

Abstract

Tamoxifen remains the first line therapy for estrogen receptor positive (ER+) breast cancer. However, polymorphisms of the gene encoding P450 2D6 could result in no protein expression or no CYP2D6 enzymatic activity and may significantly reduce the benefit of the hormone therapy. To address this issue, we designed and synthesized three 4-hydroxytamoxifen bioisosteres utilizing a boron-aryl carbon bond that can be oxidized under physiological conditions to yield 4-hydroxytamoxifen. We show that the bioisosteres inhibit the growth of two ER+ breast cancer cell lines, MCF-7 and T47D, with potencies comparable to or greater than that of 4-hydroxytamoxifen. We further demonstrate that after incubation with breast cancer cells, the majority of the bioisosteres has been converted to 4-hydroxytamoxifen. Our study suggests that boron-based 4-hydroxytamoxifen bioisosteres may be an effective therapeutic remedy for intrinsic tamoxifen resistance in breast cancer patients deficient in CYP2D6 metabolism.
Keywords: 4-hydroxytamoxifen bioisosteres/4-hydroxytamoxifen prodrugs, CYP2D6, boron–carbon bond, breast cancer
Tamoxifen has been the mainstay hormonal therapy for breast cancer that expresses the estrogen receptor (ER+) since the 1980s. Nearly 70% of all diagnosed breast tumors are classified as ER+, most of which initially respond to tamoxifen treatment.1 However, approximately 8% of breast cancer patients present intrinsic resistance to tamoxifen2 mainly because these patients do not have a functional P450 2D6 enzyme (CYP2D6) that is responsible for converting tamoxifen to its more potent metabolites, 4-hydroxytamoxifen (4-OHT) and endoxifen.3 Indeed, breast cancer mortality was significantly increased in patients with CYP2D6-null allele as compared to wild-type patients.4 Jordan and co-workers first reported that high first-pass metabolism of tamoxifen led to a significant increase in its activity and characterized the first active primary metabolite, 4-OHT,5,6 which showed 30–100-fold greater potency than tamoxifen in inhibiting estrogen-dependent cell proliferation.7–9
The predominant biotransformation route, demethylation of tamoxifen to form N-desmethyltamoxifen, is catalyzed by the P450 enzyme CYP3A4/5.10 This major primary metabolite has a potency similar to tamoxifen. It is then converted into the more potent secondary metabolite endoxifen exclusively by CYP2D6. On the other hand, the same CYP2D6 is required for the formation of 4-OHT, which is equally or more potent than endoxifen. While the hydroxylation at the 4C-position depends on CYP2D6, the biotransformation from 4-OHT to endoxifen does not. In fact, it has been shown that CYP3A4/5 is primarily responsible for this metabolic route of 4-OHT demethylation leading to endoxifen formation. Thus, both endoxifen and 4-OHT represent desirable alternatives to tamoxifen for breast cancer patients lacking the active form of the P450 enzyme CYP2D6.11–15
To achieve increased bioavailability of 4-OHT in CYP2D6 deficient breast cancer patients as well as enhanced drug selectivity toward tumor cells, we sought to explore the utility of a boron-based prodrug strategy. This strategy takes into consideration the need to have an effective level of the active form of tamoxifen and the preferential uptake of 4-OHT by cancer cells. To this end, we have designed three boronic derivatives of tamoxifen as 4-OHT prodrug candidates. Boronic acid has been previously used in imaging and medicinal chemistry offering unique advantages associated with its low toxicity and stability.16 It is well-known that the boron-aryl carbon bond is susceptible to oxidative cleavage by hydrogen peroxide to form a phenol compound.17–20 Because tumor cells have elevated concentration levels of hydrogen peroxide,21–23 it is anticipated that upon entering breast cancer cells, these prodrugs will undergo facile oxidization reaction to form 4-OHT (Figure 1). This approach may thus have the potential to increase the efficacy of hormone therapy by (1) increasing the concentration of 4-OHT in cancer cells, (2) providing the active and more potent form (4-OHT) of tamoxifen for patients with defective CYP2D6 polymorphism, and (3) allowing the formation of endoxifen as a downstream metabolic product with equivalent potency as 4-OHT independent of CYP2D6.
Figure 1.

Design of boron-based 4-OHT bioisosteres.
The three designed boron-containing derivatives of tamoxifen share the common boron-aryl carbon bond and are all expected to be transformed to 4-OHT upon oxidization, but each of them has its own feature. The boronic acid pinacol ester (4) was designed for attenuated polarity as compared to 4-OHT. The potassium trifluoroborate derivative (5) was intended to afford greater solubility of the prodrug. The boronic acid prodrug (6) was included for its increased stability.
The synthesis of boron-based 4-OHT bioisosteres is outlined in Scheme 1. 4,4′-(2-Phenylbut-1-ene-1,1-diyl)diphenol (1) was obtained by McMurry reaction of 4,4′- dihydroxybenzophenone with propiophenone. The monoalkylaton of 1 by 2-(dimethylamino)ethylchloride hydrochloride in the presence of cesium carbonate in DMF gave 4-OHT (2).24 Then, 4-OHT was converted into the triflate 3 by reacting with trifluoromethanesulfonic anhydride in CH2Cl2. The PdCl2(dppf)-catalyzed borylation of the triflate 3 gave the pinacolyl boronate ester of tamoxifen derivative 4 using a diboron reagent in the presence of KOAc.25,26 The reaction of the pinacolyl boronate ester 4 with potassium bifluoride formed the potassium trifluoroborate of tamoxifen 5. The free boronic acid prodrug 6 was obtained successfully via hydrolysis of 5 using trimethylsilyl chloride as an effective fluorophile.27
Scheme 1.

Preparation of Boron-Based 4-OHT Bioisosteres
Reagents and conditions: (a) Propiophenone, TiCl4, Zn, THF, reflux. (b) 2-(Dimethylamino)ethylchloride hydrochloride, Cs2CO3, DMF, 70–80 °C. (c) (CF3SO2)2O, pyridine, CH2Cl2, 0 °C–rt. (d) 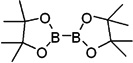 , PdCl2(dppf), KOAc, dioxane, reflux. (e) KHF2, MeOH/H2O, rt. (f) (CH3)3SiCl, CH3CN, H2O, rt.
, PdCl2(dppf), KOAc, dioxane, reflux. (e) KHF2, MeOH/H2O, rt. (f) (CH3)3SiCl, CH3CN, H2O, rt.
To determine the antiestrogenic effects of the prodrugs, we performed breast cancer cell survival assays in which the ability of the prodrugs to inhibit growth of MCF-7 and T47D cells was measured. The 4-OHT treated cell survival data were used as positive controls. MCF-7 and T47D breast cancer cells both express the estrogen receptor, and their growth and proliferation depend on estrogen. Antiestrogenic compounds such as tamoxifen and ICI 182780 (Fulvestrant) are used to treat hormone responsive breast cancer. Thus, MCF-7 and T47D cell lines have been widely used as cellular models for ER+ breast tumors.
As shown in Figure 2A, tamoxifen inhibited MCF-7 cell survival and growth by 18 and 52% at 10−7 and 10−6 M, respectively. 4-OHT, the active metabolite of tamoxifen, more strongly inhibited growth of MCF-7 cells at these two doses (by 56 and 63%, respectively). In comparison, the pinacolyl boronate ester prodrug (4) showed 52% growth inhibition at 10−7 M and 63% at 10−6 M, similar to the efficacy of 4-OHT. The trifluoroborate prodrug (5) demonstrated growth inhibition on MCF-7 cells with 59% at 10−7 M and 76% inhibition at 10−6 M concentration. The boronic acid prodrug (6) was found to inhibit cell growth with efficacies comparable to 5 at the two doses (51 and 75% inhibition, respectively). Additional survival assays using T47D cells confirmed the efficacies of the prodrugs (Figure 2B). Consistent with previous reports on the sensitivity of T47D and MCF-7 to tamoxifen,28 T47D cells exhibited slightly higher survival ratios than MCF-7 cells when treated with 10−7 and 10−6 M 4-OHT except tamoxifen at 10−7 M. For example, growth of T47D cells was inhibited by 51% in 10−7 M 4-OHT as compared to the 56% for MCF-7 cells. While the prodrugs were equally effective in T47D as compared to 4-OHT, their overall potency was also slightly lower than in MCF-7 cells. At 10−6 M, the prodrug 6 achieved 54% growth inhibitions of the T47D cells, lower than the 75% inhibition of MCF-7 cells; treatment of T47D cells with prodrugs 5 and 4 at 10−6 M dose resulted in 50 and 60% inhibition, respectively, versus 76 and 63% in MCF-7 cells. These results clearly demonstrate the effectiveness of the 4-OHT prodrugs as antiestrogens in inhibiting the growth of ER+ breast cancer cells represented by MCF-7 and T47D cell lines.
Figure 2.

Prodrugs of 4-OHT inhibit growth of (A) MCF-7 cells and (B) T47D cells.
Dose–response studies were performed to evaluate the antiestrogenic effects of the prodrugs on MCF-7 and T47D cells as compared with tamoxifen and 4-OHT. A dose–response curve for each drug was obtained yielding IC50 values as listed in Table 1. Consistent with previous studies,6 the IC50 concentration of 4-OHT in MCF-7 cells was observed at 0.029 µM, nearly 30 times more potent than that of tamoxifen (0.79 µM). In comparison, the boron prodrug 4 exhibited an IC50 concentration of 0.15 µM, about five times higher than 4-OHT but five times lower than tamoxifen. The other two prodrugs, 5 and 6, had IC50 values of 0.0063 and 0.042 µM, respectively, again comparable or more active than 4-OHT (0.029 µM).
Table 1.
IC50 Values of Prodrugs in MCF-7 and T47D Cells
| IC 50 (µM) | ||
|---|---|---|
| drug | MCF-7 | T47D |
| tamoxifen | 0.794 | 1.13 |
| 4-OHtamoxifen | 0.0291 | 0.210 |
| boron prodrug (4) | 0.148 | 0.144 |
| boron prodrug (5) | 0.00626 | 0.260 |
| boron prodrug (6) | 0.0420 | 0.228 |
To determine if the prodrugs undergo oxidative cleavage by intracellular hydrogen peroxide present at elevated concentration levels in the breast cancer cells, we analyzed the cell culture media for concentrations of the prodrugs and the active form of tamoxifen, 4-OHT. Using HPLC coupled to a linear trap mass spectrometer, we were able to separate, identify, and quantify the prodrugs and its active product, 4-OHT. As shown in the total ion chromatogram (TIC) in Figure 3A, the boronic acid prodrug of 4-OHT (6) was observed at 17.3 min, while the desired active drug form, 4-OHT, eluted at 17.6 min. The relative peak areas were measured at 30.4 (prodrug 6) and 69.6% (4-OHT), respectively, indicating that the majority of the prodrug has been converted to 4-OHT in the media after incubation with MCF-7 cells. This transformation was more complete in T47D cells where the % peak area of 4-OHT reached 79.1 versus 20.9% for the prodrug 6 (Figure 3B). For prodrugs 4 and 5, the % peak areas of 4-OHT were 73.3 and 81.6%, respectively, in MCF-7 cells. Similar results were observed when T47D cells were treated with prodrugs 4 and 5, where 69.9% of 4 and 86.3% of 5 were transformed into 4-OHT.
Figure 3.

Relative concentrations of prodrugs and their common oxidative product, 4-OHT, as determined by LC-MS/MS after incubation with (A) MCF-7 cells and (B) T47D cells.
We next sought to investigate the in vitro stability of the boron prodrugs using MCF-7 cells. By monitoring the prodrug and 4-OHT concentrations in the media at 11 time points (0–144 h), the half-life of each prodrug, namely, the point where half of the prodrug has been converted to 4-OHT, was determined. The time-dependent oxidative conversion to 4-OHT for each boron prodrug yielded a half-life of 96 h, or 4 days, for prodrug (6) as illustrated in the Supporting Information, Figure 1S. Similar half-life data were obtained for prodrug (4) (89 h) and prodrug (5) (84 h).
The modification of 4-OHT structure by incorporation of boron may have improved the uptake of the prodrugs by breast cancer cells. To compare the relative uptake of the prodrugs versus 4-OHT, we measured the concentrations of the remaining drugs in media after a 6 day period of incubation with MCF-7 cells (Supporting Information, Figure 2S). Results show that the concentration of 4-OHT in media is approximately 2–4 times higher than those of prodrugs 4–6. While it is also possible that the boron prodrugs may have their own antiestrogenic activities, our experimental data suggest that the prodrugs were more efficiently uptaken by the cancer cells.
Finally, to confirm that the boron prodrugs, like their resulting active form 4-OHT, exert the growth inhibitory effects on breast cancer cells through the estrogen receptor expressed in both MCF-7 and T47D, we tested the in vitro activity of the prodrugs in an ER negative breast cancer cell line, MDA-MB-231. Results (Supporting Information, Figure 3S) show that at 10−7 M, tamoxifen and 4-OHT both had minimal effect on cell growth with greater than 95% survival ratio. The prodrugs 4–6 showed no inhibition of the growth of MDA-MB-231 cells while appearing to stimulate growth to various degrees. In contrast, the boron prodrugs all inhibited the growth of MCF-7 cells by over 60%. These data suggest that the inhibitory effect of boron prodrugs on breast cancer cells is ER dependent.
In summary, we have synthesized three 4-OHT prodrugs by linking a cleavable aryl carbon–boron bond to the tamoxifen moiety. The utility of such prodrugs is based on the premise that they can be converted rapidly to the active form of the antiestrogen under favorable oxidative conditions in breast cancer cells with elevated levels of hydrogen peroxide. The cell survival assay showed that the prodrugs were equally or more effective in inhibiting the growth of MCF-7 and T47D breast cancer cells, as compared to 4-OHT. Analysis of the prodrugs and 4-OHT concentrations in the cell culture media suggests that inhibition of the ER expressing breast cancer cells by the prodrugs may be attributable to their increased uptake by the cancer cells and efficient conversion into the active form of tamoxifen, 4-OHT. Our study demonstrated the feasibility of using boron chemistry as a prodrug strategy for 4-OHT. These 4-OHT prodrugs may significantly improve response to antiestrogen therapy in patients with pharmacogenetic variations or drug interactions that affect the activity of CYP2D6. Further studies of the anticancer mechanism by boron-based 4-OHT prodrugs are in progress.
Supplementary Material
Acknowledgments
Funding
This work was supported by a NIH RCMI program through Grant 5G12RR026260-03, the Louisiana Cancer Research Consortium (LCRC), Department of Agriculture Grant 58-6435-7-019, and Office of Naval Research Grant N00014-99-1-0763.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures for the synthesis and characterization of the 4-OHT bioisosteres, cell culture and survival assay, and analysis of 4-OHT and 4-OHT bioisostere concentrations in culture media. Additional data of drug uptake in breast cancer cells, time-dependent conversion from prodrug to 4-OHT, and the in vitro activities of the prodrug in an ER negative breast cancer cell line. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999;17:1474. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 2.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 3.Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat. Rev. Cancer. 2009;9:576–586. doi: 10.1038/nrc2683. [DOI] [PubMed] [Google Scholar]
- 4.Bijl M, van Schaik R, Lammers L, Hofman A, Vulto A, van Gelder T, Stricker B, Visser L. The CYP2D6*4 polymorphism affects breast cancer survival in tamoxifen users. Breast Cancer Res. Treat. 2009;118:125–130. doi: 10.1007/s10549-008-0272-2. [DOI] [PubMed] [Google Scholar]
- 5.Jordan VC, Collins MM, Rowsby L, Prestwich G. A Monohydroxylated metabolite of tamoxifen with potent antiestrogenic activity. J. Endocrinol. 1977;75:305–316. doi: 10.1677/joe.0.0750305. [DOI] [PubMed] [Google Scholar]
- 6.Jordan VC. Metabolites of tamoxifen in animals and man: identification, pharmacology, and significance. Breast Cancer Res. Treat. 1982;2:123–138. doi: 10.1007/BF01806449. [DOI] [PubMed] [Google Scholar]
- 7.Robertson DW, Katzenellenbogen JA, Long DJ, Rorke EA, Katzenellenbogen BS. Tamoxifen antiestrogens. a comparison of the activity, pharmacokinetics, and metabolic activation of the cis and trans Isomers of tamoxifen. J. Steroid Biochem. 1982;16:1–13. doi: 10.1016/0022-4731(82)90137-6. [DOI] [PubMed] [Google Scholar]
- 8.Coezy E, Borgna J-L, Rochefort H. Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res. 1982;42:317–323. [PubMed] [Google Scholar]
- 9.Furr BJA, Jordan VC. The pharmacology and clinical uses of tamoxifen. Pharmacol. Ther. 1984;25:127–205. doi: 10.1016/0163-7258(84)90043-3. [DOI] [PubMed] [Google Scholar]
- 10.Desta Z, Ward BA, Soukhova NV, Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. J. Pharmacol. Exp. Ther. 2004;310:1062–1075. doi: 10.1124/jpet.104.065607. [DOI] [PubMed] [Google Scholar]
- 11.Pujol H, Girault J, Rouanet P, Fournier S, Grenier J, Simony J, Fourtillan J-B, Pujol J-L. Phase I study of percutaneous 4-hydroxy-tamoxifen with analyses of 4-hydroxy-tamoxifen concentrations in breast cancer and normal breast tissue. Cancer Chemother. Pharmacol. 1995;36:493–498. doi: 10.1007/BF00685799. [DOI] [PubMed] [Google Scholar]
- 12.Rouanet P, Linares-Cruz G, Dravet F, Poujol S, Gourgou S, Simony-Lafontaine J, Grenier J, Kramar A, Girault J, Le Nestour E, Maudelonde T. Neoadjuvant percutaneous 4-hydroxytamoxifen decreases breast tumoral cell proliferation: A prospective controlled randomized study comparing three doses of 4-hydroxytamoxifen gel to oral tamoxifen. J. Clin. Oncol. 2005;23:2980–2987. doi: 10.1200/JCO.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 13.Mansel R, Goyal A, Nestour E, Masini-Etévé V, O’Connell K. A phase II trial of afimoxifene (4-hydroxytamoxifen gel) for cyclical mastalgia in premenopausal women. Breast Cancer Res. Treat. 2007;106:389–397. doi: 10.1007/s10549-007-9507-x. [DOI] [PubMed] [Google Scholar]
- 14.Wu X, Hawse JR, Subramaniam M, Goetz MP, Ingle JN, Spelsberg TC. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor α for degradation in breast cancer cells. Cancer Res. 2009;69:1722–1727. doi: 10.1158/0008-5472.CAN-08-3933. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad A, Ali S, Ahmad M, Sheikh S, Ahmad I. Orally administered endoxifen is a new therapeutic agent for breast cancer. Breast Cancer Res. Treat. 2010;122:579–584. doi: 10.1007/s10549-009-0704-7. [DOI] [PubMed] [Google Scholar]
- 16.Trippier PC, McGuigan C. Boronic acids in medicinal chemistry: Anticancer, antibacterial and antiviral applications. Med. Chem. Commun. 2010;1:183–198. [Google Scholar]
- 17.Ainley AD, Challenger F. Studies of the boron-carbon linkage. Part I. The oxidation and nitration of phenylboric acid. J. Chem. Soc. 1930;2171 [Google Scholar]
- 18.Kuivila HG, Armour AG. Electrophilic displacement reactions. ix. effects of substituents on rates of reactions between hydrogen peroxide and benzeneboronic acid. J. Am. Chem. Soc. 1957;79:5659–5662. [Google Scholar]
- 19.Chang MC, Pralle A, Isacoff EY, Chang CJ. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. J. Am. Chem. Soc. 2004;126:15392–15393. doi: 10.1021/ja0441716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du L, Li M, Zheng S, Wang B. Rational design of a fluorescent hydrogen peroxide probe based on the umbelliferone fluorophore. Tetrahedron Lett. 2008;49:3045–3048. doi: 10.1016/j.tetlet.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 22.Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, Petros JA, Arnold RS. Increased nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62:200–207. doi: 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
- 23.Miguel L-L. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007;252:1–8. doi: 10.1016/j.canlet.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 24.Yu DD, Forman BM. Simple and efficient production of (Z)-4-hydroxytamoxifen, a potent estrogen receptor modulator. J. Org. Chem. 2003;68:9489–9491. doi: 10.1021/jo035164n. [DOI] [PubMed] [Google Scholar]
- 25.Ishiyama T, Murata M, Miyaura N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]
- 26.Thompson ALS, Kabalka GW, Akula MR, Huffman JW. The conversion of phenols to the corresponding aryl halides under mild conditions. Synthesis. 2005:547–550. [Google Scholar]
- 27.Yuen AKL, Hutton CA. Deprotection of pinacolyl boronate esters via hydrolysis of intermediate potassium trifluoroborates. Tetrahedron Lett. 2005;46:7899–7903. [Google Scholar]
- 28.McClaine RJ, Marshall AM, Wagh PK, Waltz SE. Ron receptor tyrosine kinase activation confers resistance to tamoxifen in breast cancer cell lines. Neoplasia. 2010;12:650–658. doi: 10.1593/neo.10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.