

Abstract
Oxidative stress and Ca++ toxicity are mechanisms of hypoxic-ischemic (HI) brain injury. This work investigates if partial inhibition of mitochondrial respiratory chain protects HI-brain by limiting generation of oxidative radicals during reperfusion. HI-insult was produced in p10 mice treated with complex-I (C-I) inhibitor, pyridaben (P), or vehicle. Administration of P significantly decreased extent of HI injury. Mitochondria isolated from the ischemic hemisphere in P-treated animals showed reduced H2O2 emission, less oxidative damage to the mitochondrial matrix, and increased tolerance to Ca++ triggered opening of permeability transition pore. Protective effect of P administration was also observed when the reperfusion-driven oxidative stress was augmented by the exposure to 100% O2 which exacerbated brain injury only in V-treated mice. In vitro, intact brain mitochondria dramatically increased H2O2 emission in response to hyperoxia, resulting in substantial loss of Ca++ buffering capacity. However, in the presence of C-I inhibitor, rotenone, or antioxidant, catalase, these effects of hyperoxia were abolished.
Our data suggest that the reperfusion-driven recovery of C-I dependent mitochondrial respiration contributes not only to the cellular survival, but also causes an oxidative damage to the mitochondria, potentiating a loss of Ca++ buffering capacity. This highlights a novel neuroprotective strategy against HI-brain injury where the major therapeutic principle is a pharmacological attenuation, rather than an enhancement of mitochondrial oxidative metabolism during early reperfusion.
Keywords: Mitochondria, Hypoxia-ischemia, Brain, Complex I, Oxidative stress
Introduction
Hypoxia-ischemia severely inhibits mitochondrial oxidative phosphorylation in the brain of newborn animals (Gilland et al., 1998; Caspersen et al., 2008). Reoxygenation/reperfusion restores mitochondrial phosphorylating respiration, normalizing ATP content in the post-ischemic brain. However, following several hours of reperfusion mitochondria again exhibit a profound secondary decline in their ability to generate ATP (Lorek et al., 1994; Gilland et al., 1998; Ten et al., 2010), the event is known as a secondary energy failure (Halestrap, 2010). Structural damage to mitochondrial membranes during reperfusion has been proposed as a major molecular mechanism for secondary energy failure (Kuroda et al., 1996; Kristian, 2004; Halestrap and Pasdois, 2009). This reperfusion-associated damage is triggered by over-accumulation of mitochondrial Ca++, resulting in opening of mitochondrial permeability transition pore (mPTP) and subsequent loss of proton-motive force. This renders mitochondria incapable of ATP production and eventuates in mitochondrial swelling and release of pro-apoptotic proteins, initiating cell death mechanisms. In the presence of Ca++ an opening of mPTP can be triggered by oxidative alteration to the mitochondrial matrix (Kim et al., 2006), however, the sources of reactive oxygen species (ROS) responsible for mitochondrial and cellular oxidative damage in HI-reperfusion are not well defined. Abramov and co-authors have identified three distinct sources of ROS during oxygen-glucose deprivation and reoxygenation in cultured neurons; mitochondrial respiratory chain (MRC), xanthine oxidase and NADPH oxidase (Abramov et al., 2007). In vivo an inhibition of xanthine oxidase with oxypurinol or allopurinol failed to reduce lipid peroxidation, and did not protect the brain in a rat model of HI (Feng et al., 2003) or in human neonates with perinatal HI (Chaudhari and McGuire, 2008). Genetic or/and pharmacological inhibition of NADPH oxidase also did not exert neuroprotection in different models of perinatal HI brain injury (Doverhag et al., 2008). In mature animal models of ischemia-reperfusion injury in the brain and heart, MRC has been increasingly recognized as important source for the reperfusion-driven generation of ROS responsible for an oxidative injury (Ambrosio et al., 1993; Piantadosi and Zhang, 1996; Chen et al., 2008). In the developing brain, however, a potential deleterious effect for ROS originating from the mitochondria has not been studied.
This study demonstrates that in the developing HI-brain, the ROS generated by reverse electron transport (RET) flow in the C-I segment of MRC contribute to a reperfusion-driven oxidative damage to the mitochondria. This is associated with significant reduction in mitochondrial Ca++ buffering capacity, the condition known to promote mPTP opening, secondary energy failure and cell injury.
Materials and Methods
The model of unilateral HI brain injury and study design
The research protocol was reviewed and approved by the institutional animal care and use committee. We used the Vannucci’ model of HI-brain injury adapted to p9–10 neonatal mice of both sexes (Ten et al., 2003; Ten et al., 2004). The model consisted of a permanent ligation of the right carotid artery followed by hypoxic exposure. Briefly, surgical intervention was performed under isoflurane anesthesia. At 1.5 hours of recovery pups were exposed to hypoxia (8% O2 balanced N2) for 20 minutes. The ambient temperature during hypoxia was maintained at 37.0 – 37.5°C by placing the hypoxic chamber in a neonatal isolette (Airshield Inc. NC). Following hypoxic exposure pups were returned to their dams. To minimize a temperature-related variability in the extent of brain injury, during initial 12 hours of reperfusion mice were kept in an isolette at the ambient t = 32°C. To highlight a pathogenic role of the C-I in HI-brain injury mice were exposed to a C-I specific inhibitor, pyridaben. Pyridaben was injected intra-peritoneally (IP, 2 μlg/g) at 60 minutes prior to HI and a second dose (2 μlg/g) was given immediately after HI. Tested at 2 hours after initiation of treatment in naïve p10 mice, this dose and regimen of pyridaben exposure resulted in a moderate (~ 25%) inhibition of C-I dependent (substrate - malate-glutamate) mitochondrial phosphorylating respiration (Vehicle = 469 ± 23 nmol O2/mg/min vs Pyridaben = 358 ± 19 nmol O2/mg/min, n = 4, p = 0.001) associated with significant (p = 0.006) decrease in C-I enzymatic activity (Vehicle = 276.8 ± 25.2 nmol NADH/min/mg and Pyridaben = 194.1 ± 10.5 nmol NADH/min/mg, n = 3). A stock solution of pyridaben was 0.2 mg/ml (2% Dimethyl sulfoxide -DMSO in normal saline - NS). The 2% DMSO in NS was used as a vehicle.
To determine if the ROS originating from the C-I contribute to the oxidative injury during early reperfusion, in separate cohort of HI-mice an oxidative stress was intentionally augmented by the exposure of animals to 100% oxygen for the initial 60 minutes of reperfusion at the ambient t = 32°C. This experimental maneuver was based on reports that the rate of mitochondrial ROS generation increases dramatically in response to elevation of environmental O2 content (Boveris and Chance, 1973; Hoffman et al., 2007). In a similar mouse model of neonatal HI supra-physiological hyperoxemia maintained for 60 minutes of reperfusion significantly exacerbated oxidative brain damage and neurological deficit compared to the normoxemia-reperfused littermates (Koch et al., 2008). Thus, if the ROS from the C-I contribute to the oxidative injury during reperfusion, then partial inhibition of C-I in the pyridaben-exposed mice should limit the exacerbation of brain damage caused by post-HI hyperoxia. In contrast, in the vehicle-treated mice hyperoxia should result in a significant exacerbation of brain injury.
Assessment of the HI-brain injury
At 24 hours and at seven days of reperfusion mice were sacrificed by decapitation. Brains were harvested, sectioned into 1 mm thick coronal slices and stained with 2% triphenyl-tetrazolium chloride (TTC). Digital images of infarcted (pale-white) and viable (brick-red) areas of brains were traced (Adobe Photoshop 4.0.1) and analyzed (NIH image 1.62) by an investigator “blinded” to a study groups. The extent of brain injury (direct infarct volume) was expressed as a percentage of the hemisphere ipsilateral to the carotid artery ligation side. At seven days after HI, in a separate cohort of animals, brains were harvested, fixed in 4% paraformaldehide, paraffin-embedded, coronally sectioned (10 μm every 500 μm) and Nissl stained. Digital images were traced and processed as described above. The extent of injury was defined by the preservation of cerebral tissue in the ipsilateral hemisphere and expressed as % in relation to the corresponding contralateral hemisphere (100%).
The extent of oxidative brain damage was analyzed by visual detection and semi-quantification of markers for lipid peroxidation (4-hydroxy-nonenal, 4HNE) and protein nitration (3-nitrotyrosine, 3NT). In brief, at 5 hours of reperfusion brains were harvested from the randomly selected mice treated with pyridaben or vehicle, fixed in 4% paraformaldehyde and soaked in 30% sucrose overnight. 20 μm thick coronal sections were blocked (10% donkey serum) and incubated with rabbit polyclonal anti-4-HNE (1:500) and anti-3NT antibodies (1:100) as described (Zhu et al., 2007). Samples were examined using Bio-Rad 2000 confocal laser-scanning device attached to a Nikon E800 microscope. The images were captured at the resolution 1024×1024 pixels with x40 objective, counting frame 295 × 295 μm. Z-sectioned merged images were obtained from a stack of adjacent five sections (step = 1 μm). The 4-HNE and 3-NT immunoreactivity was analyzed by the count of immunopositive cells in five nonadjacent fields of injured cortex at three different bregma-levels (−1.0, 0, +1.0 mm). Fifteen areas of cortex were analyzed for each mouse and a mean number of the immunopositive cells per mm2 in each mouse were used for analysis.
Assessment of mitochondrial functions
Ex-vivo study
At 0, 30 minutes and 5 hours of reperfusion cerebral non-synaptosomal mitochondria were isolated from the ipsilateral hemisphere in pyridaben or vehicle-treated HI-mice as described (Caspersen et al., 2008; Ten et al., 2010). Because the mean infarct volume comprised ≈ 40% of the ipsilateral hemisphere, mitochondria were isolated only from the posterior-lateral cortex and sub-cortical structures (part of the hippocampus, striatum and thalamus) of the ipsilateral hemisphere, the regions that reproducibly damaged in this model. Cerebral specimens from 3–4 mice were pulled for mitochondrial isolation in order to obtain the sample sufficient for measurements of mitochondrial respiration rates using NAD or FAD-linked substrates, ROS emission rates and Ca++ buffering capacity. All data were compared between study groups and matched to that in naïve littermates.
In-vitro study
Cerebral non-synaptosomal mitochondria were isolated from naïve p10 mice and exposed to hyperoxia (incubation in the buffer pre-bubbled with 100% O2) for 2 minutes. The mean value of partial O2 tension (pO2) in the hyperoxic buffer (5 ml) measured following 20 seconds of bubbling (60 ml/min) with 100% O2 reached 472 ± 34 mmHg. The buffer (10 mM MOPS-Tris, pH 7.4, 120 mM KCl, KH2PO4 1 mM, EGTA 10 μM) contained 5 mM succinate and glutamate as substrates. Mitochondrial ROS emission rate and Ca++ threshold for mPTP opening were assessed in normoxic (control) and hyperoxic conditions in the presence or absence of rotenone (C-I inhibitor, 1μM) or catalase (150U/ml). Rotenone is the most commonly used C-I inhibitor and, like pyridaben, has the same inhibitory effect on C-I in cerebral mitochondria (Sherer et al., 2007). Hyperoxia results in accelerated generation of ROS from the respiratory chain in the brain and heart mitochondria (Castello et al., 2007; Hoffman et al., 2007), which mimics post-ischemic, re-oxygenation-accelerated mitochondrial ROS generation.
Mitochondrial respiration was measured using a Clark-type electrode (Oxytherm, Hansatech, UK). Mitochondria (0.05 mg of protein) were added to 0.5 ml of respiration buffer composed of 200 mM sucrose, 25 mM KCl, 2 mM K2HPO4, 5 mM HEPES, pH 7.2, 5 mM MgCl2, 0.2 mg/ml of BSA, 30 μM Ap5A (P1,P5-di(adenosine 5′)-pentaphosphate - an inhibitor of adenylate kinase), 10 mM glutamate, and 5 mM malate or 5mM succinate and 5mM glutamate at t = 32°C. To initiate a phosphorylating respiration (State 3) 100 nmol of ADP was added to the mitochondrial suspension. Rates of O2 consumption were expressed in nmol O2/mg mitochondrial protein/min. The respiratory control ratio (RCR) was calculated as the ratio of the State 3 respiration rate to the resting respiration rate (State 4) recorded after the phosphorylation of ADP has been completed. 35nM 2′-4′dinitro-phenol (DNP) was used to initiate uncoupled respiration which directly defines an activity of the respiratory chain.
Measurement of mitochondrial H2O2 emission
The rate of H2O2 emission from mitochondria was estimated by a fluorescence assay with Hitachi 7000 spectrofluorimeter set at 555 nm excitation and 581 nm emission as described earlier (Starkov and Fiskum, 2003). Briefly, mitochondria (0.05 mg) were placed in 1 ml of respiration buffer as described above and supplemented with 5 mM succinate, 10 μM Amplex® Ultrared (Invitrogen, USA) and 4 U/ml of horse radish peroxidase (HRP). After recording the fluorescence for 400 sec, samples were supplemented with 1 μM rotenone and after another ~200 sec with 1 μg/ml Antimycin A. The calibration curve was obtained by adding several 100 nmol aliquots of freshly made H2O2 to the cuvette containing the respiration buffer, Amplex® Ultrared and HRP. The rates (total and originated from C-I) of H2O2 emission in pmolH2O2/mg/min were expressed as a % of that measured in control (naïve mice) organelles tested in each experiment. The rate of H2O2 generation by C-I was estimated by subtraction of the rate recorded after supplementation of rotenone from the spontaneous (total) rate of H2O2 emission.
The rationale for assessment of mitochondrial ROS emission on FAD-linked substrate, succinate, was based on studies that demonstrated ~300% increase in succinate concentration in the rat brain following five minutes of ischemia (Folbergrova et al., 1974; Benzi et al., 1979). In contrast, the same ischemia resulted in a profound (8 –10 folds) decrease in the concentration of all mitochondrial NAD-linked substrates. In mature rats forebrain ischemia and six hours of reperfusion resulted in significant inhibition of mitochondrial respiration tested on NAD-linked substrates. However, no significant differences from the control values were detected when the same mitochondria respired on succinate (Sims, 1991), suggesting that following HI the activity of C-II is better preserved compared to the C-I activity. In order to determine how critical is a succinate-driven (C-II dependent) mitochondrial respiration for post-HI cerebral recovery a separate cohort of mice was subjected to either C-II inhibitor, 3-nitropropionic acid (3-NP, 20 μg/g at 60 minutes prior to and immediately after HI-insult), or vehicle (0.9% NaCl), followed by a comparison of the extent of brain injury. The selected dose and regimen of 3NP-exposure in naïve p10 mice resulted in ~30% inhibition of succinate-supported mitochondrial phosphorylating respiration rate (Vehicle = 674 ± 41.2 nmol O2/mg/min and 3NP = 476.5 ± 30.6 nmol O2/mg/min, n = 4, p = 0.001) which was comparable to the inhibitory effect of pyridaben on NAD-linked respiration.
Mitochondrial aconitase activity was measured as described (Morrison, 1954). Frozen-thawed mitochondria were mixed with the reaction buffer (50 mM Tris HCl, pH 7.4, 0.6 mM MnSO4, 5 mM Na citrate, 0.5 mM NADP, 1U/ml iso-citrate dehydrogenase) in a 96-well plate and the absorbance changes at 340 nm were followed for 10 min with a plate reader (Tecan, Infinity 200). The aconitase activity was expressed in mU/min/mg of mitochondrial protein.
Mitochondrial C-I activity was measured spectrophotometrically as rotenone- sensitive NADH:Q1 reductase. Reaction buffer was composed of 20 mM HEPES, pH 7.8, 75 μM NADH, 40 μM coenzyme Q1, 1 mM KCN (final concentration). Frozen-thawed mitochondria were mixed with the reaction buffer in a 96-well plate and the absorbance changes at 340 nm were followed for 15 min with a plate reader (Tecan, Infinity 200). In control the buffer was supplemented with 10 μM rotenone (final concentration). The activity of Complex I was calculated as difference between the rates of NADH oxidation (E340mM=6.22 cm−1) in the absence and in the presence of rotenone and presented in nmol NADH/min/mg of protein.
Mitochondrial Ca++ buffering capacity was measured as described (Fontaine et al., 1998; Wang et al., 2009) with minimal modifications. In brief, mitochondria (0.05 mg/ml) were incubated in 10mM Tris-MOPS buffer, pH 7.4, containing 120 mM KCl, 1mM KH2PO4, 10 μM EGTA, 10 μM Calcium green, 5 mM succinate and glutamate. The buffer for mitochondrial Ca++ buffering capacity assay was changed, because it has been shown that in the sucrose-based buffer, as opposite to the KCl-based media, isolated mitochondria are insensitive to the cyclosporine-A during mPTP opening (Chavez et al., 2003). Following 30 sec of a steady-state fluorescence, organelles were repeatedly supplemented with 10 nmoles of CaCl2 every 50 seconds until Ca++ was spontaneously released, indicating an opening of mPTP. The amount of Ca++ required to open mPTP was normalized per mg of mitochondrial protein and expressed as mitochondrial Ca++ buffering capacity. An opening of a cyclosporine A-sensitive mPTP was confirmed by the detection of cytochrome-C release from mitochondria into a buffer following Ca++ challenge (data not shown).
Assessment of cytochrome C release was performed using western blot analysis. Cerebral samples from the injured ipsilateral hemispheres were collected into isolation buffer (50 mM Tris-HCl, pH 7.4, 320 mM sucrose, 1 mM dithiothreitol, 3 mM EDTA, 0.5% protease inhibitor cocktail), homogenized and centrifuged at 1000 g for 5 min at 4 °C. The supernatant was further centrifuged at 10000 g for 15 min at 4 °C. Cytosolic and crude mitochondrial fraction were collected and stored at −80 °C. Samples were run on 10% BisTris gels (NuPAGE, Invitrogen) and transferred to nitrocellulose membranes. After blocking the membranes were incubated with mouse anti-cytochrome C (BD Pharmigen, 1:2000), mouse monoclonal anti-β-actin (Sigma, 1:50000), mouse monoclonal [20E8] anti-COX IV (Abcam, 1:5000) primary antibodies. Peroxidase-conjugated donkey anti-mouse antibodies was added for 1 hr and bands were visualized using ECL plus western blotting detection system (GE, Healthcare).
Statistical analysis
The difference in the extent of brain infarct volumes was analyzed by Student’ t-test. One-way ANOVA with Fisher’s post-hoc analysis was used to detect statistical differences in mitochondrial functions, enzymatic activities among naïve and both experimental groups. All data are mean ±SE. Difference was considered statistically significant if p-value ≤0.05.
Results
Inhibition of C-I limits a reperfusion-driven recovery of mitochondrial respiration and acceleration in ROS release from mitochondria
In the mice treated with pyridaben or vehicle at the end of HI-insult (0 minutes of reperfusion), mitochondria isolated from the ischemic brain exhibited significant decrease in their C-I dependent phosphorylating (state 3) and DNP-accelerated (not shown) respiration rates compared to that in organelles isolated from naïve littermates (Fig. 1A and B). This inhibition of mitochondrial NAD-linked respiration was associated with markedly reduced emission of H2O2 from the same mitochondria energized with succinate (Fig. 1E and F). At 30 minutes of reperfusion the activity of the respiratory chain in mitochondria isolated from the vehicle-treated mice recovered, exhibiting near-normal C-I dependent phosphorylating and DNP-accelerated (not shown) respiration rates (Fig. 1A and C). This coincided with significant acceleration in mitochondrial H2O2 emission rates compared to that detected at 0 minutes of reperfusion (Fig. 1E). At the same time of reperfusion organelles isolated from the HI-mice exposed to pyridaben demonstrated significantly poorer recovery of the C-I linked respiration (Fig. 1A, C) and this was associated with significantly reduced H2O2 release rates compared to that in the vehicle-treated littermates (Fig. 1E, F). When the same mitochondria were compared using succinate as a substrate no difference was found between naïve mice and vehicle or pyridaben treated HI-animals (Fig. 1C, D). At these time-points of reperfusion (0 and 30 min) no morphological signs of brain damage could be observed (Fig. 1A). At five hours of reperfusion, when brain injury become detectable, mitochondria isolated from HI brains in both groups of mice exhibited secondary decline in their phosphorylating (Fig. 1A) and DNP-accelerated (not shown) respiration rates. However, organelles isolated from the pyridaben-treated mice preserved their ADP-phosphorylating activity significantly better then mitochondria from the vehicle-treated littermates (Fig. 1A). This was associated with significantly greater ROS emission rate compared to that in the vehicle-treated littermates (Fig. 1E).
Fig. 1.

(A) Changes in NAD-linked (state 3) mitochondrial respiration rates in response to HI and reperfusion in HI-mice treated with vehicle (black bar) or pyridaben (gray bar) in comparison to naives (white bar, n = 21). N = 6 in each group examined at 0 minutes of reperfusion, n = 9 in each group at 30 minutes of reperfusion and n = 10 (vehicle) and 8 (pyridaben) at 5 hours of reperfusion. TTC-stained representative coronal sections of brains obtained from the same HI-mice (vehicles) used for mitochondrial respiration studies. Dashed outlining indicates areas used for isolation of mitochondria. * p < 0.01 compared to naives. (B) Representative tracings of mitochondrial NAD-linked (C-I dependent) respirations recorded at 0 minutes of reperfusion in naïve (N), vehicle (V) or pyridaben (P) treated mice. Numbers are state 3 respiration rates in N and V-treated mice. (C) Representative tracings of C-I and C-II dependent mitochondrial respirations recorded at 30 minutes of reperfusion in the vehicle (V) and pyridaben (P) treated mice compared to naïve (N) littermate. Numbers are state 3 respiration rates in V and P-treated mice. (D) FAD-linked (C-II dependent) state 3 respiration rates recorded at 30 minutes of reperfusion in the vehicle (black bar, n = 9) and pyridaben (gray bar, n = 9) treated mice compared to naives (white bar, n = 9). (E) C-I originated H2O2 emission rates in mitochondria isolated at 0, 30 min and 5 hrs of reperfusion from the vehicle (black bar, n = 7 for 0 and 30 min and n = 6 for 5 hrs of reperfusion) and the pyridaben (gray bar, n = 7 for 0 and 30 min and n = 5 for 5 hrs of reperfusion) treated mice compared to naïve littermates (open bar, n = 14). * p < 0.02 compared to naives, # p < 0.005 compared to that at 0 minutes of reperfusion. (F) Representative tracing of H2O2 emission recorded in brain mitochondria isolated from naïve and experimental mice (indicated) at 0 and 30 minutes of reperfusion (indicated).
Treatment with pyridaben protected brain against oxidative mitochondrial injury and reduced cerebral infarct volume
At 24 hours of reperfusion mice treated with pyridaben exhibited significantly decreased cerebral infarct volumes compared to the vehicle-treated littermates (Fig. 2A and B). Post-HI exposure to 100% oxygen for the initial 60 minutes of reperfusion increased the extent of HI-brain injury in both groups of mice. However, only in the vehicle-treated mice the hyperoxic reperfusion resulted in significant (p = 0.02) exacerbation of brain injury (the infarct volume has increased by 19%). In contrast, in the mice treated with pyridaben the same hyperoxic reperfusion was associated with a mild increase in the infarct volume (by 10%) which did not reach statistical significance compared to the normoxic reperfusion (Fig. 2A and B). Importantly, compared to the vehicle-treated mice, the neuroprotection afforded by the treatment with pyridaben in both experiments (normoxic reperfusion and hyperoxic reperfusion) was associated with significantly better-preserved activity of aconitase, a marker of oxidative damage to the mitochondrial matrix, (Fig. 2C). Hyperoxic reperfusion resulted in significant further reduction in mitochondrial aconitase activity in both, vehicle and pyridaben-treated mice (Fig. 2C). However, this decrease in aconitase activity in response to the hyperoxic reperfusion was significantly (p = 0.008) less in the pyridaben-treated mice (29.6 ± 18.3% decrease) compared to the vehicle-treated mice (49.3 ± 9.9% decrease). At this time-point of reperfusion (five hours after HI-insult), in addition to attenuated severity of oxidative injury to the mitochondrial matrix, HI-mice treated with pyridaben demonstrated significantly fewer 3NT and 4HNE immunopositive cells in their ischemic cortex compared to their vehicle-treated counterparts (Fig. 2D). At the same time-point of reperfusion mitochondrial Ca++ buffering capacity was significantly reduced in both groups of experimental mice in relation to their naïve counterparts (Fig. 2E and F). However, mitochondria from the ischemic brains of pyridaben-treated mice demonstrated significantly greater Ca++ buffering capacity compared to that in their vehicle-treated littermates (Fig. 2E and F). This was associated with significantly decreased release of cytochrome C from organelles into cytosol, although, the amount of cytochrome C remaining in mitochondria did not differ (Fig. 3A and B). However, at 24 hours of reperfusion, compared to the vehicle-treated littermates mice treated with pyridaben exhibited significantly greater preservation of cytochrome C in their mitochondria associated with limited release into cytosol (Fig. 3C–E). At five hours of reperfusion, similarly to the mitochondria fueled with succinate, mitochondria from pyridaben treated HI-mice supported with malate-glutamate exhibited significantly (p = 0.03) better Ca++ buffering capacity (1200 ± 365 nmoles/mg, n = 4) compared to the vehicle-treated animals (700 ± 365 nmoles/mg, n = 4), although, both groups of HI-mice exhibited significantly (p < 0.0001) reduced Ca++ buffering capacity compared to naives (2533 ± 305 nmoles/mg, n = 4).
Fig. 2.

(A and B) Cerebral infarct volume and representative TTC-stained brain sections from HI-mice exposed to either normoxia or hyperoxia (indicated) and treated with vehicle (black dots) or pyridaben (gray dots). (C) Mitochondrial aconitase activity measured at 5 hours of normoxic or hyperoxic (indicated) reperfusion in vehicle (black bar, n = 12 in normoxic and n = 10 in hyperoxic reperfusion groups) and pyridaben (gray bar, n = 13 in normoxic and n = 10 in hyperoxic reperfusion groups) treated mice compared to naïves (white bar, n = 22). * p < 0.05 compared to naives, # p < 0.02 compared to corresponding normoxia-reperfused mice. (D) The number of 3NT and 4HNE immunopositive cells (red) per mm2 and representative images of injured cortex obtained at 5 hours of reperfusion in the vehicle (black bar, n = 6) and pyridaben (gray bar, n = 6) mice. Indents show representative Nissl (blue) and 4-HNE and 3-NT (red) stained cells. Scale bar = 70 μm. (E) Representative tracing of measurement of Ca++ buffering capacity in naïve and experimental (indicated) mice. (F) Mitochondrial Ca++ buffering capacity measured at 5 hours of reperfusion in naïve (white bar, n = 9), vehicle (black bar, n = 14) and pyridaben (gray bar, n = 15) treated mice.
Fig. 3.

(A and B) Immunoblot for detection of cytochrome-C in the cytosol and corresponding mitochondria at five hours of reperfusion in naïve (n = 5) and HI-mice exposed to vehicle (n = 9) or pyridaben (n = 10). * p < 0.05 compared to naives. (C–E) Cytochrome C in cytosol and mitochondria at 24 hours of reperfusion in naïve (n = 3) and HI-mice treated with vehicle (n = 6) and pyridaben (n = 6).
An inhibition of C-II exacerbates HI-injury in neonatal mice
To this point our study demonstrates that the pyridaben-induced inhibition of the mitochondrial C-I dependent respiration during reperfusion is associated with protection of the developing brain against oxidative damage that occurs at the early (initial five hours) stage of reperfusion. However, this inhibition of C-I with pyridaben, as expected, caused a significant delay in post-ischemic recovery of NAD-linked ADP-phosphorylation rates. This contradicts the biological principle of tissue recovery following ischemia. We hypothesized that upon reperfusion post-HI mitochondria preferentially utilize succinate. Then, the inhibition of C-I recovery should not significantly affect mitochondrial ADP-phosphorylating ability. In contrast, an inhibition of the C-II should deleteriously affect the HI-brain, because succinate-supported ATP-generation depends on activity of C-II. Indeed, when instead of the C-I the activity of C-II was suppressed by 3NP, the HI-mice exhibited significant exacerbation of HI-injury compared to the vehicle-treated littermates (Fig. 4A). In contrast, inhibition of C-I not only significantly reduced cerebral infarct volume assessed at 24 hours of reperfusion, but, compared to the vehicle-treated counterparts, resulted in significantly greater preservation of brain tissue assessed at seven days after HI (Fig. 4B).
Fig. 4.

(A) Cerebral infarct volumes and representative TTC-stained coronal sections obtained from HI-mice treated with vehicle (black dots) or C-II inhibitor, 3-nitropropionic acid, 3NP (gray dots). (B) Residual cerebral tissue volume (% of the contralateral hemisphere) at seven days of reperfusion in HI-mice treated with vehicle (n = 10) and pyridaben (n = 8) and representative Nissl-stained cerebral coronal sections.
Inhibition of C-I prevents hyperoxia-induced loss in mitochondrial Ca++ buffering capacity
To dissect-out a pathogenic contribution of the ROS generated by C-I to mitochondrial oxidative damage and loss of Ca++ buffering capacity, organelles isolated from naive p10 mice were exposed to hyperoxia. In the buffer with excessive oxygen content, mitochondria dramatically increased their H2O2 emission rates (Fig. 5A). However, co-incubation with catalase or rotenone, markedly abrogated this surge in H2O2 release from the MRC (Fig. 5A). In the hyperoxic environment the same cerebral mitochondria demonstrated substantial loss in their Ca++ buffering capacity and opened mPTP at significantly lower levels of Ca++ load compared to controls (Fig. 5B, C). In the presence of rotenone or catalase, however, the same organelles exhibited significant resistance to the hyperoxia-promoted Ca++ mPTP opening (Fig. 5B, C). In all our experiments Ca++ buffering capacity assay was verified for its sensitivity to cyclosporine A, and for a release of cytochrome C following an opening of mPTP (data not shown). Because in vitro and in vivo experiments we used different C-I inhibitors, rotenone or pyridaben, we compared the effects of both these agents on inhibition of the reverse electron transport flow-dependent generation of ROS in isolated mitochondria (Fig. 5E). As expected, in isolated brain mitochondria pyridaben and rotenone decreased succinate-supported H2O2 emission rate to the same extent (pyridaben, by 74.9 ± 0.26% and rotenone, by 75.7 ± 0.25%). Furthermore, to verify that pyridaben does not act as a direct ROS scavenger, we measured changes in H2O2 fluorescence in the presence or absence of pyridaben and compared with antioxidant effect of catalase (Fig. 5F). As expected, no direct antioxidant effect of pyridaben was detected.
Fig. 5.

(A) Tracings of H2O2 emission from isolated mitochondria exposed to hyperoxia (O2) in the presence or absence of rotenone or catalase (indicated). Control is H2O2 emission in the same intact mitochondria. (B and C) Representative tracings for measurements of Ca++ buffering capacity in mitochondria isolated from naïve p10 mice and exposed to a different experimental conditions (indicated). Control represents natural Ca++ buffering capacity in the same mitochondria, O2 − hyperoxia, O2 + catal/rot is hyperoxia in the presence of catalase (150U/ml) or rotenone (1 μM). (D) Ca++ buffering capacity in mitochondria isolated from naïve mice (white bar, n = 9) and HI-mice exposed to a hyperoxia (black bar, n = 10), hyperoxia + catalase (striped bar, n = 5) and hyperoxia + rotenone (gray bar, n = 7). (E) Changes in H2O2 fluorescence rate in response to rotenone (Rot, 1 μM) or pyridaben (Pyr, 10 μM). (F) H2O2 fluorescence in the presence or absence (control) of pyridaben (10 μM) or catalase (150U/ml).
Discussion
This study demonstrates that inhibition of C-I protects brain against HI injury. As normalization of ATP generation is crucial for post-ischemic recovery, this result seems paradoxical, because during reperfusion mitochondria from pyridaben-mice exhibited slower recovery of C-I-dependent ADP-phosphorylation than in vehicle-treated counterparts. However in the same mitochondria, C-II-dependent (succinate-supported) ADP-phosphorylation was not affected by pyridaben, as C-II-linked respiration does not require C-I for ATP generation. In contrast, in the mitochondria respiring on succinate the activity of C-I is critical for the generation of superoxide, as the RET supports a highest rate of ROS formation (Cino and Del Maestro, 1989; Votyakova and Reynolds, 2001; Andreyev et al., 2005; Murphy, 2009). HI-insult inhibits C-I-dependent electron transport which partially (Puka-Sundvall et al., 2000) or near-fully recovers upon reperfusion (Ten et al., 2010). In our study this reperfusion-driven recovery of C-I-dependent respiration was associated with accelerated emission of ROS in the mitochondria respiring on succinate, suggesting that the recovery of C-I activity contributes not only to the generation of ATP, but also to the acceleration of ROS production. There are lines of evidence that succinate is a primary substrate at the onset of reperfusion. C-I is the most sensitive complex to ischemic inhibition in the brain (Allen et al., 1995; Almeida et al., 1995). Following forebrain ischemia in rats C-I-dependent respiration is significantly greater inhibited compared to that on C-II-linked substrates (Sims, 1991) and the concentrations of all C-I-linked substrates is decreased by 8–10 folds (Folbergrova at al., 1974). In contrast, the concentration of succinate increases by ~300% (Folbergrova et al., 1974; Benzi et al., 1979). In our study, in opposite to the mice with inhibited C-I, the mice with inhibited C-II demonstrated exacerbation of HI brain injury, suggesting the priority of FAD-linked over NAD-linked phosphorylation in the brain recovery. An inhibition of C-I with amytal significantly prolonged recovery of NADH oxidation in murine hippocampal slices during reperfusion, yet the functional (population spike amplitude) recovery was improved compared to controls (Huber et al., 2004).
The MRC (C-I and C-III) has been recognized as a major source of ROS in ischemia-reperfusion injury (Cino and Del Maestro, 1989; Ambrosio et al., 1993; Loor et al., 2010). Attenuation of the oxidative stress induced by the reperfusion-accelerated ROS generation in C-I is a plausible explanation for the neuroprotection observed here. The inhibition of C-I limited exacerbation of injury and oxidative damage to the mitochondria during intentionally augmented (hyperoxia) oxidative stress. Because the hyperoxia was applied only during initial hour following HI, our data suggest that C-I activity contributes to oxidative injury at the early reperfusion. The inhibition of C-I at the onset of reperfusion was associated with a better-preserved aconitase activity and reduced expression of 3NT and 4HNE in the injured cortex assessed four hours later. Importantly, at this delayed time-point of reperfusion, mitochondria from pyridaben-treated mice exhibited significantly greater ROS emission compared to the vehicle-treated mice. Because at five hours of reperfusion HI-mice demonstrate irreversible brain damage, this result reflects a better-recovered C-I activity evidenced by a better-preserved C-I-dependent oxidative-phosphorylation, rather than exacerbation of an oxidative stress.
We have reported that upon reperfusion mitochondria release H2O2 at the rate exceeding that in naïve littermates (Ten et al., 2010). In this study the mean value of mitochondrial H2O2 emission rate during reperfusion was similar to that in naives. This discrepancy could be explained by the difference in sampling. Earlier, for the isolation of organelles we used the entire hemisphere, including an ischemic core (the tissue that ultimately dies) and peri-ischemic regions (the tissue that ultimately survives). In the current study, because the average infarct volume was ≈40% we sampled only the portion of the hemisphere, corresponding to the ischemic-core (Fig. 1A). Thus, we studied mostly those organelles that experienced a lethal ischemia, which dose-dependently (dose = duration of ischemia) increases susceptibility of ischemic brain to an oxidative stress (Selakovic et al., 2011). Compared to the end of HI, reperfusion caused a dramatic increase in mitochondrial ROS emission rate, and the inhibition of this reperfusion-accelerated ROS emission with pyridaben reduced an oxidative damage. Inhibition of the C-I by rotenone or haloperidol reduced accumulation of hydroxyl radicals and reperfusion-driven surge in products of lipid peroxidation in rats with cerebral ischemia (Piantadosi and Zhang, 1996). C-I inhibitor, amytal, also decreased ROS level and lipid peroxidation in rabbit hearts subjected to ischemia-reperfusion (Ambrosio et al., 1993).
A reperfusion-driven oxidative stress has been implicated as a mechanism of secondary energy failure which mechanistically viewed as a formation of Ca++ triggered mPTP. mPTP renders organelles incapable of ATP production due to a loss of proton-motive force and NAD, and combined with a release of pro-apoptotic proteins eventuates in necrotic and apoptotic cell death (Vinogradov et al., 1972; Takeyama et al., 1993; Baines et al., 2005; Nakagawa et al., 2005; Halestrap and Pasdois, 2009). Although, it is well demonstrated in mature animals, the pathogenic significance of the cyclosporine A-sensitive mPTP in the developing HI-brain is uncertain. As opposite to adult, neonatal cyclophilin-D knock-out mice were susceptible to HI-injury (Wang et al., 2009). However, antagonist of cyclophilin-D, cyclosporin-A, injected after HI-insult protected developing brain, attenuating necrotic and apoptotic cell death in neonatal rats (Hwang et al., 2010). Similar results were obtained in neonatal rats subjected to a mild focal cerebral ischemia-reperfusion (Leger et al., 2010). In our study, at the time-point of reperfusion when secondary mitochondrial dysfunction occurs in this model (Ten et al., 2010) mitochondrial tolerance to cyclosporine A-sensitive Ca++ mPTP opening and ADP-phosphorylation were better preserved in HI-mice treated with pyridaben compared to their vehicle-treated littermates. This was associated with significantly reduced cytochrome-C release. In the developing brain the release of 10–20% of pro-apoptotic proteins from mitochondria is sufficient for cell death following HI (Zhu et al., 2005). ROS can initiate an opening of mPTP during ischemia (Loor et al., 2010) and reperfusion (Di Lisa et al., 2009; Lemasters et al., 2009) even in the absence of cyclophilin-D or Ca++ overload (Basso et al., 2005; Kim et al., 2006). Our data suggest that upon reperfusion ROS emission from C-I significantly increases, due to recovery of the RET. This causes an oxidative alteration to the mitochondria, decreasing tolerance to Ca++-triggered mPTP opening (Fig. 6A, B). Partial inhibition of C-I alters this pathogenic sequence and attenuates the extent of damage (Fig. 6C)
Fig. 6.
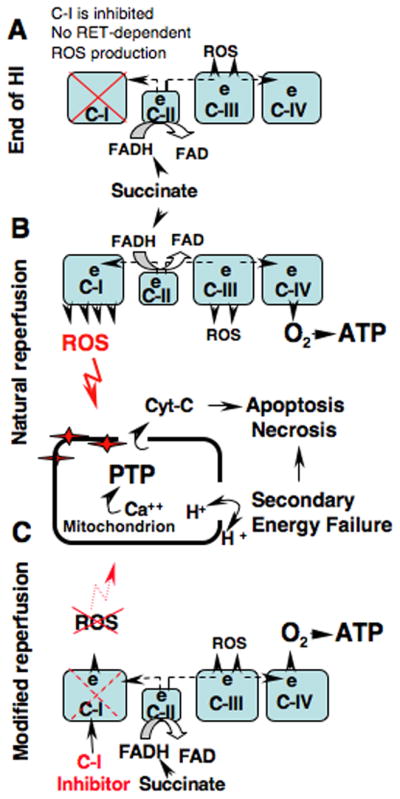
The proposed mechanism of mitochondrial oxidative injury during reperfusion by the ROS originated from the C-I and therapeutic intervention to limit this injury. (A) Succinate-supported generation of ROS from mitochondrial respiratory chain at the end of HI (0 minutes of reperfusion) is reduced due to inhibited reverse electron transport flow in C-I. (B) At the initial hours of natural reperfusion the reverse electron transport flow recovers leading to acceleration in production of ROS which causes an oxidative damage to the mitochondrial matrix. This augments the formation of Ca++ triggered mPTP, leading to the loss of proton motive force, secondary energy failure, necrosis and apoptosis. (C) Pharmacological inhibition of C-I modifies reperfusion by the limiting the recovery of reverse electron transport flow without alteration of ATP production. This attenuates oxidative damage to the mitochondrial matrix and improves cellular survival.
In vitro experiments support the proposed sequence of mechanistic events. In isolated mitochondria fueled with succinate an inhibition of C-I with rotenone prevented a burst in mitochondrial ROS emission and subsequent loss of Ca++ buffering capacity triggered by excessive oxygenation. The same effect was observed in the presence of H2O2 converting enzyme, catalase. Because hyperoxia induces an oxidative stress and acceleration of ROS generation from the MRC (Boveris and Chance, 1973; Castello et al., 2007; Hoffman et al., 2007), these data suggest that RET-generated ROS sensitize organelles to opening of Ca++ mPTP. In succinate-supported U-937 and KB cells an inhibition of C-I with rotenone decreased cell-death caused by Ca++-induced mitochondrial permeabilization (Chauvin et al., 2001). In this and other (Fontaine et al., 1998) reports authors proposed a structural role for C-I in formation of mPTP. In our study inhibition of C-I with rotenone closely mimicked the effect of the antioxidant, catalase. Therefore, we propose a functional (generation of ROS → oxidative stress) contribution of C-I to post-HI mitochondrial permeabilization. We cannot rule-out alternative mechanisms; functional participation of the C-I in assembly of mPTP or a short-term preconditioning effect of pyridaben. C-I inhibitor, NS1619, exerted immediate and delayed preconditioning mediated by mitochondrial ROS, protecting cultured neurons against OGD or glutamate excitotoxicity (Busija et al., 2008; Gaspar et al., 2008 Busija, 2008). However, in our study HI-pyridaben mice demonstrated neuroprotection against artificially-augmented oxidative stress applied only during reperfusion. This argues against significant contribution of preconditioning to neuroprotection.
The use of pesticide is considerable limitation for translation. However, several FDA-approved agents inhibit mitochondrial C-I. For example, similarly to pyridaben, isoflurane inhibits C-I activity at the distal site of the complex (Kayser et al., 2011), and reduces RET-dependent ROS generation (Hirata et al., 2011). An inhibition of C-I recovery should be limited only to the initial reperfusion, when RET contributes to ROS burst. At the later time points of reperfusion, once succinate level is normalized, MRC may mostly oxidize NAD-linked substrates. Then this strategy could become detrimental as the emission of ROS accelerates with inhibition of C-I. Another limitation; we used two different buffers, sucrose buffer for the respiration and ROS production and KCl buffer for Ca++ buffering capacity assays. Given that the same buffers were used in both controls and experimental mice, the experimental outcome should not be affected.
In conclusion, our data suggest that the reperfusion-driven recovery of C-I-dependent mitochondrial respiration contributes not only to the cellular survival, but also to mitochondrial oxidative damage, leading to a loss of mitochondrial Ca++ buffering capacity. Our work highlights a novel target and strategy against HI-brain injury where the major therapeutic principle is a pharmacological attenuation, rather than an enhancement of oxidative metabolism during immediate and early reperfusion.
Acknowledgments
This work was supported by NIH grants: NS056146, NS071121 (V.S.T.), and AG014930 (A.A.S.). Authors are thankful to Richard Polin M.D. for editorial assistance.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KL, Almeida A, Bates TE, Clark JB. Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischaemia. J Neurochem. 1995;64:2222–2229. doi: 10.1046/j.1471-4159.1995.64052222.x. [DOI] [PubMed] [Google Scholar]
- Almeida A, Allen KL, Bates TE, Clark JB. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J Neurochem. 1995;65:1698–1703. doi: 10.1046/j.1471-4159.1995.65041698.x. [DOI] [PubMed] [Google Scholar]
- Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Benzi G, Arrigoni E, Marzatico F, Villa RF. Influence of some biological pyrimidines on the succinate cycle during and after cerebral ischemia. Biochem Pharmacol. 1979;28:2545–2550. doi: 10.1016/0006-2952(79)90024-8. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Gaspar T, Domoki F, Katakam PV, Bari F. Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev. 2008;60:1471–1477. doi: 10.1016/j.addr.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen CS, Sosunov A, Utkina-Sosunova I, Ratner VI, Starkov AA, Ten VS. An isolation method for assessment of brain mitochondria function in neonatal mice with hypoxic-ischemic brain injury. Dev Neurosci. 2008;30:319–324. doi: 10.1159/000121416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello PR, Drechsel DA, Patel M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem. 2007;282:14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari T, McGuire W. Allopurinol for preventing mortality and morbidity in newborn infants with suspected hypoxic-ischaemic encephalopathy. Cochrane Database Syst Rev. 2008:CD006817. doi: 10.1002/14651858.CD006817.pub2. [DOI] [PubMed] [Google Scholar]
- Chauvin C, De Oliveira F, Ronot X, Mousseau M, Leverve X, Fontaine E. Rotenone inhibits the mitochondrial permeability transition-induced cell death in U937 and KB cells. J Biol Chem. 2001;276:41394–41398. doi: 10.1074/jbc.M106417200. [DOI] [PubMed] [Google Scholar]
- Chavez E, Garcia N, Zazueta C, Correa F, Aviles C, Garcia G, Balam EO. The composition of the incubation medium influences the sensitivity of mitochondrial permeability transition to cyclosporin A. J Bioenerg Biomembr. 2003;35:149–156. doi: 10.1023/a:1023798120239. [DOI] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- Cino M, Del Maestro RF. Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Biophys. 1989;269:623–638. doi: 10.1016/0003-9861(89)90148-3. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Kaludercic N, Carpi A, Menabo R, Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol. 2009;104:131–139. doi: 10.1007/s00395-009-0008-4. [DOI] [PubMed] [Google Scholar]
- Doverhag C, Keller M, Karlsson A, Hedtjarn M, Nilsson U, Kapeller E, Sarkozy G, Klimaschewski L, Humpel C, Hagberg H, Simbruner G, Gressens P, Savman K. Pharmacological and genetic inhibition of NADPH oxidase does not reduce brain damage in different models of perinatal brain injury in newborn mice. Neurobiol Dis. 2008;31:133–144. doi: 10.1016/j.nbd.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Feng Y, Shi W, Huang M, LeBlanc MH. Oxypurinol administration fails to prevent hypoxic-ischemic brain injury in neonatal rats. Brain Res Bull. 2003;59:453–457. doi: 10.1016/s0361-9230(02)00963-2. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Ljunggren B, Norberg K, Siesjo BK. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res. 1974;80:265–279. doi: 10.1016/0006-8993(74)90690-8. [DOI] [PubMed] [Google Scholar]
- Fontaine E, Ichas F, Bernardi P. A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J Biol Chem. 1998;273:25734–25740. doi: 10.1074/jbc.273.40.25734. [DOI] [PubMed] [Google Scholar]
- Gaspar T, Katakam P, Snipes JA, Kis B, Domoki F, Bari F, Busija DW. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J Neurochem. 2008;105:1115–1128. doi: 10.1111/j.1471-4159.2007.05210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilland E, Puka-Sundvall M, Hillered L, Hagberg H. Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature rat brain: involvement of NMDA-receptors. J Cereb Blood Flow Metab. 1998;18:297–304. doi: 10.1097/00004647-199803000-00008. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38:841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Hirata N, Shim YH, Pravdic D, Lohr NL, Pratt PF, Jr, Weihrauch D, Kersten JR, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane differentially modulates mitochondrial reactive oxygen species production via forward versus reverse electron transport flow: implications for preconditioning. Anesthesiology. 2011;115:531–540. doi: 10.1097/ALN.0b013e31822a2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol. 2007;292:H101–108. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- Huber R, Spiegel T, Buchner M, Riepe MW. Graded reoxygenation with chemical inhibition of oxidative phosphorylation improves posthypoxic recovery in murine hippocampal slices. J Neurosci Res. 2004;75:441–449. doi: 10.1002/jnr.10868. [DOI] [PubMed] [Google Scholar]
- Hwang JH, Lee JH, Lee KH, Bae EJ, Sung DK, Chang YS, Park WS. Cyclosporine A attenuates hypoxic-ischemic brain injury in newborn rats. Brain Res. 2010;1359:208–215. doi: 10.1016/j.brainres.2010.08.047. [DOI] [PubMed] [Google Scholar]
- Kayser EB, Suthammarak W, Morgan PG, Sedensky MM. Isoflurane selectively inhibits distal mitochondrial complex I in Caenorhabditis elegans. Anesth Analg. 2011;112:1321–1329. doi: 10.1213/ANE.0b013e3182121d37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Jin Y, Lemasters JJ. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;290:H2024–2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- Koch JD, Miles DK, Gilley JA, Yang CP, Kernie SG. Brief exposure to hyperoxia depletes the glial progenitor pool and impairs functional recovery after hypoxic-ischemic brain injury. J Cereb Blood Flow Metab. 2008;28:1294–1306. doi: 10.1038/jcbfm.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristian T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium. 2004;36:221–233. doi: 10.1016/j.ceca.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Katsura KI, Tsuchidate R, Siesjo BK. Secondary bioenergetic failure after transient focal ischaemia is due to mitochondrial injury. Acta Physiol Scand. 1996;156:149–150. doi: 10.1046/j.1365-201X.1996.449170000.x. [DOI] [PubMed] [Google Scholar]
- Leger PL, De Paulis D, Branco S, Bonnin P, Couture-Lepetit E, Baud O, Renolleau S, Ovize M, Gharib A, Charriaut-Marlangue C. Evaluation of cyclosporine A in a stroke model in the immature rat brain. Exp Neurol. 2010 doi: 10.1016/j.expneurol.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loor G, Kondapalli J, Iwase H, Chandel NS, Waypa GB, Guzy RD, Vanden Hoek TL, Schumacker PT. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamcr.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorek A, Takei Y, Cady EB, Wyatt JS, Penrice J, Edwards AD, Peebles D, Wylezinska M, Owen-Reece H, Kirkbride V, et al. Delayed (“secondary”) cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: continuous 48-hour studies by phosphorus magnetic resonance spectroscopy. Pediatr Res. 1994;36:699–706. doi: 10.1203/00006450-199412000-00003. [DOI] [PubMed] [Google Scholar]
- Morrison JF. The activation of aconitase by ferrous ions and reducing agents. Biochem J. 1954;58:685–692. doi: 10.1042/bj0580685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. discussion 332. [DOI] [PubMed] [Google Scholar]
- Puka-Sundvall M, Wallin C, Gilland E, Hallin U, Wang X, Sandberg M, Karlsson J, Blomgren K, Hagberg H. Impairment of mitochondrial respiration after cerebral hypoxia-ischemia in immature rats: relationship to activation of caspase-3 and neuronal injury. Brain Res Dev Brain Res. 2000;125:43–50. doi: 10.1016/s0165-3806(00)00111-5. [DOI] [PubMed] [Google Scholar]
- Selakovic V, Korenic A, Radenovic L. Spatial and temporal patterns of oxidative stress in the brain of gerbils submitted to different duration of global cerebral ischemia. Int J Dev Neurosci. 2011 doi: 10.1016/j.ijdevneu.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J Neurochem. 2007;100:1469–1479. doi: 10.1111/j.1471-4159.2006.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J Neurochem. 1991;56:1836–1844. doi: 10.1111/j.1471-4159.1991.tb03438.x. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- Takeyama N, Matsuo N, Tanaka T. Oxidative damage to mitochondria is mediated by the Ca(2+)-dependent inner-membrane permeability transition. Biochem J. 1993;294 (Pt 3):719–725. doi: 10.1042/bj2940719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten VS, Bradley-Moore M, Gingrich JA, Stark RI, Pinsky DJ. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav Brain Res. 2003;145:209–219. doi: 10.1016/s0166-4328(03)00146-3. [DOI] [PubMed] [Google Scholar]
- Ten VS, Wu EX, Tang H, Bradley-Moore M, Fedarau MV, Ratner VI, Stark RI, Gingrich JA, Pinsky DJ. Late measures of brain injury after neonatal hypoxia-ischemia in mice. Stroke. 2004;35:2183–2188. doi: 10.1161/01.STR.0000137768.25203.df. [DOI] [PubMed] [Google Scholar]
- Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, Sivasankar B, Morgan BP, Silverstein S, Stark R, Polin R, Vannucci SJ, Pinsky D, Starkov AA. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci. 2010;30:2077–2087. doi: 10.1523/JNEUROSCI.5249-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A, Scarpa A, Chance B. Calcium and pyridine nucleotide interaction in mitochondrial membranes. Arch Biochem Biophys. 1972;152:646–654. doi: 10.1016/0003-9861(72)90261-5. [DOI] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, Forte MA, Hagberg H. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci. 2009;29:2588–2596. doi: 10.1523/JNEUROSCI.5832-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005;12:162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- Zhu C, Xu F, Fukuda A, Wang X, Fukuda H, Korhonen L, Hagberg H, Lannering B, Nilsson M, Eriksson PS, Northington FJ, Bjork-Eriksson T, Lindholm D, Blomgren K. X chromosome-linked inhibitor of apoptosis protein reduces oxidative stress after cerebral irradiation or hypoxia-ischemia through up-regulation of mitochondrial antioxidants. Eur J Neurosci. 2007;26:3402–3410. doi: 10.1111/j.1460-9568.2007.05948.x. [DOI] [PubMed] [Google Scholar]