

Abstract
Aim
To investigate the effect of the axon guidance cue Slit2 on the density of blood vessels in the mouse brain and the permeability of the blood-brain barrier.
Methods
A Slit2 transgenic mouse line was constructed, and the phenotypes of the mice were compared according to the lateral ventricle (LV), ventricle pressure, and the choroids plexus. An in vivo Miles permeability assay and an amyloid-β permeability assay were used to assess the permeability of blood vessels in the brain. Then brain vessel casting and intracerebral hemorrhage models were built to investigate vessel density in the transgenic mice. An in vitro permeability assay was used to test whether Slit2 could change the permeability and tight junctions of blood vessel endothelial cells.
Results
Hydrocephalous occurred in some transgenic mice, and a larger lateral ventricle area and higher ventricle pressure were observed in the transgenic mice. The transgenic mice also displayed a change in the construction of the choroids plexus, which had more micro vessels, dilated vessels, gaps between epithelial cells and endothelial cells. Slit2 improved brain vessel density and also increased the permeability of brain vessels to large molecules. These blood vessels were also more sensitive to cues that induce brain hemorrhage. At the cellular level, Slit2 disturbed the integrity of tight junctions in blood vessel endothelial cells and improved the permeability of the endothelial cell layer. Thus, it promoted the entry of amyloid-β peptides from the serum into the central nervous system, where they bound to neurons.
Conclusion
Slit2 improves vessel density and permeability in the brains of transgenic mice. Thus, Slit2 induces numerous changes in brain vessels and the barrier system.
Keywords: Slit2, blood-brain barrier, permeability, vessel endothelial cell, tight junction
Introduction
Vasogenic brain edema, which is defined as the translocation of proteins and fluid from the vascular space across the blood-brain barrier (BBB)[1], is a major life-threatening complication of various injuries to the central nervous system (CNS)[2, 3]. Endothelial cells of the brain vasculature form the BBB and maintain the homeostasis of the central nervous system (CNS). Pathological conditions such as brain tumors and head injuries increase the permeability of the brain microvasculature and destroy the BBB[4]. Vascular endothelial growth factor (VEGF) is well known as the major inducer of angiogenesis, and it also increases the permeability of the microvasculature and stimulates endothelial cell growth[5–8]. Increased VEGF expression may cause vascular leakage in the CNS in vivo[9]. However, the underlying molecular and pathogenic mechanisms behind edema and blood-brain vessel leakage are poorly understood.
The Slit family of guidance cues interacts with the Roundabout (Robo) family of transmembrane receptors in physiological and pathological processes requiring cell migration[10–13]. During development of the nervous system, Slit-Robo signaling regulates the repulsion or attraction of projecting axons and migrating neurons[14, 15]. Vascular endothelial cells secrete Slit2, which binds to Robo1 on leukocytes and acts as an endogenous inhibitor of leukocyte chemotaxis[16, 17, 19–21]. Additionally, Slit2 mediates directional migration of malignant cells[22–24]. We and others have previously reported that Slit proteins secreted by solid tumors bind to Robo1, which is expressed on vascular and lymphatic endothelial cells, to stimulate angiogenesis and lymph angiogenesis[25–31]. Slit2 is expressed in the CNS while Robo1 is expressed in blood vessel endothelial cells. However, whether expression of these proteins can change the permeability of blood vessels and whether abnormal expression can induce vessel leakage and edema remain to be determined.
In this study, we constructed a transgenic mouse line that over-expresses human Slit2 and observed that hydrocephalous occurs in some of these transgenic mice. The transgenic mice also had larger lateral ventricles and higher ventricle pressure. Comparison of the choroids plexus, where cerebrospinal fluid (CSF) is secreted, revealed that there was a change in the construction of the choroids plexus, with the transgenic mice having more micro vessels, dilated vessels, gaps between epithelial cells and endothelial cells. We also found that Slit2 could improve brain vessel density and promote the permeability of brain vessels to allow large molecules. These blood vessels were also more sensitive to cues that induced brain hemorrhage. At the cellular level, Slit2 disturbed the integrity of tight junctions in blood vessel endothelial cells and improved the permeability of the endothelial cell layer. The ability of Slit2 to increase the permeability of the BBB resulted in an increase in the transfer of amyloid-β peptides from the serum to the CNS, where they bound to neurons.
Material and methods
Generation of hSlit2 transgenic mice and detection of hSlit2 over-expression
hSlit2 transgenic mice were generated according to standard procedures. The transgene was constructed by cloning cDNA encoding full-length human Slit2 between the BamH I and Xbo I restriction sites of the MCS of pCEP4F. Genotypes were confirmed by Southern blot and PCR analysis. PCR screening of hSlit2 heterozygotes was performed on standard tail genomic DNA preparations using a pair of primers specific for human Slit2 cDNA (forward: 5′-GGTGACGGATCCCATATCGCGGTAGAACTC-3′; reverse: 5′-GGACACCTCGAGCGTACAGCCGCACTTCAC-3′). PCR cycles were as follows: 95 °C, 4 min (1 cycle); 94 °C, 45 s; 55 °C, 45 s; and 72 °C, 1 min (63 cycles); and 72 °C, 10 min (2 cycles). PCR products were analyzed on 1% agarose gels. Slit2 homozygosity was confirmed by genetic methods based on the principle that the progeny of Slit2 homozygotes mated to wild-type C57 mice should all be heterozygotes. The brains from C57 control littermate mice and hSlit2 transgenic mice from founder #9 were snap frozen in liquid N2 and pulverized. The brain powder was homogenized in 1 mL RIPA lysis buffer [50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 1% Nonidet P-40, 0.5% deoxycholid acid, 0.1% sodium dodecyl sulfate (SDS), 5 mmol/L EDTA, 2 mmol/L phenylmethylsulfonyl fluoride (PMSF), 20 μg/mL aprotinin, 20 μg/mL leupeptin, 10 μg/mL pepstanin A, and 150 mmol/L benzamidine] in a Dounce tissue homogenizer. After homogenization, the samples were centrifuged at 12 000×g for 10 min to remove tissue debris and boiled in SDS sample buffer for 5 min. They were then subjected to 7% SDS-PAGE electrophoresis, transferred to blotting membranes, probed with 1 μg/mL anti-Slit2 monoclonal antibody (5A5) and detected with the horseradish peroxidase-conjugated goat anti-mIgG Ab using a chemiluminescent detection system.
Comparison of the LV area and the choroid plexus
The mouse brains were fixed in 4% PFA (Sigma) and cut into consecutive longitudinal sections. A photo was taken of every longitudinal sections from each brain with an Olympus MVX10. Then the area of the lateral ventricle was calculated by Image Tool 3 (UTHSCSA), and the average area of consecutive sections from each mouse was calculated. The data represent the means for groups of six mice. For histological examination of the choroid plexus, brains were isolated from age-matched C57 and Slit2 adult mice, cut into small coronal blocks, fixed in 4% formaldehyde, embedded in paraffin, and cut into 5-mm sections following standard procedures. Tissue sections were counterstained with hematoxylin. The same regions of the choroid plexus in all mouse brains were photographed.
Electron microscopy
The choroid plexuses used for TEM were fixed in 2% glutaraldehyde and 1% sucrose in 0.1 mol/L cacodylate buffer for 3 h. The samples were washed in 0.1 mol/L cacodylate buffer and then post-fixed in 1% osmium tetraoxide in 0.1 mol/L cacodylate buffer for 2 h, all at pH 7.0 and 25 °C. The choroid plexuses were embedded in epon after dehydration in a graded series of ethanol. Epon was polymerized at 60 °C for 48 h. Serial sections of 80 nm were cut on a Leica Super Nova ultramicrotome with a diamond knife and collected on formvar-coated nickel grids. Sections were contrasted with uranylacetate and stained with 1% toluidine blue. The sample grids were observed using a Hitachi 600 TEM.
In vivo Miles permeability assay and amyloid-β permeability assay
To investigate the effects of over-expression of hSlit2 in the brain on vascular permeability, a Miles assay was performed. Mice received an IV injection of sterile 0.5% Evans blue dye (200 μL) via the tail vein. Mice were killed 20 min after the injections by cervical dislocation after anesthesia. The brains (weighted) were cut into small pieces and incubated in 500 mL of formamide at 37 °C for 48 h to extract the Evans blue dye. The absorbance of the extracts was read at 630 nm in a spectrophotometer (Beckman DU 640). For the amyloid-β permeability assay, mice received an IV injection of 6.9 μmol/L FITC-aβ (100 μL) via the tail vein. Mice were killed 48 h after the injections by cervical dislocation after anesthesia. The brains of the mice were prepared on crystal slides, and the nuclei were stained with DAPI.
LV pressure assay
Mice (12 weeks of age) were anesthetized by IP injection of sodium pentobarbital (70 mg/kg). A small hole 0.5 mm posterior and 1.0 mm lateral of the bregma was drilled to perforate the skull. A pressure transducer linked to a monitor was injected into the brain at a depth of 2.3 mm unilaterally. Then the LV pressure was read on a computer that was linked to the monitor (Powerlab 4/30). The pressure was measured as mmWater.
Immunohistochemical staining
Antibodies against vWF (Antibody Diagnostica Inc; a 1:200 dilution for paraffin-embedding), Slit2 (5 μg/mL for paraffin-embedding sections), and Robo1 (20 μg/mL for paraffin-embedding sections) were used for immunohistochemical staining as described previously (Liu et al., 2001).
Brain vessel casting
After systemic heparinization with 750 Iu/kg intravenous heparin, the common carotid arteries were cannulated and perfused with approximately 100 mL of 27°C saline, followed by a 2.5% buffered glutaraldehyde solution (Sigma) at pH 7.4. The casts were made by perfusion of the arteries with 100 mL of Mercox (SPI, West Chester, PA) diluted with 20% methylmethacrylate monomers (Aldrich Chemical, Milwaukee, WI). After complete polymerization, the brains were harvested and macerated in 5% potassium hydroxide, followed by drying and mounting for scanning electron microscopy. The microvascular corrosion casts were imaged after being coated with gold with a Hitachi S-450 scanning electron microscope.
Intracerebral hemorrhage (ICH) model
Mice (12 weeks of age) were prepared for surgery and anesthetized by IP injection of sodium pentobarbital (70 mg/kg)32. A small hole 1.0 mm posterior and 3.0 mm lateral of the bregma was drilled to perforate the skull. A 1-μL Hamilton syringe was used to deliver 500 nL of collagenase/saline (150 U/μL) to the caudate/putamen at a depth of 4.0 mm unilaterally. After the injection of collagenase/saline (~30 s), the needle remained in place for another 2 min to prevent reflux of fluid. Then the scalp skin was closed using 4–0 nylon sutures. Twenty-four hours later, the mice were perfused with PBS, the brains were harvested, and 14-μm sections were prepared using a cryostat and mounted on glass slides. An Olympus BX100 upright systems microscope with a digital camera was used to capture images. The hemorrhage volume was measured using the Stereologer software system.
In vitro permeability assay
We coated transwell inserts (Corning, 48-well, 3-mm pore) with collagen and seeded HUVEC cells at a density of 30,000 cells per well. Cells were then cultured for another 24 hours. One hour later, Slit2, VEGF, R5, and FITC-DEXTRAN (25 mg/mL, Sigma) were added to the top of the inserts. The absorbance of the solution in each well was measured at 492 nm (n=6 wells each).
Statistical analysis
Statistical significance was determined by Student’s t- test. P-values of 0.05 and 0.01 were considered statistically significant and very significant, respectively.
Results
Generation of hSlit2 transgenic mice and over-expression of hSlit2 in mouse brains
To study the function of Slit2 in the whole blood vessel system, we constructed a Slit2 over-expressing mouse transgenic plasmid, for which a schematic display is shown in Figure 1a. The full-length human Slit2 cDNA, which is 4689 bases, was inserted between the BamHI/Xbo1 restriction sites of the MCS of the pCEP4F vector, which has a pCMV promoter and an N-terminal flag-tag. The plasmid was linearized with restriction enzymes, injected into the pronuclei of fertilized C57 x CBA F1 oocytes and transplanted into the mother mouse. The offspring mice were analyzed by dot blot (Figure 1b). Mouse lines 9, 25, and 47 were shown to express human Slit2. The presence of the human Slit2 transgene was also confirmed by Southern blot analysis (Figure 1c). Among the three transgenic strains, we found that strain 9 had the strongest signal in dot blot and Southern blot assays, so we focused our research on strain 9. We observed that the transgenic mice expressed more Slit2 in the brain compared with C57 mice at the transcriptional and translational levels (Figure 1d, e). We used the Slit2 antibody 10D10 to IP the mouse brain lysate and detected it with anti-FLAG antibody, and we found that FLAG-Slit2 was expressed in the mouse brain (Figure 1f).
Figure 1. Generation of hSlit2 transgenic mice and over-expression of hSlit2 in Slit2 mouse brains.

a Schematic display of the human Slit2 (hSlit2) transgene. The transgene was constructed by cloning cDNA encoding full length human Slit2 between the BamHI/Xbo1 restriction sites of the MCS of pCEP4F.
b Dot blotting of DNA isolated from hSlit2 transgenic mice. The presence of the transgene in mice was verified by dot blotting using a 32P-labeled fragment of the hSlit2 plasmid.
c Southern blotting of hSlit2 transgenic mice. The presence of the transgene was further confirmed by Southern blotting of DNA isolated from mice that were shown to be positive for the transgene based on the dot blot analysis with a 32P-labeled fragment of the hSlit2 plasmid.
d Expression of the hSlit2 protein in the hSlit2 transgenic mice compared with C57 mice. The expression of Slit2 (~200 kDa) in the brains of C57 and Slit2 mice was detected with anti-Slit2 IgG 5A5.
e RT-PCR of hSlit2 in mRNA extracts from C57 and hSlit2 mouse brains.
f Western blot analysis of flag-tagged proteins. Brain lysates from C57 and hSlit2 mice that were IP injected with an anti-Slit2 antibody, 10D10, and probed with an anti-flag tag antibody.
The phenotype of Slit2 mice and comparison of the LV area
Most of the transgenic mice initially appeared normal (Figure 2a), but about 5% of the transgenic mice had an intumescent head (Figure 2b). This phenotype appeared at the age of 4–6weeks in all three of the transgenic mouse lines, line 9, line 25 and line 47. Mice with an intumescent head died within 2 weeks, at the age of 6–8 weeks. Anatomical analysis of these mice revealed high levels of encephalopathic edema. In mice where this brain phenotype was observed, the brain edema was serious and the brain tissue was destroyed to such a degree that the structure of the brain could not be recognized. Thus, we focused our research on the transgenic mice that did not have this phenotype. Brain edema is linked to the CSF system, including the production, circulation and absorption of CSF. We therefore examined brain structures related to the CSF system. In the brain, CSF is produced in the lateral ventricle, so we compared the lateral ventricle of transgenic mice and non-transgenic mice. We found that the lateral ventricle area was larger in the brains of transgenic mice (Figure 2c, d) than in control mice (Figure 2e, f). For this study, the mice were all 8 weeks old, and the same result was seen in male and female mice. Figure 2g shows a schematic display of the mouse brain. The arrow indicates the lateral ventricle. We calculated the entire area of the LV in slides created from the brains of each mouse with Image Tool 3 (UTHSCSA) and found that the LV area was larger in transgenic mice (n=6, p<0.01) (Figure 2h). LV pressure was also detected in the lateral ventricles of the mouse brains. In transgenic mice, the pressure was 128 mm water, and in control mice, it was 81 mm water (Figure 2i), (n=7, p<0.01). Thus, the LV pressure was higher in the brains of transgenic mice compared with control mice.
Figure 2. The phenotype of Slit2 mice and comparison of the LV area.

a and b Comparison of a C57 mouse (a) with a Slit2 mouse (b) with a megahead. The forebrain of the Slit2 mouse is much larger than that of the C57 mouse.
c,d and e,f Slides of longitudinal sections of brains from Slit2 mice (c, male; d, female) and C57 mice (e, male; f, female). The lateral ventricle area was larger in Slit2 mice than in C57 mice.
g Schematic diagram of the mouse brain. The arrow indicates the lateral ventricle (black).
h Measurement of the lateral ventricle area in brain longitudinal section slides of C57 and Slit2 mice. The data represent the mean results from groups of six mice; bars, SE. **, P <0.01 Scale bar: 2 mm
i LV pressure in C57 and Slit2 mice. The data represent the mean results from groups of seven mice; bars, SE. *, P <0.05
Changes in the structure and function of the choroid plexus
In mouse brains, the choroids plexus produces CSF. The structure of the choroids plexus was obviously different between transgenic and control mice. In control mice, it was a tight, mono-cell layer covered with micro vessels (Figure 3a); however, in transgenic mice, there were gaps between the epithelial cell layer and the vessels (Figure 3b, c), the vessels were enlarged and had gaps, and the epithelial cells were crenated (Figure 3c). The mice used in these experiments were all 10 weeks old. Examination under an electoral microscope revealed gaps between the epithelial tight junctions in transgenic mice (Figure 3d); such gaps were not observed in control mice (Figure 3e). ZO-1 is an important component of cell-cell tight junctions and can be used as a marker for the completeness and density of tight junctions. We used fluorescence immunostaining to detect ZO-1 in the choroid plexus. In control mice, ZO-1 had an equal and continuous distribution (Figure 3f), while in transgenic mice, the distribution of ZO-1 was disturbed (Figure 3g) and the tight junctions were destroyed.
Figure 3. Changes in the structure and the function of the choroid plexus.

a, b and c The structure of the choroid plexus of a C57 mouse (a), a Slit2 mouse (b) and a Slit2 mouse with megahead (c). Slit2 mice had structurally different choroid plexuses compared with C57 mice. Scale bar: 150 μm.
d and e Electron micrograph of choroid plexus epithelial cells from the mouse lateral ventricle. Some vacuoles appear between the junctional complexes linking the epithelial cells in Slit2 mice. Scale bar: 1.5 μm.
f and g Immunoflorescence of the choroids plexus probed with an anti ZO-1 antibody. Cell-cell adhesion was different in hSlit2 mice compared with C57 mice.
More vessels are present in the choroid plexus and the brain of transgenic mice
Staining of slides of the brain cortex with anti-CD31 antibody showed that more micro-blood vessels were present in the brains of hSlit2 transgenic mice (Figure 4b) compared with C57 mice (Figure 4a). The gray density was 1.7% in C57 mice and 4.6% in hSlit2 transgenic mice, p<0.01 (Figure 4c). We also stained the brain cortex slides with anti-VWF antibody and found that hSlit2 transgenic mice had an increased number of vessels (Figure 4e) compared with C57 mice (Figure 4d). Calculating the numbers of vessels present on the slides revealed that C57 mice had about 200 micro-vessels/mm2, while hSlit2 transgenic mice had about 450 micro-vessels/mm2 (Figure 4f), and most of these brain micro-vessels consisted of only one endothelial cell. Furthermore, we performed brain vessel casting, which also showed that hSlit2 transgenic mice had more brain vessels (Figure 4h) compared with C57 mice (Figure 4g).
Figure 4. More vessels are present in the choroid plexus and the brain of transgenic mice.

a, b and c ICH of CD31 in C57 (a) and hSlit2 mice (b). Gray density statistics of CD31 staining (c), p<0.01. (scale bar: 150 μm).
d, e and f ICH of vWF in C57 (d) and hSlit2 mice (e). The difference in the number of stained vessels in the same area was statistically significant (f), p<0.01 (scale bar: 50 μm).<vr>g and h Scanning electron microscopy of corrosion casts of the brain vessel of C57 (g) and Slit2 mice (h).
Slit2 changes the permeability of cell-cell adhesions
To evaluate whether Slit2 improves the permeability of blood vessels, we performed a Miles assay on hSlit2 transgenic mice. An increased amount of Evans blue dye was detected in the brain tissue of transgenic mice compared with C57 mice (Figure 5a), which indicates that the blood vessels in the brains of transgenic mice are more permeable, allowing the entry of Evans blue dye into the tissue. Previous research has indicated that hyperplastic vessels are incomplete, more permeable, and more sensitive to destructive effects compared with normal vessels. Thus, we operated intracerebral hemorrhage model on the transgenic mice. This was accomplished by injecting collagenase into the mouse brains, which destroys blood vessels in the brain and causes intracerebral hemorrhaging. Examination of brain slides from mice in which intracerebral hemorrhaging was induced showed that transgenic mice have a larger hemorrhage area compared with the control mice (Figure 5 c, d). Next, we measured the hemorrhage volume using the Stereologer software system and found that the hemorrhage volume also was increased in the transgenic mice (Figure 5b), n=6, p<0.01.These results indicate that the blood vessels in the brains of the transgenic mice are more permeable and much more sensitive to collagenase, which destroys the vessel structure. Thus, Slit2 may not only promote angiogenesis but could also increase the permeability of blood vessel endothelial cells, which are the most important component of blood vessels. VE-cadherin is a member of the cadherin family, which is expressed specifically in endothelial cells and plays important roles in endothelial cell cell-cell adhesion. Immunofluorescent detection of HUVECs using an anti-VE-Cadherin antibody revealed that, without incubation with Slit2, VE-Cadherin expression was equal, continuous and mostly localized to the conjunction sites of cell-cell adhesions (Figure 5e), while HUVECs incubated with the Slit2 protein had a disturbed distribution and lacked continuous expression of VE-Cadherin (Figure 5f). To verify that Slit2 can affect the permeability of blood vessels, we performed an in vitro permeability assay. We coated transwell inserts (Corning, 48-well, 3-mm pore) with collagen and seeded HUVECs at a density of 30,000 cells per well. Once these cells formed a monolayer, we added Slit2 protein and other stimulating factors to the upper well. FITC-dextran was added one hour later, and fluorescence was detected in the bottom wells. Our results show that VEGF-A can improve the permeability of HUVECs and Slit2 by 1.5 fold and 2 fold, respectively, and that a blocking antibody against the Slit2-Robo signal, R5, can block the effect of Slit2 on cell permeability, returning it to basal levels (Figure 5g).
Figure 5. Slit2 expression changes blood vessel permeability at sites of cell-cell adhesion.

a Permeability of C57 and Slit2 mouse brains. The data represent the mean results from seven mice; bars, SE. **, P <0.01.
b Measurement of the ICH area in brain longitudinal section slides from C57 and Slit2 mice. The data represent the mean results from six mice; bars, SE. **, P <0.01.
c and d Longitudinal section slides of mouse brains in which ICH was induced. The ICH area was bigger in Slit2 mice (d) than in C57 mice (c).
e and f Immunoflorescence of HUVECs with anti VE-cadherin antibody. Cell-cell adhesion was different in Slit2 stimulated HUVECs (f) compared with cells not stimulated with Slit2 (e).
g Slit2 increases the permeability of HUVECs in in vitro permeability assays. The Robo blocking antibody R5 could block this effect.
The presence of amyloid-β peptide in the brains of Slit2 mice
Amyloid-β 40 peptides were detected on slides of the transgenic mouse brains (Figure 6a) but not slides of C57 mouse brains (Figure 6d). In the cortex and hippocampus, amyloid-β 42 was detected in granual and pyramid cells in transgenic mice (Figure 6b, c) but not in C57 mice of the same age (Figure 6e, f). Furthermore, we injected FITC-Amyloid-β40 into the circulation by I.P. to see if the circulating peptides could enter the CNS. Florescence was observed on brain cortex slides prepared from the transgenic mice 1 hour (Figure 6g), 24 hours (Figure 6h), and 48 hours (Figure 6i) after injection. The florescence was higher with increasing time, but even 48 hours after injection, no florescence was detected on brain cortex slides prepared from C57 mice (Figure 6j, k).
Figure 6. The presence of the Aβ peptide in the brain of Slit2 mice.

a and d Detection of Aβ 40 in Slit2 and C57 mouse brains. IHC of Slit2 mouse brain slides (a) stained with Aβ 40 antibody. No staining was observed in slides prepared from C57 mouse brains (d).
b, c and e, f Detection of Aβ 42 in Slit2 and C67 mouse brains. IHC of Slit2 mouse brain slides (b, c) stained with Aβ 42 antibody. No staining was observed in slides prepared from C57 mouse brains (e, f).
g, h, i and j, k Blood-borne FITC-labeled Aβ42 crosses the blood–brain barrier, enters into the brain tissue and binds selectively to neurons in Slit2 mice (g). Within 1 h post-injection, FITC-labeled Aβ42 was leaked from local vessels and bound to the surfaces of neurons (h, i). At 24 and 48 h post-injection, neurons bound with FITC-labeled Aβ42 were abundant in the indicated brain regions. NO FITC-labeled Aβ42 bound to neurons in C57 mice.
hSlit2 transgenic mice have normal aqueduct and subarachnoid space, and Slit2 over-expression does not alter VEGF expression levels
To find the course of the enlarged lateral ventricles observed in Slit2 transgenic mice, we compared the aqueduct and the subarachnoid space of transgenic and C57 mice. However, there were no obvious differences in these two areas between transgenic and C57 mice. The aqueduct was smooth and clear, and the aqueduct tube had the same inside diameter and did not contain any clogs (Figure 7a, b, c, d). The SAS also had a complete and clear structure with no clogs or signs of collapse in the transgenic mice (Figure 7e, f, g, h). We also found that Slit2 over-expression did not alter VEGF expression at either the protein or mRNA level (Figure 7i, j). These results show that the structures linked to circulation and absorption of CSF were complete and normal in the transgenic mice and that the enlargement of the lateral ventricle is caused by the abnormal production of CSF in the choroid plexus. On the other hand, the normal circulation and absorption of CSF could compensate for the abnormal production of cerebrospinal fluid, which could explain why edema was only observed in a small percentage of the transgenic mice.
Figure 7. Structure of the aqueduct and SAS and expression of VEGF in transgenic and C57 mice.
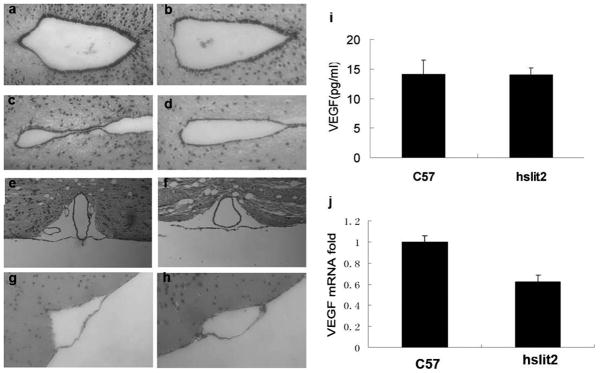
a, b and c, d: Structure of the aqueduct in Slit2 mice (a, c) and C57 mice (b, d). No difference was observed in the aqueduct between Slit2 and C57 mice.
e, f and g, h: Structure of the SAS in Slit2 mice (e, g) and C57 mice (f, h). No difference was observed in the SAS between Slit2 and C57 mice.
i VEGF concentrations in the blood of C57 and Slit2 mice. Serum VEGF levels of C57 and Slit2 mice were detected with a VEGF ELISA kit (Quantikine Immunoassay Lot: 234711). The data represent the mean results for groups of eight mice; bars, SE. P =0.8
j Q-PCR analysis of VEGF mRNA extracted from C57 and hSlit2 mice. VEGF expression was not increased by hSlit2 in mouse brains.
Discussion
The brain barriers, including the blood-brain barrier, the blood-CSF barrier and the ventricular wall, provide a stable micro-environment for the proper functioning of the central nervous system. At the bases of the barrier structures are the junction structures, such as adherence junctions and tight junctions between endothelial cells, epithelial cells and pericytes[33]. These junction structures are dynamic structures that consist of transmembrane proteins, cytoplasmic accessory proteins and scaffold proteins. Under different physiological and pathological conditions, changes occur in the expression, distribution, modification and interaction of these proteins[34]. These changes are regulated by several cell signaling pathways, and to date, the calcium channel pathway, the phosphorylation signaling pathway and the G-protein signaling pathway have been shown to change the expression and distribution of junction proteins, further affecting the function of barrier structure[35]. The brain blood vessel system is the structural basis of the blood-brain barrier, and the junctions between vessel endothelial cells have the most important effect on the permeability of the BBB. Many molecules, such as small chemical molecules, signaling proteins and inflammatory factors, can change the permeability of blood vessels. Vascular endothelial growth factor (VEGF) is an important angiogenic cue, and some reports have shown that VEGF improves the permeability of blood vessels and the BBB. VEGF binds to its receptor on blood vessel endothelial cells and triggers signaling pathways in the cell plasma. This, in turn, alters the expression, phosphorylation and distribution of VE-cadherin and thus changes the permeability of blood vessels by disturbing the junction structures[36]. However, barrier structures are very complex and have many components. Therefore, whether other cues that induce angiogenesis have some effect on the permeability of the blood-brain barrier should be investigated.
The guidance cue Slit2 has been reported to regulate a number of physiological processes, mostly in the central nervous system, by controlling cell migration. Previous research from our lab indicated that Slit2 promotes tumor angiogenesis in a manner similar to VEGF[25]. In this paper, we found that Slit2 improved blood vessel density in the brain and promoted the permeability of brain blood vessels to large molecules. In addition, these blood vessels were more sensitive to cues that can induce brain hemorrhage. At the cellular level, Slit2 disturbed the integrity of blood vessel endothelial cell tight junctions and improved the permeability of the endothelial cell layer, thus promoting the entry of amyloid-β peptides from the serum into the central nervous system, where they bind to neurons. We also found that hydrocephalous occurred in some of the hSlit2 transgenic mice. In addition, we observed a larger lateral ventricle area and higher ventricle pressure in the transgenic mice. A comparison of the choroids plexus, where CSF is secreted, revealed that transgenic mice have changes in the structure of the choroids plexus, including more micro-vessels, dilated vessels, and gaps between epithelial cells and endothelial cells. Thus, Slit2 could bind to its receptor Robo1 on endothelial cells and affect the junction through plasma signaling. It has been reported that Slit2 modifies the activity of cytoplasmic GTP enzymes, which affect tight junction structures by regulating adherence proteins and cell scaffold proteins[37]. However, the signaling pathway by which Slit2 affects cell-cell adhesion structures requires further investigation. Because adherent structures consist of a large number of proteins, it is unclear which proteins are affected by Slit2. For example, which changes in protein expression or function are the direct results of Slit2 signaling, and which protein changes are the result of subsequent disturbances of junction structures? These are all important questions that need to be answered to understand the molecular mechanism by which Slit2 alters the permeability of barriers.
In the brains of transgenic mice, at a certain age, we observed binding of the amyloid-β peptide to neurons. This suggested that Slit2 promotes the entry of amyloid-β peptides from the serum into the central nervous system, where they then bind to neurons. The binding of amyloid-β peptides to neurons is a phenotype of the early stage of Alzheimer’s disease. However, whether Slit2 is over-expressed in patients with Alzheimer’s disease and the relationship between Slit2 and Alzheimer’s disease need to be further investigated and will be the focus of future work in our lab.
Slit2 improves the permeability of the blood-brain barrier and thus may have some medical application for the delivery of drugs to the central nervous system, a problem that has puzzled many researchers because drugs targeting the central nervous system often cannot penetrate the blood-brain barrier at sufficient therapeutic doses[38]. In our study, we found that Slit2 promotes the entry of a fluorescent-labeled peptide into the central nervous system. This result suggests that Slit2 may promote the penetration of large-molecule drugs from the peripheral circulation into the central nervous system by increasing the permeability of the blood-brain barrier. Whether manipulation of Slit2 levels or activity can be applied to the field of central nervous system drug delivery requires further investigation, which we plan on pursuing in the future.
Acknowledgments
We thank Dr. Biao Wang for the construction of the pCMV-hSlit2 expression plasmid. This work was supported by grants from the National Science Foundation of China (30811120438, 30721065, 30700409, and 30630036), the Ministry of Science and Technology of China (2007CB914501, 2007CB947102, 2009ZX09103-685, and 2010CB529700), Shanghai Municipal Commission for Science and Technology (08JC1421400), and the National Institute of Health (RO1AI064743 and RO1CA126897).
References
- 1.Hackett PH. High altitude cerebral edema and acute mountain sickness: a pathophysiology update. Adv Exp Med Biol. 1999;474:23–45. doi: 10.1007/978-1-4615-4711-2_2. [DOI] [PubMed] [Google Scholar]
- 2.Murakami K, Kondo T, Yang G, Chen SF, Morita-Fujimura Y, Chan PH. Cold injury in mice: a model to study mechanisms of brain edema and neuronal apoptosis. Prog Neurobiol. 1999;57:289–99. doi: 10.1016/s0301-0082(98)00047-1. [DOI] [PubMed] [Google Scholar]
- 3.Pilitsis JG, Rengachary SS. Complications of head injury. Neurol Res. 2001;23:227–36. doi: 10.1179/016164101101198389. [DOI] [PubMed] [Google Scholar]
- 4.Wang W, Dentler WL, Borchardt RT. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am J Physiol Heart Circ Physiol. 2001;280:H434–40. doi: 10.1152/ajpheart.2001.280.1.H434. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N. The role of vascular endothelial growth factors in pathological angiogenesis. Breast Cancer Res Treat. 1995;36:127–37. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 6.Senger DR, Van de Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, et al. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev. 1993;12:303–24. doi: 10.1007/BF00665960. [DOI] [PubMed] [Google Scholar]
- 7.Stephan CC, Brock TA. Vascular endothelial growth factor, a multifunctional polypeptide. P R Health Sci J. 1996;15:169–78. [PubMed] [Google Scholar]
- 8.Thomas KA. Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem. 1996;271:603–6. doi: 10.1074/jbc.271.2.603. [DOI] [PubMed] [Google Scholar]
- 9.Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125:2549–57. doi: 10.1093/brain/awf257. [DOI] [PubMed] [Google Scholar]
- 10.Kidd T, Brose K, Mitchell KJ, Fetter RD, Tessier-Lavigne M, Goodman CS, et al. Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell. 1998;92:205–15. doi: 10.1016/s0092-8674(00)80915-0. [DOI] [PubMed] [Google Scholar]
- 11.Brose K, Bland KS, Wang KH, Arnott D, Henzel W, Goodman CS, et al. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell. 1999;96:795–806. doi: 10.1016/s0092-8674(00)80590-5. [DOI] [PubMed] [Google Scholar]
- 12.Li HS, Chen JH, Wu W, Fagaly T, Zhou L, Yuan W, et al. Vertebrate Slit, a secreted ligand or the transmembrane protein Roundabout, is a repellent or olfactory bulb axons. Cell. 1999;96:807–18. doi: 10.1016/s0092-8674(00)80591-7. [DOI] [PubMed] [Google Scholar]
- 13.Plachez C, Andrews W, Liapi A, Knoell B, Drescher U, Mankoo B, et al. Slit1 and Slit2 cooperate o prevent premature midline crossing of retinal axons in the mouse visual system. Neuron. 2002;33:219–32. doi: 10.1016/s0896-6273(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 14.Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS. Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell. 2000;101:703–15. doi: 10.1016/s0092-8674(00)80883-1. [DOI] [PubMed] [Google Scholar]
- 15.Dickson BJ, Gilestro GF. Regulation of commissural axon path finding by Slit and its Robo receptors. Annu Rev Cell Dev Biol. 2006;22:651–75. doi: 10.1146/annurev.cellbio.21.090704.151234. [DOI] [PubMed] [Google Scholar]
- 16.Wu JY, Feng L, Park HT, Havlioglu N, Wen L, Tang H, et al. The neuronal repellent Slit inhibits leukocyte chemotaxis induced by chemotactic factors. Nature. 2001;410:948–52. doi: 10.1038/35073616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan H, Zu G, Xie Y, Tang H, Jonhson M, Xu X, et al. Neuronal repellent Slit2 inhibits dendritic cell migration and the development of immune responses. J Immunol. 2003;171:6519–26. doi: 10.4049/jimmunol.171.12.6519. [DOI] [PubMed] [Google Scholar]
- 18.Kanellis J, Garcia GE, Li P, Parra G, Wilson CB, Rao Y, et al. Modulation of inflammation by Slit protein in vivo in experimental crescentic glomerulonephritis. Am J Pathol. 2004;165:341–52. doi: 10.1016/S0002-9440(10)63301-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prasad A, Qamri Z, Wu J, Ganju RK. Slit2/Robo1 modulates the CXCL12/CXCR4-induced chemotaxis of T cells. J Leukoc Biol. 2007;82:465–76. doi: 10.1189/jlb.1106678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altay T, McLaughlin B, Wu JY, Park TS, Gidday JM. Slit modulates cerebrovascular inflammation and mediates neuroprotection against global cerebral ischemia. Exp Neurol. 2007;207:186–94. doi: 10.1016/j.expneurol.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tole S, Mukovozov IM, Huang YW, Magalhaes MA, Yan M, Crow MR, et al. The axonal repellent, Slit2, inhibits directional migration of circulating neutrophils. J Leukoc Biol. 2009;86:1403–15. doi: 10.1189/jlb.0609391. [DOI] [PubMed] [Google Scholar]
- 22.Schmid BC, Rezniczek GA, Fabjani G, Yoneda T, Leodolter S, Zeillinger R. The neuronal guidance cue Slit2 induces targeted migration and may play a role in brain metastasis of breast cancer cells. Breast Cancer Res Treat. 2007;106:333–42. doi: 10.1007/s10549-007-9504-0. [DOI] [PubMed] [Google Scholar]
- 23.Mertsch S, Schmitz N, Jeibmann A, Geng JG, Paulus W, Senner V. Slit2 involvement in glioma cell migration is mediated by Robo1 receptor. J Neurooncol. 2008;87:1–7. doi: 10.1007/s11060-007-9484-2. [DOI] [PubMed] [Google Scholar]
- 24.Yuasa-Kawada J, Kinoshita-Kawada M, Rao Y, Wu JY. Deubiquitinating enzyme USP33/VDU1 is required for Slit signaling in inhibiting breast cancer cell migration. Proc Natl Acad Sci U S A. 2009;106:14530–5. doi: 10.1073/pnas.0801262106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang B, Xiao Y, Ding BB, Zhang N, Yuan X, Gui L, et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003;4:19–29. doi: 10.1016/s1535-6108(03)00164-8. [DOI] [PubMed] [Google Scholar]
- 26.Wang LJ, Zhao Y, Han B, Ma YG, Zhang J, Yang DM, et al. Targeting Slit–Roundabout signaling inhibits tumor angiogenesis in chemical-induced squamous cell carcinogenesis. Cancer Sci. 2008;99:510–7. doi: 10.1111/j.1349-7006.2007.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urbich C, Rössig L, Kaluza D, Potente M, Boeckel JN, Knau A, et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113:5669–79. doi: 10.1182/blood-2009-01-196485. [DOI] [PubMed] [Google Scholar]
- 28.Shen F, Liu X, Geng JG, Guo SW. Increased immunoreactivity to Slit/Robo1 in ovarian endometriomas. Am J Pathol. 2009;175:479–88. doi: 10.2353/ajpath.2009.090024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang B, Dietrich UM, Geng JG, Bicknell R, Esko JD, Wang L. Repulsive axon guidance molecule Slit3 is a novel angiogenic factor. Blood. 2009;114:4300–9. doi: 10.1182/blood-2008-12-193326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma S, Liu X, Geng JG, Guo SW. Increased SLIT immunoreactivity as a biomarker for recurrence in endometrial carcinoma. Am J Obstet Gynecol. 2010;202:68.e1–68.e11. doi: 10.1016/j.ajog.2009.07.040. [DOI] [PubMed] [Google Scholar]
- 31.Yang XM, Han HX, Sui F, Dai YM, Chen M, Geng JG. Slit-Robo signaling mediates lymphangiogenesis and promotes tumor lymphatic metastasis. Biochem Biophys Res Commun. 2010;396:571–7. doi: 10.1016/j.bbrc.2010.04.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu F, Previti ML, Nieman MT, Davis J, Schmaier AH, Van Nostrand WE. AbetaPP/APLP2 family of Kunitz serine proteinase inhibitors regulate cerebral thrombosis. J Neurosci. 2009;29:5666–70. doi: 10.1523/JNEUROSCI.0095-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31:497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- 34.Pottiez G, Flahaut C, Cecchelli R, Karamanos Y. Understanding the blood-brain barrier using gene and protein expression profiling technologies. Brain Res Rev. 2009;62:83–98. doi: 10.1016/j.brainresrev.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Terry S, Nie M, Matter K, Balda MS. Rho signaling and tight junction functions. Physiology (Bethesda) 2010;25:16–26. doi: 10.1152/physiol.00034.2009. [DOI] [PubMed] [Google Scholar]
- 36.Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–82. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu D, Hou J, Hu X, Wang X, Xiao Y, Mou Y, et al. Neuronal chemorepellent Slit2 inhibits vascular smooth muscle cell migration by suppressing small GTPase Rac1 activation. Circ Res. 2006;98:480–9. doi: 10.1161/01.RES.0000205764.85931.4b. [DOI] [PubMed] [Google Scholar]
- 38.Alam MI, Beg S, Samad A, Baboota S, Kohli K, Ali J, et al. Strategy for effective brain drug delivery. Eur J Pharm Sci. 2010;40:385–403. doi: 10.1016/j.ejps.2010.05.003. [DOI] [PubMed] [Google Scholar]