

Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic, relentlessly progressive fibrosing disease of the lung of unknown etiology. Significant progress has been made in recent years in elucidating key aspects of the pathobiology of IPF. Insights into disease pathogenesis have come from studies of cell biology, growth factor/cytokine signaling, animal models of pulmonary fibrosis, and human IPF cells and tissue. A consistent finding in the ultrastructural pathology of IPF is alveolar epithelial cell injury and apoptosis. Another consistent finding in the histopathology of human IPF, described as usual interstitial pneumonia, is the accumulation of aggregates of myofibroblasts in fibroblastic foci. The extent or profusion of fibroblastic foci in lung biopsies is strongly correlated with increased mortality in patients with IPF. There is emerging evidence that myofibroblasts in IPF/usual interstitial pneumonia, both in the in vivo microenvironment and during the process of differentiation in vitro, acquire resistance to apoptosis. Here, we review the current evidence and mechanisms for this apparent “apoptosis paradox” in the pathogenesis of IPF.
Keywords: epithelial cells, fibroblasts, oxidative stress, transforming growth factor β, wound healing
Orchestrated cell death is critical for maintenance of postnatal adult tissue homeostasis as it eliminates “abnormal” resident cells that may be irreparably damaged, infected, or genetically defective. In addition, during the resolution phase of tissue injury or infection, elimination of both recruited inflammatory cells and mesenchymal cells that participate in tissue repair functions is necessary for restoration of normal cellular homeostasis and the maintenance of tissue architecture and organ function. Thus, apoptosis and subsequent phagocytic clearance of inflammatory cells is required for the resolution of inflammation. Similarly, the orchestrated death and removal of reparative cells, such as myofibroblasts, is expected to be necessary for the normal resolution of tissue repair responses (1). Resistance to apoptosis of inflammatory cells may result in a chronic inflammatory process; impaired apoptosis of myofibroblasts may result in a persistent and dysregulated repair process that culminates in tissue fibrosis. Indeed, myofibroblast accumulation and activation are key features of the pathobiology of usual interstitial pneumonia (UIP), the pathologic correlate of idiopathic pulmonary fibrosis (IPF) (1, 2). Exaggerated apoptosis of certain cell types can also result in deranged tissue homeostasis and contribute to disease pathogenesis (e.g., myocyte loss in cardiomyopathies and neuronal cell loss in chronic neurodegenerative diseases). Re-epithelialization is well recognized to be a prerequisite for epithelial regeneration during normal wound repair. Increased apoptosis of alveolar epithelial cells (AECs) may result in ineffective re-epithelialization of a damaged alveolar wall, thus promoting a fibrotic tissue-repair response. Interactions with inflammatory cells and endothelial cells may also occur and contribute to this fibrotic response (1); our discussion here is primarily focused on apoptotic phenotypes of alveolar/bronchiolar epithelial cells and fibroblasts/myofibroblasts and their interactions.
Three major types of cell death have been described—apoptosis, autophagy, and necrosis—that may all contribute to disease pathogenesis in different contexts. Apoptosis and autophagy are morphologically distinct forms of programmed cell death (type I and type II, respectively) that occur in the absence of an inflammatory response, typically associated with necrotic cell death. Apoptosis is distinguished from autophagy by the early loss of cytoskeletal elements, relative preservation of intracellular organelles (until late), and a central role for proteolytic enzymes known as caspases. Although much of accumulated data to date on pulmonary fibrosis relate to apoptotic cell death, it must be recognized that there is significant overlap between these different modes of cell death and clear-cut distinctions are not always evident and may not always be possible. Apoptosis regulation is likely to be important in a wide spectrum of injury and repair processes that constitute the idiopathic interstitial pneumonias (IIPs). The focus of this review will be on IPF/UIP, the most common of the IIPs; the extent to which regulation of apoptosis in specific lung cells differentiates IPF/UIP from other IIPs, such as nonspecific interstitial pneumonia (NSIP), is currently not known.
APOPTOSIS SIGNALING—AN OVERVIEW
Apoptosis is the classical (type I) form of programmed cell death that results in the dismantling of the entire cell within the context of membrane-enclosed vesicles that are recognized and engulfed by phagocytes; this prevents the release of intracellular components from dying cells and the activation of innate immune responses. Apoptosis is executed by the concerted action of a set of cysteine proteases known as caspases. Distinct pathways to apoptosis converge on the activation of effector caspases. Apoptosis can be induced by the stimulation of those members of the tumor necrosis factor–receptor (TNF-R) super family that have a death domain—this is mediated by the extrinsic pathway; alternatively, apoptosis may be induced by “internal” cellular stressors such as DNA damage or growth factor withdrawal—this is mediated by the intrinsic pathway.
An exhaustive review of the intrinsic and extrinsic pathways to cellular apoptosis is beyond the scope of this discussion (for recent reviews, see References 3–5). In brief, the extrinsic pathway is triggered by extracellular signaling “death” ligands such as Fas, which binds to the Fas receptor, CD95, a member of the TNF-R family. Receptor ligation results in multimerization/activation of the receptor that recruits the adaptor protein, Fas-associated death domain. This leads to the assembly of a death-inducing signaling complex that includes an initiator caspase, procaspase-8 or -10. Activated caspase-8 cleaves/activates the effector caspases, caspase-3 and -7, which execute the apoptotic cell death program. The intrinsic pathway is triggered by intracellular “stress” signals, such as DNA damage or starvation that typically activate the BH3-only (proapoptotic) members of the Bcl-2 family proteins. This induces mitochondrial release of cytochrome c, which directly activates apoptosis protease-activating factor 1 (Apaf-1) and forms the multimeric complex known as the “apoptosome.” The apoptosome mediates the activation of the initiator caspase, caspase-9, which subsequently activates caspase-3 and -7, thus converging with the extrinsic pathway at the level of these effector caspases to execute apoptosis. Activated caspases are subject to negative regulation by the inhibitor of apoptosis (IAP) family of proteins, including the X-linked IAP (XIAP) (6). The antiapopotic activities of IAP proteins are, in turn, counterregulated by the second mitochondria-derived activator of caspase (Smac/DIABLO), another protein that is released by mitochondria during apoptosis. Finally, convergence and crosstalk between the extrinsic and intrinsic pathways can also occur at an upstream level; this is typically mediated by Bid, a BH3-only protein that is a physiologic target of activated caspase-8 (of the extrinsic pathway); activation of Bid can then lead to activation of the intrinsic (mitochondrial) pathway.
Loss of cell adhesion or adhesion signaling can lead to a form of apoptosis called anoikis. Anoikis is likely to represent an important apoptotic mechanism in structural cells, including epithelial cells and mesenchymal cells, which are normally adherent to the underlying basement membrane or other extracellular matrix (ECM) substrates. Mechanisms of anoikis have not been fully elucidated but may operate via different pathways in a cell-specific manner. Such mechanisms typically involve loss of “tonic” or constitutive adhesion signals—for example, focal adhesion kinase (FAK) and integrin-linked kinase (ILK). Detachment of cells from the ECM may also induce alterations in cell shape and actin/microtubular cytoskeleton reorganization that allow for translocation and activation of proapoptotic proteins (Bim, Bmf) of the Bcl-2 family (7, 8). Thus, in many ways, anoikis is mechanistically akin to an intrinsic form of apoptosis regulation and induction.
SURVIVAL (ANTIAPOPTOTIC) SIGNALING—AN OVERVIEW
Cellular microenvironmental cues may also promote cell survival and the acquisition of an apoptosis-resistant phenotype. Acquisition of an antiapoptotic/prosurvival phenotype is classically mediated by extracellular signaling by soluble trophic factors as well as by signaling from cell–cell or cell–matrix adhesion complexes. Two major pathways for cell survival signaling have emerged: (1) the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (activated primarily by soluble growth factors) and (2) the integrin/FAK pathway (activated primarily by cell adhesion).
The PI3K–Akt Pathway
The PI3Ks constitute a diverse family primarily responsible for conveying growth and survival signals from activated growth factor/cytokine receptors (9, 10). PI3K contains an 85-kD regulatory subunit that recruits the 110-kD catalytic subunit of PI3K into close proximity with the plasma membrane. Here, PI3K catalyzes the phosphorylation of inositol phospholipids, producing phosphoinositol (3–5)-triphosphate (PtdIns[3,4,5]P3) (PIP3). PIP3 is a lipid mediator that recruits proteins that contain a pleckstrinhomology domain, such as Akt, and kinases that activate Akt, the PIP3-dependent kinases, to the plasma membrane. This allows PIP3-dependent kinase to phosphorylate and activate Akt. Phosphorylation of Akt is primarily mediated at two regulatory sites: a threonine (residue 308) and a serine (residue 473). Ser-473 phosphorylation parallels the activation of PI3K and is required for activation of Akt.
The PI3K–Akt pathway mediates prosurvival/antiapoptotic signaling by several pathways. Phosphorylation of Bad by Akt promotes survival by preventing Bad from binding and inhibiting antiapoptotic Bcl-XL. Akt phosphorylates a member of the forkhead family of transcription factors, forkhead (Drosophila) homolog (rhabdomyosarcoma)-like 1 (FKHRL1), promoting its association with 14−3−3 proteins. This retains FKHRL1 in the cytoplasm and prevents the transcriptional activation of genes that promote apoptosis, including Fas ligand. Akt promotes the activation of nuclear factor (NF)-κB by phosphorylation/activation of IκB kinase α. This augments the transcriptional activity of p65 Rel, a subunit of NF-κB, and regulates the expression of genes important in cell survival. Akt also phosphorylates and inhibits the activity of caspase-9, a key initiator in the enzyme cascade that promotes cell death. Akt has recently been shown to phosphorylate XIAP, reducing its ubiquitination/degradation and thereby sustaining higher cellular levels of XIAP (11).
The Integrin–FAK Pathway
The ECM/integrin–FAK pathway may mediate prosurvival signals in addition to its roles in cellular proliferation, migration, and differentiation (12–14). Integrins are the major cell adhesion receptors for ECM ligands that mediate adhesion-dependent signaling in diverse biological processes, including cell proliferation, migration, and survival. Adhesion-dependent integrin activation recruits FAK and activates it at focal adhesions. FAK activation is mediated primarily by autophosphorylation of Tyr-397, which creates a binding site for the Src homology 2 (SH2) domain of Src and PI3K.
The ECM transmits survival signals to adherent cells; the absence of such signals elicits apoptosis or anoikis. Inhibition of FAK can induce apoptosis and overexpression of FAK can prevent anoikis and apoptosis induced in response to other stimuli. The mechanisms by which FAK mediates prosurvival signals are incompletely understood. Because FAK autophosphorylation at Tyr-397 appears to be necessary for protection against anoikis, FAK association with Src or PI3K is thought to be necessary.
APOPTOSIS OF AECS IN IPF
What Is the In Vivo Evidence for Apoptosis of AECs in IPF?
AEC injury and death are a consistent finding in human IPF/UIP. Early ultrastructural studies described damage to the epithelium with reactive epithelial cells (15–18). More recently, increased apoptosis of type II AECs has been demonstrated both in areas of IPF/UIP that appear histologically normal, without established fibrosis (19), and in epithelial cells overlying myofibroblasts (20). Increased expression of proapoptotic proteins and decreased expression of antiapoptotic proteins has also been reported (21). Thus, there is increasing evidence that at least certain subpopulations of AECs in IPF/UIP are undergoing apoptosis in association with inadequate or dysfunctional reparative responses of the alveolar and bronchiolar epithelium.
Is Apoptosis of AECs Sufficient to Induce Pulmonary Fibrosis?
Although AEC injury/apoptosis is evident in IPF/UIP, cause–effect relationships have not been clearly established. Data from animal models have provided some insights into whether excessive AEC apoptosis—as the primary event—may lead to lung fibrogenesis. Induction of AEC apoptosis by aerosolized anti-Fas (activating) antibody is associated with the development of pulmonary fibrosis in mice (22). Targeted transgenic overexpression of bioactive transforming growth factor (TGF)-β1 to the murine lung produces a transient wave of epithelial apoptosis that is followed by mononuclear-rich inflammation, tissue fibrosis, myofibroblast hyperplasia, and honeycombing (23). This study also demonstrated that a null mutation of early growth response gene (EGF)-1 or caspase inhibition rescues the fibrotic phenotype, suggesting a role for EGR-1 in TGF-β1–induced epithelial apoptosis. Moreover, the commonly used murine model of bleomycin-induced pulmonary fibrosis is characterized by early induction of reactive oxygen species (ROS)-dependent AEC apoptosis, which is inhibited by caspase inhibitors (24).
What Are the Cellular and Molecular Mechanisms for Injury/Apoptosis to AECs?
Despite the observation that AECs in IPF/UIP appear to be injured with high rates of apoptosis, the causative factors are not known and mechanisms of cell death remain unclear. Both the intrinsic (25) and extrinsic pathways (26–28) have been implicated (Figure 1). Type II AECs express high levels of p53 in association with DNA strand breaks in patients with IPF (21, 29). p53 is a key regulator of the checkpoint response that typically mediates cell cycle arrest, DNA repair, and apoptosis in response to DNA damage. The up-regulation of p53 expression may be further influenced by attenuated conjugation of p53 with Mdm2, which typically triggers p53 degradation (30). Oxidative stress has been proposed as a mechanism for the alveolar epithelial cell injury in IPF (31, 32). Evidence of oxidative DNA damage, specifically by the formation of 8-hydroxy-deoxyguanosine, and the expression of an enzyme that prevents this DNA modification are detected in AECs of patients with IIPs (33). The cellular sources for ROS generation that induce AEC injury/death in IPF have traditionally been thought to represent alveolar inflammatory cells (31, 34); however, more recent studies suggest that structural cells of lung, in particular activated myofibroblasts, produce significant concentrations of extracellular ROS that is sufficient to induce injury/apoptosis of adjacent epithelial cells (35).
Figure 1.
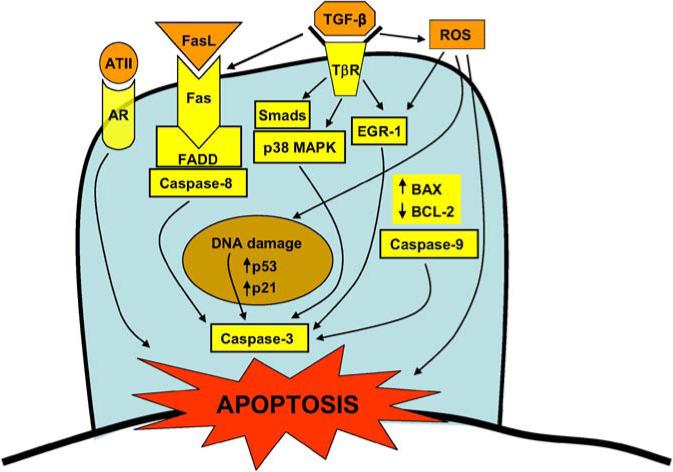
Proposed proapoptotic mediators and pathways activated in alveolar epithelial cells of idiopathic pulmonary fibrosis (IPF). Additional proapoptotic mechanisms in epithelial cells that may need to be considered include latent viral infections and acquired defects in protein processing and trafficking (not depicted—see text for details). AR = angiotensin receptor (subtype I); ATII = angiotensin II; BAX = BCL-2–associated protein; BCL-2 = B-cell lymphoma 2; EGR-1 = early growth response protein-1; FADD = Fas-associated death domain; Fas = apoptosis-mediating surface antigen (aka Apo-1 and CD95); FasL = Fas ligand; p38 MAPK = p38 mitogen-activated protein kinase; ROS = reactive oxygen species; Smads = mothers against decapentaplegia in Drosophila (Mad) gene and the related Sma gene in Caenorlabditis elegans.
Epithelial–mesenchymal interactions are important in normal developmental organogenesis as well as in the repair/regeneration of postnatal adult tissues. Dysregulated communication between these cellular compartments may result in fibrotic tissue responses (1). Several pericellular factors may contribute to this dysregulated communication. AEC apoptosis/necrosis adjacent to underlying α-smooth muscle actin–expressing myofibroblasts has been demonstrated (20); this effect may involve the secretion of angiotensin-II by myofibroblasts (36). In addition, myofibroblast-differentiated cells are potent generators of extracellular ROS, specifically hydrogen peroxide (37). Myofibroblasts isolated from IPF lung tissue, when activated by exogenous TGF-β1, secrete large amounts of hydrogen peroxide that may function in a paracrine manner to induce death of overlying lung epithelial cells (35). Moreover, TGF-β1 itself has been shown to be an enhancer of Fas-mediated AEC apoptosis (38). Thus, myofibroblast-secreted products (angiotensin-II, Fas, and ROS) may function, directly or indirectly, to induce apoptosis of adjacent AECs (Figure 1).
Are there other potential extrinsic or intrinsic factors that may induce AEC apoptosis in susceptible hosts? Environmental triggers such as latent viral infections have been proposed in the initiation or exacerbation of IPF (39). Viral hepatitis B and C have been linked to hepatocyte apoptosis with subsequent liver fibrosis (40). Moreover, viruses may induce or contribute to oxidative stress (41). Intrinsic factors specific to cellular metabolism and function, including protein folding and trafficking, may contribute to cell injury and apoptosis. An inherited mutation in surfactant protein C (SP-C), a type II AEC secreted protein, that predicts a glutamine to leucine residue appears to hinder processing of the precursor protein; this SP-C precursor protein displays aberrant subcellular localization with formation of numerous abnormal lamellar bodies and cellular atypia (42). Interestingly, mutations in the surfactant SP-C gene are associated with familial desquamative and nonspecific interstitial pneumonitis in children and UIP in adults. A similar defect in surfactant processing and secretion appears to result in “giant lamellar body degeneration” of type II AECs observed in the Hermansky-Pudlak syndrome, which is associated with mutations in the Hermansky-Pudlak syndrome-1 gene and interstitial pneumonia (43). The potential role of defects in protein processing and trafficking in sporadic cases of IIPs is unknown; these observations, nonetheless, reinforce the importance of gene–environment interactions in the expression of varied host tissue reactions and phenotypes.
SURVIVAL (APOPTOSIS RESISTANCE) OF MYOFIBROBLASTS IN IPF
What Is the In Vivo Evidence for Resistance to Apoptosis of Myofibroblasts in IPF?
There is currently no conclusive evidence that resistance to apoptosis is responsible for the persistence of myofibroblasts in fibroblastic foci of IPF/UIP. However, studies of human IPF/UIP tissues demonstrating high rates of apoptosis and increased levels of apoptosis-related markers in AECs generally show relatively little evidence of apoptosis in adjacent fibroblasts/myofibroblasts (20, 21, 44). Moreover, myofibroblasts in fibroblastic foci of IPF/UIP have been reported to undergo less apoptotic activity in comparison to myofibroblasts in the fibromyxoid lesions of bronchiolitis obliterans organizing pneumonia (BOOP) (45). Few studies have explored altered apoptosis/survival signaling in fibroblasts/myofibroblasts of human IPF. In patients with persistent/nonresolving acute lung injury, a disorder that is often associated with fibroproliferative responses, constitutive activation of prosurvival signaling pathways and an antiapoptotic phenotype in alveolar mesenchymal cells have been demonstrated (46). This supports the concept that the acquisition of an apoptosis-resistant phenotype of mesenchymal cells, which participate in the lung injury/repair process, may contribute to persistent or progressive fibrosis.
Are Alterations in Mesenchymal Cells Sufficient to Induce Fibrogenic Tissue Responses?
A serendipitous finding in a transgenic animal model does suggest that genetic alterations in mesenchymal cells, at least in principle, might be sufficient to initiate and promote a fibrogenic tissue response. Denton and colleagues expressed a kinase-deficient type II TGF-β receptor (TβRIIΔk) in fibroblasts of transgenic mice, using a potent fibroblast-specific transcriptional enhancer within the far upstream region of the mouse pro-α2(I) collagen gene (47). This led to paradoxical ligand-dependent activation of downstream TGF-β signaling and the development of spontaneous skin and lung fibrosis (48). Subsequent analysis of the fibroblasts derived from these transgenic mice suggested activation of an autocrine TGF-β1 loop with low-level transgene expression, whereas a dominant-negative effect was observed only with high-level expression of the transgene (49). The “autoactivated” fibroblast phenotype was associated with heightened type I TGF-β receptor (ALK5) activity, myofibroblast differentiation and altered production of matrix metalloproteinases. These studies provide proof-of-concept that a mesenchymal cell–specific genetic alteration—in this case, sustained activation of TGF-βsignaling in myofibroblasts—is sufficient to promote, perpetuate, or even initiate the fibrotic tissue phenotype.
What Are the Cellular and Molecular Mechanisms for Apoptosis Resistance in Mesenchymal Cells?
Elimination of myofibroblasts by apoptosis is essential during normal cutaneous wound healing (50), a process that may be hindered in fibrotic disorders. The combined effects of reduced growth factor expression, increased extracellular matrix turnover, and nitric oxide generation were proposed as the mechanism for fibroblast apoptosis seen during remodeling of skin-flap-induced regression of granulation tissue (51). Moroever, strong correlation between declining levels of TGF-β1 expression, reduced myofibroblast presence, and histopathological improvement in fibrosis is observed later in the course of bleomycin-induced pulmonary fibrosis in rodents (52, 53). Thus, altered or dysregulated cellular microenvironmental cues may permit the persistent survival of myofibroblasts in progressive fibrotic disorders.
The alveolar microenvironment in IPF/UIP is characterized by the persistent and high concentrations of fibrogenic cytokines/growth factors, ROS, and other reactive biomolecules in association with damaged basement membranes and altered extracellular matrices. Why mesenchymal cells survive while epithelial cells succumb to apoptosis within the same alveolar microenvironment is not entirely clear. Inherent differences between these cell types exist. Human lung fibroblasts have been shown to be inherently resistant to Fas-mediated apoptosis, an effect that is associated with increased expression of inhibitors of the apoptotic pathway, specifically XIAP and Fas-like interleukin (IL)-1β–converting enzyme-inhibitory protein (FLIPL) (54) (Figure 2). In addition, differences between the senescent phenotypes of fibroblasts versus those of other cell types may help to explain this “paradox”: human diploid fibroblasts enter a stable growth arrest phenotype at the end of their life span and are resistant to apoptosis, whereas senescent endothelial cells acquire a proapoptotic phenotype (55).
Figure 2.
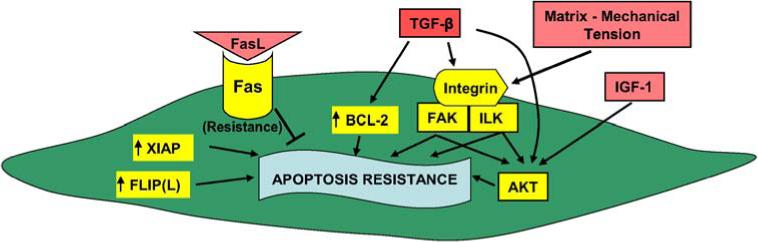
Proposed antiapoptotic (survival) mediators and pathways activated in fibroblasts/myofibroblasts of IPF. Akt = protein kinase B; FAK = focal adhesion kinase; FLIP(L) = Fas-like interleukin-1β–converting enzyme-inhibitory protein; IGF-1 = insulin growth factor-1; ILK = integrin-linked kinase; XIAP = X-linked inhibitor of apoptosis.
Fibroblasts/myofibroblasts isolated from lungs of patients with fibrotic lung disease show significant variability in proliferative and apoptotic properties when cultured in ex vivo conditions (56–59). It is difficult to draw definitive conclusions from these studies because the in vivo microenvironment, composed of a complex three-dimensional matrix and soluble factors, cannot be precisely reproduced ex vivo.TGF-β1, a profibrotic growth factor that is expressed at high levels in IPF tissues, has been shown to protect myofibroblasts from IL-1 (60) and serum-deprivation–induced apoptosis (46) when cells are grown on rigid, two-dimensional substrates. In this context, the acquisition of an apoptosis-resistant phenotype of myofibroblasts induced by TGF-β1 is, at least in part, due to the autocrine secretion of a soluble growth factor that activates the PI3K–Akt pathway (46). Interestingly, a recent study by Kobayashi and colleagues showed that TGF-β1 stimulates apoptosis of fibroblasts cultured in a contractile three-dimensional collagen gel, whereas no induction of apoptosis was noted in gels that were constrained from contraction (61). TGF-β1–induced myofibroblast differentiation is also dependent on the resistance of the substrate to deformation (62), suggesting that biomechanical signaling is a key regulator of this phenotype. On rigid substrates, TGF-β1 is capable of inducing the activation of cell adhesion/integrin-dependent FAK, a pathway that is required for the differentiation of fibroblasts to myofibroblasts (14); thus, this TGF-β1–activated FAK pathway may integrate biochemical and biomechanical signals for induction of myofibroblast differentiation and survival (Figure 2). Cumulatively, these studies suggest that the cellular context, particularly the nature of ECM, may dictate divergent signaling/physiologic responses to the soluble growth factor/morphogen TGF-β1.
There is an urgent need to understand alterations in the biochemical and biomechanical properties of the ECM in progressive fibrotic diseases such as IPF/UIP. Few studies have addressed this question in vivo. In a model of myofibroblast differentiation in vitro, biochemical cross-linking reactions in the matrix overlying myofibroblasts are induced in the context of oxidative stress (63). Further studies are required to determine if such matrix modifications occurs in vivo and how they might alter biomechanical properties of and signaling from the ECM. A better understanding of how such signals are transduced to generate apoptotic and survival signals is also required. Tian and colleagues showed that fibroblasts transfected with constitutively active PI3K display increased Akt phosphorylation and are protected from anoikis and collagen gel contraction-induced apoptosis (64). Furthermore, integrin β1 may activate FAK signaling upstream of PI3K-Akt to transduce viability signals to fibroblasts within collagen matrices (65). Thus, there appears to be significant crosstalk between the FAK and Akt signaling pathways that promote fibroblast/myofibroblast survival.
Strategies that induce fibroblast/myofibroblast apoptosis may prove to be an effective approach to the treatment of fibrotic lung diseases (66). Induction of fibroblast apoptosis may be achieved by blockade of cell surface–associated CD44 activity (67). Recent studies from our laboratory demonstrate that activation of FAK and Akt in fibrotic foci correlates with the emergence and persistence of myofibroblasts after bleomycin-induced lung injury in mice; systemic administration of protein kinase inhibitor that inhibits the activity of FAK and Akt reduces the accumulation of myofibroblasts and abrogates fibrotic responses to lung injury (68). This study provides proof-of-concept that the in vivo modulation of prosurvival protein kinase signaling pathways, highly activated in apoptosis-resistant myofibroblasts, may represent an effective antifibrotic therapeutic strategy.
CONCLUSIONS
Accumulating evidence supports the emergence of an “apoptosis paradox” in IPF—apoptosis susceptibility in epithelial cells and apoptosis resistance in myofibroblasts. Although progress has been made, we have much to learn of the underlying mechanisms for this paradox. For example, the profibrotic cytokine/morphogen TGF-β1 mediates primarily proapoptotic effects on epithelial cells and antiapoptotic effects on myofibroblasts—despite ligand activation of presumably the same cell surface TGF-β receptor complex. Postreceptor signaling mechanisms in these different cell types need to be elucidated. Another contrasting feature is the susceptibility of epithelial cells to ROS-induced injury and apoptosis whereas myofibroblasts appear to be relatively resistant to similar levels of oxidative stress. Epithelial cells that escape apoptosis appear to do so by adopting a mesenchymal cell fate—a process referred to as epithelial–mesenchymal transition (69); this likely further amplifies the fibrotic response and impedes alveolar re-epithelialization. Finally, epithelial–mesenchymal interactions may induce certain genetic or epigenetic alterations that permit the emergence of a fibroblast phenotype that is resistant to apoptosis (70). Deciphering the biochemical and molecular bases for differential apoptosis susceptibilities, cellular fate decisions, epigenetic changes, and the acquisition of apoptosis-resistant mesenchymal phenotypes will, ultimately, lead to more specific and targeted therapies for IPF.
Acknowledgment
The authors apologize for not citing potentially relevant articles by their colleagues in the field that may have been unintentionally overlooked; space limitations prohibited an exhaustive coverage of the literature pertinent to the subject matter in this short review. The authors thank the anonymous referees for their insightful thoughts and helpful suggestions.
Supported in part by National Institutes of Health grants R01 HL67967 and P50 HL74024 (V.J.T.) and K08 HL81059 (J.C.H.).
Footnotes
Conflict of Interest Statement: V.J.T. has a U.S. patent pending related to protein kinase inhibitor therapy for fibrotic diseases. J.C.H. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Thannickal VJ, Toews GB, White ES, Lynch JP, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 2.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 3.Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 4.Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol. 2005;21:35–56. doi: 10.1146/annurev.cellbio.21.012704.131040. [DOI] [PubMed] [Google Scholar]
- 5.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 6.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 7.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 8.Valentijn AJ, Zouq N, Gilmore AP. Anoikis. Biochem Soc Trans. 2004;32:421–425. doi: 10.1042/BST0320421. [DOI] [PubMed] [Google Scholar]
- 9.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 10.Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 11.Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang HG, Tsang BK, Cheng JQ. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J Biol Chem. 2004;279:5405–5412. doi: 10.1074/jbc.M312044200. [DOI] [PubMed] [Google Scholar]
- 12.Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ilic D, Almeida EA, Schlaepfer DD, Dazin P, Aizawa S, Damsky CH. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 15.Brody AR, Craighead JE. Interstitial associations of cells lining air spaces in human pulmonary fibrosis. Virchows Arch A Pathol Anat Histol. 1976;372:39–49. doi: 10.1007/BF00429715. [DOI] [PubMed] [Google Scholar]
- 16.Coalson JJ. The ultrastructure of human fibrosing alveolitis. Virchows Arch A Pathol Anat Histol. 1982;395:181–199. doi: 10.1007/BF00429611. [DOI] [PubMed] [Google Scholar]
- 17.Corrin B, Dewar A, Rodriguez-Roisin R, Turner-Warwick M. Fine structural changes in cryptogenic fibrosing alveolitis and asbestosis. J Pathol. 1985;147:107–119. doi: 10.1002/path.1711470206. [DOI] [PubMed] [Google Scholar]
- 18.Myers JL, Katzenstein AL. Epithelial necrosis and alveolar collapse in the pathogenesis of usual interstitial pneumonia. Chest. 1988;94:1309–1311. doi: 10.1378/chest.94.6.1309. [DOI] [PubMed] [Google Scholar]
- 19.Barbas-Filho JV, Ferreira MA, Sesso A, Kairalla RA, Carvalho CR, Capelozzi VL. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J Clin Pathol. 2001;54:132–138. doi: 10.1136/jcp.54.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol. 1998;275:L1192–L1199. doi: 10.1152/ajplung.1998.275.6.L1192. [DOI] [PubMed] [Google Scholar]
- 21.Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 22.Hagimoto N, Kuwano K, Miyazaki H, Kunitake R, Fujita M, Kawasaki M, Kaneko Y, Hara N. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am J Respir Cell Mol Biol. 1997;17:272–278. doi: 10.1165/ajrcmb.17.3.2893. [DOI] [PubMed] [Google Scholar]
- 23.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wallach-Dayan SB, Izbicki P, Cohen PY, Gerstl-Golan R, Fine A, Breuer R. Bleomycin initiates apoptosis of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290:L790–L796. doi: 10.1152/ajplung.00300.2004. [DOI] [PubMed] [Google Scholar]
- 25.Kuwano K, Hagimoto N, Maeyama T, Fujita M, Yoshimi M, Inoshima I, Nakashima N, Hamada N, Watanabe K, Hara N. Mitochondria-mediated apoptosis of lung epithelial cells in idiopathic interstitial pneumonias. Lab Invest. 2002;82:1695–1706. doi: 10.1097/01.lab.0000045084.81853.76. [DOI] [PubMed] [Google Scholar]
- 26.Kuwano K, Miyazaki H, Hagimoto N, Kawasaki M, Fujita M, Kunitake R, Kaneko Y, Hara N. The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am J Respir Cell Mol Biol. 1999;20:53–60. doi: 10.1165/ajrcmb.20.1.2941. [DOI] [PubMed] [Google Scholar]
- 27.Maeyama T, Kuwano K, Kawasaki M, Kunitake R, Hagimoto N, Matsuba T, Yoshimi M, Inoshima I, Yoshida K, Hara N. Upregulation of Fas-signalling molecules in lung epithelial cells from patients with idiopathic pulmonary fibrosis. Eur Respir J. 2001;17:180–189. doi: 10.1183/09031936.01.17201800. [DOI] [PubMed] [Google Scholar]
- 28.Kuwano K, Maeyama T, Inoshima I, Ninomiya K, Hagimoto N, Yoshimi M, Fujita M, Nakamura N, Shirakawa K, Hara N. Increased circulating levels of soluble Fas ligand are correlated with disease activity in patients with fibrosing lung diseases. Respirology. 2002;7:15–21. doi: 10.1046/j.1440-1843.2002.00369.x. [DOI] [PubMed] [Google Scholar]
- 29.Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, Hara N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996;154:477–483. doi: 10.1164/ajrccm.154.2.8756825. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima N, Kuwano K, Maeyama T, Hagimoto N, Yoshimi M, Hamada N, Yamada M, Nakanishi Y. The p53-Mdm2 association in epithelial cells in idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. J Clin Pathol. 2005;58:583–589. doi: 10.1136/jcp.2004.022632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cantin AM, North SL, Fells GA, Hubbard RC, Crystal RG. Oxidant-mediated epithelial cell injury in idiopathic pulmonary fibrosis. J Clin Invest. 1987;79:1665–1673. doi: 10.1172/JCI113005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1989;139:370–372. doi: 10.1164/ajrccm/139.2.370. [DOI] [PubMed] [Google Scholar]
- 33.Kuwano K, Nakashima N, Inoshima I, Hagimoto N, Fujita M, Yoshimi M, Maeyama T, Hamada N, Watanabe K, Hara N. Oxidative stress in lung epithelial cells from patients with idiopathic interstitial pneumonias. Eur Respir J. 2003;21:232–240. doi: 10.1183/09031936.03.00063203. [DOI] [PubMed] [Google Scholar]
- 34.Strausz J, Muller-Quernheim J, Steppling H, Ferlinz R. Oxygen radical production by alveolar inflammatory cells in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1990;141:124–128. doi: 10.1164/ajrccm/141.1.124. [DOI] [PubMed] [Google Scholar]
- 35.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005;19:854–856. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 36.Wang R, Ramos C, Joshi I, Zagariya A, Pardo A, Selman M, Uhal BD. Human lung myofibroblast-derived inducers of alveolar epithelial apoptosis identified as angiotensin peptides. Am J Physiol. 1999;277:L1158–L1164. doi: 10.1152/ajplung.1999.277.6.L1158. [DOI] [PubMed] [Google Scholar]
- 37.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 38.Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, Maeyama T, Hara N. TGF-beta 1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J Immunol. 2002;168:6470–6478. doi: 10.4049/jimmunol.168.12.6470. [DOI] [PubMed] [Google Scholar]
- 39.Egan JJ, Woodcock AA, Stewart JP. Viruses and idiopathic pulmonary fibrosis. Eur Respir J. 1997;10:1433–1437. doi: 10.1183/09031936.97.10071433. [DOI] [PubMed] [Google Scholar]
- 40.Hayashi N, Mita E. Involvement of Fas system-mediated apoptosis in pathogenesis of viral hepatitis. J Viral Hepat. 1999;6:357–365. doi: 10.1046/j.1365-2893.1999.00175.x. [DOI] [PubMed] [Google Scholar]
- 41.Buffinton GD, Christen S, Peterhans E, Stocker R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic Res Commun. 1992;16:99–110. doi: 10.3109/10715769209049163. [DOI] [PubMed] [Google Scholar]
- 42.Thomas AQ, Lane K, Phillips J, III, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 43.Nakatani Y, Nakamura N, Sano J, Inayama Y, Kawano N, Yamanaka S, Miyagi Y, Nagashima Y, Ohbayashi C, Mizushima M, et al. Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch. 2000;437:304–313. doi: 10.1007/s004280000241. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida K, Kuwano K, Hagimoto N, Watanabe K, Matsuba T, Fujita M, Inoshima I, Hara N. MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J Pathol. 2002;198:388–396. doi: 10.1002/path.1208. [DOI] [PubMed] [Google Scholar]
- 45.Lappi-Blanco E, Soini Y, Paakko P. Apoptotic activity is increased in the newly formed fibromyxoid connective tissue in bronchiolitis obliterans organizing pneumonia. Lung. 1999;177:367–376. doi: 10.1007/pl00007654. [DOI] [PubMed] [Google Scholar]
- 46.Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z, Thannickal VJ. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denton CP, Zheng B, Shiwen X, Zhang Z, Bou-Gharios G, Eberspaecher H, Black CM, de Crombrugghe B. Activation of a fibroblast-specific enhancer of the proalpha2(I) collagen gene in tight-skin mice. Arthritis Rheum. 2001;44:712–722. doi: 10.1002/1529-0131(200103)44:3<712::AID-ANR121>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 48.Denton CP, Zheng B, Evans LA, Shi-wen X, Ong VH, Fisher I, Lazaridis K, Abraham DJ, Black CM, de Crombrugghe B. Fibroblast-specific expression of a kinase-deficient type II transforming growth factor beta (TGFbeta) receptor leads to paradoxical activation of TGFbeta signaling pathways with fibrosis in transgenic mice. J Biol Chem. 2003;278:25109–25119. doi: 10.1074/jbc.M300636200. [DOI] [PubMed] [Google Scholar]
- 49.Denton CP, Lindahl GE, Khan K, Shiwen X, Ong VH, Gaspar NJ, Lazaridis K, Edwards DR, Leask A, Eastwood M, et al. Activation of key profibrotic mechanisms in transgenic fibroblasts expressing kinase-deficient type II Transforming growth factor-β receptor (TβRIIΔk). J Biol Chem. 2005;280:16053–16065. doi: 10.1074/jbc.M413134200. [DOI] [PubMed] [Google Scholar]
- 50.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 51.Darby IA, Bisucci T, Pittet B, Garbin S, Gabbiani G, Desmouliere A. Skin flap-induced regression of granulation tissue correlates with reduced growth factor and increased metalloproteinase expression. J Pathol. 2002;197:117–127. doi: 10.1002/path.1074. [DOI] [PubMed] [Google Scholar]
- 52.Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis: a combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114–125. [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang K, Flanders KC, Phan SH. Cellular localization of transforming growth factor-beta expression in bleomycin-induced pulmonary fibrosis. Am J Pathol. 1995;147:352–361. [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- 55.Hampel B, Malisan F, Niederegger H, Testi R, Jansen-Durr P. Differential regulation of apoptotic cell death in senescent human cells. Exp Gerontol. 2004;39:1713–1721. doi: 10.1016/j.exger.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 56.Raghu G, Chen YY, Rusch V, Rabinovitch PS. Differential proliferation of fibroblasts cultured from normal and fibrotic human lungs. Am Rev Respir Dis. 1988;138:703–708. doi: 10.1164/ajrccm/138.3.703. [DOI] [PubMed] [Google Scholar]
- 57.Torry DJ, Richards CD, Podor TJ, Gauldie J. Anchorage-independent colony growth of pulmonary fibroblasts derived from fibrotic human lung tissue. J Clin Invest. 1994;93:1525–1532. doi: 10.1172/JCI117131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramos C, Montano M, Garcia-Alvarez J, Ruiz V, Uhal BD, Selman M, Pardo A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol. 2001;24:591–598. doi: 10.1165/ajrcmb.24.5.4333. [DOI] [PubMed] [Google Scholar]
- 59.Moodley YP, Scaffidi AK, Misso NL, Keerthisingam C, McAnulty RJ, Laurent GJ, Mutsaers SE, Thompson PJ, Knight DA. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin-6/gp130-mediated cell signaling and proliferation. Am J Pathol. 2003;163:345–354. doi: 10.1016/S0002-9440(10)63658-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang HY, Phan SH. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1). Am J Respir Cell Mol Biol. 1999;21:658–665. doi: 10.1165/ajrcmb.21.6.3720. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi T, Liu X, Kim HJ, Kohyama T, Wen FQ, Abe S, Fang Q, Zhu YK, Spurzem JR, Bitterman P, et al. TGF-beta1 and serum both stimulate contraction but differentially affect apoptosis in 3D collagen gels. Respir Res. 2005;6:141. doi: 10.1186/1465-9921-6-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arora PD, Narani N, McCulloch CA. The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am J Pathol. 1999;154:871–882. doi: 10.1016/s0002-9440(10)65334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larios JM, Budhiraja R, Fanburg BL, Thannickal VJ. Oxidative protein cross-linking reactions involving L-tyrosine in transforming growth factor-beta1-stimulated fibroblasts. J Biol Chem. 2001;276:17437–17441. doi: 10.1074/jbc.M100426200. [DOI] [PubMed] [Google Scholar]
- 64.Tian B, Lessan K, Kahm J, Kleidon J, Henke C. beta 1 integrin regulates fibroblast viability during collagen matrix contraction through a phosphatidylinositol 3-kinase/Akt/protein kinase B signaling pathway. J Biol Chem. 2002;277:24667–24675. doi: 10.1074/jbc.M203565200. [DOI] [PubMed] [Google Scholar]
- 65.Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279:33024–33034. doi: 10.1074/jbc.M313265200. [DOI] [PubMed] [Google Scholar]
- 66.Tan A, Levrey H, Dahm C, Polunovsky VA, Rubins J, Bitterman PB. Lovastatin induces fibroblast apoptosis in vitro and in vivo: a possible therapy for fibroproliferative disorders. Am J Respir Crit Care Med. 1999;159:220–227. doi: 10.1164/ajrccm.159.1.9802104. [DOI] [PubMed] [Google Scholar]
- 67.Henke C, Bitterman P, Roongta U, Ingbar D, Polunovsky V. Induction of fibroblast apoptosis by anti-CD44 antibody: implications for the treatment of fibroproliferative lung disease. Am J Pathol. 1996;149:1639–1650. [PMC free article] [PubMed] [Google Scholar]
- 68.Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, Standiford TJ, Thannickal VJ. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166:367–375. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in lveolar epithelial cells by transforming growth factor-β1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]