

Abstract
Glutamate is the primary excitatory transmitter in adult brain, acting through synapses on dendritic spines and shafts. Early in development, however, when glutamatergic synapses are only beginning to form, nicotinic cholinergic excitation is already widespread; it is mediated by acetylcholine activating nicotinic acetylcholine receptors (nAChRs) that generate waves of activity across brain regions. A major class of nAChRs contributing at this time is a species containing α7 subunits (α7-nAChRs). These receptors are highly permeable to calcium, influence a variety of calcium-dependent events, and are diversely distributed throughout the developing CNS. Here we show that α7-nAChRs unexpectedly promote formation of glutamatergic synapses during development. The dependence on α7-nAChRs becomes clear when comparing wild-type mice with mice constitutively lacking the α7-nAChR gene. Ultrastructural analysis, immunostaining, and patch-clamp recording all reveal synaptic deficits when α7-nAChR input is absent. Similarly, nicotinic activation of α7-nAChRs in wild-type organotypic culture, as well as cell culture, increases the number of glutamatergic synapses. RNA interference demonstrates that the α7-nAChRs must be expressed in the neuron being innervated for normal innervation to occur. Moreover the deficits persist throughout the developmental period of major de novo synapse formation and are still fully apparent in the adult. GABAergic synapses, in contrast, are undiminished in number under such conditions. As a result, mice lacking α7-nAChRs have an altered balance in the excitatory/inhibitory input they receive. This ratio represents a fundamental feature of neural networks and shows for the first time that endogenous nicotinic cholinergic signaling plays a key role in network construction.
INTRODUCTION
Glutamatergic pathways comprise the principal excitatory networks in the central nervous system, employing a combination of synapses on dendritic spines and shafts to embed neurons in circuits (Fiala et al, 1998; Sheng and Hoogenraad, 2007). How synaptogenesis is coordinated across neuronal populations to form networks remains poorly understood. An intriguing possibility is that nicotinic cholinergic signaling plays a key role. Nicotinic acetylcholine receptors (nAChRs) reach their highest relative levels early in development (Zhang et al., 1998; Adams et al., 2002) when glutamatergic synapses are initially forming (Fiala et al., 1998; Hennou et al., 2002). Activation of nAChRs by the neurotransmitter acetylcholine produces spontaneous waves of excitation throughout much of the nervous system at this time (Bansal et al., 2000; Hanson and Landmesser, 2003; Le Magueresse et al., 2006). Early nicotinic signaling is known to influence specific aspects of local circuit formation and pathfinding (Hanson and Landmesser, 2003, 2006; Myers et al., 2005; Grubb et al., 2003).
A major nAChR subtype in brain is the α7-containing homopentamer (α7-nAChR), which has a high relative permeability to calcium (Bertrand et al., 1993; Seguela et al., 1993). The location of α7-nAChRs (Jacob and Berg,1983; Fabian-Fine et al., 2001) and their calcium permeability enable the receptors to influence diverse events, ranging from modulation of transmitter release and synaptic plasticity (McGehee et al., 1995; Gray et al., 1996; Alkondon and Albuquerque, 2001; Dickinson et al, 2008; Zhong et al., 2008; Albuquerque et al., 2009; Gu and Yakel, 2011) to control of gene expression (Chang and Berg, 2001; Hu et al., 2002; Dajas-Bailador and Wonnacott 2004). The α7-nAChRs also help determine when GABAergic transmission converts from excitation to inhibition during development (Liu et al., 2006), and they help drive survival and maturation of adult-born neurons in the hippocampus (Campbell et al., 2010). Not surprisingly, α7-nAChRs have been implicated in a variety of neurological functions and disorders (Freedman et al., 2001; Liu et al., 2001; Picciotto and Zoli, 2002; Grassi et al, 2003; Harrison and Weinberger, 2005; Hoyle et al., 2006; Levin et al., 2009).
Here we show that early endogenous nicotinic activity through α7-nAChRs is a major determinant promoting the development of glutamatergic pathways formed in the brain. Transgenic mice lacking α7-nAChRs (α7 knockouts; α7KOs) acquire fewer glutamatergic synapses during early postnatal life. Activation of α7-nAChRs by chronic exposure to nicotine in culture increases the number of functional glutamatergic synapses. Receptor knockdown by RNA interference (RNAi) in vivo demonstrates that α7-nAChRs must be expressed by the postsynaptic neuron to receive proper glutamatergic innervation. GABAergic synapses remain unchanged both in culture and in vivo; this causes a shift in glutamatergic/GABAergic input to α7KO neurons. The changes produced during development by disrupted nicotinic cholinergic input represent substantial network aberrations and may account for lasting behavioral deficits reported for mice lacking the receptors (Fernandes et al., 2006; Hoyle et al., 2006; Young et al., 2007; Levin et al., 2009). This is an unexpected and important role for endogenous nicotinic cholinergic signaling.
MATERIALS AND METHODS
Viral injections
High-titer lentiviral stocks containing short-hairpin RNAs representing either RNAi or scrambled control sequences were obtained from the Viral Vector Core (Salk Institute). For lentiviral injections, postnatal day (P) 1-P2 mice of either sex were anesthetized and then stabilized in a custom clay mold before receiving 70 nl of high titer lentiviral stock intracranially at each of 5 sites using a Nanoject (Drummond Scientific Company, Broomall, PA) and beveled glass injection pipette (Adesnik et al., 2008). Animals were allowed to recover and returned to the dam immediately after injection; 10–11 days later they were perfusion-fixed with 4% paraformaldehyde (PFA). Analyses were performed blind to genotype and viral construct.
Perfusion-fixed tissue slices
Perfusion-fixed tissue slices were obtained by anesthetizing animals of either sex with ketamine/xylazine, transcardially perfusing with 4% PFA, and postfixing for 1–2 hours. Brains were cryoprotected in 0.1 M phosphate buffer, pH 7.4, containing 25% sucrose and sectioned at 40 μm with a Leica cryostat. The α7KO mouse C57BL/6J line was purchased from Jackson Laboratories (Bar Harbor, ME). A mouse line lacking the β2-nAChR gene (β2KO) was provided by Marla Feller, University of California, Berkeley) and crossed with the α7KO line to generate double knockout mice (dKO). In all cases, results were based on pups from ≥2 litters per genotype.
Mouse hippocampal slice cultures
Fresh brain tissue for slice cultures was obtained by rapidly excising P0–P1 mouse brains into ice-cold artificial cerebrospinal fluid (ACSF) saturated with 95% O2/5% CO2, containing in mM, NaCl 119, KCl 2.5, CaCl2 2.5, MgSO4 1.3, NaH2PO4 1.0, NaHCO3 26.2 and glucose 11. Vibratome sections (150–200μm) were plated onto Millicell inserts (Millipore, Bedford MA) in culture medium (Neurobasal plus 10% horse serum), and incubated at 37°C in a humidified chamber with 5% CO2 for 4–7 days (Stoppini et al.,1991).
Rat hippocampal dissociated cell cultures
Rat dissociated hippocampal cell cultures were prepared from embryonic day 18 embryos of either sex and grown 10–21 days as described (Zhang and Berg, 2007).
Immunohistochemistry
Fixed 40 μm-thick brain sections were dried at room temperature and washed 3 times in PB-saline (PBS), pH 7.4, with 0.3% Triton X-100 (PBST). Sections (and slices from organotypic culture) were incubated overnight at 4°C in PBST with 10% normal donkey serum containing rabbit anti-PSD-95 (1:500, Life Technologies, Grand Island, NY) or rabbit anti-phospho GluR1 (glutamate receptor 1; 1:500, Millipore, Billerica, MA) and guinea pig anti-vesicular glutamate transporter (anti-VGluT; 1:2500, for VGluT1, Bioscience Research Reagents, Temecula, CA) or guinea pig anti-vesicular GABA transporter (anti-VGAT; 1:250, Synaptic Systems, Goettingen, Germany) and rabbit anti-GABAA α1 receptor (1:500, Alamone Labs, Jerusalem, Israel). Sections were then washed 3 times with PBST and incubated for 2 hours with PBST containing secondary CF dye-labeled antibodies (1:500, goat anti-guinea pig CF633 and donkey anti-rabbit CF555; Biotium, Inc., Hayward, CA). Slides were mounted with medium (Vectashield, Vector Laboratories, Burlingame, CA) and imaged using a Leica TCS SP5 confocal microscope (Leica Microsystems CMS Gmbh, Mannheim, Germany). Nonspecific immunostaining was assessed by substituting non-immune IgG for the primary antibody and found to be negligible.
Surface staining of live cells in culture was achieved by incubating with rabbit anti-GluR1 antibody (1:20, Calbiochem, Gibbstown, NJ) or rabbit anti-GABAA α-1 antibody (1:50, Alomone Labs, Jerusalem, Israel) in media on ice. Cells were then washed 5 times with ice-cold ACSF saturated with 95% O2/5% CO2 and fixed with 2% PFA for 10 minutes. Fixed cells were then incubated with mouse anti-PSD 95 (1:1,000, Affinity BioReagents, Golden, CO) or mouse anti-GAD-65 monoclonal antibody (mAb; 1:20, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) for costaining, and with donkey anti-rabbit Cy3 and anti-mouse FITC or Cy5 as secondary antibodies (all at 1:500, Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hours at room temperature. Another primary antibody used on fixed cells was rabbit anti-VGluT (1:1,000, Synaptic Systems, Goettingen, Germany.
Fluorescence imaging
Fluorescence images were obtained with a Leica TCS SP5 confocal microscope using ImageJ for analysis of brain slices. For cells in culture, images were obtained with a Zeiss Axiovert S100 TV microscope and Slidebook software (Intelligent Imaging Innovations, Inc., Denver, CO) applied to three vertical stacks of contiguous optical sections. To visualize dendrites in hippocampal and cortical slices from perfusion-fixed animals, green fluorescent protein (GFP) was introduced in vivo via lentiviral constructs. Results were equivalent in the two cases and therefore combined. Brain sections (36–45 per animal) and hippocampal slices from organotypic culture were stained independently for pre- and postsynaptic markers, and confocal scans were performed in the CA1 region of the hippocampus or layer 5/6 in the visual cortex. Imaging parameters were identical for all wild-type (WT) and transgenic animals, as well as for slice and cell cultures exposed to drugs. WTs (or controls for viral construct and drug exposure where appropriate) were always included in all experiments to normalize for expected variations in antibody staining intensity performed on different days. To minimize antibody penetration problems, only 3 merged optical sections localized close to the tissue surface were used per slice (9–15 images per animal). The 3 merged optical sections were analyzed using the ImageJ puncta analyzer program. Thresholds were set at 3 standard deviations above the mean staining intensity of 6 nearby regions in the same visual field. Thresholded images present a fixed intensity for all pixels above threshold after having removed all those below. Labeled puncta were defined as areas containing at least 4 contiguous pixels after thresholding. Co-localization of puncta (e.g. VGluT and PSD-95) required that they have at least one pixel in common as revealed by Image J applied to the 3 merged optical sections or as revealed by Slidebook software in the case of cell cultures. Quantifications were performed blind to condition.
Electron microscopy
Hippocampal slices from P8 mice of either sex were obtained by perfusing the animals transcardially with 4% PFA and 0.2% picric acid in 0.1 M sodium cacodylate buffer, pH 7.6, and then removing the brains, storing them in the same solution overnight at 4°C, and sectioning at 100 μm in the sagittal plane with a Vibratome. Hippocampal slices from organotypic cultures (P4 in culture 4 days) were obtained by fixing the cultures in 4% PFA and 0.2% picric acid in CB overnight at 4°C, washing in CB, and fixing an additional 15 minutes with 0.2% glutaraldehyde in CB. The fixed slices were incubated in 1% OsO4 and 1.5% potassium hexacyanoferrate in cacodylate buffer for 15 minutes, then dehydrated in a graded series of ethanol concentrations, and embedded in Epon. Semi-thin sections stained with toluidine blue were used for orientation. Ultrathin sections of the CA1 region were counterstained with uranyl acetate and lead citrate and examined with a JEM-1200EXII electron microscope (Tokyo, Japan) operated at 80 kV and Tietz Video Image Processing Systems (TVIPS) 4K with a 4K F415 CCD camera. Alternatively, negatives were digitized with a CanonScan D 1230 U scanner at 1200 dpi. For drug treatments, slices from a common pool were divided among conditions. In all cases, separate tissue blocks were prepared for each slice (2–6 blocks per condition), and a single section from each block was used for synapse quantification so that possible overlap between sections would be avoided. Synapses were identified by ultrastructural specializations including alignment of pre- and postsynaptic membranes, pre- and postsynaptic thickenings, and clusters of synaptic vesicles. Counts were performed blind to condition (65–207 images per condition).
Electrophysiology
Whole-cell patch-clamp recording was performed on CA1 pyramidal neurons in acute hippocampal P12 and P25 slices as described (Zhang and Berg, 2007; Campbell et al., 2010). Before recording from slices (300 μm thick), a 30 minute recovery was allowed at 37.5°C, followed by 30 minutes at room temperature. The bath contained (in mM), NaCl 119, KCl 2.5, CaCl2 2.5, MgSO4 1.3, NaH2PO4 1.0, NaHCO3 26.2, glucose 11, and was saturated with 95% O2/5% CO2 (to yield pH 7.4). Patch pipettes (4–5 MΩ) were filled with (in mM) 130 Cs-methylsulfonate, 3 CsCl, 10 HEPES, 1 EGTA, 10 Na2-phosphocreatine, and 2 MgATP (adjusted to pH 7.4). For recordings in cell culture (Conroy et al., 2007) the bath contained (in mM) 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH 7.4, with NaOH) at room temperature; the patch pipettes (3–4 M) were filled with (in mM) 122.5 gluconic acid, 122.5 CsOH, 10 CsCl, 5 NaCl, 1.5 MgCl2, 5 HEPES, 1 EGTA, 10 Na2-phosphocreatine, 3 MgATP, and 0.3 NaGTP (pH 7.25, with CsOH), and the recordings were made from neurons in 10–13-day-old cultures. Pipette capacitance was minimized in all recordings; series resistance was below 25 MΩ. Data were acquired at 10 kHz and filtered at 1 kHz. When recording spontaneous miniature excitatory postsynaptic currents (mEPSCs), 1 μM tetrodotoxin (TTX) was used to block action potentials and 20 μM gabazine to block GABAA receptor-mediated postsynaptic currents (PSCs). No difference was found in mEPSC frequencies recorded from WT littermates vs. C57BL/6J WTs purchased from Jackson Laboratories (0.49 ± 0.08 vs. 0.51 ± 0.04 Hz, respectively; n = 9, 10 cells from 3, 4 animals), so values for the two were combined. For evoked PSCs, a bipolar stimulating electrode was positioned over the Schaffer collaterals about 400 μm from the patch-clamped neuron. Blockade of AMPA and NMDA receptors with APV and NBQX, respectively, confirmed that all of the evoked PSC detected at −70 mV was due to glutamatergic transmission under these conditions (Atallah and Scanziani, 2009). Blockade of GABAA receptors with gabazine confirmed that all of the evoked PSC detected at 0 mV was due to GABAergic transmission. When needed, methyllycaconitine (MLA, 100 nM) and dihyro-β-erythroidine (DHβE, 10 μM) were used to block α7-nAChRs and β2-containing heteromeric nAChRs (β2*-nAChRs), respectively. For mEPSCs, events were analyzed by Mini Analysis Program software (Synaptosoft, Inc., Decatur, GA). Evoked PSCs were analyzed with clampfit 9.2.
RNAi constructs
The β2-RNAi construct (generously provided by Uwe Maskos; Avale et al., 2008) targeting the mouse β2-nAChR transcript was tested for effectiveness by using Lipofectamine™ 2000 (Life Technologies, Grand Island, NY) to transfect HEK293T cells on glass coverslips with plasmids encoding α4-nAChR subunit from chicken, cyan fluorescent protein (CFP)-tagged β2-nAChR subunit from mouse (Addgene plasmid 15106, developed by H. Lester, Caltech), and GFP together with either β2-RNAi or a scrambled version of the sequence (β2-Scr) as nonspecific control (1:1:3 transfection ratio). Two days after transfection, the cells were fixed with 2% PFA, permeabilized, mounted, and imaged with a Leica SP5 confocal microscope. Settings for laser power and wavelength range for detection were optimized using coverslip cultures not transfected with CFP to minimize crosstalk between GFP and CFP channels. Images were taken of nearly confluent cells expressing GFP, blind to the CFP channel. CFP fluorescence was scored as the proportion of pixels above background, which was defined as the level of CFP fluorescence in cultures transfected with GFP but not CFP constructs. Transfection with the β2-RNAi construct resulted in CFP levels that were 3 ± 1% of those obtained with β2-Scr, demonstrating the effectiveness of the β2-RNAi in preventing production of β2*-nAChRs, a second major class of nicotinic receptors in the CNS. The α7-RNAi construct, targeting the mouse α7-nAChR transcript, was designed with a Genscript algorithm and shown to be homologous to both rat and mouse α7-mRNA sequences. The scrambled sequence (α7-Scr) was obtained with Genscript sequence scrambler and shown not to be homologous with any sequence in the rat and mouse genomes. The sequences were inserted into lentiviral vectors (FG12; Addgene plasmid 14884) under an H1 promoter along with GFP under a ubiquitin promoter and linked to their reverse complement by the loop sequence TCTCTTGAA to form an shRNA. Their compositions are 5′-GAGTGAGGTGATCTGCAGTGA-3′ (BP 1356-1376) and 5′-GAGAGTACGCTAAGATCCTAA-3′ for the α7-RNAi and α7-Scr, respectively. The effectiveness of the lentiviral α7-RNAi construct was tested by infecting 16-day-old rat (E18 Sprague Dawley) or mouse (E18 C57B1/6) hippocampal 3.5 cm cultures (plated at 26,000 and 62,500 cells/cm2, respectively) with 5 μl virus stock (1–5×107 IU/ml), and collecting the cells 1 week later. Solubilized α7-nAChRs were immunoprecipitated with mAb 319 (Sigma-Aldrich, St. Louis, MO) and radiolabeled with I125-αBungarotoxin (I125-αBgt: Perkin Elmer, Waltham, MA). Total protein was measured using the Bradford assay (Bio-Rad Protein Assay). Transfection with α7-RNAi reduced the amount of binding obtained to 7 ± 1% and 17 ± 11 % of that seen with α7-Scr in rat and mouse hippocampal cultures, respectively, demonstrating the α7-RNAi effectiveness. Off-target effects of β2- and α7-RNAi constructs were assessed by injecting the lentiviral constructs into the cognate KO mouse (β2KOs and α7KOs) on P2 and assessing their effects either on spine number in the case of β2-RNAi or VGluT and PSD-95 puncta in the case of α7-RNAi at P12. In each case the results were compared to those obtained with the corresponding scrambled control (β2-Scr or α7-Scr).
FM4-64 Labeling
FM4-64 staining (Pyle et al., 1999) was performed by first labeling dendrites and spines with either DiI (Life Technologies, Grand Island, NY) or GFP via a sindbis-viral construct (generous gift from Gentry Patrick, University of California, San Diego), rinsing cultures to remove nicotine, incubating 1 hour in 50 nM MLA and 1 μM DHβE to block any residual nicotinic activity, and then incubating 60 s with 5 μM FM4-64 ± 50 mM KCl prior to rinsing and imaging. Counts of FM4-64-labeled puncta along 10 μm segments of dendrites were performed blind to condition.
Statistical analysis
Data represent means ± SEMs. Unless otherwise indicated, statistical significance was assessed with Student’s t-test for unpaired values and with one-way analysis of variance (ANOVA) followed by Bonferroni post-hoc test for multiple values. *p < 0.05, **p < 0.01; ***p < 0.001.
RESULTS
Reduced numbers of glutamatergic synapses in mice lacking α7-nAChRs
To determine whether endogenous nicotinic cholinergic signaling is needed for synapse formation in the developing brain, we first carried out electron microscopic analysis (EM) comparing the abundance of synapses in WT and KO mice lacking one or more nAChR genes. Examining regions containing the apical dendrites of CA1 pyramidal neurons in P8 hippocampus revealed a significant deficit in α7KO mice, compared either to WT mice or to β2KO mice (Figure 1A,B). No further decrement was seen in dKO mice beyond that found in α7KOs. This suggested that endogenous cholinergic signaling through α7-nAChRs, but not through β2*-nAChRs, is important for synapse formation.
Figure 1.

Decrement in the number of glutamatergic synapses in α7KO mice. (A) Electron micrographs showing synapses (arrows) in regions containing apical dendrites of CA1 pyramidal neurons in P8 hippocampal slices from WT and α7KO mice. Scale bar: 0.5 μm. (B) Number of ultrastructurally identified synapses/μm2 (≥ 150 images from 3 animals/genotype). (C) Pictures showing a cartoon of the hippocampus (upper left) with a box indicating the CA1 region used for imaging and low magnification image of the CA1 region (lower left) with a box indicating the general area used for higher power images (right) of CA1 apical dendrite regions in P12 WT and α7KO mice. Slices were immunostained for glutamatergic synapses using the markers VGluT (blue), PSD-95 (red), and the images were merged (Merge) and thresholded (Thresh) to remove pixels of below-threshold intensity (arrows indicate examples of co-localization). Scale bars: 20 μm, left; 5 μm, right. (D) Number of VGluT, PSD-95, and co-localized puncta/400 μm2 for the indicated genotype in the CA1. (E) Number of VGluT, PSD-95, and co-localized puncta/400μm2 at P12 on neurons in layer 5/6 of the cortex (3–4 cells/animal; 5–8 animals/genotype from ≥2 litters).
To determine if the synaptic decrements represented a loss of glutamatergic synapses, we immunostained tissue slices for the presynaptic marker VGluT and the postsynaptic marker PSD-95. Co-localization of the two markers was used to identify glutamatergic synapses (Eroglu et al., 2009). Compared to WTs, both α7KOs and dKOs in the hippocampal CA1 region displayed clear deficits at P12 in their numbers of VGluT and PSD-95 puncta and in co-localization of the two (Figure 1C,D). No deficits were seen in β2KOs in any of these features. The same pattern was found in layer 5/6 of the visual cortex at P12, indicating that the deficits in glutamatergic synapses caused by α7-nAChR removal were not confined to the hippocampus (Figure 1E).
To determine whether the deficits in glutamatergic synapses observed by imaging in α7KOs correlated with reductions in synaptic function, we quantified mEPSCs. TTX and gabazine were included to block action potentials and GABAA receptors, respectively. In acute hippocampal slices prepared at P12, mEPSC frequency in CA1 pyramidal neurons was significantly reduced in α7KOs compared to WTs (Figure 2A,B). A similar decrement was seen in dKOs while β2KOs were indistinguishable from WTs. No change was seen in mEPSC amplitude in any of the KOs compared to WTs (Figure 2B). Adding the nicotinic blockers DHβE and MLA to WT slices to block β2*-nAChRs and α7-nAChRs, respectively, 20 minutes before measuring mEPSC frequency did not alter the frequencies observed in WT neurons (101 ± 8% in MLA/DHβE vs. control). This eliminated the possibility that differences between WTs and KOs reflected endogenous acetylcholine acting through receptors in WTs to elevate mEPSC frequency acutely.
Figure 2.
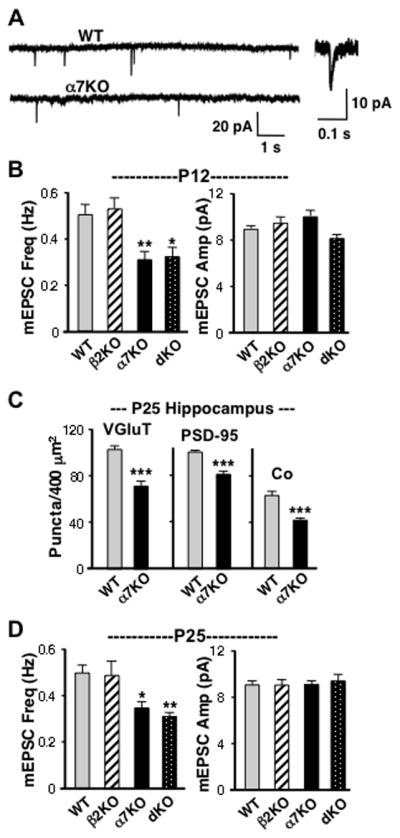
Electrophysiological analysis showing reduced mEPSC frequency in α7KO mice compared to WTs. (A) Patch-clamp recordings of spontaneous mEPSCs in CA1 pyramidal neurons from P12 WT and α7KO acute slices in TTX and gabazine. Inset: WT mEPSC with expanded time scale. (B) Quantification of mEPSC frequency (left) and amplitude (right) at P12. (C) Number of VGluT, PSD-95, and co-localized puncta/400 μm2 in the CA1 region of P25 WTs and α7KOs analyzed as in Fig 1. (D) Quantification of mEPSC frequency (left) and amplitude (right) at P25 for the indicated genotypes. Loss of the α7-nAChR gene product, but not the β2-nAChR gene product, correlates with reduced mEPSC frequency with no change in mEPSC amplitude, a result consistent with α7KOs having fewer functional glutamatergic synapses (≥ 50 events/cell, 11–19 cells from 4–7 animals/genotype).
To determine whether the synaptic deficits persisted, we examined P25 animals. Immunostaining of the hippocampal CA1 again indicated a substantial deficit in glutamatergic synapses in α7KOs compared to WTs (Figure 2C). Patch-clamp recording also showed differences in mEPSC frequencies as found at P12: α7KOs and dKOs both had lower mEPSC frequencies than did WTs, β2KOs were equivalent to WTs, and none of the KOs differed from WTs in mEPSC amplitude (Figure 2D). Immunostaining at times well into adulthood showed that the glutamatergic deficits persisted in α7KOs: at P60 VGluT puncta/400 μm2 for WT vs. α7KOs were 83.3 ± 1.7 vs. 58.1 ± 2.6 (mean values ± SEM; p < 0.0001), respectively; for PSD-95, the values were 84.1 ± 3.6 vs. 61.3 ± 3.4 (p < 0.0001), and for co-localization 74.6 ± 3.4 vs. 45.5 ± 3.9 (p < 0.0001) in the CA1. The results support those from EM and immunostaining, indicating that the α7-nAChR gene product is required to establish normal numbers of glutamatergic synapses.
Nicotine induction of functional glutamatergic synapses via α7-nAChRs
To determine whether nicotine can exploit α7-nAChR pathways to induce glutamatergic synapses and to gain additional evidence that α7-nACh-induced synapses are functional, we first turned to hippocampal cell culture. In the absence of septal input, hippocampal cultures have little, if any, endogenous cholinergic signaling (Lewis et al., 1967). Rat hippocampal cells in culture were incubated 1 week with 1 μM nicotine, a concentration in the range of peak serum levels found in smokers (Matta et al., 2007); the cultures were then co-immunostained for GluR1-containing AMPA receptors and PSD-95. The nicotine treatment increased the numbers of GluR1 and PSD-95 puncta and their co-localization (Figure 3A,B), suggesting increased numbers of synaptic contacts. Including either the α7-nAChR blocker MLA or αBgt with the nicotine prevented the increases while the β2*-nAChR blocker DHβE had no effect (Figure 3B). This indicated that α7-nAChRS were needed for the induction. Similar results were obtained when the cultures were co-stained for VGluT and PSD-95, again yielding an increase in co-localization as expected for increased synaptic contacts (Figure 3C,D). In contrast, immunostaining revealed no increase in the numbers of GAD-65 and GABAAR puncta or in their co-localization, indicating that GABAergic synapses were unlikely to be changed in number by the nicotine treatment (Figure 3E,F). The results strongly suggest that nicotine, acting through α7-nAChRs, increases the number of glutamatergic synapses in culture.
Figure 3.

Nicotine in cell culture acts through α7-nAChRs to increase glutamatergic synaptic contacts. (A) Control (Con) and nicotine-treated (Nic) cell cultures expressing GFP and surface-stained for GluR1 receptors (red) and then intracellular PSD-95 (blue). Here and below, overlap of 3 colors appears as white. Scale bar: 10 μm. (B) Quantification showing a significant increase in GluR1, PSD-95, and co-localized puncta in nicotine unless α7-nAChRs were blocked with either MLA or αBgt (n = 3–12 cultures, 5 cells/culture). (C) Cultures immunostained for VGluT (red) and PSD-95 (green). The dashed white lines indicate the edges of a neurite; spines extending from the neurite (as in panel A) are not demarcated. Scale bar: 5 μm. (D) Quantification showing increased puncta and co-localization of VGluT and PSD-95 after nicotinic activation of α7-nAChRs. (E) Cultures expressing GFP and immunostained first for surface α1-containing GABAA receptors (red) and then for intracellular GAD-65 (blue). Scale bar: 10μm. (F) Quantification showing no changes in the numbers of GAD-65 (blue) and GABAA receptor puncta (GABAR, red) or in their co-localization (n = 3–4 cultures; 5 cells/culture). Nicotinic stimulation appears to increase glutamatergic, but not GABAergic, synapses.
To determine whether the increased numbers of synaptic contacts represented functional synapses, we examined mEPSC frequency in TTX and gabazine at least an hour after replacing the nicotine with the blockers MLA and DHβE. A 7-day nicotine treatment substantially increased mEPSC frequency, and the effect was prevented by MLA but not by DHβE (Figure 4A,B); no change was seen in mEPSC amplitude (not shown). Exposure to the same concentration of nicotine for only 1 hour at the end of the 7-day period had no effect (Figure 4B), thereby excluding the possibility that increased mEPSC frequency represented residual nicotine activating receptors during the test itself. A similar outcome was obtained when functional synapses were identified by KCl-stimulated uptake of the dye FM4-64 (Pyle et al., 1999). After replacing the nicotine with MLA/DHβE for 1 hour, the cells were assayed for KCl-dependent FM4-64 uptake during a 1-minute period (Fig 4C). The nicotine treatment produced a significant increase in the number of FM4-64-identified synapses (Figure 4D). Including MLA with nicotine during the 7-day nicotine treatment completely blocked the increase, confirming that α7-nAChR activation was required. As seen above with mEPSC frequency, including DHβE with the nicotine did not prevent the nicotine-induced elevation of FM4-64 uptake, indicating that β2*-nAChR activation was not required. A 1-hour nicotine exposure at the end of the 7-day period, as a negative control, produced no increase. The results indicate that nicotine, acting on α7-nAChRs, increases the number of functional glutamatergic synapses formed in cell culture.
Figure 4.

Nicotine-induced increases in the number of functional glutamatergic synapses in cell culture. Rat hippocampal cultures were incubated 7 days (7d) in nicotine (Nic) with or without blockers (MLA, DHβE), rinsed to remove nicotine, and then incubated 1 hour in MLA/DHβE. As a control, some cultures received nicotine only the last hour of the 7-day incubation (1 hr Nic). (A) Patch-clamp recording of spontaneous mEPSCs in TTX and gabazine. (B) Quantification of mEPSC frequency (15–20 cells from 5–8 cultures per condition). (C) Images of cells in culture having received the indicated treatment, then incubated with MLA/DHβE for 1 hour followed by labeling with a one-minute incubation in FM4-64 (red) plus 50 mM KCl to reveal active synapses on dendrites and spines of cells expressing GFP. Scale bar: 10 μm. (D) Quantification of FM4-64-labeled puncta. Baseline control (7dNic/noK): KCl omitted from FM4-64 incubation (n = 3–6 cultures; 5–7 cells/culture).
To obtain additional evidence for the nicotinic effect, we examined mouse hippocampal slices in organotypic culture where ultrastructural analysis could be applied to the CA1 region. P4 slices were incubated 4–6 days with 1 μM nicotine and then examined by electron microscopy. The nicotine treatment increased the number of synapses, and the increase was prevented by including MLA (Figure 5A,B). DHβE had no effect. Evidence that the differences represented glutamatergic synapses came from immunostaining for either VGluT and PSD-95 or for GluR1 and PSD-95. Nicotine treatment increased puncta number in each case and increased co-localization of the respective pairs (Figure 5C,D). MLA again blocked the effect, implicating α7-nAChRs, while DHβE had no effect. The results indicate that extended nicotine exposure either in cell culture or in organotypic culture substantially increases the number of glutamatergic synapses formed and does so by activating α7-nAChRs. Nicotine in this way appears to mimic the effect of endogenous ACh on glutamatergic synapse formation in vivo.
Figure 5.

Nicotine induction of glutamatergic synapses in organotypic slice culture. (A) Electron micrographs showing examples of synapses (arrows) in CA1 region of P4 hippocampal slices incubated in culture 4 days with (Nic) and without (Con) 1 μM nicotine. Scale bar: 1 μm. (B) Quantification of ultrastructurally identified synapses per unit area (from 150–250 images/condition from 3–6 animals). (C) Hippocampal slices from P0–P1 mice, maintained in culture for 4–7 days, were treated with vehicle (Con), nicotine (Nic), nicotine plus MLA (Nic/MLA), or nicotine plus DHβE (Nic/DHβE) for the last 4 days and then immunostained for VGluT (blue) and PSD-95 (red), and the images merged (Merge) and thresholded (Thresh) as in Fig. 1C. Quantification of the number of indicated puncta/400 μm2 in the CA1 region was performed as in Fig. 1D for the drug conditions indicated. Scale bar: 5 μm. (D) Images and quantification obtained as in panel C except that slices were co-immunostained for VGluT and GluR1 (6 cultures/condition). Nicotinic stimulation of α7-nAChRs (blocked by MLA), but not β2*-nAChRs (blocked by DHβE), increases the number of glutamatergic synapses identified immunohistochemically, and the increases are reflected in the total number of synapses per unit area identified ultrastructurally.
Cell-autonomous role for α7-nAChRs
To determine whether the α7-nAChR requirement is cell-autonomous, i.e. needed in the postsynaptic neuron, we used RNAi constructs to decrease α7-nACR expression acutely in vivo. These included constructs specific for α7-nAChRs (α7-RNAi), for β2*-nAChRs (β2-RNAi), and the corresponding scrambled sequences (α7-Scr, β2-Scr) as controls. Mice were injected intracranially on P1–P2 with lentiviral constructs (Adesnik et al., 2008) containing the RNAi and were taken for immunostaining at P12. Two strategies were employed to evaluate the relative numbers of glutamatergic synapses. In the first, regions were chosen in which heavy infection had occurred, causing most of the local neurons to express GFP. In these cases, the total numbers of VGluT, PSD-95, and co-localized puncta were counted in a 400 μm2 area and compared to values obtained in a nearby control region lacking any detectable GFP. Normalizing the values to those obtained in adjacent control regions in this manner corrected for possible differences in staining among tissue sections. We found that expression of α7-RNAi in the hippocampal CA1 caused a significant reduction in the numbers of VGluT and PSD-95 puncta and a reduction in their co-localization, indicating fewer glutamatergic synapses (Figure 6A,B). The adjacent uninfected control sites had consistent labeling across conditions for individual experiments, eliminating concerns about variation among sections (data not shown). Similar results were found in layer 5/6 of the visual cortex where again α7-RNAi, and only α7-RNAi, produced significant decrements (Figure 6C). The α7-RNAi displayed no off-target effects as assessed by expression in the cognate KO (Fig, 6D). Both the β2-RNAi and α7-RNAi were effective in reducing their cognate receptor in cell culture (see Methods), and both have been used previously (Avale et al., 2008; Campbell et al., 2010).
Figure 6.

Relevance of local α7-nAChRs for glutamatergic synapse formation. (A) Apical dendrite regions of P12 hippocampal CA1 expressing either lentiviral α7-Scr (upper) or α7-RNAi (lower) after intracranial injection in vivo (GFP) and immunostaining for VGluT (blue) and PSD-95 (red) shown together (Merge) and after thresholding (Thresh; arrows indicate examples of co-localization). Scale bar: 5 μm. (B) Quantification of VGluT, PSD-95, and co-localized puncta/400 μm2 in P12 hippocampal CA1. (C) Quantification in P12 visual cortex layer 5/6. Results are normalized to values obtained in adjacent regions lacking viral infection and demonstrate that α7-nAChRs within the local area are required for neurons to acquire WT levels of glutamatergic synapses. (D) Quantification of VGluT, PSD-95, and co-localized puncta in α7KOs expressing the indicated constructs, showing that the α7-RNAi construct has no off-target effects as defined by changes induced in α7KOs (3 fields/animal; 4–8 animals/condition).
As a second strategy to assess cell-autonomous actions of α7-nAChRs, we quantified puncta on individual isolated dendrites expressing the viral constructs on the perimeter of the infected region in the CA1. The same outcome was obtained: α7-RNAi prevented the neuron from receiving normal numbers of glutamatergic synapses, judging by the number of VGluT and PSD-95 puncta co-localized on the dendrite (Figure 7A,B). The results demonstrate that neurons must express α7-nAChRs to acquire WT levels of glutamatergic synapses.
Figure 7.

Cell-autonomous requirement for α7-nAChRs to promote glutamatergic synaptogenesis. (A) Isolated dendrites of P12 CA1 pyramidal neurons on the periphery of the virally infected area, expressing either α7-Scr (left) or α7-RNAi (right) and immunostained for VGluT (blue), PSD-95 (red), merged (Merge), and thresholded (Thresh). Arrows indicate examples of co-localized puncta. Scale bar: 10 μm. (B) Quantification of puncta on isolated dendrites (from 3–4 cells/animal, 4–8 animals/condition). Down-regulation of the α7-nAChR gene product in isolated neurons reduces the number of glutamatergic synapses formed on the neuron, demonstrating a requirement for expression of the receptors by the postsynaptic cell for normal levels of glutamatergic innervation.
Diminished ratio of glutamatergic/GABAergic input in α7KO neurons
The finding that nicotine increases the number of glutamatergic, but not GABAergic, synapses in culture raised the question of whether α7-nAChR deficits in vivo might generate a sustained imbalance of excitatory/inhibitory input to neurons. To examine this, we first used immunostaining to quantify GABAergic synapses in P12 α7KOs, as we had glutamatergic synapses. The presynaptic marker VGAT and the postsynaptic marker GABA α1 receptor were chosen for this purpose in hippocampal slices. No differences were found between WTs and either α7KOs or β2KOs in the numbers or co-localization of the two kinds of puncta (Figure 8A,B). Considering the reduced numbers of glutamatergic synapses, the results suggest that α7KO neurons have a reduced ratio of glutamatergic/GABAergic contacts, compared to WTs and β2KOs.
Figure 8.

Decreased ratio of glutamatergic/GABAergic input in α7KO neurons. (A) CA1 apical dendrite regions in P12 WT and α7KO mice immunostained for VGAT (blue), GABAA α1 receptor (red), merged (Merge), and thresholded (Thresh). Scale bar: 5 μm. (B) Number of VGAT, GABAA α1 receptor, and co-localized puncta/400 μm2 in P12 (3 fields/animal; 3 animals/genotype). (C) Evoked glutamatergic postsynaptic currents (PSCs) recorded at −70 mV and GABAergic PSCs recorded at 0 mV (aligned by stimulus artifact) in WT and α7KO CA1 at P12. (D) Ratios of evoked PSC peak amplitudes mediated by glutamatergic and GABAergic synapses for individual neurons at P12 (left) and P25 (middle; Glut/GABA PSC; 5–11 cells from 3–6 animals/genotype). Right: Ratios in WT neurons before (Con) and after adding MLA (MLA), showing that the responses are not influenced by acute endogenous α7-nAChR signaling during the recordings (5 cells from 3 animals). (E) No differences were seen in PPR for evoked EPSCs amplitudes in α7KO vs. WT neurons. Two EPSCs evoked in a WT CA1 pyramidal neuron clamped at −70 mV while stimulating the Schaffer collateral input twice, 100 ms apart (vertical line indicates stimulus artifact). (F) Ratio of the second EPSC amplitude divided by the first (PPR) for the indicated genotype at the indicated age (5–11 cells from 3–6 animals per genotype and age).
To test for functional differences in the ratio of excitatory/inhibitory synaptic input to a neuron, we stimulated the Schaffer collaterals and adjacent regions while recording from CA1 pyramidal neurons first at the reversal potential for GABA (−70 mV) to quantify glutamatergic responses and then at the reversal potential for glutamate (0 mV) to quantify GABAergic responses. The reversal potentials were established by using the recording electrode to dialyze the interior of the cell (Atallah and Scanziani, 2009). Gabazine blocked the response at 0 mV, confirming it was GABAergic; APV and NBQX blocked the response at −70 mV, confirming it was glutamatergic (data not shown). Comparing the evoked responses at 0 and −70 mV showed that α7KO CA1 pyramidal neurons receive less glutamatergic transmission, normalized for GABAergic transmission, than do WTs (Figure 8C,D). This difference remains at P25 (Figure 8D).
The reduced glutamatergic input to α7KO relative to WT neurons did not reflect an acute effect of α7-nAChRs on WT neurons during the recordings because repeating the stimulation in the presence of MLA did not alter the ratio of evoked responses in WT neurons (Figure 8D, right; 1.45 ± 0.14 vs. 1.41 ± 0.12 for EPSC(glutamatergic)/EPSC(GABAergic) in MLA vs. control). Nor did the reduced glutamatergic input in α7KO neurons appear to arise from differences in release probability at synapses onto the cells: comparing the peak amplitudes of two EPSCs evoked 100 ms apart yielded a paired-pulse ratio (PPR) that was equivalent in α7KOs and WTs (Figure 8E,F). No change in PPR is generally interpreted as reflecting no change in release probably (Mao et al., 2011). Taken together, the results demonstrate a selective decrement in glutamatergic synapses on α7KO neurons and a decrease in the amount of glutamatergic input compared to GABAergic input.
DISCUSSION
Nicotinic cholinergic signaling occurs early in development throughout the nervous system (Zhang et al., 1998; Adams et al, 2002; Bansal et al., 2000; Hanson and Landmesser, 2003; Myers et al., 2005; Le Magueresse et al., 2006). The results presented here show that endogenous nicotinic signaling plays an unexpected and important role in shaping glutamatergic networks during development. Preventing expression of α7-nAChRs by neurons reduces the number of glutamatergic synapses it would otherwise receive. This dependence does not extend to GABAergic synapses, causing an imbalance: in the absence of α7-nAChRs, neurons receive a diminished ratio of functional glutamatergic/GABAergic input. The sustained decrements seen in glutamatergic synapses and glutamatergic/GABAergic input to neurons represent important features of neural networks and likely account for a number of behavioral deficits found in mice lacking α7-nAChRs (Fernandes et al., 2006; Hoyle et al., 2006; Le Magueresse et al., 2006; Young et al., 2007; Levin et al., 2009).
The effects of α7-nAChRs on glutamatergic synapse formation are substantial. Ultrastructural analysis revealed a clear deficit early on in total number of synapses in α7KOs compared to WTs. Immunostaining for pre- and postsynaptic markers expected at glutamatergic synapses indicated a pronounced deficit in the number of glutamatergic synapses in α7KOs at all ages examined (P12–P60). No deficits were found in GABAergic synapses by immunostaining. Care was taken to compare across tissue samples at equivalent depths and exposure to antibody. Similar immunostaining criteria and quantification of co-localized puncta of pre- and postsynaptic markers have been widely used to quantify synapses (Christopherson et al., 2005; Kayser et al., 2006; Stevens et al., 2007; Eroglu et al., 2009).
The electrophysiological analysis also suggested that α7KOs have fewer glutamatergic synapses. Under conditions where GABAergic synapses were blocked, patch-clamp recording found reduced mEPSC frequency in α7KOs compared to WTs with no change in mEPSC mean amplitude. Moreover, the relative amplitude of evoked glutamatergic PSCs normalized for GABAergic PSCs in the same neuron was reduced in α7KOs compared to WTs. No change was found in PPR under these conditions, arguing against a change in release probability. Though the mEPSC analysis alone would not exclude possible presynaptic changes, all of the data taken together - EM, immunostaining, and patch-clamp results - strongly support the conclusion that fewer functional glutamatergic synapses are found in α7KOs.
Studies on hippocampal neurons in organotypic culture and in cell culture further corroborated the importance of α7-nAChRs. Nicotinic stimulation of the receptors increased the number of glutamatergic synapses formed on hippocampal neurons as assessed by immunostaining. This conclusion was further supported by the increases seen in mEPSC frequency and the increased number of puncta labeled by potassium-induced FM4-64 uptake. Ultrastructural analysis again confirmed an increase in total synapses, while immunostaining detected no changes in number of GABAergic synapses. Control cultures largely, if not completely, lacked endogenous nicotinic cholinergic signaling; cholinergic input to the hippocampus is provided almost entirely by the septum (Lewis et al., 1967) which was not included in the cultures. As a result, blockade of α7-nAChRs with MLA during nicotine treatment consistently held glutamatergic synapse numbers at values seen in controls; MLA would not have been expected to reduce the values below those controls.
The nicotine experiments confirmed that α7-nAChR activation is likely essential for the beneficial effect of the receptors, rather than simply requiring their presence. Nicotine was used as an agonist to avoid activation of muscarinic receptors and to minimize degradation through hydrolysis. The concentration chosen was physiological in the sense that it represented the upper range found in heavy smokers. Notably nicotine exposure generated larger increases in the number of glutamatergic synapses than predicted from a comparison of WTs with α7KOs. This presumably reflects the chronic excessive stimulation of α7-nAChRs achieved in culture.
Deficits in dKOs were equivalent to α7KOs whether quantifying total synapses by EM or glutamatergic synapses by immunostaining or mEPSC frequency by patch-clamp recording. No deficits were found in β2KOs by these criteria. Though adult β2KOs have altered patterns of dendritic spines (Ballesteos-Yanez et al. 2010) and unique behavioral deficits (Picciotto et al., 1995; Levin et al., 2009; Wang et al., 2009), absence of the β2-nAChR gene does not appear to reduce the total number of glutamatergic synapses a neuron receives. While β2*-nAChRs represent a major class of nicotinic receptors in the CNS, comparable in number to α7-nAChRs (Albuquerque et al., 2009; Gotti et al., 2009; Changeux, 2010), they do not appear to be required for glutamatergic synapse formation.
The fact that equivalent decrements in synapse number were found in the hippocampal CA1 and cortical layer 5/6 of α7KOs compared to WTs signals the breadth of the effect. It is distinct from the rapid action of presynaptic α7-nAChRs reported to activate “silent” glutamatergic synapses acutely in developing hippocampus (Maggi et al., 2003). In the present work, both immunostaining and EM revealed fewer synapses in α7KOs. Moreover, the RNAi knockdown experiments implicated α7-nAChRs on the postsynaptic neuron as being critical to achieve WT levels of glutamatergic synapses. Interestingly, α7RNAi-induced deficits were substantially greater than those seen in α7KOs, perhaps reflecting a competitive disadvantage of the α7RNAi-expressing neurons compared to “normal” neighbors. All neurons would have the same disadvantage in the α7KOs. It should be noted that the results do not exclude roles for other α7-nAChRs populations, perhaps on presynaptic terminals or even on adjacent astrocytes (Fabian-Fine et al., 2001; Sharma and Vijayaraghavan, 2001; McGehee et al., 1995; Le Magueresse et al., 2006), in supporting glutamate synapse formation or stabilization.
A likely mechanism for α7-nAChR effects on glutamatergic synapse formation is the ability of the receptors to elevate intracellular calcium. They do this via their permeability to calcium and by recruitment of calcium from other sources (Bertrand et al., 1993; Seguela et al., 1993; Dajas-Bailador and Wonnacott 2004; Fayuk and Yakel et al., 2007; Albuquerque et al., 2009; Shen and Yakel, 2009). Location and time course will shape outcome. Presynaptic α7-nAChRs enhance transmitter release by elevating local calcium (McGehee et al., 1995; Gray et al., 1996; Sharma and Vijayaraghavan, 2003). Postsynaptic α7-nAChRs can produce persistent changes by altering gene expression, presumably because of the signal transduction pathways available (Chang and Berg, 2001; Hu et al., 2002; Albuquerque et al., 2009). Altered gene expression is a plausible mechanism for enhancing synapse formation.
The ability of α7-nAChRs to increase glutamate synapse formation may also explain how this receptor helps determine when GABA converts from excitation to inhibition during normal early postnatal development. Previous work demonstrated that the conversion depends on activity through α7-nAChRs, accelerating the change in chloride transporters required to produce the mature chloride gradient that renders GABA inhibitory (Liu et al., 2006). Recent studies with adult-born neurons in the dentate gyrus demonstrate that the prolonged period of depolarizing GABAergic input found in α7KOs correlates with reduced dendritic arborization and reduced glutamatergic input (Campbell et al., 2010). The extended period of GABAergic depolarization does not result in hyperinnervation. Instead, it most likely reflects a delay in development caused by the inability of synapse number in α7KOs to reach some critical threshold required to drive the GABAergic conversion.
Endogenous nicotinic cholinergic control of glutamatergic synapse formation may help coordinate innervation across large neuronal populations in specific brain areas during development. Individual cholinergic neurons can have large innervation fields in the CNS (Witten et al., 2010), and the spontaneous waves of nicotinic excitation that occur at this time extend over large regions of the nervous system (Bansal et al., 2000; Hanson and Landmesser, 2003, 2006; Le Magueresse et al., 2006; Myers et al., 2005; Grubb et al, 2003). Neurons having the appropriate receptors and receiving cholinergic input could be recruited to a common network. Lack of required receptors or excessive stimulation by nicotine at early times could alter the pathways formed. Recent evidence indicates that endogenous nicotinic signaling retains the capacity to reorganize networks in the adult cortex if relieved of naturally occurring inhibition mediated by the endogenous prototoxin lynx1 (Morishita et al., 2010). The present work indicates that α7-nAChRs are likely to be key in this process, shaping network connectivity.
The deficits found here in α7KOs are substantial, and raise questions about how the animals function. A number of behavioral aberrations and cognitive deficits have been reported for α7KOs, including attention deficits, impaired spatial discrimination, and diminished working/episodic memory (Fernandes et al., 2006; Hoyle et al., 2006; Le Magueresse et al., 2006; Young et al., 2007; Levin et al., 2009). Many of these are likely to result directly or indirectly from the synaptic deficits reported here. Nonetheless, it is surprising that α7KOs have not been found to display more extreme behavioral abnormalities, given their synaptic imbalances. One possibility is that the neural network develops compensatory mechanisms to use the existing connections more effectively; homeostatic mechanisms may come into play (Ibata et al., 2008; Turrigiano, 2008) to adjust for structural deficits in glutamatergic pathways. Another possibility, however, is that the rearing conditions and behavioral tests commonly used do not adequately reveal cognitive abilities intended for a more challenging environment. These will be interesting issues to pursue in the future.
Acknowledgments
We thank Uwe Maskos and Andrew W. Halff for the β2- and α7-RNAi constructs, respectively, Xiao-Yun Wang and Jeff Schoellerman for expert technical assistance, and Gentry Patrick and Stevan Djakovic for stocks of sindbis-GFP. Grant support was provided by the US National Institutes of Health (NS012601, NS034569, and DA237482) and the Tobacco-Related Disease Research Program (17FT-0053 and 19XT-0072).
Footnotes
Conflict of interest: The authors declare no conflict of interests.
References
- Adams CE, Broide RS, Chen Y, Winzer-Serhan UH, Henderson TA, Leslie FM, Freedman R. Development of the α7 nicotinic cholinergic receptor in rat hippocampal formation. Dev Brain Res. 2002;139:175–187. doi: 10.1016/s0165-3806(02)00547-3. [DOI] [PubMed] [Google Scholar]
- Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA. NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci (USA) 2008;105:5597–5602. doi: 10.1073/pnas.0800946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Phys Revs. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic acetylcholine receptors α7 and α4β2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol. 2001;86:3043–3055. doi: 10.1152/jn.2001.86.6.3043. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Scanziani M. Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron. 2009;62:566–577. doi: 10.1016/j.neuron.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avale ME, Faure P, Pons S, Robledo P, Detheil T, David DJ, Gardier AM, Maldonado R, Granon S, Changeux JP, Maskos U. Interplay of β2* nicotinic receptors and dopamine pathways in the control of spontaneous locomotion. Proc Natl Acad Sci (USA) 2008;105:15991–15996. doi: 10.1073/pnas.0807635105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Yanez I, Benavides-Piccione R, Bourgeois J-P, Changeux J-P, DeFilipe J. Alterations of cortical pyramidal neurons in mice lacking high affinity nicotinic receptors. Proc Natl Acad Sci (USA) 2010;107:11567–11572. doi: 10.1073/pnas.1006269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A, Singer JH, Hwang B, Feller MB. Mice lacking specific nAChR subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON/OFF circuits in the inner retina. J Neurosci. 2000;20:7672–7681. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci (USA) 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through α7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci. 2010;30:8734–8744. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001;32:855–865. doi: 10.1016/s0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Changeux J-P. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Nai Q, Ross B, Naughton G, Berg DK. Postsynaptic neuroligin enhances presynaptic inputs at neuronal nicotinic synapses. Dev Biol. 2007;307:79–91. doi: 10.1016/j.ydbio.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. TIPS. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the α7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayuk D, Yakel JL. Dendritic Ca2+ signalling due to activation of alpha 7-containing nicotinic acetylcholine receptors in hippocampal neurons. J Physiol (Lond) 2007;582:597–611. doi: 10.1113/jphysiol.2007.135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes C, Hoyle E, Dempster E, Schalkwyk LC, Collier DA. Performance deficit of α7 nicotinic receptor knockout mice in a delayed matching-to-place task suggests mild impairment of working/episodic-like memory. Genes Brain Behav. 2006;5:433–440. doi: 10.1111/j.1601-183X.2005.00176.x. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Leonard S, Olincy A, Kaufmann CA, Malaspina D, Cloninger CR, Svrakic D, Faraone SV, Tsuang MT. Evidence for the multigenic inheritance of schizophrenia. Am J Med Gen. 2001;105:794–800. doi: 10.1002/ajmg.10100. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Grassi F, Palma E, Tonini R, Amici M, Ballivet M, Eusebi F. Amyloid β1-42 peptide alters the gating of human and mouse α-bungarotoxin-sensitive nicotinic receptors. J Physiol (Lond) 2003;547:147–157. doi: 10.1113/jphysiol.2002.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Rossi FM, Changeux J-P, Thompson ID. Abnormal functional organization in the dorsal lateral geniculate nucleus of mice lacking the β2 subunit of the nicotinic acetylcholine receptor. Neuron. 2003;40:1161–1172. doi: 10.1016/s0896-6273(03)00789-x. [DOI] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci. 2003;23:587–600. doi: 10.1523/JNEUROSCI.23-02-00587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Increasing the frequency of spontaneous rhythmic activity disrupts pool-specific axon fasciculation and pathfinding of embryonic spinal motoneurons. J Neurosci. 2006;26:12769–12780. doi: 10.1523/JNEUROSCI.4170-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psych. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Hennou S, Khalilov I, Diabira D, Ben-Ari Y, Gozlan H. Early sequential formation of functional GABAA and glutamatergic synapses on CA1 interneurons of the rat foetal hippocampus. Eur J Neurosci. 2002;16:197–208. doi: 10.1046/j.1460-9568.2002.02073.x. [DOI] [PubMed] [Google Scholar]
- Hoyle E, Genn RF, Fernandes C, Stolerman IP. Impaired performance of alpha7 nicotinic receptor knockout mice in the five-choice serial reaction time task. Psychopharmacol. 2006;189:211–223. doi: 10.1007/s00213-006-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Jacob MH, Berg DK. The ultrastructural localization of α-bungarotoxin binding sites in relation to synapses on chick ciliary ganglion neurons. J Neurosci. 1983;3:260–271. doi: 10.1523/JNEUROSCI.03-02-00260.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser MS, McClelland AC, Hughes EG, Dalva MB. Intracellular and trans-synaptic regulation of glutamatergic synaptogenesis by EphB receptors. J Neurosci. 2006;26:12152–12164. doi: 10.1523/JNEUROSCI.3072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Magueresse C, Safiulina V, Changeux J-P, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J Physiol (Lond) 2006;576:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Petro A, Rezvani AH, Pollard N, Christopher NC, Strauss M, Avery J, Nicholson J, Rose JE. Nicotinic α7- or β2-containing receptor knockout: Effects on radial-arm maze learning and long-term nicotine consumption in mice. Behav Brain Res. 2009;196:207–213. doi: 10.1016/j.bbr.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PR, Shute CCD, Silver A. Confirmation from choline acetylase analyses of a massive cholinergic innervation to the rat hippocampus. J Physiol (Lond) 1967;191:215–224. doi: 10.1113/jphysiol.1967.sp008246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q-S, Kawai H, Berg DK. β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci (USA) 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux J-P, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci (USA) 2003;100:2059–2063. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D, Galagher K, McGehee DS. Nicotinic potentiation of excitatory inputs to ventral tegmental area dopamine neurons. J Neurosci. 2011;31:6710–6720. doi: 10.1523/JNEUROSCI.5671-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Pyschopharmacol. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- McGehee D, Heath MJ, Gelber S, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Morishita H, Miwa JM, Heintz N, Hensch TK. Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science. 2010;330:1238–1240. doi: 10.1126/science.1195320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL. Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron. 2005;46:37–49. doi: 10.1016/j.neuron.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, Vincent P, Pich EM, Brûlet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M. Nicotinic receptors in aging and dementia. J Neurobiol. 2002;53:641–655. doi: 10.1002/neu.10102. [DOI] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Choi S, Tsien RW. Visualization of synaptic activity in hippocampal slices with FM1-43 enabled by fluorescence quenching. Neuron. 1999;24:803–808. doi: 10.1016/s0896-6273(00)81028-4. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci (USA) 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Shen JX, Yakel JL. Nicotinic acetylcholine-receptor mediated calcium signaling in the nervous system. Acta Pharmacol Sin. 2009;30:673–680. doi: 10.1038/aps.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Rangarajan KV, Lawhn-Heath CA, Sarnaik R, Wang BS, Liu X, Cang J. Direction-specific disruption of subcortical visual behavior and receptor fields in mice lacking the β2 subunit of nicotinic acetylcholine receptor. J Neurosci. 2009;29:12909–12918. doi: 10.1523/JNEUROSCI.2128-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, Finlayson K, Sharkey Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharm. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Zhang J, Berg DK. Reversible inhibition of GABAA receptors by α7-containing nicotinic receptors on the vertebrate postsynaptic neurons. J Physiol (Lond) 2007;579:753–763. doi: 10.1113/jphysiol.2006.124578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Liu C, Miao H, Gong Z-H, Nordberg A. Postnatal changes in nicotinic acetylcholine receptor α2, α3, α4, α7 and β2 subunits genes expression in rat brain. Int J Dev Neurosci. 1998;16:507–518. doi: 10.1016/s0736-5748(98)00044-6. [DOI] [PubMed] [Google Scholar]
- Zhong C, Du C, Hancock M, Mertz M, Talmage DA, Role LW. Presynaptic type III neuregulin 1 is required for sustained enhancement of hippocampal transmission by nicotine and for axonal targeting of alpha7 nicotinic acetylcholine receptors. J Neurosci. 2008;28:9111–9116. doi: 10.1523/JNEUROSCI.0381-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]