

Abstract
Alcohol abuse causes widespread changes in gene expression in human brain, some of which contribute to alcohol dependence. Previous microarray studies identified individual genes as candidates for alcohol phenotypes, but efforts to generate an integrated view of molecular and cellular changes underlying alcohol addiction are lacking. Here, we applied a novel systems approach to transcriptome profiling in postmortem human brains and generated a systemic view of brain alterations associated with alcohol abuse. We identified critical cellular components and previously unrecognized epigenetic determinants of gene co-expression relationships and discovered novel markers of chromatin modifications in alcoholic brain. Higher expression levels of endogenous retroviruses and genes with high GC content in alcoholics were associated with DNA hypomethylation and increased histone H3K4 tri-methylation, suggesting a critical role of epigenetic mechanisms in alcohol addiction. Analysis of cell type – specific transcriptomes revealed remarkable consistency between molecular profiles and cellular abnormalities in alcoholic brain. Based on evidence from this study and others, we generated a systems hypothesis for the central role of chromatin modifications in alcohol dependence that integrates epigenetic regulation of gene expression with pathophysiological and neuroadaptive changes in alcoholic brain. Our results offer implications for epigenetic therapeutics in alcohol and drug addiction.
Introduction
Brain gene expression is a critical determinant of brain function, including brain disease. Since the introduction of microarray technology, numerous studies have used transcriptome profiling to investigate the mechanisms underlying brain plasticity and brain pathology (Geschwind and Konopka, 2009). Alcohol and other drugs of abuse cause widespread changes in gene expression in human brain (Mayfield et al., 2008; Zhou et al., 2011), some of which contribute to the development and maintenance of drug dependence. Microarray studies in humans and animal models identified individual genes as mechanistic candidates for addiction phenotypes (Mayfield et al., 2008; Maze et al., 2011; Zhou et al., 2011), but an integrated view of molecular and cellular changes underlying alcohol and drug addiction is lacking. Most genomic studies to date focused on individual genes with the highest statistical significance, limiting their discoveries to a handful of candidates. In some cases this strategy resulted in mechanistic discoveries, but the bias towards most significantly regulated genes often lacks the functional foundation and contextual information for generating scientifically sound hypotheses.
Recent developments in statistical genomics and gene annotations provide a foundation for a shift from gene-centric to network- or module-centric systems approaches in data analysis. This shift is warranted by three key findings from recent literature on brain transcriptomes: 1) individual populations of neurons and glia are characterized by unique transcriptional signatures that reflect current functional state of these cells, 2) transcriptomes from complex tissues, such as whole brain, are organized into networks or modules of co-expressed (correlated) genes and 3) these modules of co-expressed genes often reflect functional and structural organization of brain tissue and can be explained by known biological categories, such as cell type, cell organelles and synaptic functions (Sugino et al., 2006; Lein et al., 2007; Cahoy et al., 2008; Doyle et al., 2008; Miller et al., 2008; Oldham et al., 2008; Ponomarev et al., 2010; Day and Sweatt, 2011). These discoveries advanced our understanding of organizational principles of brain transcriptomes and provided a biologically relevant context for interpreting differential expression of individual genes associated with CNS plasticity and pathology, offering critical insight into the mechanisms of Alzheimer's disease (Miller et al., 2010), schizophrenia (Torkamani et al., 2010) and post-traumatic stress disorder (Ponomarev et al., 2010).
To generate an integrated view of the brain transcriptome in human alcoholism, we profiled gene expression levels in postmortem brains of human alcoholics and matched control cases and used a systems approach to data analysis that combined differential gene expression, gene co-expression networks, cell type – specific transcriptomes and a wide range of gene annotations. As a result, we identified critical epigenetic components in gene co-expression and proposed a central role for epigenetic regulation in alcohol-induced changes in global gene expression. Our approach allowed us to generate a unique systems hypothesis of brain changes in human alcoholism that integrated the epigenetic regulation of gene expression with previously reported cellular abnormalities. Our results offer implications for relevant treatment strategies and may serve as a prototype for analysis of the wealth of existing and emerging microarray data.
Materials and methods
Case selection and post-mortem tissue collection
Autopsy brain samples were obtained from the Tissue Resource Center (TRC) at the University of Sydney. The TRC is funded in part by NIAAA to provide brain tissue for alcoholism research. Fresh-frozen sections of tissue from the central (CNA) and basolateral nucleus (BLA) of amygdala, as well as the superior frontal cortex (CTX), were obtained from 32 cases (17 alcoholics and 15 matched controls; 30 males and 2 females). These regions are important substrates in the reward circuitry that is involved in the development of alcohol dependence and alcoholism (Koob and Volkow, 2009). Cases were matched as closely as possible by age, gender, post-mortem interval (PMI) and brain pH. Diagnoses were confirmed by physician interviews, review of hospital medical records, questionnaires to next-of-kin, and from pathology, radiology and neuropsychology reports. Cases were also chosen on the basis that agonal hypoxia did not appear to have differed significantly from the study group. Moreover, none of the brains showed evidence of hypoxic encephalopathy, further suggesting that agonal hypoxia was minimal. We did not accept cases that suffered prolonged agonal states. Cases with a history of polydrug abuse were excluded. Cases were matched for smoking history. In addition, cases with concomitant diseases such as cirrhosis of the liver, Korsakoff psychosis, or Wernicke or hepatic encephalopathies were excluded. The concentration and quality of all RNA samples was determined, and degraded (RNA integrity number, RIN < 4) and/or contaminated RNA samples were excluded from the analysis. The Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) diagnosis assigned to each case was based on a detailed and standardized clinical assessment summary created by specially trained staff with a background in psychiatry and/or psychology.
Microarray profiling and qRT-PCR validation
Total RNA was extracted and used to generate biotin-labeled cRNA using the Illumina TotalPrep RNA Amplification Kit (Ambion, Austin, TX). Biotin-labeled cRNA was then hybridized to Illumina HumanHT-12 whole genome expression beadchips (Illumina, San Diego, CA). The quality of the Illumina bead summary data was assessed using the Bioconductor package Lumi. Data preprocessing included variance stabilization and quantile normalization. To eliminate potentially confounding effects of RNA quality on gene expression, we calculated residuals from the regression analysis of RIN values on gene expression and used them for statistical analysis and WGCNA network construction. We next removed outlier values for each gene within a group using Grubbs’ test (p<0.05). Statistical analysis comparing alcoholic and control groups was performed using the Bioconductor package Limma to carry out a Bayesian two-tailed t-test. A false discovery rate (FDR) for each list of significantly regulated genes with nominal P values < 0.05 was estimated using the method of Benjamini and Hochberg (1995). Our systems approach to prioritizing individual genes is based on integration of nominal statistical significance, gene network information and functional relevance. Therefore, to avoid omitting true positives, all genes with nominal P values < 0.05 were considered. After initial data processing, microarray data from three brain regions of 15 controls and 17 alcoholics were used for network construction.
qRT-PCR validation
Several genes were selected for technical validation using qRTPCR. qRT-PCR was conducted using amplified RNA from the same samples used for microarray experiments. Expression of GIPC1 and DNMT1 was examined in BLA and CTX and expression of MBD3, MLL4 and SETD1 was examined in BLA. All real-time TaqMan® assays are pre-designed by Applied Biosystems (Foster City, CA) and labeled with FAM as a reporter and a non-fluorescent quencher. Detailed TaqMan® protocols are available on the manufacturer's website: (http://www.appliedbiosystems.com/). GUSB was used as endogenous control and the ΔΔCt method was used to analyze data. Outlier values for each gene within a group were then removed based on the Grubbs’ test (p<0.05) and alcoholic and control groups were compared using a one-tailed t-test.
Weighted Gene Co-expression Network Analysis (WGCNA)
Network Construction
The general framework of WGCNA has been described in detail (Zhang and Horvath, 2005). We constructed a signed network for each brain region. Briefly, Pearson correlations were calculated for all pairs of genes, and then a signed similarity (Sij) parameter was derived: Sij = (1+cor(xi,xj))/2, where gene expression profiles xi and xj consist of the expression of genes i and j across multiple microarray samples. In the signed network, the similarity between genes reflects the sign of the correlation of their expression profiles. The signed similarity (Sij) was then raised to power β to represent the connection strength (aij): aij = Sijβ. This step aims to emphasize strong correlations and reduce the emphasis of weak correlations on an exponential scale. Here we chose a power of β = 12 for all brain regions so that the resulting networks exhibited approximate scale-free topology. Next, all genes were hierarchically clustered based on a dissimilarity measure of topological overlap which measures inter-connectedness for a pair of genes (Zhang and Horvath, 2005). The resulting gene dendrogram was used for module detection with the Dynamic Tree Cut method (minimum module size = 100, cutting height = 0.99 and deepSplit = True). Gene modules corresponding to the branches cut-off of the gene tree were labeled in unique colors. Unassigned genes were labeled in grey.
Functional annotation of genes and over-representation analysis of modules
We used several complementary approaches to characterize gene modules. DAVID annotation: For each module, all genes in the module were submitted to the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/). Cell type annotation: Gene sets known to be preferentially expressed in mouse oligodendrocytes, astrocytes, and neurons were obtained from Tables S4-S6 of Cahoy et al. (2008). We restricted our analysis to genes with at least four-fold enrichment in a given cell type. Another set of genes preferentially expressed in microglia was obtained from Table ST3 of Oldham et al. (2008). For each brain region, cell type enrichment analysis was carried out for all modules using the hypergeometric test with gene symbols as unique identifiers. TE annotation: To identify microarray probes targeting potential transposable element (TE) regions, we first obtained genomic coordinates of all Illumina's probes from the probe annotation file, HumanHT-12_V3_0_R2_11283641_A (available at http://www.switchtoi.com/annotationfiles.ilmn), and those of TEs from the UCSC Genome Bioinformatics website (http://genome.ucsc.edu, genome assembly: NCBI36/hg18, track: RepeatMasker). Then we searched for probes targeting to TE regions by requiring that such a probe should fall completely into an annotated TE region. For each brain region, TE enrichment analysis was also carried out for all modules and all major TE classes (i.e. DNA, LTR, LINE, SINE) using the hypergeometric test. GC content analysis: GC content (GC%) values for known genes were obtained from Ensembl (http://www.ensembl.org) using BioMart data management system. Gene GC content includes average GC% of the whole gene including exons, introns and untranslated regions. GC% for each 50-mer Illumina probe was calculated based on the proportion of G and C nucleotides. Effects of gene GC content on gene co-expression was examined with a one-way ANOVA for average gene GC% calculated for each of 72 co-expression modules across all three brain regions.
Identification of alcohol-responsive modules
To identify potential alcohol-responsive modules, we used an effect size - based approach and determined the direction and magnitude of alcohol-induced changes for each co-expression module. We used the t statistics from the comparison of alcohol and control groups and calculated average t values for all genes within each module. We defined a “significant” alcohol-responsive module by requiring average | t | > 1.8 and a “suggestive” alcohol-responsive module by requiring average | t | being in between 0.9 and 1.8. In addition, we used a hypergeometric test to determine the degree of over-representation of differentially expressed genes in each module. All “significant” modules and a majority of “suggestive” modules were also significantly over-represented with genes differentially regulated between the groups.
Meta-network analysis across brain regions
Module comparison between brain region networks was done following the approach described in Oldham et al. (2008). Briefly, for each pair of networks, the overlap between all possible pairs of modules was calculated, and the significance of module overlap was assessed using a one-sided hypergeometric test. The software Cytoscape (http://www.cytoscape.org/) was used to visualize the comparisons and create a meta-network of highly overlapping modules.
Control for confounding variables
In addition to correction for the effects of RIN by regression as described above, we determined if confounding variables, such as age, gender, smoking history, PMI or brain pH contributed to differential gene expression between alcoholic and control cases. We obtained the first eigengene for each module (generated by WGCNA) which could explain the largest portion of variance in gene expression and then correlated these eigengenes with the confounding variables using Pearson correlation. The resulting P values were corrected for multiple comparisons with a Bonferroni procedure. None of the modules were significantly correlated with age, gender or smoking history. Two modules were correlated with PMI (bla9, bla14) and three with brain pH (ctx4, ctx6, ctx16) and genes from these modules were excluded from consideration as alcohol-related candidate genes for future studies.
Chromatin assays
Quantification of global H3K4 histone methylation
Total histone extracts were prepared from frontal cortex tissues of 6 alcoholics and 6 controls using the EpiQuik™ Total Histone Extraction Kit (Epigentek, NY) according to the manufacturer's instructions. For global histone methylation quantification, mono-, di-, and tri-methylations of H3K4 were measured using the EpiQuik™ Global Pan-Methyl Histone Quantification Kits (colorimetric assay) (Epigentek, NY). For each sample, 1.5μg of histone extracts was used in each assay.
DNA methylation analysis of LTR retrotransposons
Genomic DNA was extracted from frontal cortex tissues of 6 alcoholics and 6 controls using the DNeasy Blood & Tissue kit (Qiagen, CA). In a total volume of 80μL, 80ng of genomic DNA was digested with 30U McrBC (New England Biolabs, MA) at 37°C for 3 hours. The same reaction was set up for the mock digestion without adding of McrBC. Then digested and undigested DNA was subjected to real-time PCR quantification using primers for specific LTRs. MLT2A1 (F: 5'-GAGAGGCAGACCCACCCTTA-3', R: 5'-CACGATCACAAGGTCCCACAA-3'), LTR8 (F: 5'-CAAGCTGTCCTTGTTCATTCCT-3', R: 5'-CTGCTTTGGGAAAGGGCTGTT-3'), THE1B (F: 5'-TCATCTTGAATTGTAGCTCCCAT-3', R: 5'-TCCCCTTTATAAAACCATCAGAT-3'). Primers were designed based on the consensus LTR sequences retrieved from the RepBase 14.04. Real-time PCR amplification was set up in triplicate in a 20μL volume composed of 5ng of digested or undigested DNA, 0.2μM of each primer, 1×Power SYBR Green PCR Master Mix (Applied Biosystems, CA) in the 7900HT Fast Real-Time PCR System (Applied Biosystems, CA). All cycling began with an initial denaturation at 95°C for 10min, followed by 40 cycles of 95°C for 15s, and 60°C for 1min. The “mock-digestion” control was used to normalize the McrBC-qPCR data. Fold change was calculated by dividing the normalized data by the mean of the control group.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed for 5 alcoholics and 5 controls using the EpiQuik™ Tissue Methyl-Histone H3-K4 ChIP kit (Epigentek, NY). The ChIP-grade antibody, anti-trimethyl-H3K4 was purchased from the same company. The normal IgG served as a negative control in the ChIP assay. The manufacturer's ChIP protocol was followed with minor changes. Briefly, for each sample ~10mg of frontal cortex tissue was smashed and cross-linked in the presence of 1% formaldehyde for 15min at room temperature. The cross-linking was then stopped by adding 1/10 volume of 1.25M glycine solution. After being washed by 1mL of ice-cold 1×PBS, tissue was homogenized using a motorized pestle (VWR, PA), and pelleted through centrifugation at 10,000rpm for 5min at 4°C. Tissue pellet was then re-suspended in a lysis buffer containing protease inhibitor cocktail. Chromatin was sheared by sonication on ice for 10min in total (20sec ON, 40sec OFF) at level 2 using the Fisher Model 60 Sonic Dismembrator (Fisher Scientific, MA). The length of sheared DNA was checked by agarose gel electrophoresis, which is usually between 200-1000bp. Next, following the manufacturer's protocol, 100μL of sheared chromatin was used for immunoprecipitation with anti-trimethyl-H3K4. For input-DNA preparation, 2μL of proteinase K was first added into 100μL of sheared chromatin, and incubated at 65°C for 15min. Then 4μL of 5M NaCl was added into the input-DNA solution, and incubated at 65°C for 2 hours. After reversal of cross-linked DNA, IP- and input-DNA were purified through fast-spin columns. IP- and input-DNA were subjected to real-time quantitative PCR using primers specific to the promoter regions of one of 6 genes: GIPC1 (F: 5'-CCCCAGAGATTGAATGCATCTT-3', R: 5'-GATTCGAACTTCCGACGTCCA-3'), BCL2L1 (F: 5'-TGAACCCCATTGAGAAGTCCCT-3', R: 5'-ACTGGGAGCCAGGAGTACTCT-3'), UBE1 (F: 5'-CTTGACAGCCTGGCTGCAACA-3', R: 5'-TGCATAAAGTTCCCTACTCGGT-3'), ARHGDIA (F: 5'-CCTCACACTGCCCCAGAGGAT-3', R: 5'-GCGCACTTCTGAGCAGGAGT-3'), CLPTM1 (F: 5'-GGAAACAAACGGGCTGGGAGA-3', R: 5'-CGCGAGATTTCACGCTTTCCTA-3'), and CALCOCO1 (F: 5'-TGCGCGCAGCCTTCTGGGAT-3', R: 5'-CAACAAAAACAGCACTCCGACT-3'). Real-time PCR amplification was set up in triplicate in a 20μL volume composed of 0.5μL of IP- or input-DNA, 0.2μM of each primer, 1×Power SYBR Green PCR Master Mix (Applied Biosystems, CA) in the 7900HT Fast Real-Time PCR System (Applied Biosystems, CA). All cycling began with an initial denaturation at 95°C for 10min, followed by 40 cycles of 95°C for 15s, and 60°C for 1min. PCR specificity was checked by melting curve analysis. The “input” control was used to normalize the ChIP-qPCR data. Fold change was calculated by dividing the normalized data by the mean of the control group.
Results
Alcohol abuse is associated with widespread changes in brain gene expression
To define alterations in the brain transcriptome produced by chronic alcohol abuse, whole-transcriptome gene expression profiling was conducted for three brain regions (basolateral amygdala, BLA; central nucleus of amygdala, CNA; and superior frontal cortex, CTX) from 17 alcoholics and 15 matched control cases. History of alcohol abuse was associated with global changes in gene expression in all three brain regions. “Global” here refers to the fact that numbers of transcripts differentially expressed at a nominal P < 0.05 in different brain regions [3,589 for BLA (FDR<20%), 2,656 for CNA (FDR<28%), and 2,716 for CTX (FDR<28%)], were statistically greater than those expected by chance (all hypergeometric P < 0.0001). Overall, our results corroborate previous studies showing widespread changes in brain gene expression in alcoholics (Mayfield et al., 2008; Zhou et al., 2011). These studies identified many candidate genes that may play a role in alcoholism, but our goal was to extend this line of research beyond the gene-centric approach and to generate and validate easily testable hypotheses at a systems level.
Construction and validation of gene co-expression networks
Our next step was to construct gene co-expression networks to gain insights into functional organization of the brain transcriptome. “Co-expression” here refers to the implication that genes whose expression co-varies (is correlated) across samples are co-expressed, i.e., regulated by similar mechanisms. Identification of gene co-expression patterns has been a fruitful approach to understanding mechanisms of transcriptional regulation in brain (Oldham et al., 2008). We used the weighted gene co-expression network analysis (WGCNA) (Oldham et al., 2008) to construct a gene co-expression network for each of the three brain regions. This method is described in detail elsewhere (Oldham et al., 2008) and its utility as a systems tool has been validated by several research groups (Saris et al., 2009; Torkamani et al., 2010; Mulligan et al., 2011).
All reliably detected genes were included in the network construction and data from both alcoholics and non-alcoholics were combined to detect co-expression patterns. In total, we identified 72 modules in three gene co-expression networks with 25 modules for BLA, 25 for CNA and 22 for CTX (Figure 1). The module size (i.e., total number of genes in a module) ranged from ~100 to ~1,600. To evaluate biological significance of the modules, we used a wide range of gene annotations, including GO, KEGG and major cell classes in brain (Cahoy et al., 2008; Oldham et al., 2008) and examined an over-representation (enrichment) of each biological category in a given module by comparing numbers of genes annotated with this biological category to chance (see Methods for details). Most modules were highly over-represented with at least one functional or structural category, thus, validating biological relevance of gene co-expression relationships. Similar to previous reports (Oldham et al., 2008; Miller et al., 2010; Ponomarev et al., 2010) the most over-represented biological categories in the present study included major cell classes, such as neurons, astrocytes, oligodendrocytes and microglia and cell organelles, such as mitochondrion, nucleus and ribosome (Table 1). This shows that cells and cellular compartments are main sources of gene co-expression and indicates that cell type – specific transcriptional signatures can be obtained from complex brain tissue without isolating cellular populations. We next asked if variation in chromatin states could contribute to gene co-expression.
Figure 1.

Network analysis of gene expression in three brain regions of human alcoholics and control cases identifies distinct modules of co-expressed genes. Shown are dendrograms produced by average linkage hierarchical clustering of transcripts (see Methods). Horizontal color bars represent different co-expression modules that are also numbered. Bar sizes correspond to the number of transcripts in each module. CTX = prefrontal cortex; BLA = basolateral amygdala; CNA = central nucleus of amygdala.
Table 1.
Biological categories critical for the organization of brain transcriptomes and gene co-expression relationships. Shown are categories highly over-represented in at least one module in each brain region. Over-representation P-values in column 3 represent the highest statistical significance detected for a given biological category.
| BIOLOGICAL CATEGORIES | P value | |
|---|---|---|
| Cell Classes | Neuron | 10-104 |
| Microglia | 10-56 | |
| Astrocyte | 10-94 | |
| Oligodendrocyte | 10-54 | |
| Cell Organelles | Ribosome | 10-57 |
| Mitochondrion | 10-16 | |
| Nucleus | 10-9 | |
| Transposable Elements | LTR (Long Terminal Repeat) | 10-17 |
| SINE (Short Interspersed Nuclear Element) | 10-151 |
Detecting epigenetic variation in gene co-expression
Understanding principles of modular organization in gene co-expression remains a challenge, because many modules of highly co-expressed genes are not readily explained by cellular identity or any of the other commonly used annotated functions. This has not been a problem of annotation availability, but rather a problem of annotation usage, as most gene expression studies do not explore expression patterns beyond traditionally used databases, such as GO and KEGG. One area where additional effort is warranted is chromatin marks at individual gene locations. Changes in chromatin structure, often termed epigenetic changes, including DNA methylation and histone modifications are critical variables affecting global gene expression. Therefore, it is reasonable to expect that co-expression of genes in some modules will be driven by chromatin changes.
To explore the effects of chromatin state on gene co-expression relationships, we used two variables that are easily obtained from microarray data: expression of genomic repeats and gene GC content. Repeated sequences, most of which are represented by transposable elements of various classes, constitute a large fraction of most eukaryotic genomes. Transposons are homologous DNA fragments that are present in multiple copies in the genome and are capable of being reproduced and randomly inserted in the host genome (Slotkin and Martienssen, 2007). Transposons are normally silenced by epigenetic mechanisms, including DNA methylation, modifications of histone tails and alterations in chromatin packing and condensation, but can be expressed when the epigenetic silencing is released (Slotkin and Martienssen, 2007). Therefore, expression of transposons may serve as a sensitive marker of changes in chromatin state.
We used the RepeatMasker program (see Methods) and found that 3,992 Illumina microarray probes could be mapped to one of four classes of transposable elements (TE), either DNA transposons or one of three types of RNA transposons (retrotransposons): long terminal repeat (LTR) – containing endogenous retroviruses, long interspersed nuclear elements (LINE) or short interspersed nuclear elements (SINE). Expression of 825 of these probes was statistically greater than the background noise in at least one brain region. We validated these results by manually checking the genomic location of ~15% of the probes using the UCSC genome browser. Most probes corresponding to TEs also mapped to untranslated regions or introns of known or predicted genes, while a relatively large fraction of LTR probes (~20%) also mapped to multiple intergenic regions (Figure 2). Our over-representation analysis of co-expression modules identified several modules that showed significant enrichment with TEs in all brain regions. Coordinated expression of Illumina probes corresponding to LTRs and SINEs was of particular interest as several modules were highly statistically over-represented with these TEs (Table 1). Many TEs have retained functional promoters and the effects of TEs on expression of adjacent individual genes have been well documented (Waterland and Jirtle, 2003). Our over-representation results suggest, for the first time, that epigenetically-controlled TEs can regulate multiple genes in a coordinated fashion.
Figure 2.

Illumina probes can detect expression of transposable elements (TE) in human genome. Shown are 7 example panels from the UCSC Genome Browser (http://genome.ucsc.edu) showing genomic locations that are perfect matches for several Illumina probes representing TEs. Each panel shows whole chromosome on top, with the small red bar representing the location of the Illumina probe. The next track (in blue) shows the location of a known gene, which is followed by the location of the Illumina probe (in brown). Four tracks at the bottom represent the location of known or predicted TEs including SINE, LINE, LTR and DNA transposons. Red arrows point to TEs to which Illumina probes map. A. Probe mapping to an LTR transposon and a 3’UTR of a known gene (IGFL3). B. Probe mapping to a SINE transposon and a 3’UTR of RAX2gene. C. Probe mapping to a LINE transposon and a 3’UTR of FCRL2 gene. D. Probe mapping to a DNA transposon and a 3’UTR of ZNF395 gene. E. Probe mapping to a SINE transposon and an intron of RCBTB2 gene. F. Probe mapping to an LTR transposon and an intron of TTY14 gene. G. Probe mapping to an LTR transposon and an intergenic region.
The second variable obtained from our microarray data was gene GC content, a measure of the nucleotide composition of the gene. Nucleotide composition of individual genes and their promoters plays a critical role in regulation of transcription; two examples include DNA methylation at CpG dinucleotides – a marker of transcriptional repression, and preferential binding of different transcription factors and other regulatory proteins to either GC- or AT-rich motifs (Dekker, 2007). This notion is consistent with studies that reported robust correlations between genomic GC content and several epigenetic marks including DNA methylation, some histone modifications and chromatin condensation (Vinogradov, 2005; Koch et al., 2007). We next examined if gene GC content contributed to gene co-expression. GC content (GC%) values for each gene were obtained from Ensembl, averages for each co-expression module were calculated and one-way ANOVA was carried out. Average GC% showed remarkable variability among modules, ranging from 40 to 56% (Figure 4A) and ANOVA resulted in a highly significant P value (F(71, 34145) = 235; P<10-500), indicating that gene GC content is a critical variable affecting gene co-expression and suggesting that genes with similar GC content are generally co-regulated. Because both TEs and GC% are mechanistically associated with chromatin marks, our data point to previously unrecognized epigenetic sources in gene co-expression and suggest that co-regulation of TEs and genes with similar GC content reflect individual variation in chromatin states and can be used as markers of epigenetic regulation of gene expression.
Figure 4.
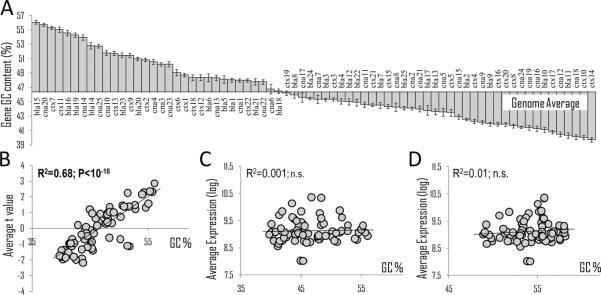
Effects of gene GC content on gene co-expression relationships and regulation by alcohol abuse. A. ANOVA of average gene GC content (GC%) for 72 co-expression modules (labeled with brain region and module number) reveals remarkable heterogeneity among modules [F(71, 34145)=235; P<10-500]. B. Average gene GC% for 72 co-expression modules plotted vs average t values. A t-value represents the magnitude and direction of alcohol effects (t < 0 - down-regulation; t > 0 - up-regulation by alcohol). Average gene GC% (C) and average GC% of Illumina probes (D) are plotted vs average gene expression values for the 72 co-expression modules. (R = Pearson correlation, n.s. = not significant).
Co-expression patterns are highly conserved across brain regions
We next inquired whether observed co-expression patterns in three networks were conserved across brain regions. Module comparison between networks was accomplished by identifying overlapping genes and calculating statistical significance of the overlap between all possible pairs of modules. We found that all modules in a given brain region have genes significantly overlapping with at least one module from a different brain region and the majority of modules were highly overlapping across all three brain regions (Figure 3), suggesting conserved patterns of gene regulation in different brain regions. This finding was consistent with the Oldham et al. study (Oldham et al., 2008) that showed similar co-expression preservation in three different regions. Similar to their study, we found that major brain cell classes that include neurons, astrocytes, oligodendrocytes and microglia as well as cellular organelles including mitochondria and ribosomes are the most conserved biological categories with respect to gene co-expression. In addition, our analysis provides the first evidence that regulation of modules enriched with LTR and SINE TEs as well as modules containing genes with high or low GC content are conserved and cluster together. We hypothesize that conserved modules representing TEs and opposite ends of the GC% spectrum reflect fundamental epigenetic influences on gene co-expression relationships.
Figure 3.
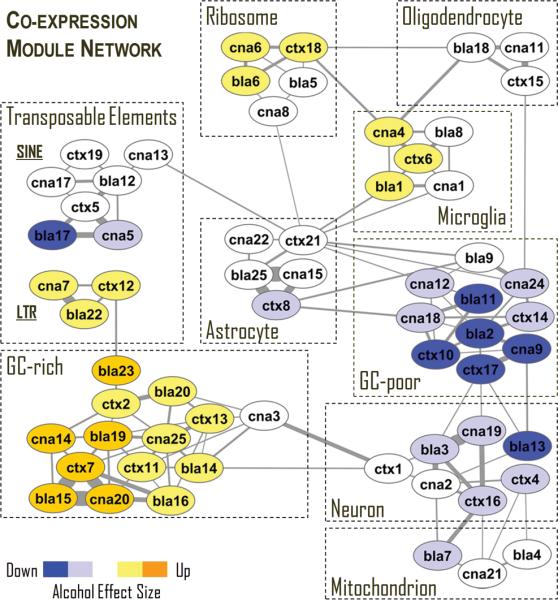
A meta-network of overlapping gene co-expression modules in human brain. Each node represents a module of co-expressed genes. Nodes are labeled with brain region and a module number. An edge between two nodes indicates a significant overlap of genes between two modules of different brain regions. Shown are only highly overlapping modules (P < 10-20). An overlap of three and more modules from different brain regions (e.g. ribosomes) indicates a cluster of highly conserved co-expression modules (represented by rectangular boxes with a dashed borderline). All overlapping modules within a cluster are over-represented with genes from a major biological category shown in Table 1, such as Neuron or Transposable Elements. In addition, several modules were clustered based on average GC content of their genes; GC-rich and GC-poor modules formed separate clusters. Several GC-poor modules were also over-represented with “nucleus” genes. Thickness of connecting edges is proportional to the significance of the overlap. Module colors represent the direction and magnitude of regulation in alcoholic brain based on average t values (see Methods) (yellow = up-regulation, blue = down-regulation in alcoholics; intense colors: |t|>1.8; light colors: |t|>0.9; bla = basolateral amygdala; cna = central nucleus of amygdala, ctx = prefrontal cortex).
Gene co-expression networks provide insight into functional changes in alcoholic brain
By constructing gene co-expression networks and identifying biological sources of co-expression modules, we created a functional framework for interpretation of differential expression between alcoholics and controls at a systems level. To examine global effects of alcohol abuse on gene co-expression networks, we used an effect size - based approach and determined the direction and magnitude of alcohol-induced changes by calculating average t values for genes of each co-expression module (Figure 3, shown in color). T-tests were conducted for every transcript in each brain region to compare gene expression between alcoholics and control cases and t values can be used as estimates of the effect size. This revealed three main findings: 1) chronic alcohol abuse differentially affected major cell types in brain; transcripts from neuronal modules were mainly down-regulated in alcoholics while several modules representing microglia were up-regulated, 2) alcohol abuse resulted in up-regulation of LTR retrotransposons and 3) most genes from GC-rich modules were up-regulated in alcohol abusers, while genes from GC-poor modules were mainly down-regulated. This last finding was especially intriguing because it suggested that gene nucleotide composition determines, at least in part, whether genes will be regulated in response to strong environmental challenges such as chronic alcohol abuse. We further investigated the relationship between gene GC content and regulation by chronic alcohol by calculating Pearson correlation between average gene GC content and t values for the 72 co-expression modules (Figure 4B). Remarkably, gene GC content accounted for ~68% (R = 0.83; P<10-18) of the differential gene expression between alcoholics and controls. This relationship was not an artifact of differential microarray probe hybridization, because neither average gene GC content nor average Illumina probe GC content correlated with average transcript expression values (Figure 4CD). Based on the rationale discussed above, the coordinated regulation of LTR retrotransposons and genes with similar GC content suggests a critical role of chromatin modifications in the modulation of gene expression in the alcoholic brain. Based on the results of the integration of co-expression networks with differential gene expression, we further investigated the effects of alcohol abuse on cellular transcriptomes and chromatin modifications.
Effects of alcohol abuse on cell type – specific transcriptomes
Neuronal and glial cells are the fundamental constituents of the CNS. Despite identical genomes, different cell types use distinct transcriptional programs that result in remarkable heterogeneity of cellular transcriptomes that are thought to reflect physiological properties and the functional state of individual cells (Sugino et al., 2006; Doyle et al., 2008). To investigate the effects of alcohol abuse on cell type – specific gene expression, we again used the effect size – based approach and analyzed distributions of t values for genes that are primarily expressed in one of the four major cell classes: neurons, microglia, oligodendrocytes and astrocytes (Figure 5). Cell type-specific genes were determined by literature (Cahoy et al., 2008; Oldham et al., 2008; see Methods for detail). We hypothesized that the shape and position of the t distributions can reveal global effects of alcohol on individual cell types. In principle, an alcohol-induced change in expression of a particular gene reflects one of two distinct possibilities: an actual change in mRNA copy number or a change in the abundance of tissue or the number of cells where this gene is preferentially expressed. For example, a significant shift of the t distribution mean or median compared to zero chance would most likely indicate a change in abundance or general activity of a cellular population, while deviation from normality as, for example, “bumps” on the distribution may indicate a coordinated expression of functionally relevant genes.
Figure 5.

Global effects of alcohol abuse on transcriptomes of four brain cell classes. Direction and magnitude of alcohol-induced changes were estimated by plotting t-distributions for genes enriched in a specific cell type (cell type specificity was determined based on Cahoy et al., 2008 and Oldham et al., 2008). A t-value represents the magnitude and direction of alcohol effects (t < 0 - down-regulation; t > 0 - up-regulation by alcohol). Left panels: Average t distributions for each cell class for three brain regions. Right panels: Corresponding average t values (± SEM) (* = P < 0.05; based on one-sample t-test comparing average t values to zero chance with a Bonferroni correction). Grey arrow points to a “bump” on cortical distribution caused by a cluster of alcohol up-regulated genes (all t > 2).
Our analysis revealed discrete effects of alcohol on different cell types in different brain regions (Figure 5). Neuronal distributions in the amygdalar regions were significantly shifted to the left while all microglial distributions were shifted to the right, suggesting a decrease in numbers of neurons and an increase in microglia. In addition, several molecular markers of activated microglia, such as CCL2 and TSPO, were significantly up-regulated in the amygdala (all P < 0.02), while neuronal markers, such as SST, VIP and GABRG2 were generally down-regulated. These results are consistent with alcohol literature showing general degeneration of neurons as well as activation and proliferation of microglia in alcoholic brain (Crews et al., 2011). This analysis also showed clear differences in regional sensitivity to alcohol, as BLA was the most affected region, while frontal cortex was the least affected.
Detailed analysis of the neuronal t distribution in cortex revealed a deviation from normality as several genes significantly up-regulated in alcoholics contributed to a “bump” on the distribution (indicated by an arrow in Figure 5). Most of these genes were clustered in the GC-rich ctx7 module (see Figure 3). A majority of the deviated genes were annotated as being involved in synaptic transmission, particularly at glutamatergic synapses; examples include dynamin (DNM1; P = 0.007), syntaxin (STX1A; P = 0.04), synapsin I (SYN1; P = 0.05), synaptophysin (SYP; P = 0.005), glutamate NMDA receptor NR1 subunit (GRIN1; P = 0.008) and vesicular glutamate transporter 1 (VGLUT1, SLC17A7; P = 0.001). Two additional up-regulated genes from the ctx7 module with roles in glutamatergic neurotransmission are GIPC1 (P = 0.005) and MIB2 (P = 0.0002) which are involved in NMDA receptor trafficking and ubiquitination of the NMDA NR2B subunit, respectively (Yi et al., 2007; Jurd et al., 2008). Another striking discovery was that GC content of all of these genes was greater than average, suggesting this played a role in coordinated up-regulation of synaptic genes in alcohol abusers.
Activation of endogenous retroviruses in alcoholic brain is associated with DNA hypomethylation
Detailed examination of three highly overlapping modules over-represented with LTR transcripts (Figure 3) revealed that the majority of the transcripts were up-regulated in alcoholics, with many up-regulated probes mapping to multiple intronic and intergenic genomic regions corresponding to LTR TEs. This pattern of expression is consistent with a genome-wide transcriptional activation of LTR retrotransposons in alcoholic brain. LTR-containing TEs represent a class of endogenous retroviruses (ERVs) most of which are non-functional remnants of ancient retroviral infections (Antony et al., 2004). However, many human ERVs (HERVs) have retained functional promoters and the potential to encode viral proteins, randomly insert their DNA in the genome and modify the expression of adjacent genes (Morgan et al., 1999; Waterland and Jirtle, 2003). Because expression of ERVs can cause genomic instability and disease (Antony et al., 2004), eukaryotic hosts developed defense mechanisms against these genomic parasites. The LTR regions of ERVs are heavily methylated in somatic cells, which was proposed as a primary mechanism of their transcriptional repression (Waterland and Jirtle, 2003). Expression of ERVs correlates with subtle changes in DNA methylation status (Waterland and Jirtle, 2003) and ERV activity can be used as a sensitive marker of global DNA hypomethylation (Schulz et al., 2006; Balada et al., 2009).
Here, we tested the hypothesis that DNA in brains of alcoholics is less methylated, which results in transcriptional activation of HERVs. We used a qPCR-based method to measure DNA methylation in frontal cortex of alcoholic and control cases for three ERV families and observed a reduction of DNA methylation in the LTR region of these retrotransposons (Figure 6A), suggesting that activation of ERVs in alcoholics was due, at least in part, to DNA hypomethylation. This finding was consistent with a 20-30% down-regulation of the DNA methyltransferase, DNMT1, in all three brain regions of alcoholics (BLA: P=0.002; CNA: P=0.05; CTX: P=0.04). DNMT1 plays an important role in the establishment and regulation of tissue-specific patterns of methylated cytosine residues and a reduction of DNMT1 activity is often observed together with global DNA hypomethylation in several types of cancer and other pathological conditions (Hervouet et al., 2010). Alcohol-induced global DNA hypomethylation has been reported in liver (Lu et al., 2000), fetal tissue (Garro et al., 1991) and colon (Choi et al., 1999), and our study is the first to report it in human brain.
Figure 6.
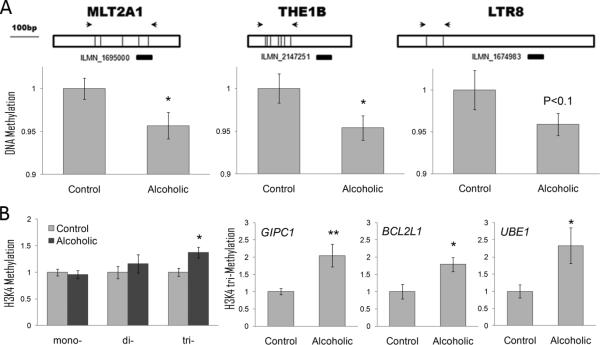
Epigenetic modifications in alcoholic cortex. A. DNA hypomethylation is associated with higher expression of LTR retrotransposons in alcoholics. Top panel: Schematic diagram of three families of LTR (MLT2A1, THE1B and LTR8). Illumina probes shown below as black bars can detect expression of these classes of LTRs and were up-regulated in alcoholic brain (Fold Change range from 20 to 48%; P value range from 0.007 to 0.03). Arrows show locations of RT-PCR primers used for DNA methylation assays. DNA methylation shown as fold change compared to control group was measured using a combination of DNA digestion with a methylation-sensitive enzyme and qRT-PCR. B. Tri-methylation of histone 3 Lysine 4 (H3K4me3) shown as fold change compared to control group is increased in alcoholics. Left panel: Global methylation of H3K4 measured using a combination of chromatin immunoprecipitation (ChIP) and qRT-PCR. Three right panels: H3K4me3 levels at the promoter of three hub genes from the ctx7 GC-rich module (gene symbols are italicized). * = P < 0.05; ** = P < 0.01, as determined by a t-test.
Up-regulation of GC-rich genes in alcoholics is associated with increased H3K4 tri-methylation
We next focused on modules containing GC-rich genes, many of which were up-regulated in alcoholics (Figure 3). Three of these genes significantly up-regulated in all three brain regions (all P < 0.05) were the histone methyltransferases MLL, MLL4 and SETD1A specific for tri-methylation of histone 3 at Lysine 4 (H3K4me3), a chromatin mark of actively transcribed genes. Because of this up-regulation and a strong positive correlation between genome GC content and the H3K4me3 mark (Koch et al., 2007), we hypothesized that up-regulation of some genes from the GC-rich modules in alcoholics is associated with increased H3K4me3. First, we found that global tri-methylation was increased in alcoholic brain (Figure 6B). Next, we used ChIP-qPCR to test H3K4 tri-methylation level at the promoter region of six hub genes from the ctx7 module that were up-regulated in alcoholics. Three out of six genes (GIPC1, BCL2L1 and UBE1) showed significantly increased levels of H3K4 tri-methylation in alcoholics (Figure 6B), which was consistent with the up-regulation of their transcripts, while H3K4me3 levels of the other three genes did not differ between the groups. These results suggest that the alcohol-induced up-regulation of genes in the GC-rich modules may, at least in part, be explained by increased levels of H3K4me3 in their promoters.
Alcohol-induced up-regulation of genes involved in transcription co-repressor complexes
Up-regulation of several functionally related genes point to another mechanism of epigenetic control. Methyl-CpG-binding protein, MBD3, and chromodomain helicase, CHD4, were significantly up-regulated in alcoholics (MBD3 in all brain regions; CHD4 in BLA; all P < 0.006). These proteins are partners in the NuRD (nucleosome remodeling and histone deacetylation) transcription co-repressor complex that is involved in transcriptional repression via coupling histone deacetylase activity with methylated DNA and establishing a repressive chromatin state (McDonel et al., 2009). Strikingly, most other members of the transcription co-repressor complexes were also up-regulated in alcoholics; these include SIN3A, SIN3B, MTA1, MTA2, RBBP4, GATAD2A and GATAD2B, suggesting that these complexes are activated and play a role in down-regulation of some genes in alcoholic brain.
Identification of candidate genes for alcohol addiction using a systems approach
Identification of candidate genes for human diseases remains the strategy of choice for genome-wide surveys, such as microarrays and genome-wide association studies. One goal of our systems approach was to establish a functional framework for prioritization of candidate genes. Gene co-expression network analysis determines a connectivity measure for individual genes based on their Pearson correlations with all of the other genes in the module. This measure provides an estimate of the gene's importance in gene networks, as highly connected hub genes proved to be functionally significant (Horvath et al., 2006). We nominated candidate genes based on two criteria: 1) differential expression between alcoholics and controls in, at least, one brain region and 2) being in the top 20% of hub genes with the highest intra-modular connectivity. We hypothesize that these genes have high functional significance in biological processes associated with alcohol addiction. Our finding that alcohol abuse changes gene expression through changes in chromatin states provided rationale for giving additional priority to genes involved in epigenetic regulation of gene expression.
For example, two histone methyltransferases, MLL4 and SETD1A, were among alcohol-regulated hub genes in CTX and BLA respectively, providing additional support for the importance of H3K4me3 in establishing patterns of gene expression in alcoholic brain. Another hub gene, TRIM28 (KRAB-associated protein 1, KAP1) is 20-40% up-regulated in alcoholics in all brain regions (all P < 0.004). The product of this gene is critical for silencing ERVs during early embryonic development (Rowe et al., 2010), and its up-regulation may indicate the cell's compensatory response to ERV activation. Methylation of DNA and other transmethylation reactions rely on the availability of SAM (S-adenosylmethionine) molecule, the primary methyl group donor in the cell. One of the hub genes down-regulated in alcoholics in all brain regions was MAT2B (methionine adenosyltransferase II, beta subunit; all P < 0.004), the enzyme involved in the synthesis of SAM from methionine. The beta subunit changes kinetic properties of the catalytic alpha subunit by rendering it more susceptible to product inhibition by SAM, and a down-regulation of MAT2B in T cells was accompanied by a 6–10-fold increase in intracellular SAM levels (LeGros et al., 2001). Because SAM levels are decreased in alcoholics (Blasco et al., 2005), the down-regulation of MAT2B in alcoholic brain may indicate a compensatory response to this reduction. In addition, several cortical genes acting at glutamatergic synapse, including GRIN1, STX1A, SYP, DNM1, GRIK5, GRINA, VAMP2, GIPC1 and MIB2 were among the significantly up-regulated hub genes (all P < 0.05), suggesting a central role of glutamate neurotransmission in alcohol dependence. Differential expression of several prioritized genes including DNMT1, MBD3, MLL4, SETD1A and GIPC1 was further validated using qRT-PCR (Table 2). Overall, this analysis provides rationale for targeting epigenetic processes, glutamatergic synapse and functionally relevant individual genes to promote the development of new therapies for human alcoholism.
Table 2.
Results of qRT-PCR validation. Array P-values are based on a Bayesian two-tailed t-test and Taqman P-values are based on s Student's one-tailed t-test. FC = fold change. Positive FC = up-regulation in alcoholics.
| Brain Region | Gene Symbol | Illumina ID | Taqman Assay ID | Array P-value | Array FC | Taqman P-value | Taqman FC |
|---|---|---|---|---|---|---|---|
| BLA | GIPC1 | ILMN_1796177 | Hs00991802_m1 | 7.4E-05 | 1.65 | 4.9E-03 | 1.91 |
| BLA | SETD1A | ILMN_1714327 | Hs00986924_g1 | 4.1E-04 | 1.28 | 5.0E-02 | 1.27 |
| BLA | MLL4 | ILMN_1715968 | Hs00998464_gH | 2.7E-02 | 1.11 | 2.6E-02 | 1.26 |
| BLA | DNMT1 | ILMN_1760201 | Hs02558036_s1 | 2.4E-03 | -1.27 | 2.7E-03 | -1.44 |
| BLA | MBD3 | ILMN_1698491 | Hs00924265_g1 | 1.4E-06 | 1.42 | 3.8E-04 | 2.38 |
| CTX | GIPC1 | ILMN_1796177 | Hs00991802_m1 | 4.9E-03 | 1.55 | 1.8E-02 | 1.93 |
| CTX | DNMT1 | ILMN_1760201 | Hs02558036_s1 | 4.2E-02 | -1.21 | 3.1E-02 | -1.32 |
Discussion
We used a novel systems approach to transcriptome profiling and provided the first comprehensive assessment of gene expression changes in alcoholic brain at a systems level. This approach allowed us to generate several systems hypotheses with an emphasis on epigenetic regulation of gene expression and we obtained functional evidence for two of these hypotheses experimentally. Our results provide a functional framework for integrating data across alcohol-related studies, which we used to generate a global systems hypothesis for the role of chromatin modifications in alcohol dependence that consolidates the epigenetic regulation of gene expression and cellular changes in alcoholic brain (Figure 7). We hypothesize that neuropathology and neuroadaptations that contribute to alcohol addiction and dependence are, at least in part, mediated by alcohol-induced epigenetically-mediated changes in gene expression. Below we discuss the evidence for individual components of this hypothesis and the rationale for their integration.
Figure 7.
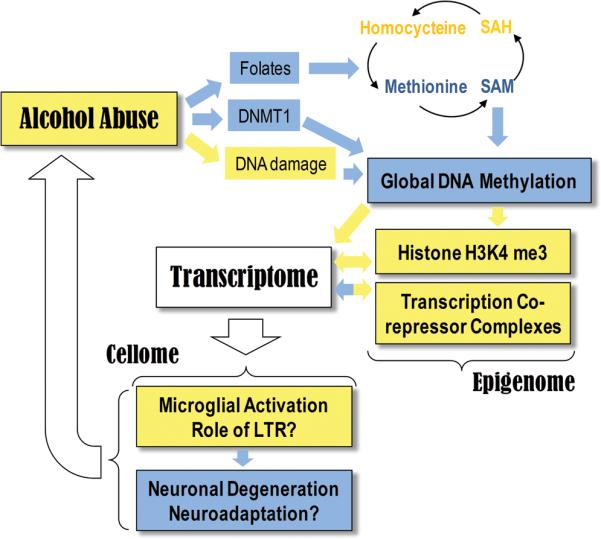
A systems hypothesis for the central role of epigenetic modifications in alcohol dependence. Yellow color indicates general increase, up-regulation or activation, while blue color indicates general decrease, down-regulation or degeneration. Chronic alcohol causes well-documented vitamin B and folate deficiencies that negatively affect one-carbon metabolism and can result in homocysteinemia and a decreased production of S-adenosylmethionine (SAM), the methyl group donor in most transmethylation reactions. Decreased SAM and other alcohol-mediated effects, such as acetaldehyde-induced inhibition of the maintenance DNA methyltransferase, DNMT1 and 5-methylcytosine demethylation induced by the DNA damage and repair can cause global DNA hypomethylation, a chromatin state associated with many pathological conditions including cancer. DNA hypomethylation may trigger a chain of events resulting in changes in chromatin state, such as increase in H3K4 tri-methylation and activation of transcription co-repressor complexes, which result in changes in global gene expression. Epigenome – mediated changes in transcriptome can determine cell type – specific functional states, such as activation of microglia, neuronal degeneration in the amygdala and neuroadaptations in the PFC. In summary, alcohol-induced epigenetically-mediated changes in gene expression may underlie brain pathology and brain plasticity associated with alcohol abuse and alcohol dependence.
Central to our hypothesis is the potentially critical role of DNA hypomethylation in alcohol-induced gene expression. We detected a small, but reliable, decrease in methylation of HERV sequences associated with a much larger increase in HERV transcript abundance, suggesting that a small decrease in DNA methylation can have profound effects on global gene expression. ERVs are heavily methylated in mammalian genomes, accounting for a large fraction of all methylated cytosines (Walsh et al., 1998) and an increase in ERV transcription is a sensitive marker of global DNA hypomethylation (Schulz et al., 2006; Balada et al., 2009). One example of the relationship between DNA methylation and ERV activity is that coat color in the yellow agouti mouse is controlled by the level of DNA methylation of LTR, upstream of the agouti locus (Morgan et al., 1999; Waterland and Jirtle, 2003). Methyl supplements including extra folates, vitamin B12, choline, and betaine fed to dams increased the level of DNA methylation in the agouti LTR and changed the phenotype of offspring from yellow to mottled to pseudoagouti (Waterland and Jirtle, 2003). This research shows that subtle changes in DNA methylation are proportional to the level of activation of ERVs, which is consistent with our findings. Two other observations provide additional support for global DNA hypomethylation in alcoholic brain. First, a down-regulation of DNMT1 in alcoholic brain is consistent with literature showing similar response in some hypomethylating states associated with cancer and other pathological conditions (Hervouet et al., 2010). And second, up-regulation of several ribosomal modules (see Figure 3) suggests a release of transcriptional repression of ribosomal DNA repeats by DNA hypomethylation (McStay and Grummt, 2008). Alcohol-induced global DNA hypomethylation has been reported in several peripheral tissues of alcohol-related animal models, where it was proposed to play a role in alcoholic liver disease, fetal alcohol syndrome and colon cancer (Garro et al., 1991; Choi et al., 1999; Lu et al., 2000; Shukla et al., 2008; Hamid et al., 2009). Although the effects of alcohol on DNA methylation and expression of individual genes in the CNS has been reported (Bleich and Hillemacher, 2009), our study is the first to demonstrate global changes in DNA methylation in alcoholic brain, where it may contribute to the development and maintenance of alcohol dependence. Chronic alcohol can result in a decrease in DNA methylation via several mechanisms (Figure 7), including vitamin B and folate deficiencies, resulting in an impairment of one carbon metabolism and a decrease in SAM (Hamid et al., 2009), acetaldehyde-mediated inhibition of the activity of DNMT1 (Garro et al., 1991) and 5-methylcytosine demethylation induced by alcohol-related DNA damage (Chen et al., 2011). Specifically, SAM is decreased, while its metabolites, S-adenosylhomocysteine (SAH) and homocysteine are increased in chronic alcoholics (Blasco et al., 2005), which may be one cause of the global DNA hypomethylation.
Many chromatin modifications are mechanistically linked (Jaenisch and Bird, 2003), resulting in a limited number of chromatin states (Ernst et al., 2011). In addition to DNA hypomethylation, we detected an increase in global and gene-specific tri-methylation of H3K4 and activation of several genes involved in transcription co-repressor complexes (TCC) in alcoholic brain, with all these chromatin modifications being associated with the methylation status of cytosines within CpGs. Both H3K4me3 and the histone deacetylase (HDAC) activity coupled to TCCs can be changed by drugs of abuse (Pandey et al., 2008; Renthal and Nestler, 2009; Zhou et al., 2011). Specifically, acute ethanol and cocaine decrease HDAC activity, while ethanol withdrawal and chronic cocaine increase it (Pandey et al., 2008; Renthal and Nestler, 2009). Consistent with these findings and our own results is an up-regulation of MBD3 in brains of alcoholics and cocaine abusers (Liu et al., 2006; Zhou et al., 2011), which is a TCC protein critical for coupling HDAC activity and chromatin remodeling. Importantly, drug effects on key epigenetic “master regulators” result in changes in chromatin state which cause changes in global gene expression, some of which are molecular determinants of functional changes in brains of drug addicts. Drugs targeting these master switches are emerging as potential therapeutics for neurodegenerative disorders and drug addiction (Abel and Zukin, 2008; Renthal and Nestler, 2009).
Another important component of our hypothesis is the transcriptional activation of HERVs in alcoholic brain induced by DNA hypomethylation. Activation of ERVs has been linked to chronic diseases including cancer, multiple sclerosis and autoimmune disorders (Balada et al., 2009). It appears that this activation is not just a marker of global DNA hypomethylation but can result in functional consequences as an ERV-encoded glycoprotein, syncytin, can directly activate microglia and astrocytes and produce neuroinflammation (Antony et al., 2004). Microglial activation can result in neuronal degeneration (Crews et al., 2011), and compounds secreted by syncytin-activated astrocytes can produce cytotoxicity to oligodendrocytes and myelin degeneration (Antony et al., 2004), which is consistent with pathologies observed in alcoholics (Harper et al., 2003; Pfefferbaum et al., 2009; Zahr et al., 2011). Alcohol-induced neuroimmune response was recently proposed to be a critical factor in alcohol addiction (Crews et al., 2011) and we propose a potential role for endogenous retroviruses in neuroinflammation and brain pathophysiology of human alcoholism. Our global profiles of cell type-specific genes were mainly consistent with microglial activation in all brain regions and neuronal degeneration in the amygdala. In addition, we detected a subset of synaptic genes highly up-regulated in cortex of alcohol abusers. This up-regulation may indicate an adaptive increase in glutamatergic transmission in response to the loss of neurons or alcohol-induced inhibition of NMDA receptors, which is consistent with literature showing general potentiation of glutamatergic synapses after chronic alcohol (Gass and Olive, 2008; Henriksson et al., 2008). The glutamatergic potentiation in the PFC may underlie the long-lasting impairment in cognitive control of goal-directed behaviors that characterizes addicted individuals (Koob and Volkow, 2009).
One unexpected finding of our study was that the nucleotide composition of a gene, measured as gene GC content, could determine, at least in part, its patterns of expression and regulation by homeostatic perturbations, such as chronic alcohol. The importance of genome GC content in regulation of gene expression is well established because many transcription factors and other DNA-binding proteins that are part of chromatin modification complexes preferentially bind to GC- or AT-rich motifs (Lorincz et al., 2004; Zhang et al., 2004; Dekker, 2007; McDonel et al., 2009). It is possible that up-regulation of many GC-rich genes and down-regulation of many GC-poor genes in alcoholic brain were mediated by some of these DNA-binding regulators. How exactly nucleotide composition of a given gene contributes to its co-expression patterns and regulation by alcohol abuse will be addressed in future studies.
Our study was not designed to address causality in our integrative view of alcohol dependence. Therefore, alternative interpretations of our results are possible. For example, the observed chromatin changes may be secondary to the primary effects of chronic alcohol on different cells. The cause and effects relationships between different components of our systems hypothesis will be addressed by validation experiments in the future. Another limitation is that we cannot distinguish gene expression changes produced by chronic alcohol abuse and those associated with pre-existing conditions, such as genetic polymorphisms or pathological states that may lead to alcoholism. Multiple studies in humans and animal models highlighted the importance of the genetic component in alcohol addiction (reviewed in Crabbe, 2008; Mayfield et al., 2008; Spanagel, 2009; Treutlein and Rietschel, 2011), In attempt to determine genes that may be regulated by genetic differences, we checked our top candidate genes against the Genetic Association Database (http://geneticassociationdb.nih.gov/) which is an archive of human genetic association studies of complex diseases, including brain pathologies and substance abuse syndromes. None of the chromatin modifying genes was genetically associated with alcoholism or other substance abuse diseases, and only the NMDA receptor NR1 subunit (GRIN1) was associated with alcoholism (Wernicke et al., 2003). Additional support for non-genetic causes of differential gene expression in our study is provided by a recent report showing that chronic intermittent alcohol consumption changes gene expression in brain of genetically homogeneous C57BL/6 mice (Wolstenholme et al., 2011). This study showed that chromatin modifications and glutamate signaling were top functional groups over-represented with alcohol-related genes, which is consistent with our findings. Finally, a correlational analysis between alcohol variables and alcohol-related modules showed that the first eigengene of the ctx12 LTR module (see Figure 3) up-regulated in alcoholics was significantly correlated with the duration of drinking (corrected P = 0.03), suggesting that DNA hypomethylation and the up-regulation of LTR retrotransposons is a consequence of chronic alcohol and not a pre-existing condition. The combined evidence suggests that in our study, global changes in gene expression in alcoholic brain are mainly caused by chronic alcohol abuse and that alcohol abuse changes gene expression via changes in chromatin states. To better understand the interplay between genetic, epigenetic and environmental causes in controlling gene expression in alcoholism, integrative approaches across studies are warranted.
In summary, our study is the first to present an integrated view of alcohol dependence using a systems approach to transcriptome profiling in human brain. The systems analysis of the transcriptome allowed us to make mechanistic predictions about the upstream epigenetic control as well as downstream cellular physiology. One implication is that epigenetic interventions may effectively correct the widespread changes in brain gene expression and functional abnormalities produced by chronic alcohol abuse. Many epigenetic therapeutics have been developed for other diseases and our study may direct some of these therapeutics toward alcoholism and drug addiction.
Acknowledgments
This work was supported by NIH NIAAA grants: AA012404 to R.A.H., INIA grants (AA013518 to R.A.H., AA016648 to R.D.M., AA013517 Pilot Projects to R.D.M. and I.P., AA013476 (Subcontract to I.P.) and a K award to I.P. (AA017234). The authors are grateful to the Australian Brain Donor Programs NSW Tissue Resource Centre for providing alcoholic and control brain tissue for analysis. The Centre is supported by the University of Sydney, National Health and Medical Research Council of Australia, Schizophrenia Research Institute, and the National Institute of Alcohol Abuse and Alcoholism. We thank Dr. Vishy Iyer for critical reading of the manuscript and Brianne Patton and Elizabeth Osterndorff-Kahanek for technical assistance.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Contributor Information
Igor Ponomarev, Waggoner Center for Alcohol and Addiction Research and the College of Pharmacy, University of Texas at Austin, Austin, Texas 78712..
Shi Wang, Waggoner Center for Alcohol and Addiction Research and the College of Pharmacy, University of Texas at Austin, Austin, Texas 78712.; Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
Lingling Zhang, Waggoner Center for Alcohol and Addiction Research and the College of Pharmacy, University of Texas at Austin, Austin, Texas 78712.; Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
R Adron Harris, Waggoner Center for Alcohol and Addiction Research and the College of Pharmacy, University of Texas at Austin, Austin, Texas 78712..
R Dayne Mayfield, Waggoner Center for Alcohol and Addiction Research and the College of Pharmacy, University of Texas at Austin, Austin, Texas 78712..
References
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony JM, van Marle G, Opii W, Butterfield DA, Mallet F, Yong VW, Wallace JL, Deacon RM, Warren K, Power C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci. 2004;7:1088–1095. doi: 10.1038/nn1319. [DOI] [PubMed] [Google Scholar]
- Balada E, Ordi-Ros J, Vilardell-Tarres M. Molecular mechanisms mediated by human endogenous retroviruses (HERVs) in autoimmunity. Rev Med Virol. 2009;19:273–286. doi: 10.1002/rmv.622. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. 57:289. [Google Scholar]
- Blasco C, Caballeria J, Deulofeu R, Lligona A, Pares A, Lluis JM, Gual A, Rodes J. Prevalence and mechanisms of hyperhomocysteinemia in chronic alcoholics. Alcohol Clin Exp Res. 2005;29:1044–1048. doi: 10.1097/01.alc.0000169265.36440.ee. [DOI] [PubMed] [Google Scholar]
- Bleich S, Hillemacher T. Homocysteine, alcoholism and its molecular networks. Pharmacopsychiatry. 2009;42(Suppl 1):S102–109. doi: 10.1055/s-0029-1214396. [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Pan CH, Chen CC, Huang MC. Increased oxidative DNA damage in patients with alcohol dependence and its correlation with alcohol withdrawal severity. Alcohol Clin Exp Res. 2011;35:338–344. doi: 10.1111/j.1530-0277.2010.01349.x. [DOI] [PubMed] [Google Scholar]
- Choi SW, Stickel F, Baik HW, Kim YI, Seitz HK, Mason JB. Chronic alcohol consumption induces genomic but not p53-specific DNA hypomethylation in rat colon. J Nutr. 1999;129:1945–1950. doi: 10.1093/jn/129.11.1945. [DOI] [PubMed] [Google Scholar]
- Crabbe JC. Review. Neurogenetic studies of alcohol addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363:3201–3211. doi: 10.1098/rstb.2008.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun. 2011 doi: 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. Epigenetic Treatments for Cognitive Impairments. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2011 doi: 10.1038/npp.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. GC- and AT-rich chromatin domains differ in conformation and histone modification status and are differentially modulated by Rpd3p. Genome Biol. 2007;8:R116. doi: 10.1186/gb-2007-8-6-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcoholism, clinical and experimental research. 1991;15:395–398. doi: 10.1111/j.1530-0277.1991.tb00536.x. [DOI] [PubMed] [Google Scholar]
- Gass JT, Olive MF. Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol. 2008;75:218–265. doi: 10.1016/j.bcp.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Konopka G. Neuroscience in the era of functional genomics and systems biology. Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid A, Wani NA, Kaur J. New perspectives on folate transport in relation to alcoholism-induced folate malabsorption--association with epigenome stability and cancer development. Febs J. 2009;276:2175–2191. doi: 10.1111/j.1742-4658.2009.06959.x. [DOI] [PubMed] [Google Scholar]
- Harper C, Dixon G, Sheedy D, Garrick T. Neuropathological alterations in alcoholic brains. Studies arising from the New South Wales Tissue Resource Centre. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:951–961. doi: 10.1016/S0278-5846(03)00155-6. [DOI] [PubMed] [Google Scholar]
- Henriksson R, Kuzmin A, Okvist A, Harper C, Sheedy D, Garrick T, Yakovleva T, Bakalkin G. Elevated synaptophysin I in the prefrontal cortex of human chronic alcoholics. Synapse. 2008;62:829–833. doi: 10.1002/syn.20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervouet E, Lalier L, Debien E, Cheray M, Geairon A, Rogniaux H, Loussouarn D, Martin SA, Vallette FM, Cartron PF. Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PLoS One. 2010;5:e11333. doi: 10.1371/journal.pone.0011333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, Lee Y, Scheck AC, Liau LM, Wu H, Geschwind DH, Febbo PG, Kornblum HI, Cloughesy TF, Nelson SF, Mischel PS. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103:17402–17407. doi: 10.1073/pnas.0608396103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jurd R, Thornton C, Wang J, Luong K, Phamluong K, Kharazia V, Gibb SL, Ron D. Mind bomb-2 is an E3 ligase that ubiquitinates the N-methyl-D-aspartate receptor NR2B subunit in a phosphorylation-dependent manner. J Biol Chem. 2008;283:301–310. doi: 10.1074/jbc.M705580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CM, et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of Addiction. Neuropsychopharmacology. 2009;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGros L, Halim AB, Chamberlin ME, Geller A, Kotb M. Regulation of the human MAT2B gene encoding the regulatory beta subunit of methionine adenosyltransferase, MAT II. J Biol Chem. 2001;276:24918–24924. doi: 10.1074/jbc.M102816200. [DOI] [PubMed] [Google Scholar]
- Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, Mayfield RD. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology. 2006;31:1574–1582. doi: 10.1038/sj.npp.1300947. [DOI] [PubMed] [Google Scholar]
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- Lu SC, Huang ZZ, Yang H, Mato JM, Avila MA, Tsukamoto H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in alcoholic rat liver. Am J Physiol Gastrointest Liver Physiol. 2000;279:G178–185. doi: 10.1152/ajpgi.2000.279.1.G178. [DOI] [PubMed] [Google Scholar]
- Mayfield RD, Harris RA, Schuckit MA. Genetic factors influencing alcohol dependence. Br J Pharmacol. 2008;154:275–287. doi: 10.1038/bjp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Feng J, Wilkinson MB, Sun H, Shen L, Nestler EJ. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci U S A. 2011;108:3035–3040. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonel P, Costello I, Hendrich B. Keeping things quiet: roles of NuRD and Sin3 co-repressor complexes during mammalian development. Int J Biochem Cell Biol. 2009;41:108–116. doi: 10.1016/j.biocel.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay B, Grummt I. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu Rev Cell Dev Biol. 2008;24:131–157. doi: 10.1146/annurev.cellbio.24.110707.175259. [DOI] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH. A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci. 2008;28:1410–1420. doi: 10.1523/JNEUROSCI.4098-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Rhodes JS, Crabbe JC, Mayfield RD, Adron Harris R, Ponomarev I. Molecular Profiles of Drinking Alcohol to Intoxication in C57BL/6J Mice. Alcohol Clin Exp Res. 2011;35:659–670. doi: 10.1111/j.1530-0277.2010.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Rosenbloom M, Rohlfing T, Sullivan EV. Degradation of association and projection white matter systems in alcoholism detected with quantitative fiber tracking. Biol Psychiatry. 2009;65:680–690. doi: 10.1016/j.biopsych.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Rau V, Eger EI, Harris RA, Fanselow MS. Amygdala transcriptome and cellular mechanisms underlying stress-enhanced fear learning in a rat model of posttraumatic stress disorder. Neuropsychopharmacology. 2010;35:1402–1411. doi: 10.1038/npp.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Chromatin regulation in drug addiction and depression. Dialogues Clin Neurosci. 2009;11:257–268. doi: 10.31887/DCNS.2009.11.3/wrenthal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–240. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- Saris CG, Horvath S, van Vught PW, van Es MA, Blauw HM, Fuller TF, Langfelder P, DeYoung J, Wokke JH, Veldink JH, van den Berg LH, Ophoff RA. Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genomics. 2009;10:405. doi: 10.1186/1471-2164-10-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol. 2006;310:211–250. doi: 10.1007/3-540-31181-5_11. [DOI] [PubMed] [Google Scholar]
- Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcoholism, clinical and experimental research. 2008;32:1525–1534. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- Torkamani A, Dean B, Schork NJ, Thomas EA. Coexpression network analysis of neural tissue reveals perturbations in developmental processes in schizophrenia. Genome Res. 2010;20:403–412. doi: 10.1101/gr.101956.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein J, Rietschel M. Genome-wide association studies of alcohol dependence and substance use disorders. Curr Psychiatry Rep. 2011;13:147–155. doi: 10.1007/s11920-011-0176-4. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. Dualism of gene GC content and CpG pattern in regard to expression in the human genome: magnitude versus breadth. Trends Genet. 2005;21:639–643. doi: 10.1016/j.tig.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernicke C, Samochowiec J, Schmidt LG, Winterer G, Smolka M, Kucharska-Mazur J, Horodnicki J, Gallinat J, Rommelspacher H. Polymorphisms in the N-methyl-D-aspartate receptor 1 and 2B subunits are associated with alcoholism-related traits. Biological psychiatry. 2003;54:922–928. doi: 10.1016/s0006-3223(03)00072-6. [DOI] [PubMed] [Google Scholar]
- Wolstenholme JT, Warner JA, Capparuccini MI, Archer KJ, Shelton KL, Miles MF. Genomic analysis of individual differences in ethanol drinking: evidence for non-genetic factors in C57BL/6 mice. PLoS One. 2011;6:e21100. doi: 10.1371/journal.pone.0021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z, Petralia RS, Fu Z, Swanwick CC, Wang YX, Prybylowski K, Sans N, Vicini S, Wenthold RJ. The role of the PDZ protein GIPC in regulating NMDA receptor trafficking. J Neurosci. 2007;27:11663–11675. doi: 10.1523/JNEUROSCI.3252-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Kaufman KL, Harper CG. Clinical and pathological features of alcohol-related brain damage. Nat Rev Neurol. 2011;7:284–294. doi: 10.1038/nrneurol.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kasif S, Cantor CR, Broude NE. GC/AT-content spikes as genomic punctuation marks. Proc Natl Acad Sci U S A. 2004;101:16855–16860. doi: 10.1073/pnas.0407821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yuan Q, Mash DC, Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci U S A. 2011;108:6626–6631. doi: 10.1073/pnas.1018514108. [DOI] [PMC free article] [PubMed] [Google Scholar]