

Abstract
A novel combination of experimental data and extensive computational modeling was used to explore probable protein-protein interactions between photoactivated rhodopsin (R*) and experimentally determined R*-bound structures of the C-terminal fragment of α-transducin (Gtα(340–350)) and its analogs. Rather than using one set of loop structures derived from the dark-adapted rhodopsin state, R* was modeled in this study using various energetically feasible sets of intracellular loop (IC loop) conformations proposed previously in another study. The R*-bound conformation of Gtα(340–350) and several analogs were modeled using experimental transferred nuclear Overhauser effect data derived upon binding R*. Gtα(340–350) and its analogs were docked to various conformations of the intracellular loops, followed by optimization of side-chain spatial positions in both R* and Gtα(340–350) to obtain low-energy complexes. Finally, the structures of each complex were subjected to energy minimization using the OPLS/GBSA force field. The resulting residue-residue contacts at the interface between R* and Gtα(340–350) were validated by comparison with available experimental data, primarily from mutational studies. Computational modeling performed for Gtα(340–350) and its analogs when bound to R* revealed a consensus of general residue-residue interactions, necessary for efficient complex formation between R* and its Gtα recognition motif.
INTRODUCTION
The photoreceptor of the eye, rhodopsin, is the prototypical member of the vast family of G-protein coupled receptors (GPCRs). More than 16,000 GPCRs across many different genomes are known (GPCRDB; http://www.gpcr.org), and GPCRs are the largest protein superfamily in humans (1). GPCRs are integral membrane proteins that include seven transmembrane helical stretches (TM helices) connected by loops that form the intracellular (IC) and extracellular (EC) domains, together with the fragments containing the N- and C-termini. Generally, GPCRs are activated by extracellular agonists. Binding an agonist leads to a conformational change in the receptor that exposes an intracellular binding site within the IC domain for its G-protein. Upon binding to an activated GPCR, the heterotrimeric G-proteins dissociate and generate complexes of β- and γ-subunits, as well as complexes of α-subunit with adenosine triphosphate (ATP), which trigger a signaling cascade downstream (2). Currently, over 50% of the drugs used in clinics target GPCRs (3), making an understanding of the molecular interactions at atomic resolution between GPCRs and their ligands, both intra- and extracellular, extremely valuable.
The conformational change from the inactive (R) to activated state (R* or metarhodopsin II (MII)) in rhodopsin is initiated by a single photon of light of the correct wavelength, rather than by binding an extracellular molecular ligand. The photon is absorbed by the chromophore, cis-11 retinal, which is covalently bound to the side chain of K-296 in helix 7 (TM7). Isomerization of cis-11 retinal to the all-trans isomer initiates a conformational change of the trans-membrane helices that generates a binding site among the IC loops for the heterotrimeric G-protein, transducin (4). Transducin may be considered an intracellular ligand of rhodopsin, as the C-terminal ends of transducin’s α- and γ-subunits (Gtα and Gtγ) directly interact with rhodopsin (5–8).
Some forms of visual impairment, such as congenital night blindness (CNB) and retinitis pigmentosa, are due to mutations in rhodopsin that lead to its constitutive activation, i.e., spontaneous transition from the dark-adapted (R) to activated (R*) state (2). Unlike other GPCRs, rhodopsin cannot be targeted by blocking an extracellular ligand because its “ligand” is a photon; however, modulating the interaction between activated rhodopsin and transducin in the eye with an intracellular inhibitor, such as a small molecule peptidomimetic of the C-terminal fragment of Gtα, Gtα(340–350), could be a potential therapy for the above diseases. To lay the foundation for the molecular design of intracellular inhibitors that could prevent transducin from binding constitutively active rhodopsin mutants, one must know which specific residue-residue interactions occur between the IC regions of rhodopsin and transducin.
The photoactivated state of rhodopsin (R*, MII) is stabilized by interaction with the C-terminal undecapeptide Gtα(340–350) (6). The NMR studies of TrNOE (transferred nuclear Overhauser effect) revealed three-dimensional (3D) structure(s) of Gtα(340–350) in complex with R* (6); similar studies were also performed for several analogs of Gtα(340–350) (9–12). Therefore, the conformation of Gtα(340–350) may serve as a convenient model constraint for Gtα in complex with R*. The 3D structures of the IC segments of R* in complex with transducin are not known, however. Although the X-ray structure of rhodopsin has been solved, the structures of the IC loops and the C terminus are either poorly resolved, or contradictory, in different structures obtained by x-ray crystallography (13–16), due to the inherent flexibility of the IC region. Further, the x-ray structures of rhodopsin were originally determined for the dark-adapted state that does not interact with transducin, whereas the rhodopsin-transducin complex involves the photoactivated MII state of rhodopsin. Recently, the x-ray crystal structure of the photoactivated MII state of rhodopsin has been solved (17). This structure conflicts, however, with a large amount of biophysical data that suggest more movement of certain transmembrane helices as a result of the conformational change associated with the transition from R → R*. This discrepancy leads to ambiguities as to whether this crystal structure reflects the true state of R* that binds transducin, despite the MII spectral intermediate being observed in the crystal. Further, this x-ray crystal structure was solved at only 4.15 Å resolution with several regions on the intracellular loops unresolved, so precise positions of amino acids and some loop positions were not discernable.
At the same time, the arrangement of the TM helices in the R* state of rhodopsin was suggested by the ESR (electron spin resonance) spectroscopy (4) and by independent molecular modeling (18). Subsequent modeling studies, based on the 3D models for the TM region corresponding both to the R and R* states, revealed possible low-energy structures of the flexible EC and IC loops in rhodopsin (19). In this study, the experimental TrNOE structures of Gtα(340–350) and several of its analogs were docked to various low-energy structures for the IC region of rhodopsin in the activated R* state to determine possible residue-residue interactions between the two molecules upon forming the complex. Since all these peptides of similar structure stabilize MII and bind it with comparable affinities (Table 1), it was logical to hypothesize that they possess a common binding mode in the complex with the IC region of the R*. Accordingly, our objective in this study was to elucidate residue-residue interactions that are most important for mutual molecular recognition of R* and the peptide analogs experimentally studied. This study takes a novel prospective in considering multiple sets of low-energy loop conformations, whereas other studies have only considered one conformation of loops derived from the R state. Further, this methodology combines experimental data of the R*-bound conformation of several peptide ligands with extensive computational modeling to determine the important residue-residue interactions in the complex. Targeting these interactions should assist in the design of effective intracellular inhibitors to treat certain types of congenital visual impairment.
TABLE 1.
Intracellular ligands of rhodopsin considered in this study and their binding affinities to rhodopsin
| Peptide | Sequence | EC50 ± SE (μM) | Reference |
|---|---|---|---|
| Gtα(340–350) | IKENLKDCGLF-OH | 530 ± 90 | (9) |
| Peptide 2 | IKENLKDCGLW-OH | 540 ± 50 | (9) |
| Peptide 11 | IKENLKDCGL(2-Nal)-NH2 | 220 ± 70 | (9) |
| Peptide 14 | IKENLKDCGLF-NH2 | 320 ± 70 | (9) |
| Peptide 3 | IKENLKDCGLX* | 600 ± 90 | (10) |
| [R341, S347]-Gtα(340–350) | IRENLKDSGLF-OH | > 1000† | (42) |
X indicates a phenylethylamine derivative.
The EC50 value was measured on an acetylated peptide.
METHODS
3D models of molecular fragments
Models for the IC loops of rhodopsin
3D models of the rhodopsin loops used in this study were adopted from the intracellular (IC) loop models built by Nikiforovich and Marshall (19), where the ensemble of intracellular loops was mounted on the 3D model of the rhodopsin transmembrane domain in its activated conformation (R*). Each set of the IC loops consisted of four molecular segments, namely the three loops connecting TM1 and TM2 (IC1, fragment 61–75), TM3 and TM4 (IC2, fragment 136–153), and TM5 and TM6 (IC3, fragment 222–249) as well as the fragment 303–322 that included the part of the C-terminal helix parallel to the membrane surface (IC4). The loops included stems of TM helices (fragments 61–63, 73–75, 136–138, 151–153, 222–224, 247–249, and 303–305, respectively) that were kept in the same spatial positions as they were in the transmembrane domain by a system of parabolic potentials (see (19) for details). At the base of the TM stems, where the transmembrane domain is normally located, the artificial N- and C-termini were capped with acetyl and NHMe, respectively. In total, nine sets of the IC1+IC2+IC3+IC4 loop “packages” that differ in their backbone conformations by a root mean-square (RMS) value of at least 3.0 Å (Cα atoms only) (19) were used in subsequent calculations to simulate backbone flexibility in the IC loops. Finer sampling was done by using structures within one of the nine sets of loop conformers that were found to be significant.
Models for Gtα(340–350) and analogs
Besides Gtα(340–350), five other analogs of the undecapeptide that showed binding to R* comparable to that of Gtα(340–350) were considered in this study (Table 1). Four of the analogs, peptide 2, peptide 11, peptide 14, and peptide 3 (denoted according to Anderson et al. (9,10)), differed from Gtα(340–350) by modifications in the last residue and C-terminal end, and the fifth analog was [R341, S347]-Gtα(340–350). The TrNOE structures of native Gtα(340–350) and its analogs that were used as the 3D models in subsequent docking calculations were borrowed from the following sources: Gtα(340–350), PDB entry 1AQG (6); peptide 14 (9); peptide 3 (10); and [R341, S347]-Gtα(340–350), PDB entry 1LVZ (12). The sets of structures deduced for Gtα(340–350) and its five analogs by TrNOE were subjected to limited energy minimization using the Optimized Potential for Liquid Simulations (OPLS)-AA-L/ Generalized Born Solvation Approximation (GBSA) force field in the TINKER modeling software (20), and the local minimum energy structure found was used in subsequent docking calculations. The OPLS-AA-L force field was chosen because its parameters were optimized for use with peptides and proteins (21). Peptides 2 and 11 had similar binding affinities, but their TrNOE structures were not available. For these two analogs, the 3D structures were constructed in the SYBYL modeling package by modifying the 3D structure of Gtα(340–350) and subjected to energy minimization using the Tripos force field. Subsequently, peptides 2, 11, 14, 3, [R341, S347]-Gtα(340–350), and Gtα(340–350) were all subjected to the same docking procedures. The N- and C-termini of the peptides were modeled with an amino group (NH2) and a carboxyl group (COOH) or carboxamido group (CONH2) (Table 1), so there was no net charge at the ligand termini. In separate calculations, the N- and C-termini of the relevant peptides were modeled with charged amino (NH3+) and carboxyl groups (COO−).
Models of crystal structures used for validation
The following set of high-resolution crystal structures of complexes was used for validation of the employed docking protocol: HLA/peptide (the PDB entry 2BVO), BclXL/Bak (1BXL), MDM2/p53 (1T4F), and CheY/FLIM (1F4V). 2BVO contains the largest and most distinct binding cavity, whereas 1F4V has the shallowest cavity. To maintain uniformity and insure force-field self-consistency with the loop structures obtained using rigid-valence geometry (see (19)), the experimental crystal structures were adjusted to the same rigid-valence geometry constraints before starting the docking procedure by applying a system of harmonic potentials. The resulting structures were very close to the initial crystal structures, with the RMS values < 0.75 Å for Cα atoms only.
Docking procedures
Low-resolution docking search
The GRAMM (Global RAnge Molecular Matching) docking protocol (first described in (22)) was used to obtain the initial docked poses of the “ligand” (various NMR structures determined by TrNOE) relative to the “receptor” (the set of the IC loops of rhodopsin) (Fig. 1 A). GRAMM predicts the docking conformation by maximizing the 3D overlap of surfaces of two molecules using a fast Fourier transformation (22–24). We used the low-resolution GRAMM protocol (24) that calculates the corresponding scoring function on the six-dimensional grid with translational steps of 3 Å and rotational steps of 20°. The specific parameters for the GRAMM procedure were as follows: grid step – 3.0, repulsion – 6.5, attraction – 0.0, potential range type – grid_step, projection – gray, representation – all, angle for rotations – 20. The GRAMM module was downloaded from the Internet site http://vakser.bioinformatics.ku.edu. For each ligand-receptor complex, 999 configurations corresponding to the top values of the scoring function were retained for further consideration.
FIGURE 1.

Flow chart of the docking procedure. (A) Low-resolution docking, which involved the GRAMM program. (B) The solutions from GRAMM were clustered at 3 Å. (C) High-resolution docking, which included an iterative rigid body and side-chain optimization followed by minimization. (D) Distance cutoffs were imposed. (E) Energy cutoffs were imposed.
High-resolution docking search
The top 999 solutions (spatial positions of the rigid ligand with respect to R*) obtained by the GRAMM procedure were divided into clusters that differed by RMS values <3 Å in their spatial location (the RMS values were calculated for the backbone atoms, excluding hydrogens) (Fig. 1 B). The solution with the best GRAMM score within each cluster was optimized further in two separate steps (Fig. 1 C). First, the relative spatial positions of the ligand and IC loops of rhodopsin were adjusted and the side chains were repacked; at this step, conformations of the peptide backbone in both molecular entities were not affected. The above procedure employed a methodology similar to that used previously to optimize spatial locations of the TM helices in the heptameric bundle (18). Briefly, the methodology involved energy minimization of the two molecules to optimize their mutual spatial arrangement as rigid bodies. The rigid-body optimization allowed both the ligand and the loop entities to translate along and rotate about the x, y, and z coordinate axes. At each recalculation of the energy gradient, side-chain repacking was done by an algorithm developed earlier that employed an option of a step-wise grid search (25), with the step size of 30°. The ECEPP/2 force field, which assumed rigid-valence geometry, was used in these calculations. Only the trans-conformation of Pro was used in the calculations, and residues Arg, Lys, Glu, and Asp were modeled as charged species. The value of the dielectric constant was equal to 2, which is considered standard for a protein environment modeled with the ECEPP force field. The energy tolerance for minimization convergence was ΔE < 0.1 kcal/mol, and the tolerance for translational and rotational coordinates was 0.1 Å and 0.1° respectively.
Second, after repacking the side chains and performing rigid-body optimization, the resulting structures were subjected to energy minimization using the OPLS-AA-L/GBSA force field as implemented in TINKER (20). The structure was subjected to preliminary energy minimization to 1.0 Å RMS using the minimize function (L-BFGS minimization), followed by energy minimization to 0.1 Å RMS using the truncated Newton method in TINKER. The TINKER molecular modeling package can be downloaded from the Internet site http://dasher.wustl.edu/tinker. Spatial positions of the three Cα atoms at the bases of the TM stems were fixed during the minimization. All other atoms, including the loop and ligand backbone atoms, were allowed to move. TINKER contains bond lengths, angle bending, and improper torsion angles for amino acids with charged termini. In this study, neutral peptide termini were used, and parameters for bond lengths, angle bending and improper torsion were derived from TINKER parameters for charged termini. Some of the docked solutions were docked in very close proximity to the receptor, causing some of the Born radii to be negative. The following line was added to the born.f TINKER code (rborn(i).lt. ri) rborn(i) =100.0d0 to turn off solvent screening if the Born radii became negative. The structures were reranked according to the energies calculated using TINKER, and only those with the relative energies E – Emin < 30 kcal/mol were considered as viable solutions.
RESULTS
Validation of docking protocol at the known protein-protein complexes
The docking protocol used in our study starts from a low-resolution search by running GRAMM, a program that scans the entire surface of a protein and scores each docked ligand-protein complex. The program enumerates all docked configurations within the accuracy of the grid-step size and rotation-step size. In addition, all configurations were scored based on complementary overlap of molecular surfaces, providing a low-resolution scoring screen. The configurations of the two molecules corresponding to the top scores (~200 configurations after clustering) were subjected to a high-resolution search by optimizing their rigid-body spatial positions and re-packing their side chains, followed by a full energy minimization procedure that allows selection of configurations with relative energies <30 kcal/mol from the lowest energy structure. The advantages of this docking protocol are the systematic search of the configurational space about the ligand-protein complex; the ability to repack side chains in both ligand and protein; and the ability to perform docking fast and within rather modest available computational resources (the complete studies for the Gtα(340–350)-rhodopsin complex, as described below, required ~35 days on a single node PC with 2.8 GHz under the Linux operating system). The exact runtime is dependent on the number of CPUs available, the number of clusters resulting from the GRAMM solutions, and on the size of the proteins. The main disadvantage of the method is that the rough grid and rigid-body approximations used by the GRAMM procedure may not contain the point closest to the lowest-energy solution; however, this might be rectified by subsequent energy minimization. At the same time, employing the finer grid would exponentially increase the computer time required.
Therefore, it was important to validate the docking protocol for several known complexes between proteins and their peptide ligands (the PDB entries 2BV0, 1BXL, 1T4F and 1F4V). Generally, configurations selected by the docking protocol include those where both the placement of the docked ligand and the residue-residue interactions present in the crystal structures of the validation complexes were reconstructed. However, the degree of success varied between different test cases. The closest similarity between docked configurations and the crystallographic data was characterized by the RMS values (all ligand backbone atoms, excluding hydrogens) of 1.80 Å (2BV0), 2.58 Å (1BXL), and 3.39 Å (1T4F); in the case of 1F4V, the closest RMS value was larger than 6 Å. The RMS values when considering all heavy atoms was not very different from the analysis with just backbone heavy atoms: 2.08 Å (2BVO), 2.96 Å (1BXL), 3.70 Å (1T4F). The slight difference in RMS between only the backbone versus all heavy atom RMS is relatively small, indicating that the side chains are roughly in the same conformations as the crystal structure. To determine if this level of similarity would be sufficient for the goals of our study in which we are elucidating the system of ligand-protein residue-residue interactions, the contacts between the side chains of the ligands and proteins were determined. A contact was defined as spatial positioning of any two atoms (including hydrogen atoms) belonging to different side chains with a distance <5.5 Å. Fig. 2 illustrates similarities and differences for the systems of residue-residue interactions in the crystal structures of 2BV0, 1BXL, and 1T4F and those found by the docking protocol.
FIGURE 2.
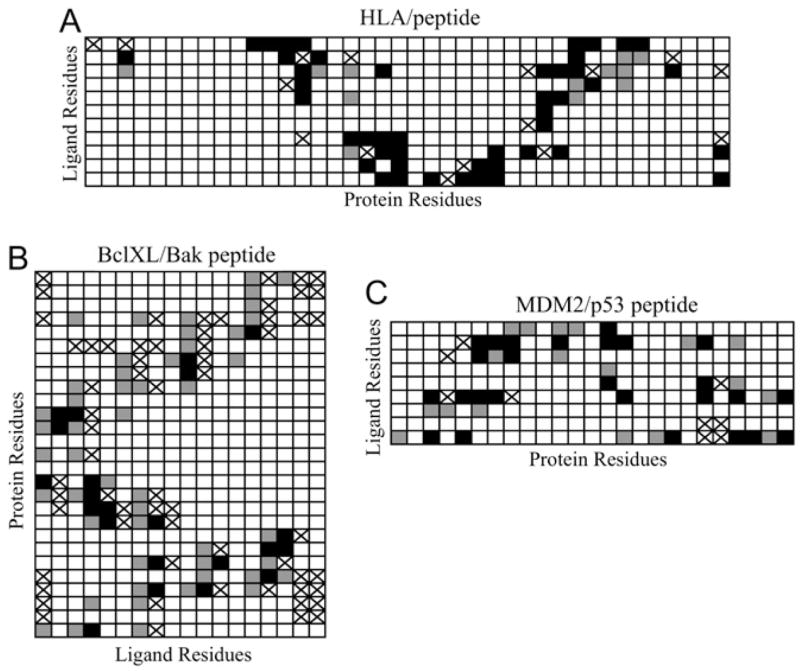
Residue-residue contacts for the three ligand-protein complexes used to evaluate docking methods. Residue-residue interactions present in both the x-ray crystal structures and in the closest docked models are shown in black. False negatives are shown with a cross, and false positives are shown in gray.
Fig. 2 shows generally good consistency in the systems of residue-residue contacts between the x-ray structures and the closest structures found by the docking protocol. In all three cases, many of the existing contacts were predicted correctly (those marked in black in Fig. 2). Notably, the false positives (contacts predicted by the protocol that were not in the crystal structure, gray in Fig. 2) are almost always located next to the correctly predicted contacts or to the false negatives (contacts present in the crystal structure that the protocol missed, cross). As one would expect, the structure with the closest RMS value to the crystal structure found for 2BV0 had more correctly predicted residue-residue interactions; in fact, out of 61 contacts existing in 2BV0, 46 (75%) were predicted correctly, 15 were missed, and 13 were false positives. Of those residue-residue interactions predicted correctly, 30 are direct interactions (65%), a direct contact being defined as side chains on both the ligand and receptor in nearly the same conformation and making a similar type of interaction (hydrogen bonding, van der Waals, etc.) based on visual inspection. In fact, if the definition of a contact is reduced to spatial positioning of any two atoms (including hydrogen atoms) belonging to different side chains with a distance <4.0 Å, we end up with only slightly more direct contacts (74%) out of those residue-residue interaction predicted correctly.
Based on the above results, one can conclude that the docking protocol works best for placing a ligand in a distinct hole or cavity, as in the case of 2BV0. Interactions in this HLA-peptide complex are somewhat analogous to those in the R*-transducin complex, because the IC region of rhodopsin also possesses a cavity into which the ligand can fit. In other cases, where the x-ray complexes did not have nearly as large a cavity or groove as did the HLA-peptide and R*-transducin complexes, the protocol reproduced crystallographic residue-residue interactions with lower, but still reasonable accuracy. The methodology failed in the case of CheY/FLIM peptide (1F4V), where the binding groove is the most shallow; given the large binding cavity within the R* loops, this was not a problem likely to be encountered, however, when Gtα(340–350) was docked to the IC loops.
Docking of Gtα(340–350) and its analogs to the IC region of R*
For Gtα(340–350) and all other ligands, the GRAMM procedure (Fig. 1 A) was run on the one representative conformation for each of the nine sets (clusters) of possible R* IC loop structures deduced previously (19). The top 999 solutions were taken from the GRAMM output of each run. For five of the sets of IC loop structures, at least some of the GRAMM solutions (7.2% to 97.8%, depending on the loop structure) were located in the cavity formed by the R* loops. However, some loop structures were too confined to yield a significant number of GRAMM solutions that located the ligand in the cavity; rather, artifact solutions corresponding to configurations of the ligand positioned where the trans-membrane domain was located were obtained. These loop/peptide configurations were eliminated from further consideration in the study of Gtα(340–350) analogs. Obviously, the remaining sets of the loop structures were the more “open” ones compared to the others. The ligand configurations corresponding to these sets of the loop structures were divided into clusters according to the RMS value of 3 Å (Fig. 1 B, see Methods), resulting in ~200 clusters for each loop structure.
The configurations with the best GRAMM score for each cluster were run through an optimization procedure, which iteratively optimized the position of the ligand and the IC region of rhodopsin as rigid bodies and then optimized side-chain positions by repacking both ligand and receptor (Fig. 1 C). The resulting configurations were then subjected to energy minimization employing the OPLS-AA-L/GBSA force field in the TINKER package. Energy minimization at this stage removed small steric clashes and also permitted the backbone to relax, providing some amount of backbone flexibility in both the receptor and ligand at this later stage of the process to further optimize the complex.
Finally, two filters were employed to select viable solutions of the entire procedure. The first filter took advantage of the rhodopsin site-directed mutagenesis data that revealed fragments important for transducin binding, namely 136–139 (Y-136, V-137, V-138, V-139 in IC2); 247–249 (E-247, K-248, E-249 in IC3); and 310–312 (N-310, K-311, Q-312 in IC4) (Fig. 1 D) (26–28). Accordingly, only configurations where the ligand made contact with one or more of these residues in each loop were retained for further consideration. A contact was defined by at least one atom belonging to the ligand and one atom belonging to protein, both of which could be a hydrogen atom, located within a maximum distance of 4 Å. Second, of the configurations from the first filter, only configurations with the relative energy E – Emin < 30 kcal/mol (where Emin was the lowest energy across all sets of the loop structures of solutions that met the first criteria, shown in Fig. 1 E) were considered viable (from 14 to 47 configurations for various ligands).
Common binding mode for Gtα(340–350) and its analogs
The main hypothesis of this study presumed the possibility of a common binding mode for all discussed ligands with the IC region of the photoactivated rhodopsin. To compare all selected configurations for each peptide to each other, the set of the loop structures was superimposed using the last three Cα atoms in the TM stems. The RMS values for all backbone atoms, excluding hydrogens, of the ligand were calculated. The most similar configurations differed in spatial positions of the ligand (relative to spatial position of Gtα(340–350)) by RMS values of 2.49–4.03 Å (Fig. 3). However, in terms of spatial positions of the IC loops, all of them corresponded to the same set of the loop structures, namely, the most “open” one. Interestingly, the same set of the loop structures persisted in the similar configurations upon increasing the RMS cutoff value up to 4.3 Å. For several analogs, the preference for this loop structure persisted with an RMS cutoff value above 6 Å. In other words, our results elucidated not only the common binding mode for the ligands in the complex with R*, but also the set of the 3D structures of the IC loops of R* characteristic for the complex. We also did finer sampling for all six loop conformations comprising this set (see (19)). The Gtα(340–350) peptide was docked onto each of these loop structures, and the common binding pose occurred on an additional loop structure with an RMS of 3.629 Å that makes the same important residue-residue interactions found for the common binding mode with the exception of the interaction between Lys-341 and Thr-70. This demonstrates that slight variations in the loop structure can still yield this common binding pose.
FIGURE 3.
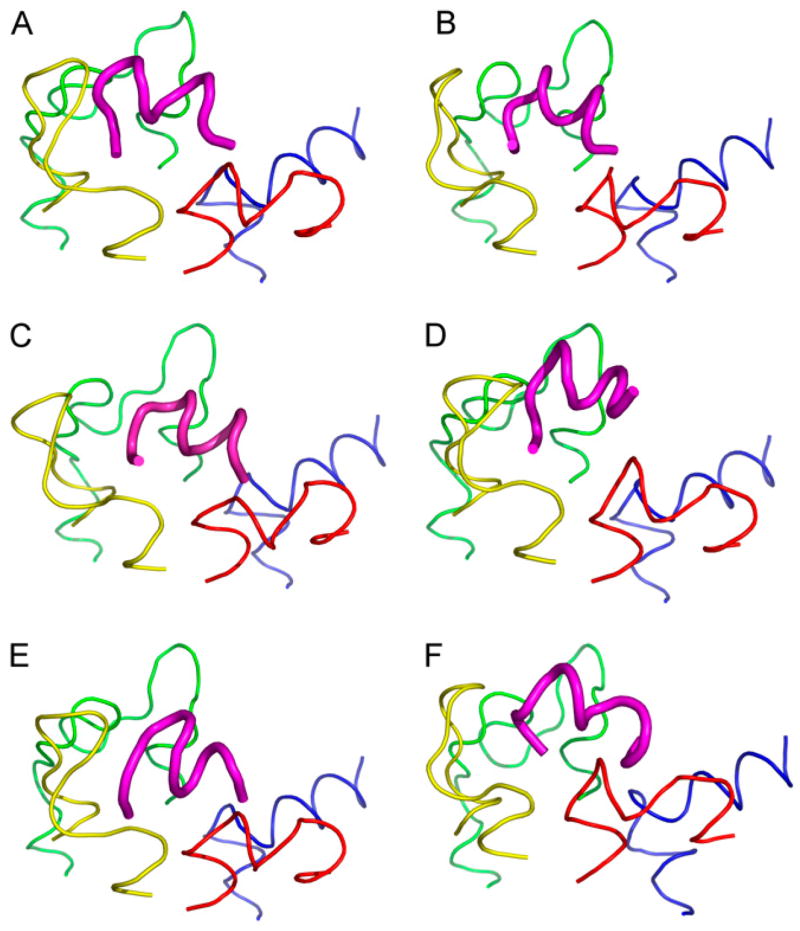
(A) Gtα(340–350) and its analogs. (B) Peptide 2, (C) Peptide 11, (D) Peptide 14, (E) Peptide 3, and (F) [R341, S347]-Gtα(340–350) are shown in the common binding mode. The first and last three Cα atoms in the loop structures were superimposed to find the common binding pose. IC1 is shown in red, IC2 in yellow, IC3 in green and IC4 in blue. Gtα(340–350) and its analogs are shown in magenta. The figure was rendered in PYMOL (DeLano Scientific, Palo Alto, CA).
Differences in spatial positions of the ligand peptide backbones shown in Fig. 3 may seem significant, but the systems of residue-residue interactions between Gtα(340–350) and its analogs with the IC loops were quite similar. Compared to 75 residue-residue contacts observed between Gtα(340–350) and R*, other ligands comprised 62–78 contacts, out of which 34–43 contacts were the same as observed for Gtα(340–350). Residues for which any loop atom was within 5.5 Å of any ligand atom were considered to be interacting. Further, some residue-residue interactions were common for all ligands (Fig. 4). These interactions were as follows: Lys-341/Thr-70, Phe-350/Leu-72, Phe-350/Val-137, Leu-349/Lys-141, Leu-349/Lys-231, Cys-347/Gln-244, Leu-344/Glu-249 (where transducin residues are shown in bold). Ile-340/Gln-312 and Ile-340/Asn-315 interactions were seen in all analogs except for Peptide 14. In our view, these particular residue-residue interactions are important targets for drug design of compounds aimed at blocking G-protein binding to constitutively active rhodopsin mutants.
FIGURE 4.
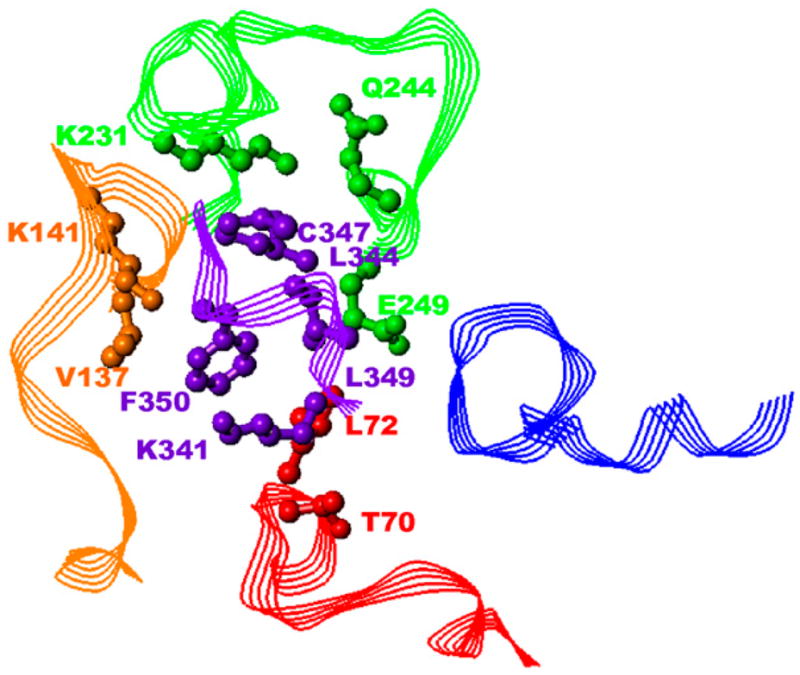
Common residue-residue interaction at 5.5 Å. Gtα(340–350) is shown in purple, IC1 in red, IC2 in orange, IC3 in green, and IC4 in blue. Only side chains are shown. The side chains of residues involved in interactions are rendered in ball-and-stick format to better illustrate the interaction. All hydrogen atoms are omitted for clarity. The residues are labeled in corresponding color.
DISCUSSION
The molecular interactions by which the α-subunit of transducin interacts with the set of the intracellular loops of photoactivated rhodopsin (R*) remain elusive. The lack of a crystal structure of the active complex has impeded the understanding of this important interaction. Furthermore, knowledge of the interaction of transducin with the IC region of R* may yield additional insights into how other G-proteins interact with their receptors in general and provide molecular targets that will aid in the development of therapeutics for eye diseases, such as retinitis pigmentosa and congenital night blindness. By combining molecular modeling and experimental data, this study aimed to elucidate these important residue-residue interactions between the C-terminal region of transducin and the IC loops of R* using the TrNOE-deduced structures of Gtα(340–350) and its analogs. Several sets of low-energy IC loop structures of R* were used in the computational docking studies, unlike previous studies (29,30) where single conformations of IC loops based on the crystal structure of R were used. Given the conformational uncertainty of the IC and EC loops in the x-ray crystal structure, we feel that this methodology provides a more complete picture of potential residue-residue interactions between R* and the C-terminal region of transducin. The computational docking techniques used in this study allowed us to explore the entire IC loop region rather than that in molecular dynamics calculations where only a small region of the receptor loop configurations could be explored.
This study determined a binding mode common for Gtα(340–350) and its analogs that have a comparable level of affinity to R*. It appeared that only the set of IC loop conformations of R* that corresponded to the most “open” conformation was relevant for this common binding mode. Also, a rather limited pattern of residue-residue interactions between R* and Gtα was shown to be important for maintaining the common binding mode. From the R* side, this pattern involved residues T-70, L-72, V-137, K-141, K-231, Q-244, E-249; from the transducin side, it involved residues K-341, L-344, C-347, L-349 and F-350 (see Fig. 4). As was mentioned above, R* fragments 136–139, 247–249, and 310–312 have been known to be important for transducin binding by previous mutagenesis experiments (26–28). These fragments were used in the filtering procedure and have some common residue-residue interactions among Gtα(340–350) and its analogs, namely V-137(IC2)-Phe-350 and E-249(IC3)-L-344. The residue K-141 located in IC2 was not used in the filtering procedure and, therefore, was predicted as being involved in the R*-transducin interaction; some experimental evidence confirms that K-141 (31–33) and Q-244 (34) are important for transducin binding. So far, there is no specific experimental evidence of involvement of R*-residues T-70, L-72 (IC1), and K-231 (IC3) in interactions with transducin, which are predicted by this study.
Our selection procedure did not use any experimental information as to which transducin residues are important for interaction with R*. The complete site-directed mutagenesis study of transducin has been performed by obtaining a series of single alanine mutations to the α-subunit of transducin (35). Some of the mutations that impaired the interaction between transducin and R* were located on two different fragments of Gtα, namely on a region that binds the βγ-subunit and on a region that directly binds R*. Specifically, seven residues on the C-terminal end of Gtα impaired the interaction with R* when they were mutated to alanine (I-340, K-341, N-343, L-344, G-348, L-349, and F-350). In the current study, all but one of the interactions that were seen in the common binding mode with R* involved residues (K-341, L-344, L-349, and F-350); these residues closely correspond to the above experimental data. An interaction with C-347 was also present in the common binding mode (see Fig. 4), but mutation of C-347 to alanine was found to have the overall wild-type phenotype in the discussed experimental study (35). However, in another set of experiments performed with Gtα(340–350) and its analogs, mutating C-347 to serine caused transducin to completely lose its ability to bind and stabilize the MII state of rhodopsin (36). Further, when the sulfhydryl group on C-347 was blocked by alkylation, very little stabilization of the MII state was seen. In the same study, L-349 was also shown to be a residue critical for interaction with rhodopsin, as synthetically changing this residue to Ile or tertleucine caused a significant reduction in MII stabilization. One can conclude, therefore, that all residues of Gtα(340–350) predicted by our study as involved in direct interaction with the IC region of R* have been shown as important by experimental studies (35,36). At the same time, none of the C-terminal transducin residues experimentally found as not significant for interaction with R* (E-342, K-345, and D-346 (35)) were predicted by these studies as involved in direct interaction with R*.
On the contrary, two 3D models of the transducin/rhodopsin complexes developed by other authors suggested rather different systems of residue-residue interactions between R* and Gtα(340–350) (29,30). In the first model, the photoactivated state of rhodopsin was modeled by switching the retinal chromophore to the all-trans conformation and applying target-driven molecular dynamics with strict constraints to satisfy experimental distances between TM helices in R* derived from spectroscopic data (29). Then, the 3D crystal structure of trimeric transducin (utilizing the TrNOE-deduced structure of Gtα(340–350) to obtain coordinates for residues 344–350) was manually docked to rhodopsin and subjected to a long molecular dynamics simulation (~10 ns) in a system that included the fully hydrated lipid bilayer. Recently, the model was developed further to account for possible oligomerization of the rhodopsin molecules (37). The authors suggested the pattern of rhodopsin/transducin residue-residue interactions that involved seven transducin residues, K-341, L-344, K-345, D-346, C-347, L-349, and F-350 (29), two of them (K-345 and D-346) were shown with wild-type phenotypes when mutated to alanine (35). The second model was built based on the assumed R* state of the TM region of rhodopsin obtained by rotation of TM6 by ~120° along the long axis (30). The model was packed in an oligomeric structure that included four rhodopsin molecules, one of them being R*. After manual docking of the 3D model of the heterotrimeric transducin to R*, the entire system was subjected to energy minimization (the model was recently updated to accommodate the newly published x-ray structures for dark-adapted rhodopsin (38)). In this model, Gtα(340–350) interacts with one R* molecule; according to the authors, interaction involves residues K-341, E-342, K-345, D-346, L-349, and F350 (30); this list, again, includes residues K-345 and D-346 shown with wild-type phenotypes when mutated to alanine (35). One more computational model, which specifically addressed binding of Gtα(340–350), suggested that only the very C-terminal transducin residues C-347- F-350 may directly contact residues of R* (36); the contacts to R* of the last residues, C-347, L-349, and F-350 were used in this model as requirements for selecting possible binding modes of Gtα(340–350) to R*. The inclusion of these contact constraints in the predictions of residue-residues contacts in the R*-transducin complex in this study, not surprisingly, agree more closely to available experimental data than the contacts suggested by other models.
In this study, we employed a combination of modeling techniques, starting from a low-resolution search of possible ligand-protein configurations by GRAMM, which exhaustively samples the interface between the two molecules, to a high-resolution search, which contains an optimization feature (rigid-body optimization and side-chain repacking) and a more accurate energy function implemented in the TINKER package. Our modeling techniques possessed some important advantages that allowed us to correctly predict residue-residue interactions between Gtα(340–350) and the IC region of R*, as well as other experimental features of the peptide(transducin)-R* complex. In our opinion, one of the advantages was that the GRAMM low-resolution search method used in this study exhaustively evaluates possible ligand positions about the protein, rather than employing a stochastic search algorithm to sample configurational space, as do most other docking techniques (39–41). Also, many docking algorithms do not accommodate ligand/receptor induced fit by allowing the residues on the ligand and receptor to repack after docking (40), whereas our docking methodology optimized both rigid-body position and side-chain orientations in both the ligand and receptor. This process did not significantly perturb the starting NMR structure of Gtα(340–350), as the common binding pose retained a conformation with a heavy atom backbone RMS of 1.003 Å and an all heavy atom RMS of 1.712 Å. Throughout the docking process, the crude-to-refined docking method significantly reduced the number of docked structures that must be considered and evaluated, allowing a more computationally intensive energy function to be used on the remaining structures. This methodology allowed us to explore the entire surface of the receptor, rather than just a small region that would have been explored with molecular dynamics calculations.
Other docking methods that provide ligand and/or receptor flexibility were tried. We tried using RosettaDock to dock the Gtα(340–350) TrNOE structure (Supplementary Material). RosettaDock (39) uses a Monte Carlo sampling method and allows flexible side chains on both the ligand and receptor. A global run failed to yield solutions that docked within the receptor loops. The transmembrane domain was added to our loop model, and the global run was repeated. A clear energy funnel did not form, and there were no clear clusters of ligand configurations that resulted from this calculation. In addition, we also tried Autodock3.0, which allows for varying degrees of flexibility on the ligand, while keeping the receptor side chains rigid (40). After trying many variations of Autodock parameters using the Lamarkian Genetic Search Algorithm (Supplementary Material), nearly all the solutions from Autodock for Gtα(340–350) and its analogs failed to yield contacts between transducin and loop residues of R* determined to be important in previous mutational experiments.
Different options of the force field parametrization (as applying charges to the peptide termini to produce a zwitterionic molecule) also failed to determine the common binding mode for all six ligands. Rather, five possible common binding modes were seen in three of the analogs. One of the five binding modes determined using the zwitterionic model was the common binding mode found with the neutral peptide termini. Using the zwitterionic model, this common binding mode was seen for Gtα(340–350), peptide 2, peptide 11, and 1LVZ. The effective dielectric within the loop region of the rhodopsin receptor is not known, and therefore, adding charges to the termini could yield artificially large electrostatic interactions that could potentially bias our model, so we feel the neutral peptide model is more reliable.
The most unique aspect of this approach, however, was not in the molecular modeling techniques. First, a variety of possible conformations were considered for the IC loops of rhodopsin in the photoactivated R* state (19), rather than focusing on one single conformation for the IC loops adopted from the x-ray structure(s) for the R state as did others (29,30,36–38). Given the fact that several low-energy conformations exist, all must be explored when building an accurate model. Further, the conformations of IC loops in the R x-ray crystal structure are unresolved, may not represent the solution structure of the loops, and most likely do not represent the loop structure of R* to which transducin or Gtα(340–350) binds. Second, our conclusions regarding the 3D structure of the peptide-R* complex were based on the combined results obtained for several ligands; such consensus may effectively increase the signal/noise ratio and cancel minor errors that could occur in the case of any particular ligand. Third, we focused on the pattern of residue-residue interactions between Gtα(340–350) and R*, which, on the one hand, provides accuracy of predictions sufficient for further use in drug design, and, on the other hand, does not require overinterpretation of site-directed mutagenesis data in more rigorous (but often inappropriate) structural terms. We believe that the novel elements of this computational paradigm may be useful for elucidating the patterns of residue-residue interactions between extracellular ligands and their receptors for other GPCRs belonging to the rhodopsin family.
CONCLUDING REMARKS
In this study, we used a novel combination of computational modeling, docking techniques, and experimental biophysical data to elucidate the residue-residue interactions between the possible structures of the flexible intracellular loops of rhodopsin in the photoactivated state and the C-terminal fragment of the α-subunit of transducin, Gtα(340–350). Further, the binding mode common for Gtα(340–350) and its analogs was identified. For the first time, many low-energy intracellular loop structures of R* were explored, making this study more extensive than studies that only considered one intracellular loop conformation based on R. Based on this study, it was established that only one type of conformation of the IC loops of R*, that corresponding to the most open structure, bound Gtα(340–350). This binding mode predicted a set of residue-residue interactions between the two molecules that was validated by previous data of site-directed mutagenesis and other experimental studies. Since blocking the interaction between R* and transducin could be important for treating certain forms of visual impairment, these results may be used to guide design of peptidomimetics or small molecule drugs. Generally, the overall approach may also be useful for studies of the interaction of other GPCRs with their specific extracellular ligands.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors are grateful to Drs. Slawomir Filipek, Rieko Arimoto, and Jerzy Ciarkowski who kindly provided us with the atomic coordinates of the models developed by Filipek and colleagues (30), Arimoto and colleagues (36), and Slusarz and Ciarkowski (29). Dr. Jerzy Ciarkowski is also acknowledged for making reference (29) and the atomic coordinates of the 3D model available before their publication. We thank Michael Schnieders for help with the TINKER package.
This work was partly supported by National Institutes of Health, Institutional National Research Service Award 5-T32-EY13360-06, from the National Eye Institute and a W. M. Keck Fellowship in Molecular Medicine (C.M.T.) and National Institutes of Health grants GM 68460, GM53630, and EY1211301 (G.V.N. and G.R.M.).
References
- 1.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 2.Bosch L, Iarriccio L, Garriga P. New prospects for drug discovery from structural studies of rhodopsin. Curr Pharm Des. 2005;11:2243–2256. doi: 10.2174/1381612054367436. [DOI] [PubMed] [Google Scholar]
- 3.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 4.Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 5.Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science. 1988;241:832–835. doi: 10.1126/science.3136547. [DOI] [PubMed] [Google Scholar]
- 6.Kisselev OG, Kao J, Ponder JW, Fann YC, Gautam N, Marshall GR. Light-activated rhodopsin induces structural binding motif in G protein alpha subunit. Proc Natl Acad Sci USA. 1998;95:4270–4275. doi: 10.1073/pnas.95.8.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kisselev OG, Downs MA. Rhodopsin-interacting surface of the transducin gamma subunit. Biochemistry. 2006;45:9386–9392. doi: 10.1021/bi060806x. [DOI] [PubMed] [Google Scholar]
- 8.Kisselev OG, Ermolaeva MV, Gautam N. A farnesylated domain in the G protein gamma subunit is a specific determinant of receptor coupling. J Biol Chem. 1994;269:21399–21402. [PubMed] [Google Scholar]
- 9.Anderson MA, Ogbay B, Arimoto R, Sha W, Kisselev OG, Cistola DP, Marshall GR. Relative strength of cation-pi vs salt-bridge interactions: the Gtalpha(340–350) peptide/rhodopsin system. J Am Chem Soc. 2006;128:7531–7541. doi: 10.1021/ja058513z. [DOI] [PubMed] [Google Scholar]
- 10.Anderson MA, Ogbay B, Kisselev OG, Cistola DP, Marshall GR. Alternate binding mode of C-terminal phenethylamine analogs of Gtα (340–350) to photoactivated rhodopsin. Chem Biol Drug Design. 2006;68:295–307. doi: 10.1111/j.1747-0285.2006.00460.x. [DOI] [PubMed] [Google Scholar]
- 11.Koenig BW, Mitchell DC, Konig S, Grzesiek S, Litman BJ, Bax A. Measurement of dipolar couplings in a transducin peptide fragment weakly bound to oriented photo-activated rhodopsin. J Biomol NMR. 2000;16:121–125. doi: 10.1023/a:1008378523816. [DOI] [PubMed] [Google Scholar]
- 12.Koenig BW, Kontaxis G, Mitchell DC, Louis JM, Litman BJ, Bax A. Structure and orientation of a G protein fragment in the receptor bound state from residual dipolar couplings. J Mol Biol. 2002;322:441–461. doi: 10.1016/s0022-2836(02)00745-3. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 14.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 15.Okada T, Fujiyoshi Y, Silow M, Navarro J, Landau EM, Shichida Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci USA. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teller DC, Okada T, Behnke CA, Palczewski K, Stenkamp RE. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs) Biochemistry. 2001;40:7761–7772. doi: 10.1021/bi0155091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salom D, Lodowski DT, Stenkamp RE, Trong IL, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci USA. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikiforovich GV, Marshall GR. Three-dimensional model for meta-II rhodopsin, an activated G-protein-coupled receptor. Biochemistry. 2003;42:9110–9120. doi: 10.1021/bi034586o. [DOI] [PubMed] [Google Scholar]
- 19.Nikiforovich GV, Marshall GR. Modeling flexible loops in the dark-adapted and activated States of rhodopsin, a prototypical G-protein-coupled receptor. Biophys J. 2005;89:3780–3789. doi: 10.1529/biophysj.105.070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren P, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107:5933–5947. [Google Scholar]
- 21.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparameterization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 22.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vakser IA, Aflalo C. Hydrophobic docking: a proposed enhancement to molecular recognition techniques. Proteins. 1994;20:320–329. doi: 10.1002/prot.340200405. [DOI] [PubMed] [Google Scholar]
- 24.Vakser IA. Low-resolution docking: prediction of complexes for underdetermined structures. Biopolymers. 1996;39:455–464. doi: 10.1002/(SICI)1097-0282(199609)39:3%3C455::AID-BIP16%3E3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 25.Nikiforovich GV, V, Hruby J, Prakash O, Gehrig CA. Topographical requirements for delta-selective opioid peptides. Biopolymers. 1991;31:941–955. doi: 10.1002/bip.360310804. [DOI] [PubMed] [Google Scholar]
- 26.Acharya S, Saad Y, Karnik SS. Transducin-alpha C-terminal peptide binding site consists of C-D and E-F loops of rhodopsin. J Biol Chem. 1997;272:6519–6524. doi: 10.1074/jbc.272.10.6519. [DOI] [PubMed] [Google Scholar]
- 27.Marin EP, Krishna AG, Zvyaga TA, Isele J, Siebert F, Sakmar TP. The amino terminus of the fourth cytoplasmic loop of rhodopsin modulates rhodopsin-transducin interaction. J Biol Chem. 2000;275:1930–1936. doi: 10.1074/jbc.275.3.1930. [DOI] [PubMed] [Google Scholar]
- 28.Ernst OP, Meyer CK, Marin EP, Henklein P, Fu WY, Sakmar TP, Hofmann KP. Mutation of the fourth cytoplasmic loop of rhodopsin affects binding of transducin and peptides derived from the carboxyl-terminal sequences of transducin alpha and gamma subunits. J Biol Chem. 2000;275:1937–1943. doi: 10.1074/jbc.275.3.1937. [DOI] [PubMed] [Google Scholar]
- 29.Slusarz R, Ciarkowski J. Interaction of class A G protein-coupled receptors with G proteins. Acta Biochim Pol. 2004;51:129–136. [PubMed] [Google Scholar]
- 30.Filipek S, Krzysko KA, Fotiadis D, Liang Y, Saperstein DA, Engel A, Palczewski K. A concept for G protein activation by G protein-coupled receptor dimers: the transducin/rhodopsin interface. Photochem Photobiol Sci. 2004;3:628–638. doi: 10.1039/b315661c. [DOI] [PubMed] [Google Scholar]
- 31.Natochin M, Gasimov KG, Moussaif M, Artemyev NO. Rhodopsin determinants for transducin activation: a gain-of-function approach. J Biol Chem. 2003;278:37574–37581. doi: 10.1074/jbc.M305136200. [DOI] [PubMed] [Google Scholar]
- 32.Franke RR, Konig B, Sakmar TP, Khorana HG, Hofmann KP. Rhodopsin mutants that bind but fail to activate transducin. Science. 1990;250:123–125. doi: 10.1126/science.2218504. [DOI] [PubMed] [Google Scholar]
- 33.Franke RR, Sakmar TP, Graham RM, Khorana HG. Structure and function in rhodopsin. Studies of the interaction between the rhodopsin cytoplasmic domain and transducin. J Biol Chem. 1992;267:14767–14774. [PubMed] [Google Scholar]
- 34.Yang K, Farrens DL, Hubbell WL, Khorana HG. Structure and function in rhodopsin. Single cysteine substitution mutants in the cytoplasmic interhelical E-F loop region show position-specific effects in transducin activation. Biochemistry. 1996;35:12464–12469. doi: 10.1021/bi960848t. [DOI] [PubMed] [Google Scholar]
- 35.Onrust R, Herzmark P, Chi P, Garcia PD, Lichtarge O, Kingsley C, Bourne HR. Receptor and betagamma binding sites in the alpha subunit of the retinal G protein transducin. Science. 1997;275:381–384. doi: 10.1126/science.275.5298.381. [DOI] [PubMed] [Google Scholar]
- 36.Arimoto R, Kisselev OG, Makara GM, Marshall GR. Rhodopsin-transducin interface: studies with conformationally constrained peptides. Biophys J. 2001;81:3285–3293. doi: 10.1016/S0006-3495(01)75962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciarkowski J, Witt M, Slusarz R. A hypothesis for GPCR activation. J Mol Model. 2005;11:407–415. doi: 10.1007/s00894-005-0270-9. [DOI] [PubMed] [Google Scholar]
- 38.Filipek S. Organization of rhodopsin molecules in native membranes of rod cells–an old theoretical model compared to new experimental data. J Mol Model. 2005;11:385–391. doi: 10.1007/s00894-005-0268-3. [DOI] [PubMed] [Google Scholar]
- 39.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 40.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian Genetic Algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 41.Fernandez-Recio J, Totrov M, Abagyan R. Identification of protein-protein interaction sites from docking energy landscapes. J Mol Biol. 2004;335:843–865. doi: 10.1016/j.jmb.2003.10.069. [DOI] [PubMed] [Google Scholar]
- 42.Aris L, Gilchrist A, Rens-Domiano S, Meyer C, Schatz PJ, Dratz EA, Hamm HE. Structural requirements for the stabilization of metarhodopsin II by the C terminus of the alpha subunit of transducin. J Biol Chem. 2001;276:2333–2339. doi: 10.1074/jbc.M002533200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.