

Abstract
Endogenous adenosine is an essential protective agent against neural damage by various insults to the brain. However, the therapeutic potential of adenosine receptor-directed ligands for neuroprotection is offset by side effects in peripheral tissues and organs. An increase in adenosine receptor responsiveness to endogenous adenosine would enhance neuroprotection while avoiding the confounding effects of exogenous ligands. Here we report novel regulation of adenosine-evoked responses by a neural tissue-specific protein, neurabin. Neurabin attenuated adenosine A1 receptor (A1R) signaling by assembling a complex between the A1R and the regulator of G protein signaling 4 (RGS4), a protein known to turn off G protein signaling. Inactivation of the neurabin gene enhanced A1R signaling and promoted the protective effect of adenosine against excitotoxic seizure and neuronal death in mice. Furthermore, administration of a small molecule inhibitor of RGS4 significantly attenuated seizure severity in mice. Notably, the dose of kainate capable of inducing an ~50% rate of death in WT mice did not affect neurabin null mice or WT mice co-treated with an RGS4 inhibitor. The enhanced anti-seizure and neuroprotective effect achieved by disruption of the A1R/neurabin/RGS4 complex is elicited by the on-site and on-demand release of endogenous adenosine, and does not require administration of A1R ligands. These data identify neurabin-RGS4 as a novel tissue-selective regulatory mechanism for fine-tuning adenosine receptor function in the nervous system. Moreover, these findings implicate the A1R/neurabin/RGS4 complex as a valid therapeutic target for specifically manipulating the neuroprotective effects of endogenous adenosine.
Introduction
The severity of neural damage is a key factor determining the mortality and morbidity under pathological conditions such as hypoxia, ischemia and excitotoxin exposure. Extracellular adenosine released under these conditions acts as a potent protective agent to reduce neural damage and to confine the development and progression of accompanying seizures that result from hyperexcitability (Cunha, 2001; Dunwiddie and Masino, 2001; Stone, 2002; Fredholm et al., 2005). Among the four subtypes (A1, A2A, A2B and A3) of adenosine receptors, the A1 subtype (A1R) is the primary mediator of adenosine-evoked anticonvulsant and neuroprotective effects (Cunha, 2001; Dunwiddie and Masino, 2001; Stone, 2002; Fredholm et al., 2005; Sebastiao and Ribeiro, 2009). However, targeting the A1R for treatment of neurological diseases has been extremely challenging due to its broad expression in multiple peripheral tissues (Stone, 2002). Thus, using A1R-directed ligands likely evokes simultaneous peripheral actions that result in severe side effects and thereby confound therapeutic outcomes.
The A1R belongs to the G protein-coupled receptor (GPCR) superfamily. Cumulative evidence has demonstrated that the magnitude and duration of GPCR signaling is tightly regulated by non-G protein GPCR-interacting partners (reviewed in Lefkowitz, 2007; Ritter and Hall, 2009; Bockaert et al., 2010; Allen et al., 2008). The complexity of GPCR signaling regulation was first revealed by identification of a group of proteins called regulator of G protein signaling (RGS), which function as GTPase activating proteins to terminate G protein signaling (Ross and Wilkie, 2000; Hollinger and Hepler, 2002; Sjogren and Neubig, 2010). Association of RGS proteins with GPCRs, either directly or indirectly through adaptor proteins, represents the primary mechanism that determines the selectivity and effectiveness of RGS-mediated attenuation of GPCR signaling (Neitzel and Hepler, 2006; Neubig and Siderovski, 2002; Xie and Palmer, 2007). RGS4 belongs to the R4 subfamily of RGS proteins, and is highly expressed in brain and heart (Bansal et al., 2007). To date, involvement of RGS4 in adenosine-evoked signaling responses and neuroprotection remains largely unexplored.
Neurabin is a multi-domain scaffolding protein specifically expressed in neural tissues (Nakanishi et al., 1997; Burnett et al., 1998). Like the A1R (Reppert et al., 1991), a high level expression of neurabin has been found in cortex, hippocampus and cerebellum (Nakanishi et al., 1997; Burnett et al., 1998). Distribution of neurabin in both dendrites and presynaptic terminals (Muly et al., 2004) positions this protein as a potential regulator for both pre- and post-synaptic activities of neurons. Whether neurabin can directly interact with GPCRs to regulate receptor functions has not been determined.
Given the importance of non-G protein interacting partners in regulation of GPCR activity, we propose that modulating interactions between the A1R and neural-specific accessory proteins would provide an effective way to increase A1R responsiveness to endogenous adenosine. Thus, enhanced neuroprotection through the A1R can be achieved while avoiding confounding effect of exogenous A1R ligands. Here, we identify a direct interaction between the A1R and neurabin, which attenuates A1R-mediated responses both in vitro and in vivo by scaffolding interaction between the active A1R and RGS4. Significantly, endogenous adenosine-elicited neuroprotection against excitotoxic seizures through the A1R is enhanced in mice with a neurabin gene deficiency or when RGS4 is blocked by a small molecule inhibitor.
Materials and Methods
Reagents
Rat anti-HA monoclonal antibody was purchased from Roche Applied Science. Mouse HA.11 monoclonal antibody was from Covance. Mouse anti-Myc monoclonal antibody was from Clontech. Goat anti-RGS4 polyclonal antibody was from Santa Cruz Biotechnology. Rabbit anti-RGS4 polyclonal antibody was purchased from Abcam. Rabbit anti-A1R polyclonal antibody was from Pierce. Rabbit anti-GPF polyclonal antibody was from Santa Cruz Biotechnology. TNT in vitro translation kit was from Promega. The Biotrak cAMP competitive enzymeimmunoassay system and [35S]Methionine were purchased from GE Healthcare. CCG-4986 (methyl N-((4-chlorophenyl)sulfonyl)-4-nitrobenzenesulfinimidoate) was from the Chembridge screening collection. Fluoro-Jade B was purchased from Histo-chem Inc. Hematoxylin solution and Eosin were from ZYMED Laboratories of Invitrogen and Surgipath, respectively. The DeadEnd ™ Fluorometric TUNEL System was from Promega. Kainate was from Milestone Pharm Tech. All other chemicals were from Sigma-Aldrich or Fisher.
Animals
Neurabin−/− (Nrb−/−) mice (Allen et al., 2006) were backcrossed for 10 generations to the C57BL/6 background. Nrb+/− mice were then mated to generate WT and Nrb−/− littermates for behavioral experiments. 12-week-old male mice were used for in vivo studies. All mice were maintained at the University of Alabama at Birmingham Animal Care Facility in accordance with procedures of the Animal Welfare Act and the 1989 Amendments to this Act.
Cells and transfection
CosM6 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% Fetal Bovine Serum (FBS, Atlanta Biologicals) and 100 unit/ml of penicillin and 10 μg/ml streptomycin. CHO-K1 cells were cultured in DMEM/F-12 (Invitrogen) with 10% FBS, penicillin and streptomycin, and 2 mM glutamine. Cells were transfected with Lipofectamine 2000 (Invitrogen).
Plasmid construction
cDNA encoding neurabin aa 146–330 and 331–453 were amplified by PCR and cloned into GST fusion vector pGEX4T (GE Healthcare) through EcoRI and XhoI sites to obtain pGEX4T-Nrb146-330 and pGEX4T-Nrb331-453. cDNA encoding A1R aa 202–326, 202–292 and 236–326 were amplified by PCR and cloned into pCMVTNT vector (Promega) through XhoI and KpnI sites to obtain pCMVTNT-A1R202-326, pCMVTNT-A1R202-292, and pCMVTNT-A1R236-326.
In vitro GST pull-down assay
Preparation of GST fusion proteins, synthesis of [35S]-labeled in vitro translated probes, and pull-down assays were performed as described previously (Wang and Limbird, 2002).
Co-immunoisolation assay
CosM6 cells were co-transfected with cDNA encoding HA-A1R together with cDNA encoding Myc-Nrb or Myc-Sp or RGS4, or both Myc-Nrb and RGS4. At 48 hrs post-transfection, cells were stimulated with 1 μM R-PIA for 5 min. Cells were lysed in immunoisolation buffer containing 20 mM HEPES (pH 7.4), 2 mM EDTA, 5 mM NaF, 0.5% NP-40, 10% glycerol, and protease inhibitors. Rat anti-HA antibody and goat anti-RGS4 antibody were used to immunoisolate HA-A1R and RGS4, respectively, following a procedure described previously (Xu et al., 2008). Immunoisolates were separated by SDS-PAGE and analyzed by Western blot.
For co-immunoisolation of endogenous neurabin and A1R from brain lysate, adult male mice aged 12 weeks were injected with saline or R-PIA (1mg/kg) and sacrificed 30 min post-injection. Whole brain was removed by dissection and homogenized on ice in lysis and immunoisolation buffer as described above. The detergent extract was then centrifuged at 100,000 × g at 4°C for 30 min, and the resulting supernatant was subjected to immunoprecipitation assays with a rabbit polyclonal antibody against either the A1R or GFP (as a negative control).
Antisense knock-down of RGS4 and RT-PCR
CHO-K1 cells were transfected with the RGS4 antisense oligo (5′-GAATCCAGGTTCACC-3′) and scrambled oligo (5′-GATCGA ACT CGC TCA-3′) (Witt-Enderby et al., 2004). 48 hrs post transfection, total RNA was prepared using TRIZOL reagent (Invitrogen). RT-PCR was performed with Qiagen™ One-Step RT-PCR kit (Qiagen) following the manufacturer’s instructions. PCR primers for application of RGS4 were 5′-GCTTTCAAAGCTTTCCTGAAGT-3 ′ and 5′-TGTGAGAATTAGGCACACTG-3′, the product of which covers partial sequences of exon3 and exon4.
Intact cell surface ELISA
Cell surface HA-A1R expression in transfected CHO-K1 cells was examined by the cell-surface ELISA methods as described previously (Brady et al., 2003).
Radioligand binding assays
Crude membrane fraction of brains from WT and Nrb−/− mice was prepared as described previously (Maemoto et al., 1997). In brief, brain homogenates in 0.32 M sucrose were centrifuged at 1000×g for 10 min, and the resulting supernatants were centrifuged again at 17,000×g for 20 min. The pellet was then resuspended in a buffer containing 10 mM HEPES, 10 mM EDTA (pH7.4) with protease inhibitors and aspirated/expelled through a 20-gauge needle 10 times. After centrifugation at 40,000×g for 15 min, the membrane pellet was washed twice in the HE buffer (10 mM HEPES, 1 mM EDTA, pH 7.4, with protease inhibitors). Membrane pellet was then resuspended in the HE buffer and protein concentrations were determined with the Bio-Rad Dc Protein Assay kit.
Saturation binding was performed to assess the A1R receptor density in brains of WT and Nrb−/− mice using a method described earlier (Scholl and Wells, 2000). Membrane proteins (150 μg/reaction) were incubated with increasing concentrations of [3H]DPCPX in a total volume of 250 μl. Prior to the binding assay, membrane preparations were incubated with adenosine deaminase (ADA, 5 unit/mg protein) at 25°C for 60 min to remove endogenous adenosine.
Competition binding was performed as described previously (Scholl and Wells, 2000) to evaluate the intrinsic affinity of agonist (R-PIA) and antagonist (theophilline) at the A1R in brains of WT and Nrb−/− mice. 0.5 nM [3H]DPCPX was used as the radioligand. Assays were performed in the presence of 100 μM 5′-guanylimidodiphosphate (Gpp(NH)p) to eliminate effects of G protein regulation of receptor affinity for agonist. All binding data were analyzed using GraphPad Prism software (GraphPad Software Inc).
Primary culture of cortical neurons
Primary culture of cortical neurons from newborn mouse pups was performed following a protocol described earlier (Cottingham et al., 2011). In brief, newborn mouse cortices were dissected out, minced, and digested with papain for 15 min at 37°C. Neurons were dissociated by trituration. Dissociated cortical neurons were plated in Neurobasal™-A medium supplemented with 5% FBS, 2% B27, 2% glutamax, 0.2% gentamycin at 5×104 cells/well in 24-well plates. From the second day, cells were fed with Neurobasal™-A medium supplemented with 2% B27, 2% glutamax, 0.2% gentamycin, which were changed every 4–6 days.
Measurement of cAMP levels
CHO-K1 cells were co-transfected with cDNA encoding HA-A1R together with or without cDNA encoding Myc-Nrb or Myc-Sp. For RGS4 knock-down experiments, cells were also co-transfected with antisense or scrambled oligos. Cells were then seeded into 96-well plates (4×104 cells/well). 48 hrs post-transfection, cells were stimulated with 10 μM forskolin with or without 1 μM R-PIA for 20 minutes at 37 °C. cAMP levels in cells were measured using the Amersham cAMP Biotrak Enzymeimmunoassay (EIA) system following the manufacturer’s instructions. To examine the effect of RGS4 inhibition on cAMP production, cells were pretreated with 30 μM CCG4986 for 10 min, and then stimulated with forskolin and R-PIA in the presence of CCG4986.
cAMP levels were also measured in cortical neurons cultured for 9–11 days in vitro following a method described previously (Murphy et al., 1991) with slight modifications. Prior to agonist stimulation, neurons were incubated with ADA (2 unit/ml) for 1 hr to remove adenosine in medium. Neurons were then pre-treated with 1μM R-PIA or vehicle for 10 min, followed by co-treatment with 10 μM forskolin (to activate adenylate cyclase) and 25 μM Ro-20–1724 (to inhibit cAMP-specific phosphodiesterase) in the presence or absence of 1 μM R-PIA for another 10 min. cAMP levels were measured using the EIA system as described above. To examine the effect of RGS4 inhibition, 30 μM CCG4986 was added to cells together with forskolin and Ro-20–1724 in the presence or absence of R-PIA.
Immunofluorescence staining of cultured neurons
Cortical neurons cultured for 11–13 days in vitro were used for immunofluorescence studies. Prior to treatment, cells were washed in Neurobasal-A medium containing 0.5 unit/ml adenosine deminase for 30min. Cells then were treated with 1 μM R-PIA or solvent for 5 min, and fixed with 3% paraformaldehyde and 3% sucrose following treatment. Immunofluorescence staining was performed as described previously (Richman et al., 2001) using a Myc antibody or RGS4 antibody. Fluorescent images were taken under a confocal Laser scanning microscope, and analyzed with MetaMorph software. Measurement of sedative response. 12 week-old WT and Nrb−/− mice were injected intraperitoneally with increasing doses of R-PIA or UK14,304. Sedative response was assessed by rotarod latency as described previously (Wang et al., 2004).
Induction and evaluation of seizure
12 week-old WT and Nrb−/− male mice were given one of the following treatments by i.p. injection: (1) kainate at 20 or 25 mg/kg (dissolved in saline); (2) kainate (25 mg/kg in saline) together with 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX, 0.5mg/kg in saline); (3) with DMSO; (4) with CCG4986 (20 mg/kg in DMSO); (5) with both DMSO and DPCPX (0.5 mg/kg in saline); (6) with both CCG4986 (20 mg/kg in DMSO) and DPCPX (0.5 mg/kg in saline). Mice were observed for 2 hrs after kainate injection by trained observers blind to genotype or treatment. The severity of seizure behaviors was scored 0 to 7 with 0 representing normal behavior and 7 representing death according to reference descriptions (Schauwecker and Steward, 1997; Yang et al., 1997). Seizure scores were recorded every 20 min for 2 hrs.
Assessment of cell death in hippocampal slices
Seven days after initial kainate exposure, mice were deeply anaesthetized with isoflurane and perfused with 4% paraformaldehyde in phosphate buffered solution (pH 7.4). Hippocampal slices (7 μm) were stained with Hematoxylin & Eosin. Neuronal death was detected by Fluoro-Jade B staining and by the DeadEnd™ Fluorometric TUNEL System (Promega) following the manufacturer’s instructions.
Electroencephalography (EEG) Recordings
Fourteen weeks after kainate injection, mice were anesthetized with i.p. injection of a ketamine/xylazine mixture. Electrodes were placed onto the dura mater bilaterally. After an overnight recovery, EEG activity of freely moving mice was monitored and recorded using a DSI NeuroScore CNS Analysis Software (Data Sciences International) for 24 hrs.
Statistical Analysis
All quantitative data were analyzed using GraphPad Prism software (GraphPad Software Inc). Seizure scores were analyzed by two-way ANOVA with Bonferroni post tests to compare replicate means. Genotype or treatment was considered as between subject factor and time was considered as within subject factor. Other data were analyzed with Student's t-tests (2-tail, unpaired). p< 0.05 was considered statistically significant.
Results
Neurabin interacts with the adenosine A1R in an agonist-regulated manner
In our effort to search for A1R interacting proteins, we identified a direct interaction between the A1R and neurabin using a GST pull-down assay (Fig. 1A). The neurabin sequences that interacted with the A1R were mapped to aa 331–453 (Fig. 1B), within the region least conserved between neurabin and its close homolog, spinophilin (Fig. 2A). While both the third intracellular (3i) loop and C-terminal tail of A1R appeared to interact with neurabin, the 3i loop was required for the maximal level of interaction (Fig. 1C). Interaction between neurabin and the A1R in intact cells was markedly enhanced with stimulation by the A1R agonist R-PIA for 5 (Fig. 2B left) and 30 min (Fig. 2C), indicating that the A1R-neurabin interaction is regulated by receptor activation. Thus, a ~3 fold increase in the amount of neurabin co-immunoisolated with HA-tagged A1R was measured in cells stimulated with R-PIA compared to cells receiving no stimulation (Fig. 2D). Moreover, systemic injection of R-PIA enhanced the amount of endogenous neurabin co-immunoisolated with the A1R in brain (Fig. 2E). Consistent with the fact that neurabin sequences interacting with the A1R are distinct from its homolog, spinophilin, an interaction between the A1R and spinophilin was not detected in cells co-expressing these two proteins (Fig. 2B right).
Figure 1.

The A1R directly interacts with neurabin. A, Direct interaction between A1R 3iloop and C-tail and neurabin aa 146–453. GST or GST-Nrb146-453 was incubated with in vitro translated [35S]-labeled probe as indicated. B, Neurabin aa 331–453 is involved in the direct interaction with the A1R. GST or GST-fused neurabin constructs with [35S]-labeled probe containing A1R 3iloop and C-tail. C, The 3iloop of A1R is required for a full level of interaction with neurabin. GST or GST-Nrb146-453 was incubated with [35S]-labeled probe containing A1R 3iloop, C-tail, or both, as indicated. Comparable amounts of GST fusion proteins in each reaction were confirmed by coomassie staining. Free probe represents 1/20 of the input in each reaction.
Figure 2.

Agonist exposure specifically promotes interaction of the A1R with neurabin but not its homolog spinophilin in intact cells. A, Sequence homology between domains in neurabin and spinophilin. B, Interaction of the A1R with neurabin (left) or spinophilin (right) in cells treated with or without A1R agonist. Cells expressing neurabin alone or co-expressing HA-A1R with Myc-Nrb or Myc-Sp were stimulated with or without 1μM R-PIA for 5 min, and cell lysates were subjected to immunoisolation assay using an antibody against HA. C, The A1R-neurabin interaction detected with prolonged R-PIA treatment for 30 min. Cells co-expressing HA-A1R and Myc-Nrb were stimulated with 1 μM R-PIA or vehicle for 30 min, and cell lysates were subjected to immunoisolation assay. D, Quantitation of A1R-neurabin interaction representing 3–6 independent co-immunoisolation experiments. Data are expressed as fold change of neurabin in complex with the A1R over no stimulation control (defined as 1.0 fold). Value = mean ± SEM. *, p<0.05, R-PIA stimulated vs. control. E, Endogenous interaction between neurabin and A1R in mouse brain. Mice were given i.p. injections of saline or R-PIA (1mg/kg), with whole brains isolated and homogenized 30 min post-injection. Detergent-solubilized fractions were then prepared and subjected to co-immunoisolation using equal concentrations of an antibody against either GFP (negative control) or the A1R. Representative blots from three independent experiments are shown. A degradation product of the endogenous neurabin was also detected.
Neurabin attenuates A1R-mediated responses in vitro and in vivo
To determine effects of the adenosine-evoked A1R-neurabin interaction on A1R signaling, we measured A1R-mediated inhibition of cAMP production stimulated by R-PIA. In CHO cells expressing the A1R, R-PIA stimulation caused inhibition of forskolin-induced cAMP production (Fig. 3A). When exogenous neurabin was expressed in CHO cells (which do not express endogenous neurabin), A1R-mediated inhibition of cAMP production was abolished (Fig. 3A) while cell surface expression of the A1R was not changed (Fig. 3B). These data suggest that neurabin interaction with the A1R leads to suppression of receptor signaling. Consistent with our inability to detect A1R-spinophilin interactions, overexpression of spinophilin had no effect on A1R-mediated inhibition of cAMP production (Fig. 3A).
Figure 3.

Neurabin attenuates A1R-mediated G protein signaling in cells. A, Neurabin expression attenuates A1R-mediated inhibition of cAMP production. Cells expressing HA-A1R alone or in combination with either neurabin or spinophilin were treated with 10 μM forskolin alone or forskolin plus 1 μM R-PIA. Surface receptor density in each experimental group was comparable, as verified by intact cell ELISA using an anti-HA antibody. n=11 for cells expressing the A1R alone; n=8 for cells expressing the A1R and neurabin; n=5 for cells expressing the A1R and spinophilin. Data are expressed as fold change in cAMP production over forskolin alone control (defined as 1.0 fold). *, p<0.05, R-PIA stimulated vs. control. B, R-PIA induces similar levels of receptor internalization from the cell surface in cells either with or without exogenous expression of neurabin or spinophilin. Surface receptor density in cells described in (A) was examined by intact cell ELISA using an anti-HA antibody. *, p<0.05 when comparing cell surface A1R in R-PIA treated cells vs. nontreated controls.
Since the A1R-neurabin interaction attenuates A1R signaling, A1R-mediated in vivo responses should be enhanced in the absence of neurabin expression. To test this prediction, we assessed the ability of the A1R agonist R-PIA to evoke sedation mediated by the central A1R in wildtype (WT) versus neurabin deficient (Nrb−/−) mice (Allen et al., 2006). Nrb−/− mice were more sensitive to R-PIA-evoked sedation, as manifest by a leftward shift of the dose-response curve (Fig. 4A). These data suggest that when neurabin is endogenously expressed, it negatively regulates A1R-mediated responses in the brain, and this suppression is relieved in Nrb−/− mice. We next measured the sedation response in these mutant mice evoked by the central α2 adrenergic receptor (AR), which does not interact with neurabin (Richman et al., 2001). As expected, the dose-response curve for sedation evoked by the α2AR agonist, UK 14304, was indistinguishable in WT and Nrb−/− mice (Fig. 4B), suggesting that perturbation of A1R responses in Nrb−/− mice is not generalized or nonspecific in nature.
Figure 4.

Neurabin attenuates A1R-mediated responses in vivo. A, Neurabin-deficient mice are more sensitive to R-PIA-elicited sedation, as assessed by rotarod latency. The EC50 values for sedation in Nrb−/− (n=8) and corresponding WT littermates (n=5) are 1.29 and 2.64 mg/kg, respectively. *, p<0.05; **, p<0.01. B, The α2AR-agonist, UK14,304, induces comparable sedation responses in both WT and Nrb−/− mice as assessed by rotarod latency. n=5 for both WT and Nrb−/− mice. Error bars represent mean±SEM. C, The density of A1Rs is indistinguishable in brain membrane preparations obtained from WT and Nrb−/− mice as measured by saturation binding assays. Values represent mean ± SEM; n=3 for each genotype. The Bmax values predicted by nonlinear regression fit for the A1R in WT and Nrb−/− brain homogenates are 1547 ± 90 and 1515 ± 76 fmole/mg protein, respectively. D, The intrinsic affinity of the A1R for R-PIA in brains of WT mice is similar to that in brains of Nrb−/− mice. Competition binding assays were performed in the presence of 100 μM Gpp(NHp). Binding of the [3H]DPCPX radioligand is given as a percentage of binding without competitors. Values represent mean ± SEM; n=3 for each genotype. The IC50 values predicted for R-PIA in competition for radioligand binding in WT and Nrb−/− brain homogenates are 0.41±0.11 and 0.29±0.12 μM, respectively.
The observed changes in sensitivity to A1R-evoked sedation in Nrb−/− mice may result from alterations in A1R expression density and/or intrinsic affinity of the receptor for agonists, caused by loss of neurabin in these mice. However, saturation binding analysis revealed that A1R density in the brains of WT and Nrb−/− mice was indistinguishable, as was the Kd value for binding of the radiolabeled A1R antagonist, [3H]DPCPX (Fig. 4C). Additionally, the intrinsic affinity of the A1R for its agonist R-PIA was nearly identical in brain membrane preparations from WT and Nrb−/−, as indicated by the IC50 values for competition of R-PIA with [3H]DPCPX (Fig. 4D). Taken together, our findings indicate that interactions between neurabin and the A1R lead to attenuation of A1R function in vivo without affecting A1R receptor density or its intrinsic affinity for agonists.
Functional involvement of RGS4 in neurabin-mediated inhibition of A1R signaling
Multiple molecular mechanisms could account for the ability of neurabin to suppress A1R cellular signaling through interactions with the receptor. For example, since GPCR signaling is attenuated by receptor phosphorylation and internalization (reviewed in Ferguson, 2001; Reiter and Lefkowitz, 2006; Hanyaloglu and von, 2008), neurabin interactions could enhance these processes. However, we failed to detect significant A1R phosphorylation in cells stimulated with R-PIA, even though significant agonist-stimulated α2AAR phosphorylation was detectable under the same experimental conditions (data not shown). In addition, we did not find that overexpression of neurabin altered the extent of surface receptor internalization following R-PIA stimulation (Fig. 3B). These data suggest that neurabin attenuates A1R signaling via a mechanism that is independent of both receptor phosphorylation and internalization.
Mammalian RGS4 has been shown to accelerate GTPase activity of the Gi subfamily of G proteins, thus attenuating Gi-coupled signal transduction (Berman et al., 1996; Huang et al., 1997). We therefore tested the involvement of RGS4 in neurabin-mediated attenuation of A1R signaling by examining A1R-mediated inhibition of cAMP production in cells with altered RGS4 activity or blocked RGS4 expression. As shown above (Fig. 3A), R-PIA stimulation led to inhibition of cAMP production, and heterologous expression of neurabin diminished this A1R response (Fig. 5A). However, when neurabin-expressing cells were treated with an RGS4 inhibitor, CCG4986 (Roman et al., 2007; Roman et al., 2010), neurabin-dependent loss of A1R-mediated inhibition of cAMP production was blocked (Fig. 5A). Suppression of RGS4 expression by antisense oligonucleotides (Fig. 5B) or small interfering RNA (Fig. 5C) also diminished neurabin-mediated attenuation of A1R signaling. These data suggest that RGS4 is functionally involved in neurabin-mediated attenuation of A1R signaling in cells. Intriguingly, in cells without exogenous neurabin expression, the effect of RGS4 inhibition on cAMP production is negligible (Fig. 5A), indicating that neurabin is an essential component for RGS4-induced attenuation of A1R signaling.
Figure 5.

Involvement of RGS4 in neurabin-mediated attenuation of A1R signaling. A, RGS4 inhibitor CCG4986 reverses neurabin-mediated attenuation of A1R signaling. CHO cells expressing HA-A1R alone or in combination with neurabin were treated with forskolin alone, forskolin plus R-PIA, or forskolin plus R-PIA and CCG4986 (30 μM). Data are from 4 independent experiments with cells expressing HA-A1R alone and from 10 independent experiments with cells expressing HA-A1R with neurabin, and expressed as fold change in cAMP level relative to forskolin alone (defined as 1.0 fold). Values = mean ± SEM. *, p<0.05, indicated treatment vs. forskolin alone. B, Knock-down of RGS4 expression by antisense oligo diminishes neurabin-mediated attenuation of A1R signaling. CHO cells were co-transfected with HA-A1R and neurabin, and together with antisense oligo against RGS4 mRNA or scrambled oligo control. Reduction in RGS4 mRNA level was confirmed by RT-PCR. Relative cAMP level is presented as fold change vs. forskolin alone (defined as 1.0 fold). *, p<0.05, R-PIA stimulated vs. forskolin alone; n=8 for each condition. C, Knock-down of RGS4 expression by siRNA also abolishes effect of neurabin in attenuation of A1R signaling. CHO cells were co-transfected with HA-A1R and neurabin, and together with siRNA against RGS4 or control siRNA. Reduction in RGS4 mRNA level was confirmed by RT-PCR. *, p<0.05, R-PIA stimulated vs. forskolin alone; n=6–8 for each condition. D, Attenuation of A1R signaling by neurabin and RGS4 in native neurons. Primary cortical neurons cultured from WT and Nrb−/−mice were treated as indicated. Data were expressed as fold change in cAMP level relative to forskolin alone (defined as 1.0 fold). n=7–8 for each condition. *, p<0.05, when compared to the relative cAMP level in neurons of either genotype treated with forskolin alone. #, p<0.05, when compared to the relative cAMP level in WT neurons treated with R-PIA and forskolin.
We next sought to verify that this observed regulation of A1R signaling by neurabin and RGS4 occurs in native neurons. In WT neurons, activation of endogenously expressed A1Rs by R-PIA resulted in ~30% inhibition of cAMP production, whereas the same concentration of R-PIA led to a nearly 50% inhibition of cAMP production in neurons derived from Nrb−/− mice (Fig. 5D). Blockade of RGS4 by CCG4986 also significantly enhanced R-PIA-mediated inhibition of cAMP production in WT neurons (Fig. 5D). These data strongly support the role of neurabin and RGS4 in attenuating A1R signaling in native settings. Furthermore, inhibition of RGS4 by CCG4986 had no effect on R-PIA-mediated inhibition of cAMP production in neurons derived from Nrb−/− mice (Fig. 5D), confirming that neurabin expression is required for RGS4 to induce attenuation of A1R signaling in the endogenous system.
Neurabin scaffolds complex formation between the A1R and RGS4 and promotes membrane translocation of RGS4
Though RGS proteins can interact directly with GPCRs (Neitzel and Hepler, 2006) to confer their selective regulation of GPCR signaling, we failed to detect direct interactions between the A1R and RGS4 in cells expressing these two proteins (Fig. 6A left). On the other hand, in cells co-expressing neurabin with the A1R and RGS4, both neurabin and RGS4 were co-immunoisolated with the A1R following R-PIA stimulation (Fig. 6A right). Reciprocal immunoisolations using an antibody against RGS4 also pulled down the A1R and neurabin in an agonist-dependent manner (Fig. 6B). These data are consistent with the interpretation that neurabin serves as a scaffold bridging functional interactions between the A1R and RGS4 in cells, and that agonist stimulation promotes A1R/neurabin/RGS4 complex formation. This notion also explains our findings that the RGS inhibitor, CCG4986, does not alter R-PIA-elicited inhibition of cAMP production in cells unless neurabin is also expressed (Fig. 5A). In addition, R-PIA stimulation did not enhance the interaction of neurabin with RGS2 (Fig. 6C), another member of the R4 family of RGS proteins, demonstrating the selectivity of the neurabin-facilitated A1R-RGS interactions for RGS4. Taken together, our data suggest that the A1R, neurabin, and RGS4 form a complex following agonist stimulation to attenuate A1R signaling.
Figure 6.

Neurabin scaffolds formation of the A1R-RGS4 complex. A, The A1R-RGS4 interaction is dependent upon agonist stimulation and the presence of neurabin. Cells co-expressing HA-A1R and RGS4, together with or without Myc-Nrb, were incubated in the presence or absence of R-PIA for 5 min. Cell lysates were subjected to the immunoisolation assay and analyzed using an anti-HA antibody. Representative blots of at least three independent experiments are shown. B, RGS4 forms a complex with neurabin and the A1R upon agonist stimulation. Cells co-expressing HA-A1R, RGS4 and Myc-Nrb were incubated with or without R-PIA for 5 min. Cell lysates were subjected to the immunoisolation assay using an anti-RGS4 antibody and analyzed by Western blot. Representative blots of multiple independent experiments are shown. C, R-PIA stimulation fails to promote interaction between neurabin and RGS2 in cells. Cells co-expressing A1R, Myc-Nrb and RGS2 were stimulated with R-PIA for 5 min. Cell lysates were subjected to immunoisolation assay using an anti-RGS2 antibody and analyzed by Western blot. Representative blots of multiple independent experiments are shown.
Plasma membrane localization of RGS4 is critical for its ability to inhibit G protein signaling (Srinivasa et al., 1998). While RGS4 is largely localized in the cytoplasm of neuronal cells (Chatterjee and Fisher, 2000), neurabin is mainly localized to the plasma membrane via its actin-binding domain (Oliver et al., 2002). We postulated that neurabin recruits RGS4 to the plasma membrane in order to turn off G protein signaling initiated by the A1R. To test this possibility, we examined RGS4 localization in primary cortical neurons cultured from WT or Nrb−/− mouse brains. In WT neurons, RGS4, both when exogenously transfected (Fig. 7A) and when endogenously expressed (Fig. 7B), was mainly detected in cytoplasm of the majority of cells, and R-PIA stimulation caused translocation of RGS4 to the plasma membrane. By contrast, in Nrb−/− neurons, R-PIA stimulation failed to induce efficient RGS4 translocation (Fig. 7, A&B). These data suggest that neurabin serves as a scaffold to recruit RGS4 to the plasma membrane following A1R activation.
Figure 7.

RGS4 is enriched at the plasma membrane upon A1R activation in neurons derived from WT but not Nrb−/−, mice. A, Primary cortical neurons cultured from WT or Nrb−/− mice were transfected with the Myc-RGS4 plasmid. Representative images of immunostaining with anti-Myc antibody in the presence or absence of R-PIA stimulation. B, representative images of distribution of endogenous RGS4 in primary neurons. C, Quantitation of relative RGS4 intensity (in B) at the plasma membrane over that in the cytoplasm. *, p<0.05, R-PIA treated vs. control. Error bars represent mean ± SEM. 30–40 neurons from three independent experiments were quantified for each experimental condition.
A1R-mediated anti-seizure effects of endogenous adenosine are enhanced in neurabin deficient mice
The A1R mediates endogenous adenosine-elicited neuroprotection in response to various insults to the brain (Haas and Selbach, 2000; Cunha, 2001; Dunwiddie and Masino, 2001; Stone, 2002). In order to determine the impact of the A1R-neurabin interaction on the neuroprotective effect of adenosine, we used a kainate-induced seizure model, in which adenosine is released in the brain upon kainate insult and serves as an endogenous anticonvulsant via activation of the A1R (Carswell et al., 1997; Berman et al., 2000). Indeed, we found that blockade of the A1R by the subtype-selective A1R antagonist DPCPX enhanced the severity and progression of kainate-induced seizure activity in WT mice (Fig. 8A), demonstrating a role for the A1R in dampening seizure severity in our model. By contrast, kainate-induced seizures were significantly attenuated in extent and duration in Nrb−/− mice compared to WT littermates (Fig. 8, A&C), consistent with our observation that sensitivity of endogenous A1R functions is enhanced in Nrb−/− mice (Fig. 4A). While kainate administration at 25mg/kg resulted in a nearly 50% mortality rate in WT mice, all Nrb−/− mice survived the same insult (Fig. 8B). Blockade of A1Rs by DPCPX increased the mortality rate to 100% in both WT and Nrb−/− mice (Fig. 8B), further emphasizing the importance of the A1R and its regulation by neurabin in this process.
Figure 8.

Nrb−/− mice exhibit enhanced A1R-mediated anticonvulsant effects against kainate-induced seizures. A, Seizure severity in response to kainate is attenuated in Nrb−/− mice. Seizure activity over time following kainate injection (25mg/kg, i.p., n=13 for WT and n=11 for Nrb−/−) or kainate coinjection with the A1R antagonist DPCPX (0.5mg/kg) (n=9 for WT, n=8 for Nrb−/−) was scored as described in Methods with a higher score indicating greater seizure severity. *, p<0.05; **, p<0.01, ***, p<0.001, Nrb−/− vs. WT mice. #, p<0.05; ##, p<0.01, WT mice treated with kainate alone vs. kainate plus DPCPX. B, Kainate-induced lethality is reduced in Nrb−/−mice. Data are expressed as percentage of death in WT (n= 13) and Nrb−/− (n=11) mice caused by administration of kainate (25mg/kg) alone or with DPCPX. C, The percentage of mice with generalized seizure is reduced in Nrb−/− mice. The percentage of mice with generalized seizure within 120 min post-kainate administration at 20mg/kg (WT n=8, Nrb−/− n=8) or 25mg/kg (WT n=13 and Nrb−/− n=11) was calculated for each genotype.
Electroencephalography (EEG) revealed that 80% (4 out of 5) of the WT mice that did survive the initial kainate insult developed spontaneous non-convulsive seizures approximately three months after injection, whereas only 14% (1 out of 7) of Nrb−/− mice developed spontaneous seizures under the same conditions (Fig. 9A). In addition, the single Nrb−/− mouse with spontaneous seizure activity exhibited much lower seizure magnitude as measured by EEG compared with WT (Fig. 9, B&C). Importantly, blockade of the A1R during kainate injections by co-injection of the A1R antagonist DPCPX eliminated the observed differences in severity and progression of kainate-induced seizure and death between WT and Nrb−/− mice (Fig. 8, A&B), confirming that reduced seizure response to kainate in Nrb−/− mice depends on activation of the A1R. Taken together, these findings strongly indicate an important role for A1R-neurabin interactions in endogenous adenosine-evoked suppression of seizure progression in vivo, and support the idea that disruption of these interactions, as in our Nrb−/− mice, enhances the anticonvulsant effect that protects against kainate-induced seizure activity.
Figure 9.

Fewer Nrb−/− mice than WT mice develop spontaneous seizures approximately three months after initial kainate insult. A, Quantitation of EEG recordings made about three months after the initial kainate injection at 25mg/kg in WT and Nrb−/− mice. EEG recordings were performed in mice for 24 hr and the percentage of time in which a spike train was detected (indicating seizure activity in the brain) was calculated for each mouse (represented by single dots). n=5 for WT and n=7 for Nrb−/− mice. *, p<0.05. Error bars represent mean ± SEM. B&C, Representative EEG recording traces in one WT (B) and one Nrb−/− (C) mouse with spontaneous seizures show a much less EEG magnitude in Nrb−/− mice than that observed in WT mice with spontaneous seizures. A representative 10 min period with spike train detected is shown for each mouse.
Neurons in Nrb−/− mice are protected from kainate-induced cell death
To further evaluate the role of neurabin in regulating adenosine-evoked neuroprotection at the cellular level, we examined neuronal death in the brains of WT and Nrb−/− mice that reached similar seizure scores. Severe cell death was observed in the hippocampus of WT mice 7 days after kainate-induced seizure (Fig. 10, B, E, &H). However, in hippocampal slices prepared from Nrb−/− mice exposed to similar kainate injections, no significant cell death was detected (Fig. 10, C, F, & I). These morphological findings of neuronal cell survival paralleled seizure and survival findings shown above (Fig. 8), demonstrating that the adenosine-evoked neuroprotective effect against excitotoxic cell death is greatly enhanced when neurabin is not present.
Figure 10.

Kainate-induced cell death in the hippocampus is dramatically reduced in Nrb−/− mice. Hippocampal slices were collected from WT and Nrb−/− mice 7 days after indicated treatment and stained with hematoxylin and eosin (HE, A-C). Cell death was detected by Fluoro-Jade B (FJB) staining (D-F) and Tunel assay (G-I). Significant neuronal cell death was detected in the hippocampus of WT mice that received kainate (KA) administration, but not in the hippocampus of Nrb−/− mice that underwent the same treatment.
Inhibition of RGS4 enhances the anti-seizure and neuroprotective effects of adenosine in WT but not Nrb−/− mice
The in vitro mechanistic studies suggested that neurabin attenuates A1R-mediated signaling by recruiting RGS4 following A1R activation. Therefore, we predicted that inhibition of RGS4 would enhance A1R-evoked neuroprotection in vivo. To test this, we evaluated A1R-mediated suppression of kainate-induced seizures by endogenous adenosine in WT mice with or without co-injection of the RGS4 inhibitor, CCG4986. Administration of CCG4986 alone did not cause gross behavioral changes (data not shown), consistent with a previous report that RGS4 knockout mice lack apparent behavioral defects (Grillet et al., 2005). Significantly, we observed that co-injection of CCG4986 markedly reduced seizure severity caused by kainate (Fig. 11, A&B). Moreover, co-injection of CCG and kainate resulted in no mortality, compared with a mortality rate of nearly 50% with kainate insult alone (Fig. 11C). Importantly, co-injection of the A1R antagonist DPCPX eliminated the CCG4986-dependent attenuation of seizure severity (Fig. 11, A-C), indicating that activation of the A1R is required for the impact of RGS4 inhibition on kainate-evoked seizure activity. These data demonstrate that inhibition of RGS4 enhances endogenous adenosine-evoked anticonvulsant effects through the A1R.
Figure 11.

Inhibition of RGS4 enhances the A1R-mediated anti-seizure effect in WT, but not in Nrb−/− mice. A, The RGS4 inhibitor CCG4986 attenuates kainate-induced seizure in WT mice. Seizure severity was scored over time post injection with kainate (25mg/kg) in combination with one the following: DMSO vehicle (n=11); CCG4986 (20mg/kg) (n=12); DPCPX (0.5mg/kg) (n=8); CCG4986 and DPCPX (n=10). ***, p<0.001, kainate plus CCG4986 vs. kainate alone. ##, p<0.01, ###, p<0.001, kainate alone vs. kainate plus DPCPX. B, Co-injection of CCG4986 reduces the percentage of WT mice exhibiting generalized seizure within 120 min post kainate administration. WT mice treated with kainate alone or in combination with CCG4986 or DPCPX, or both were compared. C, Co-injection of CCG4986 reduces kainate-induced lethality in WT mice. Data are expressed as a percentage of death in WT mice treated with kainate alone or in combination with CCG4986 or DPCPX, or both. D, The effect of RGS4 inhibition on kainate-induced seizure is negligible in Nrb−/− mice. Seizure severity was scored over time post injection with kainate (25mg/kg) in combination with DMSO vehicle (n=8) or CCG4986 (20mg/kg) (n=8). Error bars represent mean ± SEM.
Neurabin expression appears to be required for RGS4 to induce attenuation of A1R signaling (Fig. 5, A&D). Therefore, we expected that inhibition of RGS4 would have no effect on A1R-mediated responses in Nrb−/− mice. Indeed, co-injection of CCG4986 in Nrb−/− mice did not change seizure behavior induced by kainate in these mutant mice (Fig. 11D). Taken together with data shown above (Fig. 11, A-C), these results strongly support the interpretation that the neurabin-RGS4 complex is engaged upon agonist activation of the A1R and negatively regulates A1R activity in vivo; disturbance of this complex by one of several complementary strategies enhances the ability of endogenous adenosine to protect against excitotoxin-evoked seizures. In addition, kainate treatment did not significantly affect brain expression levels of either RGS4 or neurabin in WT mice (data not shown), indicating that the A1R/neurabin/RGS4 complex formation is not simply being modulated by altered interacting partner ratios during seizure.
Kainate-induced neuronal cell death was further evaluated in the brains of WT mice co-injected with the RGS4 inhibitor CCG4986, and compared to that observed in the brains of WT control mice injected with kainate alone. Kainate induced significant cell death in the hippocampus of control mice (Fig. 12, A,C,&E). However, no obvious cell death was detected in the hippocampus of mice co-treated with CCG4986 (Fig. 12, B,D,&F), demonstrating enhanced neuronal cell survival in these mice. Similarly, kainate failed to cause significant cell death in the hippocampus of Nrb−/− mice treated with either CCG4986 or vehicle (data not shown). These data support an enhanced function of endogenous adenosine and the A1R when RGS4 is blocked, which protects neurons from excitotoxic damage.
Figure 12.
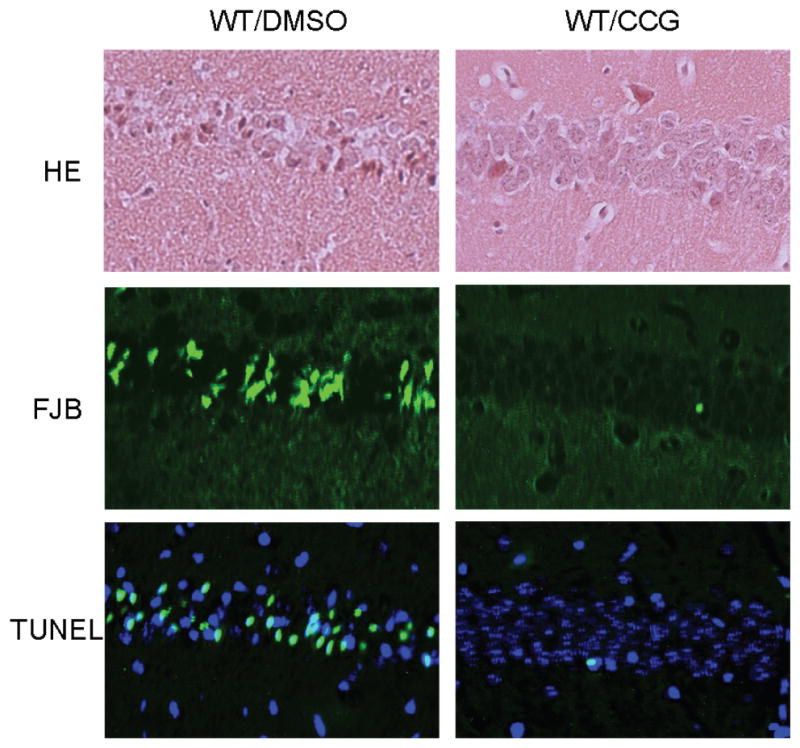
Kainate-induced cell death in the hippocampus is reduced by the RGS4 inhibitor CCG4986. Hippocampal slices of WT mice were collected 7 days after indicated treatment and stained by hematoxylin and eosin (HE, A&B). Cell death was detected by Fluoro-Jade B (FJB) staining (C&D) and Tunel assay (E&F). Significant neuronal cell death was detected in hippocampus from mice treated with kainate, but not in hippocampus from mice co-treated with kainate (KA) and CCG4986.
Discussion
In the present study, we identify a novel regulation of adenosine-evoked neuroprotection by a neural tissue-specific protein, neurabin. As illustrated in Fig. 13, neurabin directly interacts with the active A1R upon agonist stimulation and scaffolds complex formation between the A1R and RGS4, which leads to translocation of RGS4 to the plasma membrane and termination of A1R-induced G protein signaling. When the neurabin scaffold is absent (as in our Nrb−/− mice), RGS4 fails to form a complex with the A1R and cannot effectively terminate G protein signaling. As a result, adenosine-elicited anticonvulsant and neuroprotective effects through the A1R are significantly enhanced. Furthermore, direct inhibition of RGS4 with a small molecule blocker also results in increased G protein signaling and enhanced neuroprotection in response to adenosine. Taken together, our study has identified the A1R/neurabin/RGS4 complex as a key regulator in fine-tuning A1R functions in the nervous system. Disruption of this complex, either by genetic manipulation or by small molecule intervention, enhances A1R-mediated neuroprotection in response to endogenous adenosine without administration of exogenous A1R ligands, which can activate receptors in multiple organs and thereby confound therapeutic specificity.
Figure 13.

Model of neurabin/RGS4-mediated regulation of A1R signaling. Upon adenosine binding, neurabin directly interacts with both the active A1R and RGS4 and scaffolds formation of the A1R/neurabin/RGS4 complex, which attenuates A1R-induced G protein signaling and neuroprotective effects. However, when the neurabin scaffold is absent (as in our Nrb−/− mice), RGS4 cannot translocate to the plasma membrane or form a complex with the A1R, thus rendering it unable to effectively terminate G protein signaling. Also, A1R-induced G protein signaling is enhanced when RGS4 activity is inhibited (as by the small molecule blocker). In both cases, adenosine-elicited anticonvulsant and neuroprotective effects mediated through the A1R are greatly improved.
The anticonvulsant effect of adenosine has long been established and appreciated, and is mainly attributed to the inhibitory function of the A1R (Haas and Selbach, 2000; Cunha, 2001; Dunwiddie and Masino, 2001; Stone, 2002). Indeed, we observe that the severity of kainate-induced seizure is greatly exacerbated in mice co-treated with an A1R selective antagonist, DPCPX (Fig. 8). By contrast, when A1R activity is enhanced, as seen in Nrb−/− mice (Fig. 8) or mice co-treated with the RGS4 inhibitor CCG4986 (Fig. 11), kainate-induced seizure severity is greatly reduced. Significantly, the dose of kainate capable of inducing an ~50% rate of death in WT mice does not affect Nrb−/− mice (Fig. 8) or WT mice co-treated with CCG4986 (Fig. 11). These data demonstrate that enhancement of A1R function by disruption of neurabin and RGS4 provides an effective means to reduce excitotoxic seizures and improve mortality. Given the effectiveness of adenosine in confining seizures in various models, including those for pharmacoresistant seizures (Boison, 2007), several strategies have been proposed to increase extracellular adenosine levels in order to treat epileptic seizures. These strategies include inhibition of adenosine kinase (Boison, 2008), which converts adenosine to AMP, ketogenic diet, which increases global and focal adenosine levels (Masino et al., 2009), as well as transplantation of adenosine-releasing cells (Boison, 2007). Our present findings open up a new therapeutic direction for seizure control by specifically enhancing A1R activity and responsiveness through disruption of neurabin and RGS4, and provide the first example that a small molecule inhibitor of RGS proteins is effective in reducing excitotoxic seizure through potentiating A1R signaling.
It is well-appreciated that basal levels of extracellular adenosine are sufficient to tonically activate adenosine receptors, providing inhibitory purinergic tone in most systems (Dunwiddie and Masino, 2001). Given that neurabin and RGS4 attenuate A1R signaling, we suspect that such tonic activation of A1Rs by adenosine would be enhanced with neurabin deficiency or RGS4 inhibition, leading to stronger inhibition of neuronal excitability and transmission. Therefore, a selective A1R antagonist would be expected to elicit greater potentiation of baseline transmission at synapses in Nrb−/− mice compared with WT mice or under conditions when RGS4 activity is inhibited. The effects of disrupting the A1R/neurabin/RGS4 complex on synaptic transmission are currently under investigation.
To the best of our knowledge, we demonstrate for the first time that neurabin directly interacts with a GPCR to negatively regulate receptor activity through an RGS protein. The neurabin homolog spinophilin has been shown to interact with multiple GPCRs, including dopamine D2 receptor (Smith et al., 1999), α2AR (Richman et al., 2001; Wang et al., 2004; Xu et al., 2008), α1AR (Wang et al., 2005), M2 and M3 muscarinic acetylcholine receptors (Kurogi et al., 2009) and the μ opioid receptor (Charlton et al., 2008); however, direct interaction between neurabin and a GPCR has not been reported. Neurabin has been shown to sequester RGS2 from binding to α1AR, thus enhancing α1AR-mediated signaling (Wang et al., 2007). By contrast, our current study demonstrates that neurabin directly interacts with the active A1R and recruits RGS4 to the A1R complex at the plasma membrane, thereby attenuating A1R-mediated signaling. Meanwhile, the agonist-promoted interaction of the A1R with neurabin seems to have a negligible effect on A1R receptor density (Fig. 4C) and intrinsic affinity for agonist (Fig. 4D) and antagonist binding (data not shown), as these A1R properties are not changed in Nrb−/− mice.
Direct interaction with a GPCR represents a novel mechanism for neurabin to regulate neuronal activity. Neurabin has been reported to modulate glutamatergic neurotransmission and synaptic plasticity by targeting PP1 to postsynaptic compartments (MacMillan et al., 1999; Terry-Lorenzo et al., 2005; Allen et al., 2006; Hu et al., 2007). Our findings suggest that neurabin attenuates adenosine-evoked inhibition of neuronal activity by interacting with the A1R to diminish its signaling. The sequence of neurabin responsible for interacting with the A1R does not overlap with the region involved in PP1 binding and regulation (Nakanishi et al., 1997; Burnett et al., 1998), Figs. 1B&2A), making it possible to specifically target neurabin modulation of A1R signaling without altering its function in targeting PP1.
Our findings also provide strong evidence that the ability of neurabin to attenuate endogenous adenosine-elicited neuroprotection is due to the concurrent association of neurabin with both the A1R and RGS4. In vitro assays have demonstrated that neurabin is capable of interacting with several RGS proteins in the R4 subfamily (Wang et al., 2007). However, upon A1R agonist stimulation in intact cells, as shown in Fig. 6, neurabin specifically forms a complex with RGS4, but not with its close homolog, RGS2. These data not only demonstrate that the neurabin-facilitated A1R-RGS interaction is highly specific for RGS4, but also suggest that agonist-induced assembly of the A1R/neurabin/RGS4 complex provides a molecular mechanism by which neurabin expression suppresses A1R responses. Furthermore, the specificity of the interaction of neurabin with RGS4 and not other structurally close family members suggests that neurabin may serve another role in facilitating the selectivity of RGS-mediated attenuation of GPCR signaling.
Our findings that RGS4 inhibitor-dependent reduction of kainate-induced seizure severity is eliminated by co-treatment with an A1R antagonist (Fig. 11) support the hypothesis that A1R signaling is specifically regulated by RGS4 in this seizure model. Moreover, it was recently reported that RGS4 gene deficiency reduced the susceptibility to audiogenic seizures in fragile X mental retardation protein (FMR1) knockout mice, likely through an increase of GABAB receptor signaling (Pacey et al., 2009). Taken together, these studies suggest that suppression of RGS4 activity may be beneficial for seizure suppression in multiple clinical settings. It is noteworthy that RGS4 null mice are viable and fertile, and lack obvious physiological defects (Grillet et al., 2005). Therefore, pharmacological inhibition of RGS4 is unlikely to cause gross abnormality, and hence represents a particularly suitable approach to treat seizures without causing global side effects.
The small molecule CCG4986 was shown to inhibit both RGS4 interaction with Gαo and RGS4 GAP activity in vitro (Roman et al., 2007), likely through modification of multiple cysteine residues including Cys148 at an allosteric site that leads to a conformational change in RGS4 (Roman et al., 2010). Two other RGS proteins that possess a cysteine at equivalent positions, RGS8 and RGS16, cannot be inhibited by CCG4986 (Roman et al., 2010), suggesting that the allosteric modification by CCG4986 is unique for RGS4 and that the inhibitory outcome of CCG4986 is not non-selective in nature. Consistent with our working model that neurabin scaffolds A1R-RGS4 complex formation so that RGS4 can diminish A1R signaling, the effect of CCG4986 inhibition of RGS4 on A1R-mediated responses requires the presence of neurabin in both cultured cells (Fig. 5A) and in vivo (Fig. 11D). Our findings regarding CCG4986 attenuation of kainate-induced seizures provide proof of concept evidence supporting the therapeutic potential of inhibition of RGS4 in neuroprotection.
In summary, we have identified a neural tissue-specific interacting partner of the A1R, neurabin, which attenuates A1R signaling by recruiting RGS4 to form a complex with the receptor. In vivo, endogenous adenosine-evoked protective effects against cell death and kainate-induced seizure are remarkably enhanced in mice lacking neurabin expression or when RGS4 activity is blocked by a small molecule inhibitor. Significantly, all mice survive the LC50 (median lethal concentration) dose of kainate under these genetic or pharmacological manipulations. Thus, neurabin-RGS4 represents a tissue-selective regulatory mechanism for fine-tuning adenosine receptor function in neurabin-expressing target cells, i.e. in the nervous system. Disruption of this novel A1R/neurabin/RGS4 complex provides a promising therapeutic strategy for specific enhancement of neuroprotective effects in response to endogenous adenosine released on-site on the demand of various insults to the brain, and thus opens a new direction for treatment of pathological conditions that cause neural damage.
Acknowledgments
The authors are grateful to Dr. R. Colbran (Vanderbilt) for neurabin constructs, GST-Nrb146-453 and Myc-Nrb, and for neurabin antibody; Dr. J. Hablitz (UAB) for suggestions on seizure studies; the UAB Behavioral Neuroscience Core [supported by NIH grants NS 047466 (JMW) and NS 057098 (JMW)] for electroencephalography analysis. We also thank Drs. L. Limbird and R. Colbran (Vanderbilt) for critical reading of the manuscript. This research is supported by UAB faculty development grant (QW) and NIH grants MH081917 (QW) and DA10044 (PG).
Footnotes
Author contributions
Q.W. supervised the entire project. Y.C., Y.L. and C.C. performed the experiments. L.M., K.J., P.G. and Q.W. were involved in experimental design and data analysis. Y.C., C.C., L.M., K.J., P.G. and Q.W. prepared the manuscript.
Any Conflict of Interest: none
Reference List
- Allen JA, Yadav PN, Roth BL. Insights into the regulation of 5-HT2A serotonin receptors by scaffolding proteins and kinases. Neuropharmacology. 2008;55:961–968. doi: 10.1016/j.neuropharm.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen PB, Zachariou V, Svenningsson P, Lepore AC, Centonze D, Costa C, Rossi S, Bender G, Chen G, Feng J, Snyder GL, Bernardi G, Nestler EJ, Yan Z, Calabresi P, Greengard P. Distinct roles for spinophilin and neurabin in dopamine-mediated plasticity. Neuroscience. 2006;140:897–911. doi: 10.1016/j.neuroscience.2006.02.067. [DOI] [PubMed] [Google Scholar]
- Bansal G, Druey KM, Xie Z. R4 RGS proteins: regulation of G-protein signaling and beyond. Pharmacol Ther. 2007;116:473–495. doi: 10.1016/j.pharmthera.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- Berman RF, Fredholm BB, Aden U, O'Connor WT. Evidence for increased dorsal hippocampal adenosine release and metabolism during pharmacologically induced seizures in rats. Brain Res. 2000;872:44–53. doi: 10.1016/s0006-8993(00)02441-0. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Perroy J, Becamel C, Marin P, Fagni L. GPCR interacting proteins (GIPs) in the nervous system: Roles in physiology and pathologies. Annu Rev Pharmacol Toxicol. 2010;50:89–109. doi: 10.1146/annurev.pharmtox.010909.105705. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine-based cell therapy approaches for pharmacoresistant epilepsies. Neurodegener Dis. 2007;4:28–33. doi: 10.1159/000100356. [DOI] [PubMed] [Google Scholar]
- Boison D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol. 2008;84:249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady AE, Wang Q, Colbran RJ, Allen PB, Greengard P, Limbird LE. Spinophilin stabilizes cell surface expression of alpha 2B-adrenergic receptors. J Biol Chem. 2003;278:32405–32412. doi: 10.1074/jbc.M304195200. [DOI] [PubMed] [Google Scholar]
- Burnett PE, Blackshaw S, Lai MM, Qureshi IA, Burnett AF, Sabatini DM, Snyder SH. Neurabin is a synaptic protein linking p70 S6 kinase and the neuronal cytoskeleton. Proc Natl Acad Sci U S A. 1998;95:8351–8356. doi: 10.1073/pnas.95.14.8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carswell HV, Graham DI, Stone TW. Kainate-evoked release of adenosine from the hippocampus of the anaesthetised rat: possible involvement of free radicals. J Neurochem. 1997;68:240–247. doi: 10.1046/j.1471-4159.1997.68010240.x. [DOI] [PubMed] [Google Scholar]
- Charlton JJ, Allen PB, Psifogeorgou K, Chakravarty S, Gomes I, Neve RL, Devi LA, Greengard P, Nestler EJ, Zachariou V. Multiple actions of spinophilin regulate mu opioid receptor function. Neuron. 2008;58:238–247. doi: 10.1016/j.neuron.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee TK, Fisher RA. Cytoplasmic, nuclear, and golgi localization of RGS proteins. Evidence for N-terminal and RGS domain sequences as intracellular targeting motifs. J Biol Chem. 2000;275:24013–24021. doi: 10.1074/jbc.M002082200. [DOI] [PubMed] [Google Scholar]
- Cottingham C, Chen Y, Jiao K, Wang Q. The Antidepressant Desipramine Is an Arrestin-biased Ligand at the {alpha}2A-Adrenergic Receptor Driving Receptor Down-regulation in Vitro and in Vivo. J Biol Chem. 2011;286:36063–36075. doi: 10.1074/jbc.M111.261578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Grillet N, Pattyn A, Contet C, Kieffer BL, Goridis C, Brunet JF. Generation and characterization of Rgs4 mutant mice. Mol Cell Biol. 2005;25:4221–4228. doi: 10.1128/MCB.25.10.4221-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Selbach O. Functions of neuronal adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:375–381. doi: 10.1007/s002100000314. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von ZM. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- Hu XD, Huang Q, Yang X, Xia H. Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus. J Neurosci. 2007;27:4674–4686. doi: 10.1523/JNEUROSCI.5365-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Hepler JR, Gilman AG, Mumby SM. Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc Natl Acad Sci U S A. 1997;94:6159–6163. doi: 10.1073/pnas.94.12.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurogi M, Nagatomo K, Kubo Y, Saitoh O. Effects of spinophilin on the function of RGS8 regulating signals from M2 and M3-mAChRs. Neuroreport. 2009;20:1134–1139. doi: 10.1097/WNR.0b013e32832fd93e. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. Seven transmembrane receptors: something old, something new. Acta Physiol (Oxf) 2007;190:9–19. doi: 10.1111/j.1365-201X.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- MacMillan LB, Bass MA, Cheng N, Howard EF, Tamura M, Strack S, Wadzinski BE, Colbran RJ. Brain actin-associated protein phosphatase 1 holoenzymes containing spinophilin, neurabin, and selected catalytic subunit isoforms. J Biol Chem. 1999;274:35845–54. doi: 10.1074/jbc.274.50.35845. [DOI] [PubMed] [Google Scholar]
- Maemoto T, Finlayson K, Olverman HJ, Akahane A, Horton RW, Butcher SP. Species differences in brain adenosine A1 receptor pharmacology revealed by use of xanthine and pyrazolopyridine based antagonists. Br J Pharmacol. 1997;122:1202–1208. doi: 10.1038/sj.bjp.0701465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Kawamura M, Wasser CA, Pomeroy LT, Ruskin DN. Adenosine, ketogenic diet and epilepsy: the emerging therapeutic relationship between metabolism and brain activity. Curr Neuropharmacol. 2009;7:257–268. doi: 10.2174/157015909789152164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muly EC, Allen P, Mazloom M, Aranbayeva Z, Greenfield AT, Greengard P. Subcellular distribution of neurabin immunolabeling in primate prefrontal cortex: comparison with spinophilin. Cereb Cortex. 2004;14:1398–1407. doi: 10.1093/cercor/bhh101. [DOI] [PubMed] [Google Scholar]
- Murphy MG, Moak CM, Byczko Z, MacDonald WF. Adenosine-dependent regulation of cyclic AMP accumulation in primary cultures of rat astrocytes and neurons. J Neurosci Res. 1991;30:631–640. doi: 10.1002/jnr.490300406. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Obaishi H, Satoh A, Wada M, Mandai K, Satoh K, Nishioka H, Matsuura Y, Mizoguchi A, Takai Y. Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol. 1997;139:951–961. doi: 10.1083/jcb.139.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitzel KL, Hepler JR. Cellular mechanisms that determine selective RGS protein regulation of G protein-coupled receptor signaling. Semin Cell Dev Biol. 2006;17:383–389. doi: 10.1016/j.semcdb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Neubig RR, Siderovski DP. Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov. 2002;1:187–197. [Google Scholar]
- Oliver CJ, Terry-Lorenzo RT, Elliott E, Bloomer WA, Li S, Brautigan DL, Colbran RJ, Shenolikar S. Targeting protein phosphatase 1 (PP1) to the actin cytoskeleton: the neurabin I/PP1 complex regulates cell morphology. Mol Cell Biol. 2002;22:4690–4701. doi: 10.1128/MCB.22.13.4690-4701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK, Heximer SP, Hampson DR. Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol Pharmacol. 2009;76:18–24. doi: 10.1124/mol.109.056127. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- Richman JG, Brady AE, Wang Q, Hensel JL, Colbran RJ, Limbird LE. Agonist-regulated Interaction between alpha 2-Adrenergic Receptors and Spinophilin. J Biol Chem. 2001;276:15003–8. doi: 10.1074/jbc.M011679200. [DOI] [PubMed] [Google Scholar]
- Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–830. doi: 10.1038/nrm2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman DL, Blazer LL, Monroy CA, Neubig RR. Allosteric inhibition of the regulator of G protein signaling-Galpha protein-protein interaction by CCG-4986. Mol Pharmacol. 2010;78:360–365. doi: 10.1124/mol.109.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR. Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol. 2007;71:169–175. doi: 10.1124/mol.106.028670. [DOI] [PubMed] [Google Scholar]
- Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl DJ, Wells JN. Serine and alanine mutagenesis of the nine native cysteine residues of the human A(1) adenosine receptor. Biochem Pharmacol. 2000;60:1647–1654. doi: 10.1016/s0006-2952(00)00474-3. [DOI] [PubMed] [Google Scholar]
- Sebastiao AM, Ribeiro JA. Adenosine receptors and the central nervous system. Handb Exp Pharmacol. 2009:471–534. doi: 10.1007/978-3-540-89615-9_16. [DOI] [PubMed] [Google Scholar]
- Sjogren B, Neubig RR. Thinking outside of the (RGS) box: New Approaches to Therapeutic Targeting of Regulators of G protein Signaling. Mol Pharmacol. 2010 doi: 10.1124/mol.110.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Oxford GS, Milgram SL. Association of the D2 dopamine receptor third cytoplasmic loop with spinophilin, a protein phosphatase-1-interacting protein. J Biol Chem. 1999;274:19894–900. doi: 10.1074/jbc.274.28.19894. [DOI] [PubMed] [Google Scholar]
- Srinivasa SP, Bernstein LS, Blumer KJ, Linder ME. Plasma membrane localization is required for RGS4 function in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1998;95:5584–5589. doi: 10.1073/pnas.95.10.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW. Purines and neuroprotection. Adv Exp Med Biol. 2002;513:249–280. doi: 10.1007/978-1-4615-0123-7_9. [DOI] [PubMed] [Google Scholar]
- Terry-Lorenzo RT, Roadcap DW, Otsuka T, Blanpied TA, Zamorano PL, Garner CC, Shenolikar S, Ehlers MD. Neurabin/protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol Biol Cell. 2005;16:2349–2362. doi: 10.1091/mbc.E04-12-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Limbird LE. Regulated Interactions of the alpha 2A Adrenergic Receptor with Spinophilin, 14-3-3zeta, and Arrestin 3. J Biol Chem. 2002;277:50589–50596. doi: 10.1074/jbc.M208503200. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhao J, Brady AE, Feng J, Allen PB, Lefkowitz RJ, Greengard P, Limbird LE. Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science. 2004;304:1940–1944. doi: 10.1126/science.1098274. [DOI] [PubMed] [Google Scholar]
- Wang X, Zeng W, Kim MS, Allen PB, Greengard P, Muallem S. Spinophilin/neurabin reciprocally regulate signaling intensity by G protein-coupled receptors. EMBO J. 2007;26:2768–2776. doi: 10.1038/sj.emboj.7601701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zeng W, Soyombo AA, Tang W, Ross EM, Barnes AP, Milgram SL, Penninger JM, Allen PB, Greengard P, Muallem S. Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nat Cell Biol. 2005;7:405–411. doi: 10.1038/ncb1237. [DOI] [PubMed] [Google Scholar]
- Witt-Enderby PA, Jarzynka MJ, Krawitt BJ, Melan MA. Knock-down of RGS4 and beta tubulin in CHO cells expressing the human MT1 melatonin receptor prevents melatonin-induced receptor desensitization. Life Sci. 2004;75:2703–2715. doi: 10.1016/j.lfs.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Xie GX, Palmer PP. How regulators of G protein signaling achieve selective regulation. J Mol Biol. 2007;366:349–365. doi: 10.1016/j.jmb.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chen Y, Lu R, Cottingham C, Jiao K, Wang Q. Protein Kinase A Phosphorylation of Spinophilin Modulates Its Interaction with the {alpha}2A-Adrenergic Receptor (AR) and Alters Temporal Properties of {alpha}2AAR Internalization. J Biol Chem. 2008;283:14516–14523. doi: 10.1074/jbc.M710340200. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]