication) and measures of quality of life did not show important improvement with treatment. ABI findings, however, were intriguing (Figure 4). While there was virtually no change in ABI among patients in the placebo group, there was a significant improvement in this objective measure in both treatment groups at both time points. This finding corroborates the proposed mechanism of benefit, improvement in the lower extremity blood flow. Overall, TRAFFIC showed that a single infusion of FGF-2 30 μg kg−1 improved treadmill performance, but a second infusion after 30 days did not provide incremental benefit.
Figure 3.
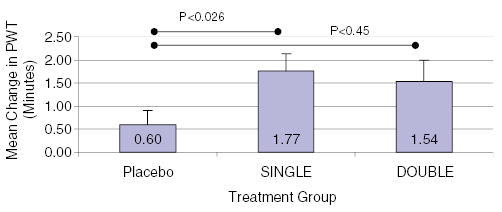
Mean change in peak walking time among the three different treatment groups in the TRAFFIC study of peripheral artery disease (Lederman et al., 2002): placebo, single bolus of FGF-2 (30 μg kg−1), or double bolus of FGF-2 (30 μg kg−1). By intention-to-treat analysis (nonparametric analysis), the difference in PWT was different between all groups (P<0.034). When compared with placebo, patients treated with a single bolus of FGF-2 had higher PWT (pairwise comparison).
Figure 4.
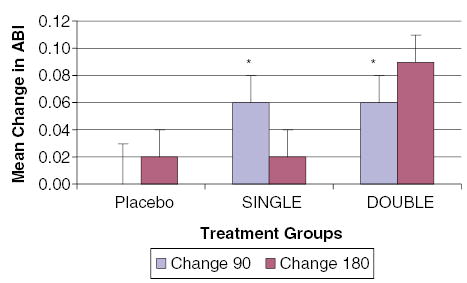
Mean change in ABI between 90 and 180 days in the TRAFFIC study of peripheral artery disease (Lederman et al., 2002). There was a small increase in ABI at 90 days among treated patients (*P<0.05).
Several aspects of the findings in TRAFFIC are noteworthy. The study adhered to a prespecified statistical analysis plan that was perhaps too specific for a phase II or proof-of-concept trial. The prespecified ANOVA (which did not capture patients not evaluable because of adverse events, surgical treatment, or loss to follow-up) did not achieve statistical significance. Statistical significance was only found using a secondary intention-to-treat nonparametric analysis that captured all subjects. More important, the observed placebo effect at 90 days was relatively low when compared with the 180-day finding. In other words, it is possible that the effect at 90 days was the result of a spuriously low placebo effect. The study, however, was powered to detect a treatment effect, assuming not only that single FGF-2 treatment was efficacious, but also that double FGF-2 treatment was superior. In this light, it was remarkable to detect any treatment effect. Similarly, TRAFFIC was not powered to detect differences at 180 days, and, therefore, findings at 180 days may reflect type II error. Indeed, analysis of ‘responder rates,’ in this case meaning the proportion of patients enjoying a 2-min overall improvement in treadmill performance, showed continued benefit in both treatment arms compared with placebo at both 90 and 180 days (unpublished observation). Another interesting observation was a relative maldistribution of current smokers, with significantly more smokers in the placebo group than in the treatment groups. A positive ‘trial effect,’ including lifestyle improvements such as smoking cessation, might therefore have been enhanced in the placebo group. If true, this would have made it even more difficult to detect benefit from FGF-2. Unfortunately, the study did not capture smoking cessation rates. The ABI finding demonstrates the importance of corroborative hemodynamic testing in clinical studies of therapeutic angiogenesis.
Future steps
Establishing stable and functional vascular networks is a complex process involving a number of growth factors and signaling peptides/pathways, and the notion that single angiogenic factor therapy can replicate the complexity of what occurs under physiologic conditions in response to ischemic and hypoxic stimuli is simplistic. In vitro and in vivo studies have demonstrated the synergistic angiogenic effect of combination therapy with VEGF and FGF-2 (Goto et al., 1993; Asahara et al., 1995; Pepper et al., 1998). As mentioned above, Cao et al. (2003) used rat and rabbit hind limb models of ischemia to demonstrate that FGF-2 and platelet-derived growth factor-BB synergistically promote the formation of new blood vessel networks, which remain stable for over a year of follow-up. Animal studies have also suggested that various stem or progenitor cell formulations, naïve or genetically modified (Nabel, 2002), can be delivered into, or perhaps can even home directly into, ischemic regions to promote new vessel formation.
If duration of treatment proves important, alternative formulation of FGF-2 may be useful to pursue in development, including sustained release or direct gene transfer formulations. Transfer of genetically modified autologous or allogeneic cell preparations also may facilitate adequate temporospatial delivery of the therapeutic agent.
Conclusion
Clearly, neovascularization requires almost a symphonic arrangement of temporal and spatial cues and responses, which is challenging to recapitulate therapeutically. The monotherapy of ischemic myocardial or peripheral artery disease may still be possible using FGF-2 after optimization of formulation and dosing strategies. Successful permutations may involve more ‘upstream’ regulatory factors, cellular agents, or suitable combinations. Careful consideration of clinical methodology may be useful in designing and conducting new clinical tests.
References
- Arras M, Mollnau H, Strasser R, Wenz R, Ito WD, Schaper J, Schaper W. The delivery of angiogenic factors to the heart by microsphere therapy. Nat Biotechnol. 1998;16:159 –162. doi: 10.1038/nbt0298-159. [DOI] [PubMed] [Google Scholar]
- Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM. Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesisin vivo. Circulation. 1995;92:II365–II371. doi: 10.1161/01.cir.92.9.365. [DOI] [PubMed] [Google Scholar]
- Baek SH, Hrabie JA, Keefer LK, Hou D, Fineberg N, Rhoades R, March KL. Augmentation of intrapericardial nitric oxide level by a prolonged-release nitric oxide donor reduces luminal narrowing after porcine coronary angioplasty. Circulation. 2002;105:2779 –2784. doi: 10.1161/01.cir.0000017432.19415.3e. [DOI] [PubMed] [Google Scholar]
- Bouche G, Baldin V, Belenguer P, Prats H, Amalric F. Activation of rDNA transcription by Fgf-2: key role of protein kinase Ckii. Cell Mol Biol Res. 1994;40:547 –554. [PubMed] [Google Scholar]
- Burger PE, Coetzee S, Mckeehan WL, Kan M, Cook P, Fan Y, Suda T, Hebbel RP, Novitzky N, Muller WA, Wilson EL. Fibroblast growth factor receptor-1 is expressed by endothelial progenitor cells. Blood. 2002;100:3527 –3535. doi: 10.1182/blood.V100.10.3527. [DOI] [PubMed] [Google Scholar]
- Bush MA, Samara E, Whitehouse MJ, Yoshizawa C, Novicki DL, Pike M, Laham RJ, Simons M, Chronos NA. Pharmacokinetics and pharmacodynamics of recombinant Fgf-2 in a phase I trial in coronary artery disease. J Clin Pharmacol. 2001;41:378 –385. doi: 10.1177/00912700122010230. [DOI] [PubMed] [Google Scholar]
- Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of Pdgf-Bb and Fgf-2. Nat Med. 2003;9:604 –613. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–661. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, Horner JW, II, Cordon-Cardo C, Yancopoulos GD, Depinho RA. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468 –472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Christian TF. Detecting the effects of angiogenic therapy: how do we measure efficacy? Curr Interv Cardiol Rep. 1999;1:310 –321. [PubMed] [Google Scholar]
- Criqui MH. Peripheral arterial disease – epidemiological aspects. Vasc Med. 2001;6:3–7. doi: 10.1177/1358836X0100600i102. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Claesson-Welsh L. Fgf and Vegf function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201 –207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- Deindl E, Hoefer IE, Fernandez B, Barancik M, Heil M, Strniskova M, Schaper W. Involvement of the fibroblast growth factor system in adaptive and chemokine-induced arteriogenesis. Circ Res. 2003;92:561 –568. doi: 10.1161/01.RES.0000061181.80065.7D. [DOI] [PubMed] [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine Fgfr-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045 –3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1 beta-converting enzyme (Ice)-like and cysteine protease protein (Cpp)-32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Zeiher AM. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ Res. 2000;87:434 –439. doi: 10.1161/01.res.87.6.434. [DOI] [PubMed] [Google Scholar]
- Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in Fgf-2-deficient mice. Embo J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Yan L, Shing Y, Moses MA. Hif-1alpha-mediated up-regulation of vascular endothelial growth factor, independent of basic fibroblast growth factor, is important in the switch to the angiogenic phenotype during early tumorigenesis. Cancer Res. 2001;61:5731 –5735. [PubMed] [Google Scholar]
- Feinberg RL, Gregory RT, Wheeler JR, Snyder SO, Jr, Gayle RG, Parent FN, III, Patterson RB. The ischemic window: a method for the objective quantitation of the training effect in exercise therapy for intermittent claudication. J Vasc Surg. 1992;16:244 –250. doi: 10.1067/mva.1992.36947. [DOI] [PubMed] [Google Scholar]
- Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500 –1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Mcmurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/Kdr activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, Abrams J, Chatterjee K, Daley J, Deedwania PC, Douglas JS, Ferguson TB, Jr, Fihn SD, Fraker TD, Jr, Gardin JM, O’Rourke RA, Pasternak RC, Williams SV, Alpert JS, Antman EM, Hiratzka LF, Fuster V, Faxon DP, Gregoratos G, Jacobs AK, Smith SC., Jr Acc/Aha 2002 guideline update for the management of patients with chronic stable angina – summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients With Chronic Stable Angina) Circulation. 2003;107:149 –158. doi: 10.1161/01.cir.0000047041.66447.29. [DOI] [PubMed] [Google Scholar]
- Goto F, Goto K, Weindel K, Folkman J. Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab Invest. 1993;69:508–517. [PubMed] [Google Scholar]
- Heeschen C, Dimmeler S, Hamm CW, Boersma E, Zeiher AM, Simoons ML. Prognostic significance of angiogenic growth factor serum levels in patients with acute coronary syndromes. Circulation. 2003;107:524 –530. doi: 10.1161/01.cir.0000048183.37648.1a. [DOI] [PubMed] [Google Scholar]
- Herity NA, Lo ST, Oei F, Lee DP, Ward MR, Filardo SD, Hassan A, Suzuki T, Rezaee M, Carter AJ, Yock PG, Yeung AC, Fitzgerald PJ. Selective regional myocardial infiltration by the percutaneous coronary venous route: a novel technique for local drug delivery. Catheter Cardiovasc Interv. 2000;51:358 –363. doi: 10.1002/1522-726x(200011)51:3<358::aid-ccd27>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Hiatt WR, Hirsch AT, Regensteiner JG, Brass EP. Clinical trials for claudication. Assessment of exercise performance, functional status, and clinical end points Vascular clinical trialists. Circulation. 1995;92:614–621. doi: 10.1161/01.cir.92.3.614. [DOI] [PubMed] [Google Scholar]
- Isner JM, Asahara T. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J Clin Invest. 1999;103:1231 –1236. doi: 10.1172/JCI6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R, Simons M. Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease. Trends Cardiovasc Med. 2003;13:116 –122. doi: 10.1016/s1050-1738(02)00259-1. [DOI] [PubMed] [Google Scholar]
- Kim RJ, Wu E, Rafael A, Chen EL, Parker MA, Simonetti O, Klocke FJ, Bonow RO, Judd RM. The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. N Engl J Med. 2000;343:1445 –1453. doi: 10.1056/NEJM200011163432003. [DOI] [PubMed] [Google Scholar]
- Klein S, Roghani M, Rifkin DB. Fibroblast growth factors as angiogenesis factors: new insights into their mechanism of action. Exs. 1997;79:159 –192. doi: 10.1007/978-3-0348-9006-9_7. [DOI] [PubMed] [Google Scholar]
- Laham RJ, Chronos NA, Pike M, Leimbach ME, Udelson JE, Pearlman JD, Pettigrew RI, Whitehouse MJ, Yoshizawa C, Simons M. Intracoronary basic fibroblast growth factor (Fgf-2) in patients with severe ischemic heart disease: results of a phase I open-label dose escalation study. J Am Coll Cardiol. 2000;36:2132 –2139. doi: 10.1016/s0735-1097(00)00988-8. [DOI] [PubMed] [Google Scholar]
- Laham RJ, Rezaee M, Post M, Sellke FW, Braeckman RA, Hung D, Simons M. Intracoronary and intravenous administration of basic fibroblast growth factor: myocardial and tissue distribution. Drug Metab Dispos. 1999a;27:821–826. [PubMed] [Google Scholar]
- Laham RJ, Rezaee M, Post M, Xu X, Sellke FW. Intrapericardial administration of basic fibroblast growth factor: myocardial and tissue distribution and comparison with intracoronary and intravenous administration. Catheter Cardiovasc Interv. 2003;58:375 –381. doi: 10.1002/ccd.10378. [DOI] [PubMed] [Google Scholar]
- Laham RJ, Sellke FW, Edelman ER, Pearlman JD, Ware JA, Brown DL, Gold JP, Simons M. Local perivascular delivery of basic fibroblast growth factor in patients undergoing coronary bypass surgery: results of a phase I randomized, double-blind, placebo-controlled trial. Circulation. 1999b;100:1865 –1871. doi: 10.1161/01.cir.100.18.1865. [DOI] [PubMed] [Google Scholar]
- Lazarous DF, Shou M, Stiber JA, Dadhania DM, Thirumurti V, Hodge E, Unger EF. Pharmacodynamics of basic fibroblast growth factor: route of administration determines myocardial and systemic distribution. Cardiovasc Res. 1997;36:78 –85. doi: 10.1016/s0008-6363(97)00142-9. [DOI] [PubMed] [Google Scholar]
- Lazarous DF, Unger EF, Epstein SE, Stine A, Arevalo JL, Chew EY, Quyyumi AA. Basic fibroblast growth factor in patients with intermittent claudication: results of a phase I trial. J Am Coll Cardiol. 2000;36:1239 –1244. doi: 10.1016/s0735-1097(00)00882-2. [DOI] [PubMed] [Google Scholar]
- Le YJ, Corry PM. Hypoxia-induced bFGF gene expression is mediated through the Jnk signal transduction pathway. Mol Cell Biochem. 1999;202:1–8. doi: 10.1023/a:1007059806016. [DOI] [PubMed] [Google Scholar]
- Lederman RJ, Mendelsohn FO, Anderson RD, Saucedo JF, Tenaglia AN, Hermiller JB, Hillegass WB, Rocha-Singh K, Moon TE, Whitehouse MJ, Annex BH. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the Traffic study): a randomised trial. Lancet. 2002;359:2053–2058. doi: 10.1016/s0140-6736(02)08937-7. [DOI] [PubMed] [Google Scholar]
- Lederman RJ, Tenaglia AN, Anderson RD, Hermiller JB, Rocha-Singh K, Mendelsohn FO, Hiatt WR, Moon T, Whitehouse MJ, Annex BH. Design of the therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (Traffic) trial. Am. J. Cardiol. 2001;88:192–195. doi: 10.1016/s0002-9149(01)01622-8. (A6-7) [DOI] [PubMed] [Google Scholar]
- Li J, Shworak NW, Simons M. Increased responsiveness of hypoxic endothelial cells to FGF2 is mediated by Hif-1alpha-dependent regulation of enzymes involved in synthesis of heparan sulfate FGF2-binding sites. J Cell Sci. 2002;115:1951 –1959. doi: 10.1242/jcs.115.9.1951. [DOI] [PubMed] [Google Scholar]
- Lopez JJ, Edelman ER, Stamler A, Hibberd MG, Prasad P, Caputo RP, Carrozza JP, Douglas PS, Sellke FW, Simons M. Basic fibroblast growth factor in a porcine model of chronic myocardial ischemia: a comparison of angiographic, echocardiographic and coronary flow parameters. J Pharmacol Exp Ther. 1997;282:385 –390. [PubMed] [Google Scholar]
- Mazue G, Bertolero F, Jacob C, Sarmientos P, Roncucci R. Preclinical and clinical studies with recombinant human basic fibroblast growth factor. Ann NY Acad Sci. 1991;638:329 –340. doi: 10.1111/j.1749-6632.1991.tb49043.x. [DOI] [PubMed] [Google Scholar]
- Mcdermott MM, Greenland P, Liu K, Guralnik JM, Criqui MH, Dolan NC, Chan C, Celic L, Pearce WH, Schneider JR, Sharma L, Clark E, Gibson D, Martin GJ. Leg symptoms in peripheral arterial disease: associated clinical characteristics and functional impairment. Jama. 2001;286:1599 –1606. doi: 10.1001/jama.286.13.1599. [DOI] [PubMed] [Google Scholar]
- Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260 –2268. doi: 10.1128/mcb.20.6.2260-2268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or Tnp-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99:1726 –1732. doi: 10.1161/01.cir.99.13.1726. [DOI] [PubMed] [Google Scholar]
- Mukherjee D, Comella K, Bhatt DL, Roe MT, Patel V, Ellis SG. Clinical outcome of a cohort of patients eligible for therapeutic angiogenesis or transmyocardial revascularization. Am Heart J. 2001;142:72 –74. doi: 10.1067/mhj.2001.115786. [DOI] [PubMed] [Google Scholar]
- Nabel EG. Stem cells combined with gene transfer for therapeutic vasculogenesis: magic bullets? Circulation. 2002;105:672–674. [PubMed] [Google Scholar]
- Okada-Ban M, Thiery JP, Jouanneau J. Fibroblast growth factor-2. Int J Biochem Cell Biol. 2000;32:263–267. doi: 10.1016/s1357-2725(99)00133-8. [DOI] [PubMed] [Google Scholar]
- Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci USA. 2001;98:6074 –6079. doi: 10.1073/pnas.111114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:3005.1–3005.12. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper MS, Mandriota SJ, Jeltsch M, Kumar V, Alitalo K. Vascular endothelial growth factor (Vegf)-C synergizes with basic fibroblast growth factor and Vegf in the induction of angiogenesis in vitro and alters endothelial cell extracellular proteolytic activity. J Cell Physiol. 1998;177:439 –452. doi: 10.1002/(SICI)1097-4652(199812)177:3<439::AID-JCP7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Post MJ, Laham R, Sellke FW, Simons M. Therapeutic angiogenesis in cardiology using protein formulations. Cardiovasc Res. 2001;49:522 –531. doi: 10.1016/s0008-6363(00)00216-9. [DOI] [PubMed] [Google Scholar]
- Powers CJ, Mcleskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165 –197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- Ritchie J, Bateman TM, Bonow RO, Crawford MH, Gibbons RJ, Hall RJ, O’Rourke RA, Parisi AF, Verani MS. Guidelines for clinical use of cardiac radionuclide imaging. A report of the American Heart Association/ American College of Cardiology Task Force on Assessment of Diagnostic and Therapeutic Cardiovascular Procedures, Committee on Radionuclide Imaging, developed in collaboration with the American Society of Nuclear Cardiology. Circulation. 1995;91:1278 –1303. [PubMed] [Google Scholar]
- Ruel M, Laham RJ, Parker JA, Post MJ, Ware JA, Simons M, Sellke FW. Long-term effects of surgical angiogenic therapy with fibroblast growth factor 2 protein. J Thorac Cardiovasc Surg. 2002;124:28 –34. doi: 10.1067/mtc.2002.121974. [DOI] [PubMed] [Google Scholar]
- Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, Udelson JE, Gervino EV, Pike M, Whitehouse MJ, Moon T, Chronos NA. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation. 2002;105:788 –793. doi: 10.1161/hc0802.104407. [DOI] [PubMed] [Google Scholar]
- Sullivan CJ, Doetschman T, Hoying JB. Targeted disruption of the Fgf2 gene does not affect vascular growth in the mouse ischemic hindlimb. J Appl Physiol. 2002;93:2009 –2017. doi: 10.1152/japplphysiol.00451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udelson JE, Dilsizian V, Laham RJ, Chronos N, Vansant J, Blais M, Galt JR, Pike M, Yoshizawa C, Simons M. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 improves stress and rest myocardial perfusion abnormalities in patients with severe symptomatic chronic coronary artery disease. Circulation. 2000;102:1605 –1610. doi: 10.1161/01.cir.102.14.1605. [DOI] [PubMed] [Google Scholar]
- Unger EF, Banai S, Shou M, Lazarous DF, Jaklitsch MT, Scheinowitz M, Correa R, Klingbeil C, Epstein SE. Basic fibroblast growth factor enhances myocardial collateral flow in a canine model. Am J Physiol. 1994;266:H1588–H1595. doi: 10.1152/ajpheart.1994.266.4.H1588. [DOI] [PubMed] [Google Scholar]
- Unger EF, Goncalves L, Epstein SE, Chew EY, Trapnell CB, Cannon RO, III, Quyyumi AA. Effects of a single intracoronary injection of basic fibroblast growth factor in stable angina pectoris. Am J Cardiol. 2000;85:1414 –1419. doi: 10.1016/s0002-9149(00)00787-6. [DOI] [PubMed] [Google Scholar]
- Wijns W, Vatner SF, Camici PG. Hibernating myocardium. N Engl J Med. 1998;339:173–181. doi: 10.1056/NEJM199807163390307. [DOI] [PubMed] [Google Scholar]
- Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125:753–765. doi: 10.1242/dev.125.4.753. [DOI] [PubMed] [Google Scholar]