

Abstract
Periodontal diseases are initiated by bacterial species living in polymicrobial biofilms at or below the gingival margin and progress largely as a result of the inflammation initiated by specific subgingival species. In the past few decades, efforts to understand the microbiota of periodontal diseases have led to an exponential increase in information about biofilms associated with periodontal health and disease. In fact, the oral microbiota is one of the best characterized microbiomes that colonize the human body. Despite this increased knowledge, one has to ask if our fundamental concepts of the etiology and pathogenesis of periodontal diseases have really changed. In this chapter we will review how our comprehension of the structure and function of the subgingival microbiota evolved over the years in search of lessons learned and unlearned in periodontal microbiology. More specifically, this review focuses on: 1) how the data obtained through molecular techniques has impacted our knowledge of the etiology of periodontal infections; 2) the potential role of viruses in the etiopathogenesis of periodontal diseases; 3) how concepts of microbial ecology have expanded our understanding of host microbial interactions that might lead to periodontal diseases; 4) the role of inflammation in the pathogenesis of periodontal diseases; and 5) the impact of these evolving concepts on treatment and preventive approaches to periodontal infections. We will conclude by reviewing how novel systems biology approaches promise to unravel new details of the pathogenesis of periodontal diseases and, hopefully, lead to a better understanding of periodontal disease mechanisms.
Introduction
The microbiology of periodontal diseases has been the focus of intense investigation for several decades. This focus is justifiable since bacteria are the etiological agents of periodontal diseases, which remain the primary cause of tooth loss in adults worldwide. In addition, therapies that predictably can treat the condition in all subjects are still missing, as evidenced by the existence of refractory cases in which disease continues to progress despite comprehensive periodontal treatment. As a result of efforts to understand the microbiota of periodontal diseases and the continuous dedication of researchers in the field, our knowledge of the structure and composition of the polymicrobial biofilms associated with periodontal health and disease has expanded exponentially in the past few decades. In fact, the oral microbiota is one of the best characterized microbiomes that colonize the human body. Despite this increased knowledge, one has to ask if our fundamental understanding of the etiology and pathogenesis of periodontal diseases has really changed.
Much of the recent knowledge regarding the composition of the subgingival microbiota has been the result of technological advances in molecular techniques that have afforded the high-throughput analysis of a large number of samples, circumventing some of the limitations of cultural techniques. These technologies have allowed us, for instance, to examine the presence of the uncultivable segment of the subgingival microbiota in greater detail, expanding our knowledge of the diversity of the supra and subgingival microbiotas. Further, molecular techniques have also afforded the examination of the potential role of viruses in periodontal disease etiology. Other recent critical changes in our knowledge of periodontal infections derive from the realization that these diseases are caused by biofilms rather than bacteria in a planktonic state and how adoption of ecological concepts for studying of the acquisition and maturation of the oral microbiota. Although these conceptual changes have helped revamp our interest in the microbiology of periodontal diseases, a review of the classic literature on this topic questions how ground-breaking these concepts really are. Revisiting the literature on the microbial etiology of periodontal diseases for the purpose of this manuscript, we will illustrate how the essence of our understanding of the etiology and pathogenesis of periodontal diseases has been in place for decades.
The study of the microbiota associated with periodontal diseases has also been impacted by changes in paradigms regarding the etiology and pathogenesis of periodontal diseases over the years. At times the focus of the scientific community has shifted from the microbial etiology of periodontal diseases to other aspects of the pathogenesis of these infections, including the impact of genetic and environmental factors on the initiation and progression of these conditions. Paul Keyes once wrote: “I am convinced that although many clinicians and investigators do not exclude the role of bacteria in periodontal lesions, at this point interest in microorganisms often dissipates and attention shifts to other areas” (173). In recent years, much attention has been given to the essential role of inflammation and other immune mechanisms in periodontal disease pathogenesis. These studies are fundamental to our understanding of the complex mechanisms involved in these multifactorial diseases. Unfortunately, at times, this change in focus has been misconstrued as “evidence” of a lesser role of the supra and subgingival microbiota in the etiology of periodontal diseases. Further, the study of the microbial etiology of periodontal diseases has also suffered the impact of vagaries of the funding agencies, compromising the continuity of efforts in this field. Therefore, it should come as no surprise that there is still much work to be done before we can fully understand how the subgingival microbiota interacts with the host to result in periodontal diseases.
In this chapter we will review how our knowledge of the structure and function of the subgingival microbiota has changed over the years in search of lessons learned and unlearned in periodontal microbiology. We will also examine certain aspects of the host-microbial interactions that are associated with periodontal health and disease and attempt to place them in a historical context. More specifically, we will focus on: 1) how the data obtained through molecular techniques has impacted our knowledge of the etiology of periodontal infections; 2) the potential role of viruses in the etiopathogenesis of periodontal diseases; 3) how concepts of microbial ecology have expanded our understanding of host microbial interactions that might lead to periodontal diseases; 4) the role of inflammation in the pathogenesis of periodontal diseases; and 5) the impact of these evolving concepts on treatment and preventive approaches to periodontal infections. We will conclude by reviewing how novel systems biology approaches promise to unravel new details of the pathogenesis of periodontal diseases and, hopefully, lead to a better understanding of periodontal disease mechanisms.
Lesson learned: periodontal diseases are infections caused by bacteria
There is overwhelming evidence in the literature to support the etiological role of bacteria in periodontal diseases. Before we elaborate on this point we should be clear about the concept of “etiology”. As defined in the Merriam-Webster dictionary, etiology is “the cause or causes of a disease or abnormal condition”. Therefore, by characterizing bacteria as the etiological agents of periodontal diseases, we are stating that they cause these diseases. Let's elaborate further on the definition of cause. Rothman & Greenland (297) defined cause as “an antecedent event, condition, or characteristic that was necessary for the occurrence of the disease at the moment it occurred, given that other conditions are fixed”. By this definition, it becomes clear that bacteria are not sufficient to cause periodontal diseases; in fact, Rothman & Greenland (297) stressed that “no specific event, condition, or characteristic is sufficient by itself to produce disease”. They went on to describe “sufficient cause” as the constellation of minimal conditions and events that produce disease. Implicit in the word “minimal” is the idea that all the conditions and events are essential for disease to occur. Onset of disease will occur when all minimal conditions of the sufficient cause have taken place.
These concepts agree with our understanding of periodontal diseases as multifactorial diseases. In 1994, Haffajee & Socransky (131) argued that periodontal disease initiation and progression required the simultaneous occurrence of a number of factors: 1) the virulent periodontal pathogen; we will elaborate further on this concept later, for now this would be the equivalent to bacteria; 2) the local environment and 3) host susceptibility. Indeed, the notion that variation in “host resistance” impacts the outcome of periodontal diseases has been recognized since at least the 1970s (332). Later, Page & Kornman (266) expanded this model to acknowledge the contributions of genetic and acquired risk factors. The concept of multiple causes is clearly not unique to periodontal diseases; although tobacco smoking is well accepted as a cause of lung cancer, it is also clear that by itself it is not a sufficient cause. The requirement of susceptibility of the host for the development of periodontal diseases has led some to refer to bacteria as a condition “required but not sufficient” to cause periodontal diseases. This is a moot point because no disease process is the result of a single isolated cause or event; i.e. no cause is necessary and sufficient in itself to produce disease. Further, for any given infection, if disease is to result from a host-microbial interaction, the host has to be susceptible. This causal model can also accommodate variations in the dose of each component in the constellation of sufficient causes. The model for sufficient causes for periodontal tissue destruction described above (bacteria, local environment, host susceptibility), can easily accommodate the notion of varying doses. For instance, if a susceptible host has an immunodeficiency, a lower bacterial challenge might be enough to complete the minimal conditions that result in periodontal disease.
Lesson unlearned: periodontal diseases are infections caused by bacteria
The infectious nature of periodontal diseases has recently been described as an example of a hypothesis rooted in “low-level evidence” (156). According to the author, the infectious nature of periodontal diseases was not supported by what he described as the “epidemiological baton of discovery”. According to this concept, observational epidemiology should serve as the basis for hypotheses of causality which would then be tested using laboratory experiments and human clinical trials. Randomized clinical trials, cohort studies and case-control studies would all be examples of high-level evidence, while case-series, biological plausibility, “pathophysiological reasoning”, animal studies, bench research and expert opinion would all be characterized as low-level evidence. The experimental gingivitis classic study by Löe et al. (213) was cited as an example of “low-level” evidence that infection leads to destructive periodontal disease. It was argued that the study was flawed because it had a small sample size of 12 subjects, used an acute model to make inferences on a chronic disease, and the findings were extrapolated to periodontitis that, according to the author, was “a huge leap”. This is not the first time that the infectious nature of periodontal diseases has been put into question and is a good example of the vicious cycle of “lessons learned and unlearned” in periodontal microbiology. As described by Socransky & Haffajee in their paper on the historical perspective of the bacterial etiology of periodontal diseases, between the mid-1920s and the early 1960s, periodontal diseases were considered the result of some constitutional defect on the part of the patient, trauma from occlusion, disuse atrophy or some combination of those factors (335). Additional theories proposed in the past included local irritation from calculus, rough restoration margins, systemic diseases and conditions, diet and nutritional deficiencies (269).
The concept of an infectious cause for periodontal diseases had, in fact, its modern resurgence (late 1950s and 1960s) partially due to cross-sectional studies that suggested a close association between the level of bacterial debris on tooth surfaces and the extent and severity of gingival inflammation (18, 121, 224, 315). Therefore, at least for gingivitis, the infectious theory of causation did follow the so called epidemiological baton. The classic studies on experimental gingivitis were pioneers in the sense that, for the first time, the reversal phase of the gingival inflammation was closely observed after subjects resumed their oral hygiene practices. This satisfies the condition of “experimental evidence” as described by Hill in 1965 (152) in his list of considerations on the determination of causality or the criterion of “elimination” according to Haffajee & Socransky's criteria for defining periodontal pathogens (131), regarding the cause-effect relationship between dental plaque and gingivitis. Since its description, “experimental gingivitis” has become a standard model to examine the effects of different anti-plaque agents and in the study of risk factors for the development of gingivitis and, therefore, reproduced hundreds of times. In fact, in 1971, Löe (210) reported that up to that time 150 students had already participated in several studies conducted by his group using the experimental gingivitis model. The criticism that the model has an acute onset quite distinct from the chronic nature of gingivitis is valid. However, human studies have documented the resolution and prevention of recurrence of naturally occurring gingivitis with mechanical plaque removal, similar to the observations by Löe and co-authors using the acute model (25, 196, 223, 351).
The understanding of periodontal disease pathogenesis in the 1960s was that, if left undisturbed, gingivitis would invariably lead to periodontitis. Therefore, since dental plaque accumulation correlated with gingival inflammation, its association with periodontal tissue destruction was a logic extension. In addition, an experimental periodontitis study in beagle dogs demonstrated that plaque accumulation could also lead to periodontitis, at least in an animal model (206). Indirect evidence for a role of plaque accumulation and periodontitis has also been provided by studies demonstrating that meticulous supragingival plaque control can arrest the progression of destructive periodontal diseases for prolonged periods of time (23, 24, 26, 27, 351). The concept that gingivitis would inevitably result in attachment loss was eventually challenged by additional epidemiological studies demonstrating that a subset of individuals would not develop periodontitis even after years of exposure to large amounts of plaque accumulation and longstanding gingivitis (212). In any case, the original experimental gingivitis studies have been presented in the periodontal literature as evidence of a cause and effect relationship between dental plaque and gingivitis, not periodontitis.
Studies that evaluated the predictive value of clinical parameters for disease progression failed to identify dental plaque accumulation as a strong risk factor for attachment loss (28, 56, 161, 211). This finding suggested a less robust association between dental plaque accumulation and attachment loss compared to gingivitis, casting doubt on the theory of the infectious nature of periodontitis. This apparent disconnect between plaque accumulation and periodontal tissue destruction can be partially explained by examining further the precision (or lack thereof) of existing methods to measure the amount of bacterial challenge (we will address differences in the nature of the bacterial challenge later in the text). The limitations of the many plaque indices that have been employed in epidemiological surveys are notorious. First, plaque accumulation varies within a period of hours and the same subject can have dramatically different scores depending on the time of the day when the measurements takes place (237). Indices of gingival inflammation have been proposed as a better assessment of the consistency of the individual's oral hygiene practices because they would reflect exposure of plaque over time. Still, the inflammatory gingival status of a subject also fluctuates over relatively brief periods of time (days), depending on the efficacy and consistency of the oral hygiene practices by the subject. Therefore, it is unreasonable to expect that cross-sectional measures of plaque accumulation, even when repeated a few times, would give a very accurate assessment of the long-term exposure to the bacterial challenge. This is particularly relevant if one considers the chronic nature of periodontal diseases and the lengthy time required for attachment loss to occur.
Analogous to this situation is the observation that bleeding on probing was found to be a poor predictor of periodontal disease progression, casting doubt on the relevance of gingival inflammation in this process (197). Data from a recent longitudinal study that followed a cohort of 223 Norwegian subjects over 26 years have clearly demonstrated that long-term, constant exposure to gingivitis was a risk-factor for attachment loss and tooth loss (313, 314). A recently published paper examined the oral condition of subjects from New Zealand over 32 years (43). The study population initially comprised 1,037 children. Oral examinations were repeated at ages 3, 5, 7, 9, 11, 13, 15, 18, 21, 26 and 35. Data for at least 4 time points, including the baseline and the last visit were available for 911 subjects. Plaque scores were assessed using Greene and Vermillion's Simplified Oral Hygiene Index and patterns of “lifetime plaque exposure” were determined using group-based trajectory modeling. Three subgroups were characterized according to their “trends” in plaque exposure over the years as: high, medium and low trajectories. Periodontal disease parameters were measured in 897 subjects at age 32 years. Using this approach the authors could examine the potential role of the cumulative exposure to dental plaque over many years as a risk factor for periodontal disease initiation and progression. The results from multivariate models indicated that higher “plaque trajectories” were, indeed, associated with poorer oral health outcomes including caries- and periodontal disease-related measurements. These outcomes were present even after adjusting for confounding variables such as childhood socioeconomic status, gender, dental visit patterns and smoking. In summary, the failure of early epidemiological studies to detect a close correlation between plaque accumulation and periodontal disease occurrence and progression might have been the result of the imprecise measurement of the exposure to the bacterial challenge. The long-term exposure to other well-established risk factors for periodontal disease progression such as cigarette smoking can be better estimated through, for instance, the use of questionnaires. Unfortunately, one cannot estimate using questionnaires how many “plaque-years” a subject has been exposed to.
Lesson relearned: periodontal diseases are infections caused by bacteria
There are many lines of evidence that support the hypothesis that periodontitis is caused by bacteria, as elegantly reviewed by Socransky & Haffajee in 1994 (335), including: 1) the fact that acute periodontal infections such as acute necrotizing ulcerative gingivitis and acute necrotizing ulcerative periodontitis can be alleviated by any of a number of antibiotics; 2) epidemiological studies demonstrating a positive correlation between the amount of bacterial plaque and the severity of gingival inflammation and of bone loss; 3) human intervention studies demonstrating the control of gingivitis by means of antibiotics or antiseptic agents; 4) human intervention studies demonstrating the control of periodontal disease progression after surgery followed by regular professional tooth cleaning; 5) studies indicating an adjunctive effect of antibiotics in the treatment of “localized juvenile periodontitis”, “refractory periodontitis”, “recurrent periodontitis”; 6) host immune response; 7) the pathogenic potential of plaque bacteria when implanted into extraoral sites; and 8) studies in experimental animals. Since then, clinical trials continue to indicate that the adjunctive use of systemic antibiotics, particularly the combination of amoxicillin and metronidazole, result in significant improvements over the clinical results obtained with mechanical therapy alone (54, 90, 125, 135, 151, 232, 240, 245, 385). These results are compelling evidence of the bacterial etiology of periodontal diseases; particularly because these agents, in contrast to other classes of antibiotics (153), have no known anti-inflammatory effects. Even more persuasive was the report by Lopez and colleagues (220) on the outcome of a randomized placebo controlled clinical trial to examine the clinical effects of amoxicillin and metronidazole as the sole therapy for chronic periodontitis. Patients were examined every two months for 12 months to detect periodontal disease progression by changes in clinical attachment level. The antibiotic regimen (amoxicillin 500 mg and metronidazole 250 mg tid × 7 days) was administered at baseline, 4 and 8 months. The antibiotics group had statistically significantly better clinical outcomes at all 2-month intervals, including a lower percentage of progressing sites. Periodontal sites that exhibited multiple cycles of disease progression and periodontal abscesses were only detected in the placebo group. This study was followed by another clinical trial that examined the clinical and microbiological effects of a single regimen of this systemic antimicrobial combination as the sole therapy, demonstrating that the clinical improvements were accompanied by statistically significant reductions in most of the 40 species investigated using checkerboard DNA-DNA hybridization (221). These intervention studies provided additional support to the bacterial etiology of periodontitis; further, they indicated that live bacteria, rather than the mere presence of their constituents, are key elements in the pathogenesis of periodontal diseases.
Lesson learned: periodontal diseases are specific bacterial infections
Several researchers had a different interpretation of the apparent lack of direct correlation between amount of plaque accumulation and periodontal tissue destruction observed in the 1970s. Their view was that this was additional evidence of the requirement of a specific microbiota for periodontal disease to occur. Keyes wrote: “Although it is rare, most dentists at some time in their careers have seen patients with so-called “dirty mouths”, yet with negligible if any caries or periodontal disturbances. One possibility is that such mouths may not harbour odontopathic microorganisms” (173). This specific plaque hypothesis was also supported by the recognition that there were differences in the composition of biofilms associated with periodontally healthy or diseased sites and subjects. In addition, cases of acute necrotizing ulcerative gingivitis and localized juvenile periodontites (now localized aggressive periodontitis), where clinically distinct periodontal diseases were associated with distinctly different subgingival microbiotas also helped create momentum for the re-introduction of the specific plaque hypothesis. Particularly, cases of localized aggressive periodontitis where very little plaque accumulation and clinical signs of gingival inflammation were accompanied by severe connective tissue destruction and bone loss challenged the notion that the mass of bacterial plaque was the key element. The accepted dogma became that specific microorganisms were associated with different periodontal diseases and several groups embarked on the search for the periodontal pathogens responsible for different periodontal conditions (87, 88, 132, 244, 246, 329, 334, 338, 340, 378). We referred to these historic events as responsible for the re-introduction of the notion of specificity in periodontal infections because, as pointed by Socransky & Haffajee in 1994 (335), scientists started to look for the etiological agents of periodontal diseases in the 1880s. It was not until the mid-1930s that the search for the microorganisms responsible for destructive periodontal diseases was interrupted. Here we have yet another example of the cycle of lessons learned and unlearned in the field of periodontal microbiology.
The idea of specificity in the etiology of periodontal diseases launched an extremely prolific era of research in periodontal microbiology focusing on understanding the role of the many species that colonized the subgingival environment in health and disease. Out of this hard work, several putative periodontal pathogens were identified, culminating with the designation of Actinobacillus actinomycetemcomitans (currently Aggregatibacter actinomycetemcomitans), Porphyromonas gingivalis and Tannerella forsythia as periodontal pathogens in the 1996 World Workshop in Periodontics (2). These efforts are still ongoing and will probably require a few decades of intense research before we have identified all periodontal pathogens or combinations of pathogens that can lead to periodontal destruction. Difficulties in determining the role of specific components on the subgingival microbiota include: 1) the diversity of microbial taxa that can be found in the subgingival environment, many of which are still uncultivable; 2) difficulties in obtaining a representative sample; 3) the identification of active sites that are undergoing tissue destruction; 4) and the understanding that periodontal diseases are mixed infections with many combinations of “pathogens” that can lead to disease. In 1994, Haffajee & Socransky (131) summarized a set of criteria that helped define subgingival bacterial species as periodontal pathogens. The criteria included: association, elimination, host response, virulence factor, animal studies, and risk assessment. Much of our current understanding of the potential role of different species as etiological agents of periodontal diseases comes from data obtained with molecular techniques that overcame many of the limitations of culture techniques. However, out of the 6 criteria listed by Haffajee & Socransky, only 3 can be fulfilled using molecular techniques alone: association, elimination and risk assessment. Further, different clonal types of the same species might have distinct pathogenic potentials and the existing techniques used in the study of virulent strains of the same species such as multiple locus sequence typing and comparative genomic hybridization require cultivation. Therefore, if we are to fully examine the pathogenic potential of yet uncultivable species, methods to grow then in the laboratory will have to be in place.
Lesson unlearned: periodontal diseases are specific bacterial infections
As described by Socransky and co-authors, “The search for the etiological agents of destructive periodontal diseases has engrossed periodontal research workers for close to 100 years” (340). They characterized the search for periodontal pathogens as another example of cycles of lessons learned and unlearned in the field of periodontal microbiology, where putative pathogens were proposed and subsequently forgotten or dismissed “in a seemingly never ending cycle”. In the manuscript they detailed a series of difficulties encountered by microbiologists in the study of periodontal pathogens including: 1) technical difficulties: a) sample taking, b) dispersion of plaque samples, c) difficulties in cultivation, d) characterization and identification of isolates; 2) conceptual problems: a) complexity of the microbiota, b) mixed infections, c) opportunistic infections, d) identifying disease activity, e) different diseases diagnosed as being the same, f) the possibility of multiple diseases within a subject; and 3) data analysis. Despite the considerable technological advances since the publication of that paper, it can be argued that none of these issues have been satisfactorily resolved and all remain as obstacles to the precise definition of the etiologic agents of periodontal diseases. If nothing else, the expansion of our knowledge regarding the complexity of the subgingival microbiota has only aggravated many of these problems, particularly the difficulties in data analysis. In spite of these challenges, the efforts of microbiologists have helped define a relatively short list of likely candidates as periodontal pathogens, including: P. gingivalis, T. forsythia, A. actinomycetemcomitans, Prevotella intermedia, Prevotella melaninogenica, Fusobacterium nucleatum, Parvimonas micra, Eikenella corrodens, Prevotella nigrescens, Capnocytophaga gingivalis, Treponema denticola, Treponema socranskii, Eubacterium nodatum, and Campylobacter rectus (126, 141, 222, 271, 273, 308, 382, 383, 405). Other species have also been reported to be associated with periodontal diseases by different groups, including: Porphyromonas endodontalis, Prevotella denticola, , Filifactor alocis and Cryptobacterium curtum (188); Eubacterium saphenum and Mogibacterium timidum (235); Prevotella corporis, Prevotella disiens and Peptostreptococcus magnus (304); Slackia exigua (39); Treponema maltophilum and Treponema sp. Smibert-3 (76); Treponema lecithinolyticum (402); Treponema putidum sp. nov. (403); and Enterococcus faecalis, Escherichia coli and Bartonella sp. (60)
Nevertheless, the relevance of certain species as etiological agents of periodontal diseases remains controversial even among microbiologists; “Whether individual bacterial species are important remains debatable” (393).
Temporality, an inconvenient criterion
A common argument used to question the specific role of certain bacterial species is the lack of evidence of what was defined by Hill (152) as the condition of “temporality”. The concept refers to the requirement that the cause precede the effect in time. Therefore, if one postulates that P. gingivalis causes periodontal tissue destruction, there should be evidence of its presence prior to the detection of attachment or bone loss. Several studies have tried to establish temporality in an effort to implicate specific bacterial species in the etiology of periodontal diseases with mixed results (87, 98, 137, 138, 322, 353, 354). Still, it can be argued, based on the studies by Fine et al. (99) that the criterion of temporality can be used in support of an etiological role for A. actinomycetemcomitans in cases of localized aggressive periodontitis. At times, it has been argued that the increased levels and proportions of certain species in diseased sites occur as a consequence of the environmental changes resulting from the disease process. Although this might well be the case, one must bear in mind that the detection of increased levels of putative periodontal pathogens after tissue destruction has occurred is not evidence against its pathogenic role either. In addition, there is also the possibility that in other circumstances the same species might cause periodontal tissue destruction, or that it might be responsible for further destruction at a later time (297).
The criterion of temporality is particularly difficult to satisfy in the study of periodontal diseases. We have alluded above to the notion of disease activity. Current models of periodontal disease progression posit that tissue destruction progresses through periods of acute exacerbations (activity) followed by periods of remission (120, 130, 133, 134). At any given time, different sites in an individual's mouth might be at different stages of disease progression. Clinically, the only way of determining that a periodontal site has undergone periodontal disease progression is by measuring longitudinal changes in clinical attachment level. This implies that in order to determine disease activity, longitudinal monitoring at close time intervals is required. This has been the approach used by many investigators in the past in order to assess risk factors for periodontal disease initiation and progression (133). Typically, study subjects are monitored clinically every two months and bacterial samples are collected at baseline and when disease progression is diagnosed and the microbial composition of the subgingival microbiota examined to determined species potentially involved in disease progression (87). Limitations of early studies were the low-throughput of existing microbiological techniques, which limited how many samples could be conveniently collected and processed. Nevertheless, these studies have helped implicate several subgingival species in the pathogenesis of periodontal diseases.
The concept of temporality is further complicated by the concept of “induction period”, defined as the time from causal action to disease initiation. (297). If, for instance, one observes that disease progression above a certain threshold occurs after the detection of an increase in levels and/or proportions of a subset of the microbiota, this observation would implicate these species as one of the causes of that event. If a sudden change in the composition of the subgingival microbiota triggers a cascade of events (e.g. the appropriate immunoinflammatory response) that eventually lead to tissue destruction of the magnitude that can be detected as changes in clinical attachment level, one still needs to determine the induction period linking the two events. As of now, we have no concept of the length of time that separates changes in the subgingival microbiota and periodontal tissue destruction (assuming these two events are linked). This simple consideration complicates considerably the determination if any given event satisfies the condition of temporality proposed by Hill (152). Further, because any disease process is caused by a constellation of causes, each one of them will have their own induction time and only when all are in place will disease occur.
Therefore, even if one successfully identifies the composition of the subgingival microbiota (there are probably many) that causes tissue destruction, it is clear that the presence of this microbiota will not be sufficient to guarantee disease occurrence. In our current model of periodontal disease pathogenesis, it is well accepted that immune pathology is a major mechanism of tissue destruction. Hence, after the climax community that causes disease has been established, the immunoinflammatory response that it triggers (which has its own induction time) needs to be in place for tissue destruction to occur. The terms initiator and promoter have been used in carcinogenesis to describe component causes of cancer that act early or late in the causal mechanism (297). In the context of periodontitis, bacteria could be described as the initiator and the inflammatory response the promoter. This is an obvious oversimplification as the immunoinflammatory response by the host encompasses many mechanisms that might be required for tissue destruction to occur, each one with its own induction time. For instance, the mechanisms involved in the early lesion described by Page & Schroeder (268) can also be thought of as initiators, while the final mechanisms that result in bone resorption such as activation of osteoclasts might be defined as promoters. Due to the chronic nature of periodontal diseases, one might anticipate that early initiators (bacteria are most likely in this category) might have long induction periods of months or years. It is likely that the longitudinal monitoring of periodontal diseases has a better change at identifying promoters with a short induction time, i.e. mechanisms that occur later in the chain of events that lead to periodontal tissue destruction.
To complicate matters further, disease initiation does not necessarily coincide with disease detection or diagnosis. The time interval between the two has been termed the “latent period” (297). This latent period can be reduced by improvements in the methods of disease detection. These concepts of causation and causal inference will have to be incorporated in any model of periodontal pathogenesis if we are to ever determine the etiological agents of periodontal disease.
Lesson learned: periodontal diseases are viral-bacterial infections
In the 1990s a potential role of viruses in the etiology of periodontal diseases was proposed. The hypothesis was based primarily on association studies that have demonstrated an increase in the load of Epstein-Barr virus type 1, human cytomegalovirus and other herpesviruses in sites and subjects with periodontal diseases compared to gingivitis and periodontally healthy controls. This hypothesis posits that subgingival bacteria and viruses infecting the adjacent periodontal tissues would form a pathogenic consortium (327). In fact, synergy between viruses and bacteria has been demonstrated in several clinically relevant infections including: respiratory infections (influenza-associated pneumonia caused by Streptococcus pneumoniae, alpha-hemolytic streptococci, Haemophilus influenzae, Staphylococcus aureus and Moraxella catarrhalis); acute otitis media (rhinovirus, adenovirus, coronavirus, influenza virus, human cytomegalovirus and other herpesviruses associated with H. influenza, S. pneumoniae, M. catarrhalis, or Prevotella and Peptostreptococcus species); bacterial sinusitis following viral colds; pharyngotonsilitis (Epstein-Barr virus induced overgrowth of P. intermedia and F. nucleatum); and gastroenteritis (associated with rotavirus, adenovirus, norovirus, astrovirus and bacterial taxa such as E. coli, Salmonella, Shigella and Campylobacter jejuni) (324). In a mouse model, co-infection with murine cytomegalovirus and P. gingivalis resulted in a significantly higher rate of mortality compared to mice mono-infected with either pathogen alone (346). Early models of combined viral-bacterial synergistic infections proposed primarily that the polymicrobial infection would enhance the virulence of bacteria or decrease their clearance. However, recent studies have suggested that this co-infection could also enhance the virus pathogenicity. For instance, P. gingivalis sonicate can re-activate Epstein-Barr virus (347). Interestingly, Barton et al. (32) reported results suggesting that herpesvirus latency can confer symbiotic protection against bacterial infections. In a mouse model, latent infection with murine gammaherpesvirus or murine cytomegalovirus provided resistance to infection with Listeria monocytogenes and Yersinia pestis thanks to an elevated expression of interferon-γ and macrophage activation.
Several mechanisms that would explain the potential role of viruses in the etiopathogenesis of periodontal diseases have been proposed such as an impaired local host response or modulation of local cytokine expression induced by viruses, increasing the levels and virulence potential of periodontal pathogens. It has been demonstrated that herpesviruses can produce virus-derived homologues of the anti-inflammatory cytokine interleukin-10 (204) and other inhibitors of a T helper cell-1 response (330). In animal models, cytomegalovirus can impair neutrophil chemotaxis, phagocytosis, oxidative burst and intracellular killing capacity (5). Periodontitis subjects with subgingival herperviruses had reduced neutrophil chemotaxis and bactericidal activity compared to herpesvirus-free individuals (324). Herpesviruses can also interfere with macrophage and complement system antibacterial functions (205, 214). In addition, human cytomegalovirus interleukin-10 can suppress tumor necrosis factor-α and interleukin-1β transcription (250). Alternatively, viruses might induce the release of catabolic inflammatory mediators or other immunopathological mechanisms, causing indirect damage to periodontal tissues. For instance, herpervirus reactivation induces a major increase in cytotoxic T-cells and pro-inflammatory cytokines (243). Herperviruses was shown to induce collagen degradation in an in vitro system (40). In fact, several of the features of periodontal disease pathogenesis can potentially be explained by viral infections. For instance, the conversion of a gingivitis lesion to a periodontitis lesion and of a stable lesion to a progressing one could reflect cycles of activity and latency in herpesvirus infection of the periodontium (326). Herpesvirus reactivation can be triggered by many immunosupressing factors which have also been implicated as risk factors or risk indicators for periodontal infections (328). In addition, the localized nature of the periodontal lesions could be associated with the tissue tropism of herpesvirus infections, while absence of viral infection or latency of the infection might help explain the presence of periodontal pathogens in periodontally healthy tissues and stable periodontal lesions (62).
Several lines of evidence support a potential etiological role for viruses in destructive periodontal diseases. Epstein-Barr virus-1, human cytomegalovirus and other herpesviruses have been detected at high frequency and levels in localized and generalized aggressive periodontitis, chronic periodontitis, and acute necrotizing ulcerative gingivitis (328). Noteworthy is the fact that the elevated frequency of detection of herpesviruses in periodontitis lesions compared to gingivitis and periodontal health has been reported by studies examining samples from different geographic locations (40, 47, 48, 160, 179, 311, 350, 401). Sites with active periodontitis also carry significantly higher numbers of lymphocytes with latent viruses than stable sites (409). Viral DNA has been detected not only in subgingival plaque samples (401) and gingival crevicular fluid samples (40), but also in samples from the adjacent inflamed periodontal pocket wall (185) and surgically removed inflamed periodontal tissues (61). Periodontal lesions infected with Epstein-Barr virus-1, human cytomegalovirus or both also harbored elevated levels of periodontal pathogens such as P. gingivalis, T. forsythia, Dialister pneumosintes, P. intermedia, P. nigrescens, C. rectus, T. denticola, and A. actinomycetemcomitans (63, 169, 310, 311, 325). Counts of human cytomegalovirus and Epstein-Barr virus correlated positively with clinical parameters of periodontal disease such as clinical attachment level, pocket depth and bleeding on probing (311). B-lymphocytes and monocytes/macrophages and T-lymphocytes present in periodontal lesions can be infected with Epstein-Barr virus-1 and human cytomegalovirus, respectively (62, 63). Intervention studies have demonstrated that periodontal mechanical therapy can reduce the levels of herpeviruses (123, 309) and that human cytomegalovirus seems to be particularly susceptible to the effects of periodontal therapy (298). The reduction in the inflammatory infiltrate in the adjacent periodontal tissues might result in lower numbers of infected immune cells (324). Sunde et al. (350) reported on the treatment of a subject with refractory periodontitis who exhibited high levels of subgingival Epstein-Barr virus using the anti-viral drug valacyclovir-HCl, 500 mg BID for 10 days. The therapy resulted in the suppression of Epstein-Barr virus for up to one year and resulted in a significant clinical improvement with reductions in pocket depth and bleeding on probing.
Recently, Garlet et al. (110) provided evidence for an association between human T lymphotropic virus-1 and periodontitis. They examined the presence of periodontal pathogens and herpesviruses in biofilm samples from human T lymphotropic virus-1-seropositive chronic periodontitis subjects (chromic periodontitis/human T lymphotropic virus-1); human T lymphotropic virus-1-seronegative chronic periodontitis subjects and human T lymphotropic virus-1-seronegative periodontally healthy control individuals using real-time polymerase chain reaction (RT-PCR). The expression of mRNA for inflammatory markers in tissue samples was also measured using RT-PCR. The results indicated a higher severity of periodontal disease in the chromic periodontitis/human T lymphotropic virus-1 group compared to the chromic periodontitis subjects. Individuals seropositive for human T lymphotropic virus-1 had significantly higher levels of interleukin-1β, interferon-γ mRNA, elevated expression of interleukin-12 and interleukin-17, and similar levels of expression of tumor necrosis factor-α and interleukin-4 compared to seronegative subjects. Conversely, expression of the regulatory T-cell marker FOXp3 and interleukin-10 was significantly decreased in the lesions from the chromic periodontitis/human T lymphotropic virus-1 subjects compared to samples from the chromic periodontitis and control groups. Interestingly, the levels of periodontal pathogens such as P. gingivalis, T. forsythia, T. denticola, A. actinomycetemcomitans and of herpes simplex virus 1, Epstein-Barr virus, human cytomegalovirus were similar in samples from the chromic periodontitis/human T lymphotropic virus-1 and chromic periodontitis groups. The authors concluded that human T lymphotropic virus-1 might alter the local cytokine milieu, resulting in an increased immunoinflammatory response to a similar bacterial challenge when compared to human T lymphotropic virus-1-seronegative subjects.
However, the etiological role of viruses in periodontal diseases is not without controversy. Sunde et al. (349) failed to find an association between human cytomegalovirus and periodontal lesions and found levels of Epstein-Barr virus to be very close to the lower detection limit of the real-time PCR assay they used. The authors argued that the association between the presence of viral particles and periodontal lesions might simply reflect a higher content of blood and virus infected lymphocytes. They noted that several viruses can be present and replicate in human cells without causing disease symptoms and cautioned against the clinical relevance of the presence of viruses in periodontal lesions and their role in the pathogenesis of periodontal infections. Further, they suggested that evidence of active viral infection such as the presence of high levels of viral particles and/or detection of cells producing viral RNA or protein would provide better support for a role of viruses in the etiology of periodontal diseases. In this context, significantly more herpesvirus genomic copies are found in progressive and untreated periodontal lesions that in stable or treated periodontitis sites (up to 8.3 × 108 Epstein-Barr virus and 4.6 × 105 human cytomegalovirus DNA copies), suggesting possible active replication. In fact, the total count of viruses in severe periodontitis lesions might approach that of bacteria (324).
Obtaining definitive evidence of the participation of viruses in the pathogenesis of periodontal infections will not be an easy task. If viruses are to be involved in periodontal disease initiation and progression, it is anticipated that viral replication would occur prior to periods of disease activity or progression. This implies that longitudinal monitoring of progressing periodontal lesions would be required to determine “temporatlity” in the causal-effect relation between viral activation and periodontal tissue destruction. To complicate matters further, oral herpervirus reactivation in immunocompetent subjects is a phenomenon that lasts only a few hours or a few days (228), making its detection quite difficult. In addition, as discussed for specific bacterial species, the induction period between viral activation and disease progression is unknown and might be rather lengthy. Large scale intervention studies with the use of anti-viral drugs will also be difficult to implement. Although these drugs can be orally administered and are well tolerated with mild side effects, they are only effective against viruses in the lytic phase, limiting their use to the time where active viral infection is occurring. Due to difficulties in select for active viral replication, it can be anticipated that expanding the experiment reported in the case report described by Sunde et al. (350) to a large scale clinical trial would be challenging.
Messenger RNA for herpervirus genes that encode structural proteins can be used as markers of active herpesvirus infection. Therefore, metatranscriptomic analysis of biofilm or tissue samples might help elucidate a potential association between active viral replication and periodontal disease onset and progression in the near future.
Lesson learned: periodontal diseases are biofilm infections
One of the main changes in our understanding of the microbial etiology of periodontal diseases comes from the concept that dental plaque associated diseases such as periodontal diseases are biofilm infections. The understanding that bacteria live in communities and have sophisticated molecular mechanisms of communication among themselves has shaped our understanding of microbial infections in recent years. It is now recognized that more than 65% of all bacterial infections in humans are associated with biofilms (201) and it is estimated that less than 0.1% of the total microbial biomass lives in the planktonic mode of growth (37). Biofilms are characterized by heterogeneous microenvironments within their structure, determined by the gradients of nutrients, oxygen and metabolic waste, providing growth conditions for multiple species and different strains of the same species with distinct phenotypic traits (36). The specialization of subpopulations within the biofilm structure, the so-called “division of labour” (15) has also been described as a common feature of the biofilm mode of growth (36). The understanding of biofilms as multispecies communities has shifted the focus from the role of individual species within the biofilm to a more holistic view of how biofilms as a whole cause and perpetuate chronic infections.
The first descriptions of clumps or aggregating bacteria associated with chronic infections in the medical field dates from 1977 (37) and it wasn't until fairly recently, 1993, that the American Society for Microbiology formally accepted that the biofilm mode of growth was important to microbiology (66). However, the idea of infections caused by “a structured community of bacterial cells enclosed in a self produced polymeric matrix, adherent to a surface”; the definition of biofilms by Costerton et al. (67), is not foreign to the field of oral microbiology. Dental plaque was first defined by James Leon Williams in 1897 who described a “gelatinous accumulation of bacteria, attached to the tooth surface and related to the development of dental caries” (46). Prior to that, in 1846, Finicius (183) had already reported on the consistent presence of clumps of microorganisms associated with dental caries. The notion of an extracellular matrix of polysaccharides produced by plaque bacteria, which “may favor the crowding and grouping of microorganisms in the plaque…” (183) has also permeated the dental literature since the 1960s. An updated description of dental plaque from 1973: “the nonmineralized microbial accumulation that adheres tenaciously to tooth surface, restorations, and prosthetic appliances, shows structural organization with predominance of filamentous forms, is composed of an organic matrix derived from salivary glycoproteins and extracellular microbial products and cannot be removed by rinsing or water spray” (207), already contained all elements of Costerton's biofilm definition. Therefore, it seems that the dental field was decades ahead of the medical field in recognizing chronic biofilm infections.
Despite the many advances in our knowledge about periodontal infections brought by the study of biofilm mode of growth, detailed description of the structural features of supra and subgingival dental plaques were already in place since the late 1960s and 1970s, long before they were understood as biofilms. Early descriptions of different stages in dental plaque development and maturation on tooth surfaces were detailed by studies using light and transmission electron microscopy. For instance, the notion that dental biofilms grow by lateral spread, followed a gain in thickness as a consequence of multiplication of early colonizers comes from those pioneer studies. Competition for space and nutrients, aggregation of bacteria from different species and the columnar patter of microbial colonies in mature supragingival plaque, with filamentous bacteria perpendicular to the tooth surface, were features readily recognized. Those early studies also demonstrated how certain bacterial species tend to thrive under specific environmental conditions and get organized into communities giving supra and subgingival biofilms their unique structural characteristics. Interestingly, none of the microscopic images of in vivo supragingival dental plaque has revealed the so-called mushroom structures often found in in vitro systems of Pseudomonas aeruginosa biofilms. In fact, in vivo biofilms of P. aeruginosa also do not seem to form the highly structured mushrooms, suggesting that they might be artificial and the result of the in vitro growth conditions (37).
The biofilm mode of growth implies that bacteria live and behave as integrated communities, rather than independent entities. This notion also puts into question the role of certain species in the pathogenesis of chronic biofilm infections such as periodontal diseases. In other words, it challenges the paradigm of the specific plaque hypothesis. The concomitant activity of many species that compose the subgingival biofilm would be required to induce disease in what was described by van Steenbergen et al. (381) as “pathogenic synergy”; a concept akin to the mixed infection concept of the 1970s. Others have suggested the idea of a “pathogenic microbial community” (35) or “the community as pathogen” (288) in contrast to the traditional “single-pathogen model”. Despite the relevance of the biofilm as a whole to the periodontal disease process, it is also clear that neither all compositions of the subgingival microbiota result in tissue destruction, nor all elements of a pathogenic biofilm have the same significance to the disease process. For instance, the supra and subgingival plaques are considered a continuum; however, at a certain point during the biofilm maturation the subgingival habitat seems to become isolated from the supragingival environment. The independence between these two ecosystems is such that, at least in deep pockets, consistent removal of supragingival plaque has a minimum impact in the composition of the subgingival biofilm and, more importantly, its capacity to sustain periodontal disease (364). This observation suggests that after a certain point in its development the subgingival microbiota no longer depends on bacterial species from the supragingival biofilms for its survival and pathogenicity. Preliminary data comparing the transcriptomic profile from healthy associated to disease associated biofilms indicate that putative periodontal pathogens seem to be more “active” in disease than in healthy associated microbial communities. In summary, the understanding of periodontal diseases as infections caused by a microbial community is not necessarily novel and was already incorporated in our appreciation of periodontal diseases as polymicrobial infections. Further, the concept of a community as pathogen, does not discard the possibility that certain components of that community have a more relevant role in the disease process.
The most striking difference between the biofilm compared to the planktonic mode of growth is the extreme tolerance of biofilms against antimicrobial agents and their elevated resistance against innate and adaptive immune mechanisms. This feature of biofilms is so important to clinical microbiology that some have proposed its inclusion in the definition of the term biofilm, which was described as “A coherent cluster of bacterial cells imbedded in a matrix – which are more tolerant to most antimicrobials and the host defence, than planktonic bacterial cells” (37). Several mechanisms have been proposed to explain this enhanced tolerance to antimicrobials, including: 1) restricted penetration of antimicrobials; 2) differential physiological activity; 3) persisters and phenotypic variants; 4) entrapment and concentration of antibiotic degrading enzymes; and 5) overexpression of resistance genes. It is noteworthy that most of these mechanisms have been identified using in vitro systems and that the extent to which they contribute to protection of bacteria during biofilm infections is still to be determined. For instance, the hypothesis that the extracellular matrix of the biofilm acts as a diffusion barrier has been refuted, with the possible exception of the negatively charged aminoglycosides (74).
It is also important to realize that biofilms are not more resistant to antibiotics, but rather more tolerant to them; once dispersed, the bacterial components of a biofilm become susceptible to antibiotics (33). In fact, the protection granted by the biofilm mode of growth to the subgingival microbiota does not render them immune to the effects of systemic antibiotics; as demonstrated by the observation of beneficial clinical and microbiological effects after their use as sole therapy in periodontitis cases (221). It has been recently postulated that the presence of a subset of persister cells within the biofilm community is the main mechanism that guarantees their survival after exposure to antibiotics. These cells are dormant cells with a low level of translation (319), which protect them from antibiotics that require cell replication to work. Persisters are considered phenotypic variants rather than mutants and have similar levels of tolerance to antibiotics compared to the wild-type strain. These rare non-growing cells pre-exist in the bacterial population and are not induced by antibiotic administration; however, they can survive the exposure to these drugs and help reconstitute the biofilm after discontinuation of the antimicrobial regimen. If the presence of persisters is confirmed in subgingival biofilms, strategies to kill these dormant cells such as the use of metabolic stimuli to potentiate antibiotics (e.g. mannitol, glucose, fructose and pyruvate) (12); newer β-lactam agents (e.g. cephalosporins and carbapenems), aminoglycosides, and fluoroquinolones that can kill nongrowing bacteria (74); drugs that can target proteins that are essential for the maintenance of persisters (e.g. G3P acyltransferase) (200); and Trp/Arg-containing antimicrobial peptides (52), might become attractive adjuncts for the treatment of periodontal infections. Interestingly, persisters can be readily killed by antiseptics, but their use in most infections is precluded by the toxic systemic effects of these compounds. However, in the case of periodontal diseases, a simple strategy could be devised where the regimen of systemic antibiotics would be followed by the local application of antiseptics such as povidone-iodine or chlorhexidine to the base of the residual pockets.
Lesson relearned: periodontal diseases result from dysbiosis
Ecological concepts have been used in recent years to help explain how the interplay between bacteria and the host might result in destructive periodontal diseases. Marsh elegantly summarized this concept in the so-called “ecological plaque hypothesis” (229, 230). The tenets of this hypothesis were that changes in the environment increase the competitiveness of the putative pathogen at the expenses of species associated with oral health and up-regulate the expression of virulence factors. The ecological plaque hypothesis also helps frame the interplay between bacteria and the immunoinflammatory host response in the etiopathogenesis of periodontal diseases. It is apparent that the maturation of the supra- and subgingival biofilms leads to gingival inflammation, fostering additional changes in the microbial composition of the adjacent biofilm, in a positive feedback loop termed “reciprocal interaction” (336). This intimate relationship between the periodontal microbiota and the host implies that in order to fully understand the sequence of events that result in periodontal tissue destruction, both processes will have to be examined at the same time. However, factors in the subgingival environment that can affect biofilm composition are not restricted to the nature of the immunoinflammatory response. Environmental conditions such as temperature; osmotic pressure; concentration of iron, magnesium or calcium; redox potential; pH and the availability of nutrients, among many others, can alter patterns of gene expression and possibly the pathogenic potential of certain bacterial strains (131, 231).
Marsh was not the first to recognize the importance of the “local environment” in the composition and function of the subgingival microbiota. The notion that properties of microhabitats impact biofilm development has permeated the literature on periodontal microbiology since the 1960s. In 1970, Socransky wrote: “The plaque microbiota is obviously quite complex; it appears to change during its development and probably alters its composition as its environment changes (although this has not been satisfactorily demonstrated)” (332). Studies by Theilade et al. (368) on the microbial shifts that accompanied experimental gingivitis and Ritz (1967) (292) on changes in microbial population composition in developing human dental plaque were cited in support of this hypothesis. In fact, Theilade et al. (368) posited that the initiation of subclinical gingivitis accompanied by an increase in gingival crevicular fluid flow would follow early changes in the bacterial composition of the supragingival plaque, while “clinical gingivitis is manifest about the time when the complex flora is established”. These early findings are in accordance with the modern concepts of “autogenic microbial succession” and “reciprocal interaction” proposed by Socransky & Haffajee in 2005 (336). In recent years the term “dysbiosis” has also been used to describe diseases that result from shifts in the resident microbiota of certain body sites (e.g. vaginosis, ulcerative colitis, otitis media and periodontal diseases) (35).
It is beyond the scope of this paper to fully cover the effects of all ecological factors that might impact the microbiota associate with periodontal health and disease and the readers are referred to excellent reviews that have been written on the subject matter (97, 231, 336). However, in the past decade a series of papers from our department and collaborators have examined the changes in the microbiota from different oral habitats over time using the checkerboard DNA-DNA hybridization technique (136, 199, 203, 299, 360, 373). These studies also examined the impact of environmental forces on microbial succession and greatly expanded our understanding of the interplay between the oral microbiota and the local environment. The outcome of these efforts will be briefly summarized below.
Microbial succession and composition of supragingival biofilms
Studies on succession of bacterial species in supragingival plaque samples have focused on different time frames, from very early events (within hours) (203) to days (360, 373) and months (136) of undisturbed plaque accumulation. Collectively, the results confirmed that adhesion mechanism provide specificity in the attachment of early colonizers from saliva to the tooth surface, with species of the yellow complex such as Streptococcus intermedius, Streptococcus oralis and Streptococcus mitis increasing in numbers and proportions within hours after plaque removal (203). Recolonization of the supragingival environment occurs fast, with counts reaching pre-cleaning levels within 2 days (373). Particularly, species of Veillonella parvula, C. gingivalis, E. corrodens, Neisseria mucosa and F. nucleatum seemed to flourish during 7-days of biofilm regrowth. Despite a significantly higher baseline mean bacterial counts of supragingival bacteria in periodontitis subjects compared to periodontally healthy subjects, the 7-day re-colonization on the supragingival habitat presented similar patterns in the two clinical groups (Fig. 1).
Fig. 1.
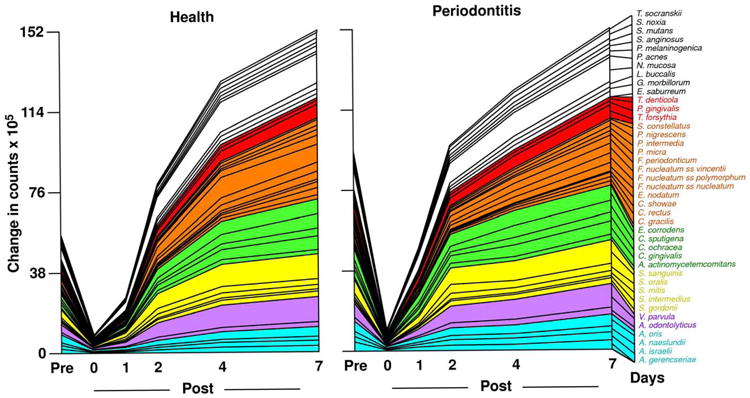
Cumulative mean counts (× 105) of 41 bacterial species in supragingival samples taken from 38 periodontally healthy and 17 subjects with periodontitis prior to professional removal of the dental biofilms, immediately after cleaning, and after 1, 2, 4 and 7 days of re-development. The subjects refrained from oral hygiene procedures for the 7 day test period. Samples were removed from the mesio-buccal aspect of each tooth (excluding third molars) pre-cleaning and immediately post-cleaning. In addition, supragingival samples were obtained from up to 7 teeth in randomly selected quadrants at 1, 2, 4 and 7 days after tooth cleaning. All samples were individually analyzed for their content of 41 taxa using checkerboard DNA-DNA hybridization. Species counts in the samples were averaged within each subject at each time point and then averaged across subjects in the 2 clinical groups. The plots present the cumulative mean values at each time point in each clinical group. The species were ordered and color-coded according to previously described microbial complexes (337). Printed with permission from Uzel et al (2011) (373).
Haffajee et al. (136) explored using cluster analysis and community ordination techniques microbial complexes present in mature biofilms, 1 to 7-day old biofilms and long-term redevelopment biofilms (i.e. sampled 3 to 24 months post periodontal therapy). The results demonstrated that the community structure of the mature and long-term regrowth biofilms were quite similar (Figs. 2 and 3), while the recently formed biofilms were a mixture of communities observed in the more mature biofilms. The typical complexes that formed in longstanding biofilms could not be identified, indicating that 7 days was probably not a sufficient time for the establishment of a climax community with late colonizers. Complexes of mainly Streptococci (yellow complex) and Actinomyces species, similar to those described for subgingival biofilms (337) could be identified. For the other complexes (i.e. green, purple, orange and red), there were a few noticeable differences from the composition of those described for the subgingival biofilms. Capnocytophaga ochracea appeared to be more associated with members of the orange complex than the green complex while V. parvula and N. mucosa appeared to comprise a new ‘purple’ complex in which N. mucosa replaced Actinomyces odontolyticus. However, in the long-term redevelopment analyses, N. mucosa and, to a lesser extent, V. parvula appeared to join the green complex. The largest complex observed in the supragingival samples in terms of number of species was once more the orange complex; however, there were distinct subsets within this community. For instance, Campylobacter gracilis, Selenomonas noxia, and C. ochracea formed a close subset within the orange complex, while P. intermedia, P. nigrescens and certain fusobacteria, formed additional distinct subsets within this larger complex. C. rectus and Campylobacter showae were closely related, possibly because of their similar nutrient requirements. The subsets within the orange complex of supragingival biofilms seemed to be more closely associated compared with subsets observed in subgingival samples. Interestingly, the core red complex species, P. gingivalis, T. forsythia and T. denticola, were joined by E. nodatum in supragingival plaque in both the mature and the long-term redevelopment biofilms. There was also a loose association of red complex species with A. actinomycetemcomitans, Eubacterium saburreum, P. micra, and P. melaninogenica in the long-term redevelopment biofilms.
Fig. 2.

Diagrammatic representation of the relationships of species within microbial complexes and between the microbial complexes in supragingival biofilm samples. This diagram was based on the results of 9 cluster and 2 community ordination analyses using the baseline data from 187 subjects. Modified with permission from Haffajee et al. (136).
Fig. 3.

Diagrammatic representation of the relationships of species within microbial complexes and between the microbial complexes in supragingival biofilm samples. This diagram was based on the results of 9 cluster and 2 community ordination analyses using the long-term plaque re-development data from 93 subjects with post-therapy microbiological data from 3-24 months. Modified with permission from Haffajee et al. (136).
The correlation between the bacterial species detected in supragingival biofilms and periodontal clinical parameters was quite strong, particularly with indices of inflammation. Sites with bleeding on probing or redness were strongly associated with members of the orange and red complexes, in accord with findings for the subgingival biofilms. Interestingly, there was also a close association between the levels of orange and red complex species in supragingival plaque samples with pocket depth. This association was true even accounting for the presence of clinical signs of inflammation. It is possible that the higher levels of these species in deeper periodontal pockets and a higher flow of gingival crevicular fluid associated with them might impact the supragingival microbiota by providing a source of cells for recolonization and nutrients for plaque regrowth. These results suggest that periodontal pocket reduction after therapy might reduce the rate of supragingival plaque accumulation, particularly of species of the orange and red complexes. The presence of gingival recessions also seemed to enhance supragingival biofilm accumulation and favor certain species such as E. corrodens and to a lesser extent C. ochracea and C. gingivalis. This was possibly due to an increased surface area for plaque retention or differential adherence of specific species to cementum and/or dentin.
In two subsequent manuscripts, Haffajee et al. (139, 140) examined the impact of plaque mass and tooth position on the composition of supragingival biofilms. The data indicated that species of the green and orange complexes increased markedly in proportion in samples with high plaque mass, while the proportions of members of Actinomyces and purple complexes decreased (Fig. 4). Interestingly, the proportion of red complex species in supragingival biofilms did not seem to be impacted by plaque biomass. Confirming their previous observations, the presence of gingival inflammation and deep pockets in the tissues adjacent to the sampled site correlated with an increased mass of biofilm. When the relation between tooth position and biofilm mass and composition was investigated, it became apparent that mean bacterial counts were higher at upper and lower molar sites, as well as at lower incisor/canine sites (Fig. 5). Plaque composition was influenced by tooth position even after adjusting for total DNA probe counts, periodontal status, smoking, age and gender. For instance, multiple linear regression demonstrated a positive association between the proportions of C. gingivalis and Streptococcus sanguinis with lower incisor/canine teeth, while Actinomyces naeslundii 2 (currently A. oris) had a positive association with upper incisor/canine teeth and a negative correlation with lower molars.
Fig. 4.

Plot of the total of the mean proportions comprised by the 7 supragingival complexes described by Haffajee et al. (136). The samples were divided according to total DNA probe counts into 10 groups using the 10, 20, 30, 40, 50, 60, 70, 80, and 90th percentile of the total counts as cut points. The x-axis values represent the mean values for total DNA probe counts of each group and the y-axis represents the sum of the proportions comprised by the species in each microbial complex. Printed with permission from Haffajee et al. (139).
Fig. 5.
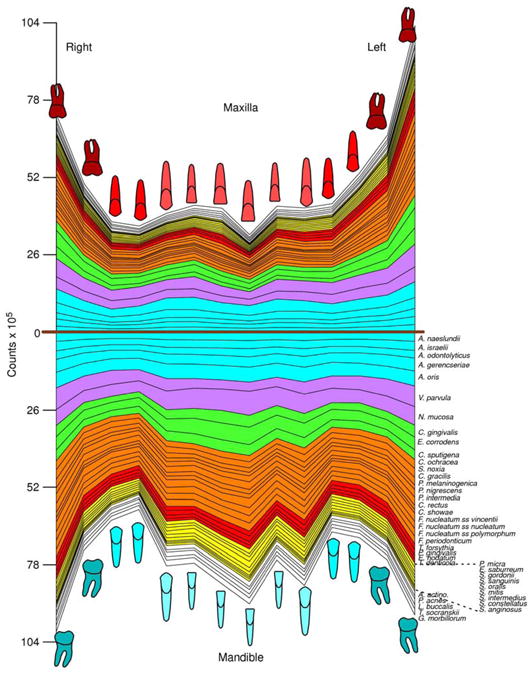
Mean counts (× 105) of 40 test species in supragingival biofilm samples from the mesiobuccal surface of each tooth of 187 subjects. The upper panel represents the maxilla and the lower panel the mandible. Species counts were averaged across subjects for each tooth separately. All species differed significantly among teeth at p < 0.001 (Kruskal Wallis test) after adjusting for 40 comparisons (338). The species were ordered and color-coded according to supragingival microbial complexes (136). In this cumulative plot, the total height at each sample location provides the mean total DNA probe count at that site. The cartoons of each tooth are presented to depict the location of each tooth sample. The full genus and species names of the 40 taxa listed in the Fig. are as follows: Actinomyces naeslundii, Actinomyces israelii, Actinomyces odontolyticus, Actinomyces gerencseriae, Actinomyces oris, Veillonella parvula, Neisseria mucosa, Capnocytophaga gingivalis, Eikenella corrodens, Capnocytophaga sputigena, Capnocytophaga ochracea, Selenomonas noxia, Campylobacter gracilis, Prevotella melaninogenica, Prevotella nigrescens, Prevotella intermedia, Campylobacter rectus, Campylobacter showae, Fusobacterium nucleatum subspecies vincentii, Fusobacterium nucleatum subspecies nucleatum, Fusobacterium nucleatum subspecies polymorphum, Fusobacterium periodonticum, Tannerella forsythia, Porphyromonas gingivalis, Eubacterium nodatum, Treponema denticola, Parvimonas micra, Eubacterium saburreum, Streptococcus gordonii, Streptococcus sanguinis, Streptococcus oralis, Streptococcus mitis, Streptococcus intermedius, Streptococcus constellatus, Streptococcus anginosus, Aggregatibacter actinomycetemcomitans, Propionibacterium acnes, Leptotrichia buccalis, Treponema socranskii, Gemella morbillorum. Modified with permission from Haffajee et al. (140) (bacterial species names were updated).
The composition of supragingival biofilms and their rate of redevelopment are also influenced by the nature of the surface being colonized and the access to gingival crevicular fluid as a source of nutrients. This was demonstrated in recent publications that have examined the composition and development patterns of biofilms that formed on denture teeth compared to natural teeth (299, 360). Teles et al. (360) found that the rate of biofilm redevelopment on natural teeth was far more rapid than on denture teeth and reasoned that this observation could be partially explained by differences in the physical and chemical properties of hydroxyapatite compared to acrylic. However, previous in vivo studies have shown that protein adsorption to surfaces and bacterial adherence are mostly determined by surface roughness rather than by other physicochemical properties of those surfaces (283, 284, 320, 366). With increased roughness more plaque was accumulated and the complexity of the composition of these biofilms also seemed to increase, with higher proportions of rods, motile organisms and spirochetes (366). Based on surface roughness, it was anticipated that the rate of biofilm regrowth would be faster on denture teeth rather than natural teeth. Therefore, other factors unique to natural teeth might have contributed to the differences in plaque redevelopment. Another important difference between these two ecosystems is the presence of the gingival crevicular fluid that bathed the crevice of natural teeth but was absent on dentures, suggesting that this extra source of nutrients might have helped the faster biofilm growth on natural teeth. Further, the proliferation of bacterial cells located in the subgingival habitat might also have contributed to a faster rate of plaque development and a more complex biofilm on natural teeth.
Besides differences in the rate of biofilm regrowth, the composition of the “supragingival” biofilms also differed considerably between denture and natural teeth. For instance, species of streptococci such as S. mitis, S. oralis and Streptococcus mutans were found in higher proportions in samples from denture teeth in pre-cleaning samples. These findings were in accord with data from other studies that used culture techniques (348, 367), checkerboard DNA-DNA hybridization (73) and cloning and sequencing (45) to examine the microbiota associated with dentures that also reported high levels of streptococci. Members of the green and orange complexes such as E. corrodens, Capnocytophaga sputigena, C. gingivalis, C. ochracea, P. intermedia and F. nucleatum subspecies could be detected in samples from denture teeth but were predominant in dentate subjects. In fact, they were present in significantly higher levels on natural teeth compared to denture teeth after just 2 days of plaque redevelopment. The fast accumulation species that have been associated with increases in plaque mass such as E. corrodens and C. gracilis, and of the bridging species of fusobacteria might have fostered the faster development of a more complex biofilm in dentate individuals compared to edentulous subjects. In addition, the higher gingival crevicular fluid volume might have favored the growth of certain facultative/anaerobic non-saccharolytic species including members of the orange and red complexes (70, 305, 362).
In summary, colonization of the supragingival environment is initiated by members of the yellow complex such as S. mitis and S. oralis, while accumulation of Actinomyces species is somewhat slower. Bridging species of the orange complex and late colonizers of the red complex require longer periods of time to establish their tight communities within the supragingival biofilm. The development and composition of the supragingival microbiota is influenced by the presence of inflammation and deep pockets in the adjacent tissues. Other factors such as the nature of the surface (i.e. enamel, cementum, dentin or acrylic), tooth position and plaque mass also influence the microbial composition of supragingival biofilms.
Microbial succession and composition of subgingival biofilms
The redevelopment of subgingival biofilms during 7 days of undisturbed plaque accumulation was also investigated by Uzel et al. (373) and Teles et al. (373). At baseline, when “mature” biofilms were present, periodontitis subjects had significantly higher mean bacterial counts (40.9 ± 7.1 × 105) than periodontally healthy subjects (19.4 ± 3.7 × 105). When the kinetics of biofilm accumulation was examined, it was noticed that there was a marked increase in biofilm species counts between 2 and 4 days; a plateau in counts between 2 and 4 days; and then a second sharp increase between 4 and 7 days (Fig. 7). These phases in subgingival biofilm redevelopment might represent a change in the environment induced by the bacterial species that grew in the first wave. The first wave of growth from 0 to 2 days was associated with a sharp increase in the levels of members of the Actinomyces, purple and green complexes, while the second wave of growth from 4 to 7 days was characterized by a sharp increase in the levels of orange complex species. Changes in the levels of oxygen, nutrients available or pH might no longer be conducive to the early colonizers but facilitate the growth of other species, suggesting a pattern of autogenic microbial species succession (336).
Fig. 7.
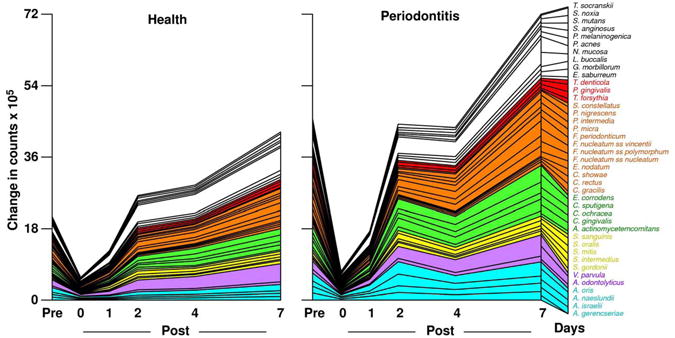
Cumulative mean counts (× 105) of 41 bacterial species in subgingival samples taken from 38 periodontally healthy and 17 subjects with periodontitis prior to professional removal of the dental biofilms, immediately after cleaning, and after 1, 2, 4 and 7 days of re-development. Calculation of species counts and description of the plot were as described in Fig. 1. Printed with permission from Uzel et al. (373).
In contrast with the supragingival biofilm (Fig. 1), the redevelopment of the subgingival biofilms was faster in periodontitis subjects compared to periodontally healthy subjects (Fig. 7). Particularly, members of the green and orange complexes such as C. gingivalis, E. corrodens, Fusobacterium subspecies and P. intermedia increased much faster in periodontitis subjects than in periodontally healthy subjects. Significant differences in the levels of these species between the two clinical groups were already present after 2 days of biofilm regrowth. In contrast, members of the red complex did not reach baseline levels in the periodontitis subjects during the 7 days of plaque regowth, indicating that they required longer periods of time to be established in the subgingival community. The faster redevelopment of subgingival biofilms in periodontitis subjects than in periodontally healthy subjects might be explained by a larger source of nutrients provided by the elevated flow of gingival crevicular fluid and a greater number of residual cells left within periodontal pockets after mechanical debridement that could contribute to the repopulation of that environment.
These observations on early subgingival biofilm redevelopment were expanded by an experimental gingivitis model where plaque accumulation was examined for 21 days using checkerboard DNA-DNA hybridization (412). The data for day 0 demonstrate low total mean counts for the 40 species tested, illustrating the high level of plaque control achieved by study subjects during the preparation phase (Fig. 8). Similar trends to those reported by Uzel et al. (373) for the first 7 days of biofilm accumulation could be observed, with levels of members of Actinomyces, green and, to a lesser extent, orange complex species increasing during this period. Further, levels of V. parvula and N. mucosa also increased in both studies during 7 days of plaque redevelopment. From day 7 to day 21, orange complex species such as Fusobacterium nucleatum ss polymorphum, P. intermedia and P. nigrescens continued to increase in numbers. Levels of members of the red complex did not change significantly even after 21 days of undisturbed biofilm accumulation, indicating that the environmental conditions that foster their growth in the subgingival habitat required periods longer than 21 days to be established.
Fig. 8.

Mean counts × 105 of subgingival taxa detected in 30 periodontally healthy subjects during experimental gingivitis. Subgingival plaque samples were collected from the mesio-buccal surface of 4 randomly selected teeth at different time points. The baseline visit occurred 21 days after a preparation phase to achieve gingival health and minimum plaque accumulation (time 0). Days 3, 7, 10, 14, 21 correspond to different time points after interruption of oral hygiene practices (experimental gingivitis phase); and day 35 corresponds to the resolution of the experimental gingivitis. Counts of 40 subgingival species were measured for each sample using checkerboard DNA-DNA hybridization. Data were averaged within a subject and then averaged across subjects in each time point separately. The species were ordered and grouped according to the complexes described by Socransky et al. (337). Figure adapted from data from Lee et al. (199).
In order to address the impact of longstanding gingivitis on the composition of the subgingival microbiota, we examined the microbial profile of 123 subjects enrolled in a 3-year longitudinal study to examine the effects of preventive programs on periodontally healthy subjects (38). Subjects had their periodontal parameters measured at 4 time points: baseline, 1, 2 and 3 yeas after therapy. Sites were grouped into 3 categories: 1) sites that did not show bleeding on probing in any of the visits (n = 1,489); 2) sites that had bleeding on probing in 1 or 2 visits (n = 1,593); and 3) sites that were bleeding on probing positive in 3 or 4 of the visits (n = 309). The levels of 40 subgingival species measured using checkerboard DNA-DNA hybridization were averaged within subjects, across subjects and then across the different visits within each site category separately, in order to obtain a summary measure of cumulative exposure to these taxa over time. The results suggested that Actinomyces species, V. parvula (purple complex), C. gingivalis, C. ochracea, C. sputigena (green complex), C. rectus, C. showae, F. nucleatum ss. polymorphum, Fusobacterium nucleatum ss vincentii, Fusobacterium periodonticum, P. nigrescens (orange complex), Leptotrichia buccalis, Propionobacterium acnes and S. noxia were associated with the presence of longstanding gingivitis (Fig. 9). Because all subjects were enrolled in preventive programs, they remained periodontally healthy over the 3 years of follow-up. The levels of red complex species were also increased in sites constantly exposed to inflammation compared to “gingivitis free” sites. However, the mean levels of P. gingivalis were below 1.3 × 105 even in constantly inflamed sites, in contrast to levels of approximately 3 × 105 cell counts for bleeding sites in periodontitis subjects and counts above 4 × 105 for deep bleeding sites in periodontitis subjects (336).
Fig. 9.

Mean counts × 105 of subgingival taxa detected in 123 periodontally healthy subjects enrolled in a 3-year preventive program. Subgingival plaque samples were collected from the mesio-buccal surface of every tooth present at baseline, 1, 2 and 3 yeas after therapy. Counts of 40 subgingival species were measured for each sample using checkerboard DNA-DNA hybridization. Sites were grouped into 3 categories: 1) sites that did not show bleeding on probing in any of the visits (n = 1,489); 2) sites that had bleeding on probing in 1 or 2 visits (n = 1,593); and 3) sites that were bleeding on probing positive in 3 or 4 of the visits (n = 309). Data were averaged within subjects, across subjects and then across the different visits within each site category separately, in order to obtain a summary measure of cumulative exposure to the 40 taxa over time. The species were ordered and grouped according to the complexes described by Socransky et al. (337). Figure generated from data from Bogren et al. (38).
In summary, it seems that for the subgingival levels of red complex species found in periodontitis subjects to be reached, additional changes in the local environment might be necessary, possibly events that result in loss of attachment and the development of periodontal pockets.
Reversing the ecological drivers that lead to dysbiosis and periodontal diseases
The ecological plaque hypothesis also provides an excellent framework to explain the effects of periodontal anti-infective therapies. For instance, this hypothesis posits that long-term stability of the clinical improvements obtained with any periodontal therapy will only be sustainable if the ecological changes that led to disease onset are prevented. If the maturation of the disease associated subgingival climax community can be retarded, initiation or recurrence of periodontal tissue destruction could be averted. A long held misconception in the field of periodontal microbiology is that recolonization of the subgingival habitat occurs fast (within weeks), resulting in the need for continuous re-instrumentation for maintenance of results obtained with therapy (226, 247, 312). As discussed above, data generated in the past decade on the study of periodontal microbial ecology have challenged this notion (364). It is clear that the maturation of the subgingival microbiota to its peak complexity (i.e. when late colonizers are established in high levels and proportions) may take months or even years to occur. The concept of exploiting our understanding of microbial succession in the subgingival habitat to improve therapies to treat and control periodontal diseases was suggested in 1998 in Socransky's benchmark study that described subgingival microbial complexes (337). The model built based on data from the microbial analysis of 13,261 plaque samples posited that colonization of the subgingival environment by orange complex species was required for the establishment of the so-called red complex species. The authors suggested that this knowledge could be used in prevention strategies where orange complex species would be targeted, resulting in inhibition of the more pathogenic species of the red complex. By examining the microbial shifts that take place after different periodontal treatments, it is apparent that the ecological changes that take place after therapy are, indeed, closely associated with the clinical outcome.
It may be speculated that the reduction in periodontal pathogens after subgingival debridement, initiates changes in the ecosystem that lead to periodontal stability. Recent data from our department suggest that such changes can last for up to two years (119). We hypothesize that the ecosystem is altered directly by reduction in pathogen numbers and indirectly by altering the status of the local host tissues. In particular, reduction in local inflammation would lead to lower levels of gingival crevicular fluid, diminishing a prominent source of nutrients for the growth of subgingival taxa (339, 360, 373). These alterations may lead to the additional slow but continued decline in a wide range of additional subgingival taxa. Further, periodontal therapy results in reduction of the deep periodontal pockets that foster colonization of red and orange complex species (337, 339). The treatment of periodontal diseases, for the most part, does not involve the reduction of a single bacterial species, but rather the alteration of an ecosystem to one in which pathogenic species remain below the threshold required for disease initiation or progression, while commensal species are favored (364). Changes in the composition of the subgingival microbiota are accompanied by changes in the habitat, particularly the adjacent host tissues. The alteration of the local environment, in turn, leads to an altered ecosystem and additional changes in the composition of the subgingival microbiota with reduction in the numbers of many other taxa. The new climax community approximates the one typical of periodontal health. Thus, the effect of therapy is to “set the clock back” for the development of the mature biofilm, requiring the subgingival microbiota to go through the time-consuming changes necessary to trigger disease recurrence.
When patients are treated with scaling and root planing alone, in time, the oral microbiota will recolonize the subgingival habitat. However, the levels and proportions of red complex species seem to be reduced for up to 12 months after therapy (68, 127). The use of adjunct systemic metronidazole and amoxicillin might have an impact on the subgingival microbiota that goes beyond the red complex species and the levels and proportions of orange complex species seems to also be affected (96, 232, 240, 321). Since these species are required for the establishment of the late colonizers, it seems that the use of this combination sets the clock further back. In this new scenario, the subgingival microbiota seems to require a longer period of time to recompose itself. It is also conceivable that a stronger anti-microbial effect might allow for a better and faster healing of the periodontal tissues, as suggested by the fewer residual pockets obtained with the use of this therapy (54, 125, 399), leading to a new environment that is less conducive for the rebuilt of the pathogenic subgingival biofilm.
In summary, when anti-infective periodontal therapy works properly, it results not simply in the reduction of certain members of the subgingival microbiota considered periodontal pathogens, but rather in a change in the composition of the climax community that colonizes the subgingival habitat to one that is more compatible with health. Further, several of the ecological drivers that resulted in the dysbiosis that led to a pathogenic subgingival microbiota in the first place such as inflammation, deep pockets and increases in the mass of supragingival are also reversed, affording a new ecosystem where non-pathogenic host-microbial interactions are favored.
Lesson learned: bacterial species are a myth
The specific plaque hypothesis seems to have reached a certain level of consensus in the scientific community since the 1970s. However, the use of molecular techniques to examine the microbiota of different body sites has raised awareness of the high degree of intra-species genetic diversity, leading some microbiologists to question the very existence of bacterial species (101). It is apparent that bacteria are profoundly different from eukaryotes in how they exchange genetic material, clouding the concept of species (198). Further, the identification of intra-species genomic variation and data suggesting that genes encoding virulence factors are only found in a subset of strains has also raised questions regarding the relevance of species-level identification (393). Therefore, one might ask - is the specific plaque hypothesis still tenable if the concept of bacterial species is in question? Traditionally, bacterial species have been defined based on morphology, gram stain and a series of phenotypic characteristics determined after a battery of biochemical tests. Indeed, the phenotype of the bacterial strain remains the primary criterion for species classification to this day. However, with the introduction of the use of molecular techniques to assist in microbial taxonomy, including sequencing of 16S ribosomal RNA (rRNA) gene, molecular approaches to distinct bacterial species have also been used. For instance, bacterial species have been defined as isolates sharing at least 70% homology after DNA-DNA hybridization under standardized conditions. However, these methods have serious limitations and cannot be used to group similar strains into species; for instance, rRNA sequences are too conserved to resolve similar species (101). Valid estimates of molecular diversity can be obtained based on variability in sequences of 16S rRNA, but that estimate does not equate with species diversity (291). In recent years 16S rRNA sequences have been favored for the determination of the closest relatives of an isolate, using phenotypic data to complement the characterization. In any event, if horizontal gene transfer (i.e. acquisition of genetic material from non-parental lineages) is as widespread as it has been suggested in the literature, how can bacterial species maintain a set of core genes and traits in face of such genomic fluidity? (291).
This is particularly relevant in the context of biofilm infections, where high cell density and close contact might enhance horizontal gene transfer. Further, horizontal gene transfer has been proposed as a mechanism that allows strains to obtain rapid access to genetic material carrying virulence factors and antibiotic resistance, impacting the pathogenic potential of the recipient cells. How relevant in this context is, for instance, the study of P. gingivalis and its association with periodontal diseases? If the many virulence mechanisms that have been identified in this species can be freely shared among other species, how important is P. gingivalis to the pathogenesis of periodontal diseases? What would preclude any other bacterial species from acquiring these genes of virulence, becoming as pathogenic as P. gingivalis? We will explore this issue by first addressing the question - how freely can genetic material be shared through the microbiome of a given habitat? Studies that have examined the mechanisms that regulate horizontal gene transfer have indicated that the phylogeny of the cells involved in the transfer play an important role. For instance, the incorporation into the genome of genetic material acquired from close relatives is favored because these genes have greater compatibility with the molecular machinery required for their processing (331). The spatial distribution of bacterial cells within the biofilm might also regulate horizontal gene transfer by restricting dispersal. Others posit that ecology rather than geography or phylogeny is more important in regulating horizontal gene transfer (331). Irrespective of the nature of the forces that regulate horizontal gene transfer, mechanisms appear to exist through which phenotypic and genotypic similarity is maintained among a large group of strains than would be expected at random (198). Periodic selection has been suggested as a mechanism to “control” genetic diversification, maintaining genotypic cohesion among bacterial strains (22). According to this concept, a beneficial mutation would allow selection of the strains that acquired this trait, eliminating genetic variability from the population. In essence, natural selection would help control the accumulation of neutral diversity that does not confer any additional benefit to the bacterial strains (i.e. with no direct effect on fitness). Ecotypes or ecospecies are an extension of this concept and refer to the selection of gene pools that confer fitness to certain strains to adapt to specific environmental conditions (57).
Horizontal gene transfer can occur through 3 mechanisms: 1) transduction, when bacteriophages carry bacterial DNA by mistake; 2) transformation, when the bacteria import naked DNA from the environment; and 3) conjugation, when plasmid DNA is transferred between cells. Once the DNA has been transferred to the recipient cell, restriction endonucleases will cleave almost all incoming DNA, while exonucleases will degrade the double-stranded DNA ends of the resulting fragments. These processes not only reduce the size of the incoming DNA, but frequently prevent the DNA fragment from integrating into the recipient chromosome. Alternatively, the incoming DNA fragment can be integrated to the chromosome through homologous recombination, replacing the corresponding resident allele. However, in order for homologous recombination to occur, a certain level of homology in nucleotide sequence between incoming and resident DNA is required. Once incorporated, the maintenance of the newly acquired genes will depend on mechanisms of natural selection of advantageous genes, as described above (198).
After a given gene is incorporated into a recipient cell, because the numbers of bacterial species in a habitat tend to be large, its prevalence in the population is miniscule. This low frequency of detection of the new gene will favor its rapid loss from the population, even if it confers a beneficial function (198). Despite these mechanisms, horizontal gene transfer occurs between bacterial species; however, lateral gene transfers involving phylogenetically distant bacteria are infrequent in comparison to the rate of DNA recombination within species. Certain species of bacteria can differentiate into a state of competence for DNA transformation, in which cells acquire single-stranded DNA through a DNA uptake complex that is specifically localized at a single cell pole (175). In fact, a recent study demonstrated that natural competence mechanisms are present in multiple strains of P. gingivalis (370). In summary, although horizontal gene transfer might result in common exchanges of DNA material within the microbiome of a given habitat, the incorporation of this genetic material into the genome of recipient cells is a much rarer event. In spite of these barriers to gene recombination, mutations will accumulate and eventually genetic isolation will result in the generation of a new species; however, gene divergence is a rather slow process and it occurs over tens of millions of years (57).
We must emphasize that the concept of bacterial species comes from our need to properly identify them in order to generate an appropriate clinical diagnosis of an infectious disease and determine the appropriate course of action, in other words, our need to identify pathogens. In the words of Lawrence & Retchless, “The response to finding spores of Bacillus subtilis versus those of Bacillus anthracis (the causative agent of anthrax) would be very different” (198). Despite the realization that the microbiome is vastly more diverse than previously suspected, microbiologists are still able to recognize clusters of bacterial isolates based on their phenotypic and genotypic characteristics and name them “bacterial species”. Some have posited that a core of genes maintains species-specific phenotypes, the so-called core genome hypothesis, arguing that bacterial species can be rationally identified and named, despite their genetic variability (291). By comparing the genomes of different isolates from the same species for which whole genome sequences were available or by using subtractive hybridization and comparative genome hybridization, scientists have demonstrated that, in fact, members of a bacterial species share large portions of their genomes, but unique, strain-specific sequences were also found. The ratio shared (core)/unique (auxiliary) portions of the genome varied greatly among different species. The core genome hypothesis reconciles the existence of dynamic genomes conferred by horizontal gene transfer with the existence of clusters of isolates that exhibit similar phenotypic traits, used by microbiologists over decades to group then into “bacterial species”. The grouping of bacteria based on phenotypic features has gained further validation by the demonstration that several well-defined phenotypic clusters correlate to genotypic clusters (101).
Here lies another lesson unlearned and learned: bacterial species do exist. In the words of Riley & Lizotte-Waniewski: “…the real argument remaining is not do they exist, but rather how can they exist in the face of potentially high levels of horizontal gene transfer” (291). The debate among molecular microbiologists regarding the concept or meaning of bacterial species will continue in the foreseeable future. However, that should not preclude the field from continuing to employ useful definitions of bacterial species, which should incorporate our expanding knowledge on the genetics of those microorganisms, while maintaining the use of well established phenotypic traits.
Lesson learned: subgingival bacterial species present high genetic diversity
During the search for the etiological agents of periodontal infections, it became apparent that, despite the colonization of the oral cavity by a large number of bacterial species, only a handful of species were consistently associated with periodontitis, particularly P. gingivalis and A. actinomycetemcomitans (1, 131, 246). The phenotypic traits of those organisms became the focus of the scientific community and their virulence mechanisms began to be characterized (114, 215). The infectious nature of the disease led to an interest in determining the epidemiology of periodontal pathogens, including their acquisition, intra-oral distribution and route of transmission (113, 280, 408). Such information would provide a better understanding of the role of bacteria in initiation and progression of periodontal diseases. Thus, during the 1990s a number of studies used restriction endonuclease analysis (113, 218) , restriction fragment length polymorphism (216), multilocus enzyme electrophoresis (217), and arbitrarily primed polymerase chain reaction (239) to measure the relatedness of bacterial strains isolated from different individuals. Related individuals, such as spouses and siblings as well as parents and offspring and unrelated individuals were studied in the search for the clonal variability of certain bacterial species (239, 280, 372, 377, 408). In most studies, the focus was the clonal variability of periodontal pathogens such as P. gingivalis and A. actinomycetemcomitans, but other relevant periodontal taxa such as E. corrodens, P. intermedia, P. nigrescens and F. nucleatum were also investigated (50, 86, 233). We will center on the studies of P. gingivalis to illustrate the implications of genomic variability of subgingival species in the study of periodontal disease etiology.
The study of clinical isolates of P. gingivalis demonstrated that that this was a highly diverse species. Very different genetic and enzymatic patterns were observed among the isolates analyzed and no predominant dominant clone was observed across samples from unrelated individuals. Instead, rather unique individual patterns were identified (11, 218, 239, 265). In addition, it was observed that the same clonal types were often found in closely related individuals, such as parents and offspring, siblings and spouses, suggesting horizontal and vertical transmission (280, 372, 377, 380). It was shown that typically only 1-2 clones would colonize any given individual (11, 20, 219). Even though the source of the clonal types and transmission could be determined with those studies, the relevance of the differences across strains remained to be elucidated. The observation that the detection of P. gingivalis was not a determinant for the occurrence of periodontitis suggested that the genetic and/or phenotypic differences observed across the isolates could perhaps explain why some P. gingivalis carriers would develop periodontitis and others would not. Most of the studies that associated clonal variability with disease status have suggested that individual clones or genetic types may express different phenotypic characteristics. The hypothesis was that clusters of clones/strains would express specific phenotypic characteristics which would confer virulence (113).
Even though specific genotypes have been associated with disease, including those enconding specific types of fimbriae and capsule (186), no particular strain appeared to predominate in disease. Most studies have failed to demonstrate a relationship between specific clusters of clones of P. gingivalis and the periodontal status of the host (217, 239). In addition, there is evidence that similar clonal types are observed in periodontal health and disease (356). It is possible that this discrepancy is due to the use of different methodological approaches and sample size. Because of the observed heterogeneity in strains and lack of a predominant clonal types associated with disease, it has been proposed that all clonal types of P. gingivalis would be equally effective in colonizing the human host and that they shared a common virulence potential (11). This argument led some authors to suggest that P. gingivalis is a commensal organism that can become an opportunistic pathogen (217, 239). However, the lack of distinct pathogenic clones within bacterial species does not define commensalism (20). In light of its widely demonstrated virulence properties and disease association, it appears unlikely that P. gingivalis is a commensal species (162, 194, 195, 396).
Besides the search for specific strains that could be involved in disease initiation and progression, different researchers also sought genotypic and phenotypic traits that could be involved in the pathogenesis of periodontal diseases. P. gingivalis has several virulence factors, like capsule, proteases and fimbria. Because fimbria is a major virulence factor of P. gingivalis, the study of the variability of fimbrial genes in P. gingivalis in periodontal health and disease seemed like a next logical step. P. gingivalis presents minor and major fimbria, which has been classified into 6 types (251). In clinical studies, the fimA II and fimA IV genes were associated with severe periodontitis and strains having these genes demonstrated greater adhesive and invasive capabilities (92, 186, 371). Other traits were also evaluated in association with disease, such as capsular antigens (193) and gingipains (34), with inconclusive results.
It can be argued that the some of the studies that aimed to identify pathogenic clones of P. gingivalis cited above employed techniques with variable discriminatory powers and reproducubilities. Techniques that utilize sequencing have become increasingly accessible and allow the molecular typing of unrelated isolates, by characterizing genetic variations in the chromosomal DNA of bacterial species. Therefore, they are more precise and reproducible than pattern-based or “fingerprinting” technologies, which simply determine distinct patterns of enzyme or DNA fragments. Molecular typing techniques, such as multi locus sequence typing, have been employed recently to study the population structure and genetic variability of P. gingivalis. Multi locus sequence typing is a nucleotide sequence-based approach for the genetic characterization of bacterial isolates, based on the genetic variations found in a pre-determined group of genes, typically 5-8 housekeeping genes, such as pepO, recA, dnaK, nah and pga (177). Using multi locus sequence typing, Enersen et al. (93) showed that periodontitis patients could harbor up to 4 clonal types of P. gingivalis and that refractory periodontitis patients could be colonized by up to 8 clonal types. This is a clear departure from the previous findings of 1 to 2 clones per patient (219). In addition, only closely related clones were observed in the same pocket, suggesting that DNA recombination may have occurred at that site. Thus, while there is considerable heterogeneity among P. gingivalis isolates, intraindividual heterogeneity is low. Such genetic plasticity seems to be indicative of efficient mechanisms to adapt to a multitude of habitats and shifts in the environment, ensuring the survival of a portion of the population. However, the question of whether different groups of clones and different sets of phenotypic traits expressed by them could be linked to periodontal disease initiation and progression remained unanswered. Because P. gingivalis colonization starts early in life (64, 270) and disease manifestation tends to occur decades later (8), it could be argued that a change into more pathogenic clonal types could lead to periodontal diseases over time. This hypothesis could explain the events of disease initiation and its bursts of activity as previously noted. In that context, the next logical question would be whether clonal stability exists in P. gingivalis colonization over time or if new (and more pathogenic) clones would arise and coincide with the onset of periodontitis.
This theory was tested, in part, by van Winkelhoff et al. (384). Using AFLP, the authors examined clonal types of P. gingivalis in a population, including families, with untreated periodontitis over 8 years. The authors observed considerable volatility in acquisition and loss of P. gingivalis genotypes. Even though P. gingivalis was detected in 105 individuals in 1994 and in 103 subjects in 2002, only 66 (46%) of those subjects remained positive in both examinations. However, the percentage of individuals presenting 1, 2 or 3 genotypes (about 70%, 20% and 5%, respectively) remained stable over time. Forty-six participants (69.7%) had at least one identical genotype in the second examination. The findings from a later study using multi locus sequence typing (374) indicated that P. gingivalis' clonal stability is a robust event, in that even in the presence of periodontal treatment consisting of scaling and root planning in combination with systemic antibiotics (amoxicillin and metronidazole), the clonal types observed post-therapy were the same ones detected prior to intervention. These results suggest at least four possibilities: 1) the incomplete eradication of P. gingivalis by treatment allowed cells to remain in the pocket and neighboring tissues and these cells recolonized the site after treatment; 2) the same strain could be re-acquired by familial transmission; 3) positive selection of clones better fit to sustain the mechanical and antimicrobial challenges imposed by periodontal treatment; and 4) combinations of all 3 mechanisms or any combination of 2 such mechanisms. Unfortunately, in the study by Winkelhoff et al. (384), the association between the stably colonizing clones and disease progression was not examined. In addition, the relation between the presence of the post-therapy isolates found by Valenza et al. (374) and response to periodontal treatment was also not tested. Therefore, inferences regarding their association with progression of periodontitis and periodontal therapy outcomes could not be made.
The studies cited above demonstrated the clonal diversity of P. gingivalis and the stability of the colonization by such clones. The genetic diversity observed in P. gingivalis seems to reflect the variability of its habitat (370), represented by different hosts and different sites, possibly representing different ecotypes (331). This is supported by the evidence that unrelated hosts harbor different clonal types and closely related hosts, like siblings, parents and spouses appear to share similar clones (384). It is also supported by the findings from Enersen et al. (92), who demonstrated that only closely related clonal types were observed in individual pockets (1 to 8 clones per pocket). In general, the variation within a site was limited to 2 genes, pepO and recA, which are involved in DNA transformation, replication, recombination and repair, suggesting DNA recombination at the periodontal site. Despite the advances in the understanding of the population structure and colonization dynamics of P. gingivalis, the relevance of the genetic differences observed across strains remained to be determined. Most of the interest in the genetic characterization of pathogens resides in their virulence mechanisms so that better diagnosis and treatments can be devised. Thus from a therapeutic standpoint, the presence of similar virulence mechanisms present in all strains of a species seems advantageous. The description of a core set of genes present in a species is a first and crucial step in that direction. Because of such high degree of genetic variation and the lack of association of genotypes with disease status, the characterization of the core genome of P. gingivalis seems especially attractive.
Studies using comparative genomic hybridization aimed at identifying such differences and led to a first draft of P. gingivalis' core genome. Comparative genomic hybridization is a microarray-based technology that allows the comparison of several variants of one genome (such as among different bacterial strains) against a reference genome (such as a well-characterized and sequenced strain). Brunner et al. (44) used comparative genomic hybridization to analyze capsular and non-capsular P. gingivalis serotypes with different levels of virulence against the well-studied and highly virulent W83. The authors observed that a conserved core genome from P. gingivalis consisted of 80% of the analyzed genes from the W83 strain and most of the virulence related genes observed in that strain could be found in the core genome. The authors argued that the genes that are not present in the core may be the determinants of the differences in virulence found between the strains. These results should be seen as a first step, as the size of the core genome is a function of the number of strains analyzed, i.e., the greater the number of strains, the smaller the core is expected to be, due to the increase in genetic variability. For instance, the core genome of E.coli has been found to be 46% of the average core genome as based on the whole genome multi-alignment of 20 E. coli strains (369).
In summary, P. gingivalis population is highly diverse, clonal colonization is stable, and many virulence genes are present across strains; however, hitherto, no set of clones harboring a set of genes has been implicated in the initiation and progression of periodontal disease. If we are to fully understand the role of P. gingivalis or any other subgingival species in the pathogenesis of periodontal diseases, this genetic diversity will have to be taken into account. This should not be a surprise to researchers in the field; as highlighted by Haffajee &Socransky in 1994 (131), “A major recognition of the last decade was that all clonal types of a pathogenic species are not equally virulent”.
Lesson learned: molecular techniques, rather than cultivation are required to study the microbiota of periodontal diseases
Benchmark studies conducted in the 1970's and 1980's defined bacterial species thought to be important in the initiation and progression of periodontal diseases as well as species thought to be host-compatible or beneficial (336). Those studies encompassed an enormous amount of work, as they entailed the cultivation and characterization of thousands of isolates from samples collected from hundreds of patients. Because culture-based techniques are slow and laborious, only a few samples could be collected per individual. Yet, studies from distinct research groups provided remarkably concordant results and provided the basis of our current understanding of the etiopathogenesis of periodontal diseases (87, 131, 246). Overall, they demonstrated that approximately 500 taxa are capable of colonizing the human oral cavity, of which 359 were frequently detected and 141 were observed only once (77, 246). Any individual may typically harbor 150-200 different species and it was estimated that between 10-30 species can initiate destructive periodontal diseases (335). A. actinomycetemcomitans, P. gingivalis E. nodatum F. nucleatum, P. intermedia, P. nigrescens appeared to be associated with periodontal diseases as the percentage of total isolates increased with increased disease severity and Actinomyces naeslundii, C. gingivalis, N. mucosa S. oralis, Streptococcus salivarius, Streptococcus sanguinis and V. parvula appeared to be associated with periodontal health or stability, as the percentage of total isolates decreased with increased disease severity (246).
Because periodontal diseases are site-specific, can occur in any of the 168 typically evaluated clinical sites and often develops in only a small subset of those sites, there was a need to improve the throughput of microbial techniques, to accommodate the analysis of a larger number of samples per individual. In addition, there was an urge to develop faster, less costly techniques that could bypass the time intensive cultivation of samples. The checkerboard DNA-DNA hybridization technique (339, 341) came to fulfill such need and allowed the enumeration of a multitude of taxa in many samples simultaneously. This technique enabled a quantum leap in our knowledge of oral microbiology and ecology as well as diagnostic, pathogenesis and treatment of periodontal diseases (85, 225, 359, 360, 365, 373, 404, 412). To put this technological advance in perspective, one must revisit the typical number of samples examined per year at Forsyth prior to the development of the checkerboard. Between 1969 and 1979, 135 subgingival plaque samples were examined by culture at Forsyth, a number that increased to 300 between 1982 and 1988 (50 samples/year). The use of a colony lift method allowed the examination of 9,600 samples between 1988 and 1993, or 1920 samples/year. The use of the checkerboard DNA-DNA hybridization technique allowed the analysis of 34,400 samples between 1993 and 1999, about 5,734 samples per year (143). The ability to bypass cultivation of clinical samples and to collect biofilm samples from large numbers of sites per patient came with the “compromise” that only a subset of the taxa known to colonize the oral cavity was included in the probe panel routinely utilized. In addition, only cultivated species could be included because of the requirement to grow bacteria for DNA extraction and probe preparation. The selection of the checkerboard probe panel was performed based on the culture studies cited above, where the relevance of bacterial species to periodontal health and disease was determined.
Two major criticisms often surrounded the checkerboard technique. The first relates to the extent and composition of the panel of probes; at times it was felt that 40 probes was to low a number in face of the more than 300 species that can colonize the periodontal pocket. Interestingly, criticism in the other direction also occurred, as 40 taxa have been suggested to be “too many” and that the panel should be narrowed further to account for the truly relevant species. The second criticism focused on the use of whole genomic DNA probes, in part because their use may increase the probability of cross-reactions between species due to common regions of DNA among closely related species. There were also concerns that they would not detect all strains of a given species and that they would have a low sensitivity in terms of the numbers of cells that they detected. Those critiques were addressed in detail in a publication in which the status of the checkerboard technique was evaluated after 10 years of routine use in clinical studies and the analysis of tens of thousands of biofilm samples (339). The authors searched for cross-reactions by hybridizing probes for the 40 typical species analyzed by checkerboard with targets from 80 bacterial species. Probes for certain species such as T. forsythia showed virtually no measurable cross-reactions to any of the test taxa. The probe for F. nucleatum ss vincentii exhibited weak cross-reactions with F. nucleatum ss nucleatum and F. nucleatum ss polymorphum as well as a weak reaction to C. rectus. The probe for S. intermedius exhibited virtually no cross-reactions except for the expected reactions with the two other members of the ‘streptococcus milleri group’, Streptococcus anginosus and Streptococcus constellatus. The authors also demonstrated that the cross-reactions observed could be eliminated with the use of subtraction hybridization and PCR probes as well as competitive hybridization, in case one was particularly interested in those species.
In addition, uncultivated and difficult to grow species could not be detected by the checkerboard technique, at least not using the traditional panel. However, if one so chooses, difficult to grow species could easily be incorporated in the checkerboard DNA-DNA hybridization technique. Still, the cultivation studies that provided the basis for the selection of the checkerboard probe panel might have overseen those difficult to cultivate species and most certainly did not detected uncultivable ones. It was known that many organisms recognized by their appearance in plaque samples viewed by light or electron microscopy, were not being cultivated (340). In fact, additional uncultivated species might not even be distinctive under the microscope, making virtually impossible to estimate the extent of the uncultivated segment of the microbiota and whether it harbors pathogenic species. The discrepancy between microscopic and plate counts had been known for many years in oral microbiology (242, 333) as well as in environmental microbiology (286), and the phenomenon was later described as the “great plate count anomaly” (343).
The work by Amman et al. (14) is often cited to support the contention that cultivation approaches detect only small fraction of the microbial content, approximately 1% of its diversity, and that molecular approaches provide a more comprehensive view. Although this number has been disputed in the field of environmental microbiology (80), it seems that environmental microbiologists face a much greater challenge than oral microbiologists. It is a testimony to the quality of the work of the pioneers in periodontal microbiology that over 50% of morphotypes present in plaque samples can be grown in the laboratory. This figure has often been preceded in several texts by the word “only”. We have a more optimistic view and see the 50% of cultivable species as a perfect example of a “glass half-full”, particularly considering the numbers suggested by Amman and co-workers for other types of samples. Due to continuous efforts to cultivate difficult to grow oral bacterial species by a small number of dedicated scientists (83, 387, 388, 390), this estimated percentage of yet uncultivable species has been recently reduced to 35% (75). In fact, Griffen et al. (124) recently used pyrosequencing to compare subgingival biofilm samples from periodontally healthy and periodontitis patients and reported that although sequences from uncultivated taxa were more abundant in samples from diseased sites, overall, 81% of the sequences identified were mapped to cultivated species.
Lesson learned: open-ended molecular techniques, rather than cultivation are required to study the microbiota of periodontal diseases
The nomenclature of oral phyla, species and phylotypes has undergone considerable revision in recent years. In the interest of clarity, in this manuscript, whenever possible, their current names will be indicated in parentheses and will follow the taxonomic anchors described in the human oral microbial database (Chen et al. (51); www.homd.org) – to be inserted as a footnote.
Open-ended culture-independent techniques were received with great enthusiasm in the field as they would be able to overcome the “bias” imposed by cultivation and a much larger set of taxa could be examined. This approach had the advantage of not focusing on a pre-determined set of bacterial species, as it had been the case for checkerboard DNA-DNA hybridization (337), PCR (19), RT-PCR (302) or in-situ hybridization (7). Rather, the amplification of conserved areas of a ubiquitous bacterial gene (16S rRNA gene) by a highly sensitive method (PCR) (122) would allow the identification of all microbial taxa present in a given sample. Ultimately, there was an expectation that new species would be identified in the uncultivated segment that would be associated with periodontal health and disease and would bring much needed answers to many of the questions about the microbiology of periodontal diseases. A few groups began using cloning and sequencing to explore the microbial diversity in the oral cavity of periodontitis patients (184, 303), but the first comprehensive description of the subgingival microbiota based on this approach was delineated by Paster et al. (276). The authors analyzed subgingival plaque samples from 31 individuals, including periodontally healthy subjects as well as chronic and refractory periodontitis, acute necrotizing ulcerative gingivitis and HIV-associated periodontitis patients. The authors estimated that 500 taxa could colonize the oral cavity and found that 347 species/phylotypes could be found in the subgingival environment. Of those, 215 were novel phylotypes (i.e. uncultivated or cultivable but unrecognized taxa) and 140 of them were detected only once. Interestingly, these numbers are strikingly similar to those obtained by Moore & Moore (246) using cultivation.
This study showed for the first time the presence of members of phyla never before detected in oral samples, such as OP11 (SR1) and TM7, for which there are no cultivated representatives, as well as Deferribacteres (Synergistetes). A number of other novel phylotypes were proposed to be associated with disease, including Desulfobulbus clone R004 (HOT [human oral taxon] 041), Eubacterium clone PUS9.170 (Peptostreptococcacea sp. HOT 103), and Megasphera clone BB166 (HOT 121, later named Anaeroglobus geminatus). Cultivated pathogenic periodontal species were also detected and associated with periodontitis, but much less frequently than other species and phylotypes. For instance, P. gingivalis and T. forsythia were detected in not more than 5 chronic or refractory periodontitis patients (8 and 11 clones, respectively), while P. endodontalis, Prevotella tannerae, E. saphenum, Filifactor alocis and Dialister pneumosintes were present in between 4 and 8 diseased patients (7-20 clones) and novel phylotypes such as TM7 clone I025 (HOT 356), Synergystetes clone BH017 (HOT 369) and Eubacterium clone PUS9.171 (Peptostreptococcacea HOT 103) were detected in 4-5 diseased pts (6-43 clones).
This benchmark study Paster et al. (276) unveiled the diversity of the subgingival microbiota in much greater breadth than ever before. Further, the authors started to make associations between certain novel taxa as well as less studied named bacterial species and periodontal health and disease. That study paved the way for other studies on the microbial diversity in the oral cavity and the potential role of the uncultivated segment of the subgingival microbiota in the development of periodontal diseases. Kumar et al. (188) expanded the associations initially proposed by Paster et al. (276) and searched for selected uncultivated/unrecognized phylotypes as well as characterized species not previously thought to be associated with periodontitis. Two taxa were associated with periodontal health, namely Deferribacteres clone W090 (Synergistetes sp HOT 363, which, until recently, was described as an unnamed cultivated taxon, but has been characterized and named Fretibacterium fastidiosum (387) and Bacteroidetes clone BU063 (Tannerella sp. HOT 286, an uncultivated taxon), which had been previously associated with health (202). Five uncultivated/unrecognized phylotypes were associated with periodontal diseases, including Deferribacteres clones D084/BH017 (Synergistetes sp HOT 360/362) and Bacteroidetes clone AU126 (HOT 274) confirming some of the findings previously reported by Paster et al. (276).
Besides expanding on the diversity of the subgingival microbiota, the most striking finding of early studies using cloning and sequencing was the infrequent detection of recognized periodontal pathogens like P. gingivalis and T. forsythia (189, 190, 276). These findings contradict the lessons learned from studies using cultivation (87), checkerboard DNA-DNA hybridization (336, 339), RT-PCR (4, 55, 260) and fluorescence in situ hybridization (FISH) (413). It seemed that decades of microbiological studies were proven incorrect or dismissed. It is quite tempting to accept novel pathogenic taxa and dispose of the old ones, as novelty tends to be rather appealing. Besides, this is the nature of science; new concepts tend to replace old dogmas as new knowledge is obtained. However, because of the overwhelming body of work that has implicated P. gingivalis, T. forsythia and other subgingival species as putative periodontal pathogens; one should exert caution in overinterpreting early findings obtained with emerging technologies.
The apparent discrepancies between data from cloning and sequencing and other microbiological techniques can be attributed to several technical issues associated with sample collection and processing. The inclusion of samples from different diseased entities might have clouded differences between periodontal health and disease. For instance, in the Paster et al. (276) study, samples from a rather heterogeneous group of disease entities were included and it is likely that each of those conditions present distinct microbiotas. Due to the low-throughput afforded by the technique, relatively small number of patients and samples from each subject were included (189, 190, 276). This can be a problem because considerable variability has been observed in the composition of host-associated microbiotas (65, 274), including subgingival biofilms (158). Further, typically 168 sites are clinically monitored in periodontal studies (28 teeth x 6 sites per tooth); therefore, 4 subgingival samples represent less than 3% of all sites being monitored. In addition, when few samples are collected from the same individual the larger one(s) tend to be overrepresented; this is particularly critical when samples are pooled together (335). A lesson learned by Socransky during his early days as a microbiologist and passed on to his many students was “thou shalt not pool”; in reference to the many problems that arrive from trying to interpret data obtained from pooled plaque samples. It is noteworthy that close-ended approaches using RT-PCR have been able to routinely detect classic periodontal pathogens, even when few samples were collected or pooled (41, 55), suggesting that other procedures associated with cloning and sequencing could be the source of the infrequent detection of periodontal pathogens.
As more studies using PCR amplification and cloning on microbial communities became available, the limitations of the procedures involved in the technique became clearer and strategies to overcome them were devised. In addition to the infrequent observation of recognized pathogenic bacterial species, members of the genera Fusobacteria and Actinomyces also appeared to be underrepresented in studies using cloning and sequencing (184, 189, 248, 249), which also contrasted with reports based on culture (246), checkerboard DNA-DNA hybridization (337) and FISH (411). One of the possible reasons for these findings was the assumption that the use of universal primers would amplify DNA from all bacterial species present in a sample with the same efficiency; this was later proven not to be the case. Hutter et al. (159) employed 2 sets of universal primers and reported that they could observe P. gingivalis in 12 of the 26 periodontitis patients examined. This was achieved even though only one sample per patient was collected and much fewer clones per patient were sequenced (22 compared to 50 to 100 routinely analyzed). Later, de Lillo et al. (72) demonstrated that “universal” PCR primers can introduce biases into the analysis of the species composition of clone libraries, because of mismatches between the primer and target organism sequences. Since then, the use of different sets of universal primers has been proposed, as well as the use of enrichment primers for specific phyla such as spirochetes (75, 159, 276).
Other sources of biases associated with the selection of primers and other procedures inherent to the PCR amplification of DNA from complex biofilm communities were explored in detail by other authors (94, 180, 227, 290, 352). Collectively, they demonstrated that several aspects of the PCR amplification of multitemplate samples including template concentration, number of amplification cycles, annealing temperature and chimera formation could bias alter the microbial profiles of the sample under study. Further, the nature of the cell wall of the species present in the sample will affect the lysis and DNA extraction protocols, and higher G+C genomes appear to be less successfully amplified (13, 78, 281, 392). In the case of cloning studies, the number of clones analyzed might also influence the resulting microbial diversity and profiles. In addition, the use of an aliquot of the DNA extracted from samples might introduce other biases analogous to the ones introduced by the dispersion and dilution necessary for cultivation studies (340) and this aliquot might not fully represent the whole sample (49). The value of validation studies testing different sources of biases at different stages of the sample processing such as collection (358) and amplification (89, 357) cannot be emphasized enough. Further, the effects of selection of primers (72, 187) and the levels and types of taxa should also be assessed using mock communities of known species (78, 157, 361). A recent paper from Diaz et al. (78) elegantly illustrates the relevance of validation studies. The authors examined the limitations associated with high throughput sequencing or next-generation sequencing and the intrinsic variability of the oral ecosystem. Next-generation sequencing is an open-ended, culture independent approach that has replaced cloning and sequencing in the study of complex ecosystems and only recently was introduced for the study of the oral microbiota (172, 410). The analysis of mock communities composed of 7 oral bacterial species showed primer and DNA isolation biases and an overestimation of diversity. The data from the clinical and mock samples allowed the authors to devise an experimental and analytical framework that should facilitate the design and interpretation of future high-throughput sequencing studies (78). The long list of biases presented above should not discourage the reader from considering the findings obtained on the composition of the periodontal microbiota using open-ended molecular techniques. Instead, it should be a reminder that all techniques have limitations; it is up to the scientist to identify them, to acknowledge them, to attempt to overcome them, and most importantly, to carefully interpret the results generated with them. This is particularly important when using new techniques or exploring new environments.
Next-generation sequencing has become more accessible in recent years, leading to an increase in the numbers of publications that uses this technology to study the oral ecosystem in health and disease (6, 78, 191, 307), including periodontal diseases (124, 209). The first study to investigate the oral microbiota via next-generation sequencing estimated that 19,000 phylotypes may be present in the human oral microbiota (172), a major increase since earlier estimates of 700 taxa (278). Meanwhile, as the technology became more routinely used, a number of potential sources of biases have been identified. For instance, Kunin et al. (192) showed that the use of low stringency filters for sequencing reads can overestimate diversity and lead to inflated numbers of taxa in a given environment. In addition, Wu et al. (400) demonstrated that PCR conditions such as the choice of polymerase, the dilution of the template and the number of PCR cycles used can have significant effects on the analysis of microbial diversity and community structure. Because these technologies provide tremendous coverage depth, it is important to determine the extent of the biases introduced on the profile of any given community. Once those sources are recognized, strategies to overcome them can be devised (157, 166, 187, 191).
When using next-generation sequencing, the selection of the targets for amplification is critical for the true representation of the microbiota under study. In a recent study, Kumar et al. (187) used 4 sets of primers, targeting different regions of the 16S rRNA gene, namely V1-V3, V4-V6 and V7-V9, to amplify subgingival biofilm samples from 10 periodontitis patients. The authors observed significant differences across the microbial communities generated by different target regions. Finally, the authors concluded that primers targeting both regions V1-V3 and V7-V9 should be used in deep sequencing efforts to characterize heterogeneous communities. In subsequent paper from the same group (124), the authors used pyrosequencing to compare the subgingival microbiota in periodontal health and disease. After using stringent methods for the selection of target regions for amplification and for data analysis, the authors observed clear differences in the microbial profiles of the 2 conditions, primarily in the levels of P. gingivalis, T. denticola and F. alocis, which were the most prominent species in diseased sites and S. mitis and S. sanguinis, which were the dominant taxa in periodontal health. Interestingly, these finding are in accord with previous investigations using checkerboard DNA-DNA hybridization (336, 337, 339) and culture (246). In addition, the authors detected a total of 692 species and the number of species per individual ranged between 100-300, which represent numbers much closer to those proposed by Paster et al. (278).
In summary, open-ended microbial techniques provide a more comprehensive view of the microbiota of a given sample beyond the confines imposed by close-ended approaches. However, care must be exercised to avoid the distraction brought about by taxa that might be transient, rare or might be the result of artifacts inherent to the chemistry of the reactions and/or the analytical pipeline.
Lesson learned: the uncultivated segment of the microbiota plays a critical role in the etiology of periodontal diseases
This is not a new lesson, uncultivated subgingival species have been implicated in the etiology of periodontal diseases since the late 1800s (335), when spirochetes were first suspected as periodontal pathogens. Early studies using microscopy identified high levels and proportions of spirochetes in deep pockets, always at the forefront of the periodontal lesion, close to inflamed tissues (104, 208, 253, 300, 301). In fact, studies in the 1980s attempted to monitor the presence of these taxa in samples of subgingival plaque using phase-contrast and dark-field microscopy and relate their levels to the severity of periodontal disease and outcome of periodontal therapy (17, 174, 285). Cross-reacting monoclonal antibodies against Treponema pallidum have also been used to identify uncultivated “pathogen-related spirochetes” in subgingival plaque samples (293-295).
In recent years as studies using culture independent molecular techniques accumulated, a number of additional uncultivated taxa began to be associated with periodontal health and disease (42, 95, 159, 264, 389, 413). The relative abundance of members of the phylum TM7 was greater in patients with periodontitis, suggesting their potential role in the pathogenesis of the disease (42, 264). Members of the phylum Synergistetes were more frequently detected in periodontitis patients and were more abundant in diseased sites than in healthy sites (389). Tannerella sp BU063 (HOT 286) was found to be more prevalent in healthy (188, 202) and gingivitis sites (413), compared to periodontitis sites. Using FISH, Zuger et al. (413) reported that this taxa was found in gingivitis sites and corresponded to <1% of the total bacteria. T. forsythia was also present in most of the gingivitis sites in low proportions (<1% of the total bacteria); however, it was detected in all periodontitis sites comprising approximately >10% of the total bacteria. These findings illustrate two important points: that phylogeny may not always be used to infer pathogenicity and that quantification is important to determine the relevance of oral microorganisms and their association with health and disease. Griffens et al. (124) used pyrosequencing to compare the subgingival microbiota of periodontitis patients and periodontally healthy individuals. The authors observed that several uncultivated taxa were significantly more abundant in deep sites from periodontitis patinents, in comparison with shallow sites from the same patients and shallow sites from periodontally healthy individulas. They included Treponema sp. oral taxon 230 (HOT 230) Desulfobulbus R004 (HOT 041), TM7 oral taxon 437 and 349 (HOT 437 and 349), and Synergistetes oral taxon G36 (not in HOMD). Many of them were present in levels comparable with those observed for bacteria previously associated with periodontitis, such as T. forsythia, P. intermedia and C. rectus.
Those associations observed in cloning and sequencing studies are encouraging and indicate that some novel taxa identified might merit further pursuit in additional studies. But there are a number of challenges in moving forward. The large diversity in subgingival biofilms; the differences in the clinical and laboratorial protocols employed by different studies; the fact that, because most of those techniques rely on DNA as the target molecule, non-viable cells could be detected; an ever growing and changing nomenclature for the novel phylotypes; as well as time and costs associated with the techniques being used were some of the obstacles facing researchers in defining the role of those organisms in periodontal health and disease. Several types of additional studies were needed in order for the role of these novel taxa to be elucidated, including, for instance, intervention studies to test whether the prevalence of those new “putative pathogens” would decrease in successfully treated patients and sites and would remain elevated in non-responding ones. Longitudinal studies were also necessary to confirm the consistent presence of that these taxa in the subgingival habitat to confirm that they are “resident” members of the microbiota, rather than transients. In addition, temporality, a criterion that is often included for the inference of causality; cannot not be established based on cross-sectional studies. As discussed elsewhere in this manuscript, longitudinal studies are also essential to examine the associations between subgingival taxa and periodontal disease progression or “risk-assessment analysis”. Further, if we are to make inferences about the role of these “new taxa” in periodontal health and disease, quantification techniques will be a must. In the past decade, 3 technological advances have occurred that permitted the implementation of intervention and longitudinal human studies examining the yet uncultivated/unrecognized segment of the subgingival microbiota, and its quantification in subgingival samples: 1) the human oral microbial identification microarray (HOMIM), 2) RNA-oligonucleotide quantification technique (ROQT) and 3) the human oral microbiome database (HOMD).
The Human Oral Microbial Identification Microarray (HOMIM)
The development of the human oral microbial identification microarray (HOMIM) (59, 277, 278) enabled the systematic investigation of the most common taxa in the oral cavity. HOMIM is a 16S rRNA-based microarray method that allows the simultaneous detection of about 300 of the most prevalent oral bacterial taxa (including yet uncultivated taxa), as determined by previous studies. The major advantage of HOMIM over sequencing and cloning is its relative high-throughput, affording the examination of a much larger number of samples. This technology has been employed in a number of studies focusing on different aspects of the oral microbiota (9, 21, 85, 263, 318, 323, 355).
In a recent study, HOMIM was used to compare the subgingival microbiota of subjects with refractory periodontitis to those in subjects with successfully treated periodontitis and periodontal health (59). A few uncultivated taxa were significantly more prevalent in refractory patients, including TM7 sp HOT 346 which was detected in >10% of all samples. Additional uncultivated taxa were more prevalent in refractory cases, but were much less common and were detected in less than 10% of all samples, namely TM7 sp HOT 349/346; Megasphaera sp HOT 123, Bulkhoderia sp HOT 406, Prevotella sp HOT 300 and Treponema sp HOT 251. The authors also compared the microbiota of sites based on gain and loss of attachment and observed that TM7 sp. HOT 346/356/437, Peptostreptococcaceae sp HOT 113, Desulfobulbus sp. HOT 041, Fusobacterium sp. HOT 203, Selenomonas sp HOT 134/442 were associated with attachment loss. Additional uncultivated taxa previously associated with disease, including Mitsuokella (Acidaminococcacea sp HOT 131), were also more prevalent in poor responders, but the differences did not reach statistical significance. The previous association of Tannerella clone BU063 (HOT 286) with healthy sites (188, 202) could not be confirmed. Several taxa listed in that publication by their genus followed by a HOT number are not uncultivated and are currently described as “unnamed cultivated”, as it will be described in the section that refers to the HOMD. Some of them were significantly associated with disease, including Bacteroidetes sp. HOT 272 and 274, Selenomonas sp. HOT 133, Capnocytophaga sp. 326 as well as Haemophilus sp. HOT 036 and Prevotella sp. HOT 299. Those that were more frequently detected in sites that presented loss of attachment in comparison with sites that gained attachment or were healthy included Selenomonas sp. HOT 146/138, Bacteroidetes sp. HOT 274, Bacteroidetes sp. HOT 272 and Selenomonas sp. HOT 133. Recognized pathogenic periodontal species such as P.gingivalis, T. forsythia, T. denticola and E. nodatum were more prevalent in refractory patients and in sites that lost attachment than in good responder subjects/sites and periodontally healthy individuals/sites. In a follow-up study from the same group (58), the authors used HOMIM to compare the changes on the subgingival microbiota of the two clinical groups of periodontitis patients described above; i.e. refractory patients and good responders, before and after periodontal therapy. The authors observed that Fusobacterium sp HOT 203, Peptostreptococcaceae sp HOT 113, a cluster of Synergistetes phylotypes, Treponema sp HOT 242/237 and TM7 sp HOT 349/346 were significantly reduced after therapy, but Fusobacterium sp HOT 203 and Peptostreptococcaceae sp HOT 113 remained elevated in the “refractory” sites. Similar findings were observed for P. gingivalis and T. forsythia.
A recently completed intervention study conducted in our department provided further evidence of the relevance of certain uncultivated taxa as well as other unnamed cultivated taxa in periodontal health and disease. In this study, subgingival biofilm samples were taken from 42 chronic periodontitis subjects (average of 10 sites per patient), and from 41 healthy subjects (average of 13 sites per patient). Samples from periodontally healthy individuals were collected at baseline while samples from chronic periodontitis subjects were collected at baseline and 3 months after periodontal therapy composed of full mouth scaling and root planing in two sessions and systemic amoxicillin (500 mg tid) and metronidazole (250 mg tid) for 2 weeks. Figures 10 to 12 present preliminary data from this clinical study. Figure 10 shows the mean prevalence of bacterial taxa detected at significantly different frequencies (P<0.05) at sites grouped according to their baseline pocket depth. It can be observed that Desulfobulbus sp HOT 041; Peptostreptococcacea sp HOT 103 and 369, Treponema sp HOT 237 and 242 were detected at greater frequencies in deep sites and Acidaminococcaceae (Veillonellacea) sp HOT 150 was more prevalent in healthy sites. The effects of therapy on the microbiota implicated a number of other taxa as candidate pathogens. Figure 11 shows the mean prevalence of bacterial taxa detected at significantly different frequencies before and after therapy (P<0.05). Data were averaged within each subject and then across subjects at each time point separately. Among the uncultivated phylotypes, it can be observed that the detection of Synegistetes sp. HOT 360, Desulfobulbus sp. HOT 041, Peptostreptococcacea sp. HOT 113, Treponema sp. HOT 237 and 242, Lachnospiracea sp. HOT 080 and a cluster of Treponema phylotypes were significantly reduced after therapy. These taxa had values of baseline detection and post therapy reductions similar in magnitude to those from known pathogenic species such as E. nodatum, T. forsythia, P. gingivalis and P. intermedia. One can also appreciate that there is a tendency for a shift to a healthier microbial profile, as a number of health-compatible taxa increase in prevalence, including S. oralis, S. sanguinis, and S. parasanguinis. Figure 12 shows the frequency of detection of taxa found in shallow and deep sites post-therapy (p<0.05). Selenomonas sp. HOT 134 and 442, Acidaminococcacea sp. (Veillonellaceae sp.) HOT 135 and Synergistetes sp. HOT 363, 453 and 452 were more prevalent in deep residual sites. Other taxa also found in higher prevalence in that category of sites included P. gingivalis and E. nodatum.
Fig. 10.

Bacterial taxa detected in sites with different pocket depth categories at baseline. Only taxa that presented significantly different prevalence across site categories were plotted (Mann- Whitney test, P<0.05).
Fig. 12.

Bacterial taxa detected in sites from 42 chronic periodontitis patients before and after therapy (site-level analysis). Only taxa that presented significantly different prevalence between the two time points categories were plotted (Wilcoxon test, P<0.05).
Fig. 11.

Bacterial taxa detected in 42 chronic periodontitis subjects (subject level analysis) before and after periodontal therapy. Only taxa that presented significantly different prevalence between the two time points categories were plotted (Wilcoxon test, P<0.05).
These intervention studies expanded our knowledge regarding the relevance of uncultivated taxa in the pathogenesis of periodontal diseases. Several of the uncultivated/unrecognized taxa identified as associated with periodontal diseases belonged to genera that already had representatives of “cultured putative periodontal pathogens”, including: Prevotella, Treponema and Fusobacterium. The data also provided additional evidence of a potential role of strains from the genera Bacteroidetes, Synergistetes, Megasphaera, Selenomonas, Desulfobulbus, Peptostreptococcacea, Acidaminococcacea and the phylum TM7, that had been previously associated with periodontal diseases based on cross-sectional studies using molecular techniques. In addition, a potential role in health and disease could be observed for cultivated taxa not typically sought in studies of the subgingival microbiota, such as D. pneumosintes, F. alocis, P. tannerae, P. denticola and Fusobacterium naviforme. Interestingly, many such species had been previously implicated in periodontal disease in predominant cultivation studies (131, 246). DNA probes targeting these cultivable species have been recently incorporated into the checkerboard DNA-DNA hybridization technique (69).
So far, HOMIM is only available in one institution (The Forsyth Institute, Cambridge, MA). Hence, one of the major advantages of studies cited above that used this technology is that they were performed using standardized techniques. Samples were collected in the same manner and their lysis, DNA extraction and amplification followed the same protocols. Further, the probe panel remained essentially the same in all analyses. This will afford a level of consistency across studies not possible for other methodologies.
RNA-oligonucleotide quantification technique (ROQT)
Most of the information regarding the association of uncultivable/unnamed cultivated taxa with periodontal health and disease is based on techniques that provide presence/absence data (58, 59, 159, 188, 276). Also, the majority of those techniques involve sample dilution, sample pooling or PCR amplification (159, 188, 276) all of which add bias to the microbial results. Until now, there has been no high-throughput technique to quantify those taxa; techniques that allow quantification of uncultivable species such as RT-PCR (42, 390) and FISH (388, 389, 413) are limited in the number of taxa and samples that that can evaluated at a time. Quantification is important in the study of microbial communities in any ecosystem (13), but it is particularly crucial in periodontal diseases because differences between periodontal health and disease and before and after therapy are quantitative, rather than qualitative (55, 128, 141, 142, 336, 337, 413). In fact, even after over 100 years of the study of the oral microbiota (including the new era of molecular techniques), no single species has been associated solely with either health or disease. Lord Kelvin, who discovered the absolute zero, once wrote about the paramount value of quantification: “I often say that when you can measure what you are speaking about, and express it in numbers, you know something about it; but when you cannot measure it, when you cannot express it in numbers, your knowledge is of a meager and unsatisfactory kind; it may be the beginning of knowledge, but you have scarcely in your thoughts advanced to the state of Science, whatever the matter may be”. High-throughput is another prerequisite because bacterial species counts are highly variable in biofilms from different subjects and sites, requiring the analysis of large number of samples from many subjects in order to detect clinically and statistically meaningful differences.
ROQT is a high-throughput method to quantify a wide range of uncultivated and unnamed taxa in oral biofilms (118, 361). It involves the use of digoxigenin labeled oligonucleotides sequences targeting the 16S ribosomal DNA gene that are hybridized with the total nucleic acids (DNA and RNA) extracted from biofilm samples. The probe sequences employed in the ROQT are the same ones used in the HOMIM, which facilitates the comparisons of results. In order to permit quantification, sequences complementary to the probes are used as standards and added to the membranes, typically at 0.004 and 0.04 picomolar (pM). Chemiluminescent signals are then visualized using a charge-coupled device (CCD) camera (Fig. 13). Besides its high-throughput and being quantitative, another advantage of the ROQT is that it targets primarily the rRNA molecule, which is far more abundant than the rRNA gene. The first benefit of this approach is that an abundant target molecule obviates the need for the PCR amplification step. Oligonucleotide probes are known to be relatively specific but lack sensitivity, therefore, an amplification step is often required if the target is the rRNA gene. Second, because of the use of rRNA as a target molecule, the metabolic activity of the target taxa can be inferred, as an actively growing cell has 103–104 rRNA molecules (316) and rRNA is more associated with cell viability than DNA (238). Hence, the use of rRNA can provide insights on the relevance of the test species in the ecosystem of interest and avoid biases in the results due to the presence of dead cells (255).
Fig. 13.

A ROQT membrane showing hybridization of clinical samples with oligonucleotide probes. Probes for uncultivated/unrecognized species (‘orange’ group of bacteria) are listed across the top. Each horizontal lane represents the total nucleic acids from a sample from the indicated numbered tooth. Standards comprised a mixture of “complementary” sequences from all the test taxa at 0.004 and 0.04 pM, respectively. Teeth marked with an asterisk (*) were absent. Printed with permission from Teles et al. (361).
It is recognized that the number of rRNA copies will differ across taxa and their stage of growth which complicates the conversion of pM units into cell counts. The absolute and relative (normalized to total rRNA) amounts of the rRNA from a specific bacterial species are no direct measures of cell counts, because rRNA content might change over at least one order of magnitude during the bacterial cell cycle (13). Therefore, we now prefer to express the results from ROQT in pM. In fact, the role of the ribosomes as protein factories is so crucial that changes in the cellular rRNA of specific populations are highly significant even though it might not be possible to discern from it specific parameters such as growth rates or cell counts; “Since an increase in activity of a certain population is usually linked to higher cellular ribosome contents and cell numbers, the monitoring of a parameter that summarizes both effects should enable reasonable correlations between population dynamics and defined function” (13).
The human oral microbiome database (HOMD)
As mentioned in earlier sections of this chapter, the diversity of the oral cavity initially demonstrated by cultivation (131, 246) was expanded by cloning studies to include the uncultivable segment of the oral microbiota. These studies indicated several novel phylotypes that were associated with periodontal health and diseases (95, 159, 189, 190, 276). One of major challenges posed by the discovery of those novel taxa is that aside from the sequence information and a clone number, not much more information was available about them. Many uncultivated/unrecognized taxa were referred to by obscure isolate or clone numbers. Because no defined, curated systematic naming scheme was in place, it was difficult to compare the results from studies using cloning. This became particularly critical when associations between specific phylotypes and certain clinical states were examined.
The human oral microbiome database (HOMD) (www.homd.com) (51) was developed to provide a provisional naming scheme where each human oral taxon (HOT) is given a unique number. This HOT number is linked to their source, curated sequence information, their synonyms (i.e., previously described clones that are genetically similar), taxonomic hierarchy, bibliographic information and their status (i.e., cultivated, unnamed cultivated/uncultivated). In addition, the estimated prevalence of each HOT in the oral cavity is also available, as determined by a comprehensive cloning and sequencing initiative (75). In this study more than 35,000 clones of clinical samples from different oral conditions as well as 1,000 clinical oral isolate from culture collections (75) were examined and sequenced. In its current format, HOMD contains 645 taxa, about 50% of which are named species, 16% are unnamed cultivated taxa and 34% represent uncultivated phylotypes. Once a taxonomic anchor (HOT number) is given to a taxon, it remains unchanged. This is a major advantage for the establishment of a foundation of the oral microbiome, particularly because of ongoing efforts to characterize oral isolates from culture collections (75, 81-83), as well as to cultivate uncultivated taxa (85, 323, 387). The success of those efforts and the value of the HOMD are highlighted by the increase in the percentage of named species and decrease in the percentage of unnamed cultivated taxa since 2010 (51). In addition to information specific to each taxon included, HOMD also offers a user friendly interface and a series of analytical tools to facilitate the query and analysis of its growing dataset.
Lesson learned: molecular and culture techniques are required to study the microbiota of periodontal diseases
Given the complexity involved in the characterization of the microbiome of any habitat, and the fact that all techniques have biases and limitations, it is likely that one technique will not be able to resolve this issue and agnostically determine the microbiota associated with periodontal health and disease. It is probable that at least two techniques combined will be necessary (e.g. culture and molecular methods). This approach has been proposed and tested by Vianna et al. (391) and Donachie et al. (80). Donachie et al. (80) challenged the dogma perpetuated by the “great plate count anomaly” that cultivation approaches detected only a fraction of the total number of bacteria in a sample and were limited in scope. They also criticized the exclusive reliance on RNA sequencing for environmental studies of microbial diversity; “overlooking a century of cultivation history and encouraging the use of only ribosomal approaches leads to significant gaps in microbial diversity data”. They reasoned that cultivation methods are critical in microbial diversity studies as they identified organisms that were undetected using molecular techniques. To test their hypothesis they examined to which extent the datasets of cultivation-dependent and independent approaches overlapped. They posited that if open-ended 16S rRNA based approaches represented better the “true” diversity present in a microbial sample, culture libraries should not detect taxa absent from datasets recovered using the molecular approach. Data from 7 studies in which cultivation and open-ended molecular techniques were used on the same samples were compared. Samples had been collected from a wide range of ecosystems, including root canals, caries lesions, lakes and oceans. It was observed that the largest overlap between the two approaches was 30% (in caries samples) and 20% in endodontic samples, which means that each approach was covering a different segment of the microbiota. In addition, in two of the ecosystems under study, there was no overlap between the two approaches. Therefore, they could not demonstrate that molecular approaches captured a wider spectrum of microbial diversity. In addition, when analyzing microscopic and plate counts, one can see at least part of what is not being detected in culture, because of the morphotypes initially observed are not recovered from agar plates. On the other hand, in molecular techniques, one does not have the opportunity to “see” what has not been amplified, unless mock communities are used.
Lesson relearned: the study of periodontal pathogens will require their cultivation
Cultivation studies appear to be experiencing a re-birth, after years of focus on molecular culture independent techniques. Recent approaches have included the pursuit of members of a specific phylum using a helper species (388), the search of culture collections (mainly the Moore's collection) for isolates of previously unnamed taxa (75, 81-83, 387) or the search for isolates in mixed bacterial populations grown in situ (168, 254, 323). In addition, an elegant approach was recently devised by Duran-Pinedo et al. (85). The authors combined molecular techniques and novel cluster analysis methods to enable the cultivation of an uncultivated taxon. HOMIM data from subgingival biofilm samples (89 healthy and 514 periodontitis sites) were evaluated using correlation network analysis, which allowed the identification of bacterial clusters, named modules that represented important microbial associations within the subgingival microbial community. The authors managed to identify in silico, a helper species that could support the growth of an uncultivated organism. By identifying hubs that could act as keystone species in the bacterial modules, they were able to cultivate Tannerella sp. HOT 286 after two rounds of enrichment by the selected helper, Prevotella oris HOT 311. Interestingly, when checkerboard DNA-DNA hybridization data from 2,565 individual subgingival plaque samples were used to validate the algorithms used in the correlation network analysis, many of the modules identified by Duran-Pinedo et al. (85) were rather similar to the complexes described by Socransky et al. (337), further validating their model 13 years after its first publication (Fig. 14).
Fig. 14.

Weighted correlation network analysis (WGCNA) results based on data obtained using checkerboard DNA-DNA hybridization. The images show the Cytoscape representation of the correlation networks for the 4 modules identified by WGCNA. Checkerboard analysis was performed for 40 species of oral bacteria on a total of 2,565 individual subgingival samples from patients with periodontitis. R2 used for scale free topology model fit was 0.40, the maximum value in the analysis. The identified modules correlated well with microbial complexes previously described by Socransky et al. (337). Printed with permission from Duran-Pinedo et al. (85).
The importance of cultivation studies for the identification of agents of health and disease cannot be overemphasized: “The increasing availability of molecular pathogen discovery methods and ease with which molecular signature are generated create a pressing problem of a different kind. How can one build a convincing body of evidence for a causative role of the putative pathogen in a disease process when the pathogen has not been isolated or purified?” (289).
The search for relevant uncultivated/unrecognized species
An ongoing study in our department is using ROQT to identify prominent as yet uncultivated or cultivable but currently unrecognized subgingival taxa associated with periodontal health or disease. The secondary objective of this study is to recover in pure culture “easy” to cultivate taxa, i.e., to isolate in pure culture cultivable but as yet unrecognized prominent taxa. In the process, we will determine which taxa are truly “uncultivable”. Because the work involving isolation and cultivation of yet uncultivable species will involve substantial effort (387, 388), ones needs to be reasonably confident of their relevance to periodontal health or disease. The goal is to identify biologically meaningful taxa currently considered to be uncultivable/unnamed; grow them in the laboratory, isolate them, characterize and properly classify them as bacterial species. These steps are crucial for virulence, antigenicity and functional genomics studies and investigation of essential growth factors and signaling molecules. In essence, we are following the recommendation of Socransky et al. (340) on how to overcome certain difficulties in the search for etiological agent of periodontal diseases: “It becomes clear that, the search for the etiological agents of destructive periodontal diseases will, of necessity, be a multistage, iterative process. (…). In the first stage, investigators will be required to lower the number of candidate pathogenic species from the over 300 found in periodontal pockets to some reasonable number (hopefully <10)”.
For instance, in the HOMD, 221 taxa are listed as uncultivated and 104 are presented as unnamed cultivated. It is unlikely that they are all equally important to health and disease. Thus if one wants to start cultivating and characterizing them, one should be reasonably confident of their relevance to health or disease. Even though efforts had been initiated for the cultivation of uncultivable taxa (254, 323, 388) and for the identification of unnamed cultivable taxa from collections of clinical isolates (75, 81, 83, 394), there has been no systematic attempt to cultivate those taxa that are likely to be more relevant to periodontal health or disease. The rationale underlying this study is to eliminate the “distraction” represented by transient and or bystander taxa and focus the search for novel prominent bacteria on common, widespread taxa. As mentioned earlier, the probes employed in ROQT share the same sequences as those included in HOMIM. Thus, we selected probes that only detected uncultivated or unnamed cultivated taxa. In order to have an estimate of their relevance, we computed their frequency of distribution, based on a subset of the data from a recent intervention study completed at the Forsyth Institute (described above under the section on HOMIM). The 92 probes included in our probe panel are listed in Table 1, in decreasing order of prevalence in periodontitis patients. It can be observed that the top 10 probes detected their targets in more than 16% of the subjects evaluated. Conversely, 25 of them never yielded a positive signal.
Table 1.
Prevalence of uncultivated/unrecognized bacterial taxa detected in 36 chronic periodontitis subjects in a recently completed clinical study using HOMIM (5R01DE017400-03). Taxa are listed in decreasing order of frequency of detection.
| Probe and Oral Taxa (OT) | UU | UC | % Positive | Probe and Oral Taxa (OT) | UU | UC | % Positive |
|---|---|---|---|---|---|---|---|
| n=36 | n=36 | ||||||
| Synergistetes sp_ot360 | 360 | 53% | Treponema sp_ot257 | 257 | 2% | ||
| Capnocytophaga sp_ot335 | 335 | 45% | Actinomyces sp_ot169 | 169 | 2% | ||
| Streptococcus sp_ot070_071 | 070, 071 | 44% | Actinomyces sp_ot448 | 448 | 2% | ||
| Acidaminococcaceae sp_ot155 | 155 | 31% | Actinomyces sp_ot178 | 178 | 2% | ||
| Peptostreptococcaceae sp _ot113 | 113 | 31% | Atopobium sp_ot416 | 416 | 2% | ||
| Bacteroidetes sp_ot274 | 274 | 28% | Bacteroidetes sp_ot281_365 | 281,365 | 1% | ||
| Desulfobulbus sp _ot041 | 41 | 28% | Capnocytophaga sp_ot326 | 326 | 1% | ||
| Peptostreptococcaceae sp_ot103_369 | 103, 369 | 21% | Prevotella sp_ot299 | 299 | 1% | ||
| Selenomonas sp_ot134_442 | 134, 442 | 17% | Prevotella sp_ot308 | 308 | 1% | ||
| Selenomonas sp_ot138_146 | 138, 146 | 16% | Prevotella sp_ot309 | 309 | 1% | ||
| Lachnospiraceae sp_ot080 | 80 | 16% | Prevotella sp_ot317 | 317 | 1% | ||
| TM7 sp_ot346_349 | 346, 349 | 16% | Neisseria sp_ot020 | 20 | 1% | ||
| Haemophilus sp_ot036 | 36 | 15% | Acidaminococcaceae sp_ot131 | 131 | 1% | ||
| Actinomyces sp_ot177 | 177 | 13% | Selenomonas sp_ot137 | 137 | 1% | ||
| Fusobacterium sp _ot210_220 | 210, 220 | 13% | Clostridiales sp_ot075 | 75 | 1% | ||
| Treponema sp_ot237_242 | 237, 242 | 12% | Clostridiales sp_ot085 | 85 | 1% | ||
| Synergistetes Cluster I_ot363_453_452 | 453, 452 | 363 | 11% | Leptotrichia sp_ot417 | 417 | 1% | |
| Treponema Cluster II_ot254_256_508_517 | 254, 256, 508, 517 | 11% | Leptotrichia sp_ot417_462 | 417,462 | 1% | ||
| Haemophilus sp_ot035 | 35 | 10% | Synergistetes sp_ot358 | 358 | 1% | ||
| Acidaminococcaceae sp_ot135_148 | 135, 148 | 10% | Actinobaculum sp_ot183 | 183 | 1% | ||
| Peptostreptococcaceae_ot106 | 106 | 10% | Capnocytophaga sp_ot324 | 324 | 0% | ||
| Bacteroidetes sp_ot272 | 272 | 9% | Capnocytophaga sp_ot336 | 336 | 0% | ||
| Eubacterium sp_ot081 | 81 | 9% | Porphyromonas sp_ot279 | 279 | 0% | ||
| TM7 sp_ot356_437 | 356, 437 | 9% | Prevotella sp_ot292_300 | 292, 300 | 0% | ||
| Bergeyella sp_ot322 | 322 | 8% | Prevotella sp_ot292 | 292 | 0% | ||
| Synergistetes sp_ot359 | 359 | 8% | Prevotella sp_ot310 | 310 | 0% | ||
| TM7 sp_ot347_350 | 347, 350 | 8% | Prevotella sp _ot376 | 376 | 0% | ||
| Megasphaera sp_ot123 | 123 | 7% | Prevotella Cluster III_ot306_310_313 | 310, 313 | 306 | 0% | |
| Actinomyces sp_ot175 | 175 | 7% | Prevotella Cluster sp_ot473_474 | 474 | 473 | 0% | |
| Tannerella sp_ot286 | 286 | 6% | Burkholderia sp_ot406 | 406 | 0% | ||
| Veillonella sp_ot780 | 780 | 6% | Neisseria Cluster IV_ot009_014_015_016 | 15 | 14, 16 | 0% | |
| Lachnospiraceae sp_ot096 | 96 | 6% | Rhodocyclus sp_ot028 | 28 | 0% | ||
| Oribacterium sp_ot078_372 | 78, 372 | 6% | Acidaminococcaceae sp _ot135 | 135 | 0% | ||
| Synergistetes sp_ot360_362_453 | 360, 362, 453 | 6% | Acidaminococcaceae sp_ot145_483 | 145, 483 | 0% | ||
| Selenomonas sp_ot133 | 133 | 5% | Acidaminococcaceae sp_ot129_150 | 150 | 129 | 0% | |
| Treponema Cluster III_ot256_508_517_518 | 256, 508, 517, 518 | 5% | Selenomonas sp_ot149_481 | 149, 481 | 0% | ||
| Acidaminococcaceae sp_ot132_150 | 132, 150 | 4% | Selenomonas sp_ot146 | 146 | 0% | ||
| Actinomyces sp clone AP064_ot170 | 170 | 4% | Oribacterium sp_ot078 | 78 | 0% | ||
| Selenomonas Cluster sp_ot136_149_478 | 478 | 136, 149 | 3% | Lactobacillus sp_ot461 | 461 | 0% | |
| Lachnospiraceae sp_ot097 | 97 | 3% | SR1 sp_ot345 | 345 | 0% | ||
| Synergistetes sp_ot363 | 363 | 3% | Synergistetes sp_ot362 | 362 | 0% | ||
| Capnocytophaga sp_ot332 | 332 | 2% | Treponema sp_ot251 | 251 | 0% | ||
| Lachnospiraceae sp_ot096 | 96 | 2% | Actinomyces sp_ot446_448 | 446, 448 | 0% | ||
| Leptotrichia sp_ot215 | 215 | 2% | Actinomyces sp_ot179 | 179 | 0% | ||
| Treponema sp_ot231 | 231 | 2% | Atopobium sp_ot199 | 199 | 0% |
Figure 15 shows the prevalence in sites from periodontally healthy individuals (n = 8) and sites from periodontitis patients (n = 8) for the taxa detected with the 92 probes selected. It can be observed that a number of uncultivated taxa were significantly more frequently detected in periodontitis sites (P<0.002), including TM7 sp. HOT 346/349, Desulfobulbus sp. HOT 041, Treponema sp. HOT 245, 256, and 508. TM7 sp. HOT 356/437 and Synergistetes sp. HOT 360/362/453. In addition, P. gingivalis and F. nucleatum, which were included as “reference” species due to their known associations with periodontal health and disease, were also more prevalent in diseased sites. This was the first phase of the study and the results have been further evaluated in a second set of 16 patients (data analysis in progress). In order to select only the most relevant taxa, we established a threshold of 10% of detection; that is, taxa that were not detected in at least 10% of the sites (depicted by the vertical red lines) will not be pursued further, due to their low probability of being relevant to health or disease.
Fig. 15.

Prevalence of uncultivated/unrecognized taxa and “reference” cultivated species (Fusobacterium nucleatum, Porphyromonas gingivalis, Veillonella parvula and Actinomyces naeslundii) examined in samples from periodontally healthy individuals (n=8) and chronic periodontitis patients (n=8). Significant differences are indicated with an asterisk (Chi Square test, P<0.002). Red vertical bars highlight taxa that were present in less than 10% of sites. Taxa present in less than 10% of sites were excluded from subsequent analyses.
Figure 16 shows the levels (pM) of the taxa selected for further analysis in sites from healthy and periodontitis individuals. It can be observed that the levels of the uncultivated taxa Haemophilus so HOT 035 and 036, Synergistetes sp. HOT 360/362/453, Eubacterium sp. HOT 081, Megaesphera sp. HOT 123, Acidaminococcaceae sp. HOT 135/148 and unnamed cultivated taxa like Veillonella sp. HOT 780 were significantly more elevated in periodontitis sites. In addition, F. nucleatum, even though not the most prevalent taxon, exhibited the highest levels and along with P. gingivalis, was significantly more abundant in sites from diseased patients. It is noteworthy that several taxa were more abundant in sites from healthy individulas, including Actinomyces sp. HOT 448, 177 and 170. The association between segments of the uncultivated/unnamed microbiota and clinical parameters was also examined. Figure 17 shows taxa present in significantly different levels in deep sites from periodontitis patients and healthy sites (pocket depth ≤3 mm) in periodontally healthy individulas. Levels of uncultivated taxa like Synergistetes sp. HOT 359, 363/453/452 and Haemophilus sp. HOT 035, as well as unnamed cultivated taxa like Capnocytophaga sp. HOT 332 and 335 as well as Bacteroidetes sp. HOT 274 were more abundant in deep periodontitis sites than in shallow sites from healthy individuals (p<0.0001). To put the differences between the 2 site categories in perspective, it is noteworthy that the magnitude of the differences was comparable to that of P. gingivalis.
Fig. 16.

Levels (pM ± SEM) of selected taxa in sites from periodontally healthy (n=8) and chronic periodontitis patients (n=8). Significant differences are indicated with an asterisk (Mann-Whitney test, p<0.05)
Fig. 17.

Levels (pM ± SEM) of selected taxa in shallow sites (pocket depth<4mm) in healthy sites in periodontally healthy individuals and deep sites (pocket depth>4 mm) in chronic periodontitis patients. Only taxa that presented significantly different prevalence between the site categories were plotted (Mann-Whitney test, P<0.0001).
The analyses described above will be repeated in a second set of individuals and the taxa most associated with health and disease (based on prevalence and levels) will be selected. We anticipate that this selection will result in a panel of 40 probes, which will be used to guide the isolation in pure culture of cultivable but as yet unrecognized taxa. The rationale for this phase of the study is that many of the uncultivated/unnamed cultivated taxa may be cultivated using standard cultivation techniques, leaving a smaller set as truly uncultivated species. We will use the oligonucleotide probes to the selected 40 prominent uncultivable/unrecognized taxa to screen isolates obtained from subgingival biofilm samples from periodontally healthy and diseased subjects. Samples will be dispersed, serially diluted, plated on 5 isolation media and grown in 3 different atmospheres: anaerobic, microaerophilic and capnophilic. After incubation, isolates will be colony lifted and hybridized with the pre-selected 40 probes. Colonies that give positive signals on each medium/atmosphere combination will be counted and the data used to select the 3 best media/atmosphere combinations that provided the greatest recovery of unrecognized taxa. In the final stage of the study, a second set of samples from periodontally healthy and diseased subjects with be grown using the 3 selected media/atmosphere combinations. After incubation, all isolates on primary isolation plates containing at least 50 colonies will be picked and spotted on 3 plates of the same medium. After incubation, two such plates will be “lifted” and hybridized with 2 sets of 20 probes, while the third one will be kept as a source of test isolates. Colonies that give positive signals will be identified using ROQT, while the unrecognized isolates will be phenotypically characterized and sequenced. We expect that the characterization of these species will be the first stage in delineating novel pathogens and will focus the search for the truly uncultivated species by providing targeting probes. Distinguishing the cultivable but unrecognized taxa will provide isolates for taxonomy, virulence and antigenicity testing and structural and physiological characterization.
Lesson learned: the study of the spatial distribution of the components of the subgingival microbiota in situ is essential to the understanding of their interactions
The interaction between bacterial taxa present in the subgingival environment in periodontal health and disease has been explored using different approaches. Unfortunately, most existing methods for biofilm sample collection disrupt the tridimentional structure of these polymicrobial structures. Even though microbial complexes have been determined by Socransky et al. (337), and corroborated by others over the years using different approaches (85, 178), the model was built based on the frequency of the simultaneous occurrence of a group of taxa, irrespective of their spatial distribution within the biofilm. During sample collection for checkerboard DNA-DNA hybridization, the physical interaction and spatial distribution of the species identified are disrupted and; therefore, they cannot be inferred using this technique. The elegant model proposed by Kolenbrander et al. (178), summarized the interactions and localization of the taxa that colonize the subgingival environment represents a compilation of findings form the authors and other researchers, deducted using reductionist in vitro and in vivo experiments. The physical interaction and biogeography of periodontal bacteria have been explored by immunohistochemistry (176, 256-259) and FISH (317, 411). Those studies have indicated that, in subgingival plaque, red complex bacteria tend to be part of the epithelium associated biofilm, the orange complex species are typically found in the “loosely attached” biofilm and that Actinomyces species, as well as green and yellow complexes species tend to localize in the coronal portions of the biofilm, mostly in the tooth associated biofilm (336). Data from Zijnge et al. (411) suggested that species of Fusobacteria and Tanerella were present in the intermediate layer of subgingival biofilms; species associated with periodontitis such as P. intermedia, P. gingivalis, P. endodontalis and P. micra localized in the outer layer of the biofilm; and a fourth layer of unattached plaque, consisting mainly of Spirochetes seemed to form on top of the outer layer of the subgingival biofilm. Noteworthy was the demonstration of the presence of species of Synergistetes in close proximity to host immune cells, indicating their potential role in host-microbial interactions (411). Recently, Schillinger et al. (317) showed a tight clustering of F. nucleatum/Fusobacterium periodonticum and T. forsythia in in vivo developed biofilms, as well as a random spatial distribution between P. gingivalis and P. intermedia in these in vivo samples.
Those studies provided valuable information for the study of the tridimentional organization of subgingival microbiota. However, limitations in current technologies have prevented a comprehensive study of microbial community organization (375). In principle, FISH probes could be designed with rRNA sequence specificity for nearly any microbial phylotype or taxon. The limitations reside in the use of the filters in fluorescence image acquisition and the excitation crosstalk and emission bleed-through of available organic fluorochromes (375). These technical constraints limit the number of fluorophores that can be differentiated simultaneously (375, 395). Valm et al. (375) overcame these limitations using a combinatorial labeling strategy coupled with spectral image acquisition and analysis, therefore, greatly expanding the number of fluorescent signatures discernible in a single image. The technique named combinatorial labeling and spectral imaging FISH (CLASI-FISH) uses genus- and family-specific probes to visualize simultaneously and differentiate 15 different phylotypes. Figure 18 presents CLASI-FISH images of semi-dispersed human dental plaque. It shows that it was dominated by early colonizers, including species of Streptococcus, Prevotella, Actinomyces and Veillonella. Proximity analysis was used to determine the frequency of inter- and intrataxon cell-to-cell associations which revealed statistically significant intertaxon pairings. Cells of the genera Prevotella and Actinomyces showed the most interspecies associations, suggesting a central role for these genera in establishing and maintaining biofilm complexity. Work is underway at The Forsyth Institute (Cambridge, MA) in collaboration with Woods Hole Marine Biological Laboratories (Woods Hole, MA) to expand the probe panel used initially and explore the spatial organization of the oral microbiome to include biofilms associated with different oral surfaces for which the microbial profiles have been described (3, 225), as well as the subgingival biofilms associated with the different surfaces of the periodontal pockets.
Fig. 18.

Representative detail images of CLASI-FISH–labeled semi dispersed human dental plaque. Color in raw spectral images represents merge of six different fluorophore channels. Color in the segmented image represents 1 of 15 taxa. (Scale bar: 10 μm). Printed with permission from Valm et al. (375).
Studies examining the in situ tridimentional structure of polymicrobial biofilms will help refine models built based on data generated using techniques that require dispersal of samples, further implicating specific species in periodontal health and disease.
Lesson learned: periodontal diseases are inflammatory diseases
At the beginning of this manuscript we described a causal model where a constellation of causal factors need to be present in order for disease to occur. Such a model can help us understand several of the apparent inconsistencies pointed out by some in the concept of periodontal diseases as specific bacterial infections. For instance, if we accept for a moment that for periodontal diseases to occur one requires periodontal pathogens, the proper local environment and a susceptible host as minimally sufficient causes; the presence of periodontal pathogens in the absence of periodontal tissue destruction can be easily explained. However, it is apparent that to fully understand the mechanisms that contribute to periodontal disease initiation, the study of the microbial challenge will not be sufficient. Among the many factors that define the so-called “host susceptibility”, the immunoinflammatory response that accompanies the bacterial challenge is clearly part of the constellation of minimal causes that lead to periodontal tissue destruction. In other words, without inflammation, there is no periodontal disease initiation and progression. Evidence in support for a role of inflammation in periodontal diseases comes from epidemiological studies that have indicated that the presence of gingival inflammation over prolonged periods of times is associated with clinical attachment loss and tooth loss (313, 314). However, at times, inflammation has taken center stage as the main event in the processes that lead to periodontal diseases: “Some individuals think of the periodontal problem essentially in terms of inflamed gingiva, pocket formation, calculus, and bone resorption. These conditions may have about the same relevance to the root infection problem as inflammation, tubercule formation, granulomatous lesions, caseation, and cavitation have to tuberculosis” (173).
Inflammation has not been traditionally considered as the etiological agent of periodontitis but rather as a mechanism of its pathogenesis, defined in the Merriam-Webster dictionary as “the origination and development of a disease”. In classical texts, bacteria have been referred to as the etiological agents of periodontal diseases, while inflammation as one of the mechanisms involved in their pathogenesis. These concepts have been the cornerstones of our understanding of periodontal diseases for decades and elegantly acknowledged the microbial challenge and the immunoinflammatory host response as key elements of periodontal disease etiology and pathogenesis. However, in recent years, this simple framework has been challenged once more by efforts to assign greater importance to the immunoinflammatory response compared to the microbiota in the pathogenesis of periodontal diseases. Such emphasis on host factors has led some authors to hypothesize that they might outweigh bacteria as determinants of periodontal disease onset and severity (266). One might argue that with so much still to be learned about the composition of the subgingival microbiota and its functions that these statements were rather premature. In addition, trying to determine the proportion of disease due to either bacteria or the host response might be counter productive as it is clear that neither factor alone will cause periodontal tissue destruction. In the words of Page and co-authors: “Saying that this process (periodontal disease) is mediated entirely by the host or entirely by the bacteria would simply propagate another decade of misunderstanding” (267).
Proponents of the primary role of the immunoinflammatory response in the tissue destruction that occurs during periodontal diseases argue that strategies aiming at dampening or modulating the host response might be better approaches for the treatment of periodontal diseases (145). We would recommend caution in these approaches; therapies that ignore the infectious nature of periodontal diseases might have disastrous consequences. In his 1970 paper, Keyes (173) expanded on his analogy between periodontal diseases and tuberculosis: “It would be interesting to anticipate the consequence to the host that would follow the suppression of inflammation in the periodontium without elimination of the radicular plaque infections. Would the response be comparable to that which follows the administration of cortocosteroids to patients with tuberculosis?”.
Some of the evidence that implicates the host response as the main mechanism in the destruction of the periodontium lies in histological observations that bacteria are not present, at least not in an obvious way, in the periodontal tissues afflicted with periodontitis. Conversely, the inflammatory infiltrate clearly results in a loss of collagen content and is adjacent to the bone resorbing activity. Although bacterial products such as proteases and lipopolysaccharides can cause direct tissue damage (282) and stimulate bone resorption (84), host derived molecules such as matrix metalloproteinases and interleukin-1β are more potent mediators of these catabolic processes (148, 342, 344, 345). Further, it is likely that the levels of inflammatory mediators that lead to tissue destruction reach much higher concentrations within the periodontal lesions than microbial products with similar functions. Hence, it is well accepted that most of the tissue destruction observed in periodontal diseases results from indirect damage, mediated by the immunoinflammatory response. However, even if inflammation is the main mechanism of destruction of periodontal tissues, this does not imply that its direct control is the best strategy for treatment and prevention of periodontal diseases. In the words of Haffajee & Socransky: “…the ultimate risk factor for an infectious disease is the causative agent of that disease. Without that agent, no disease will take place no matter what other risk factors the subject may possess” (131). In fact, the most efficient strategies in place for the prevention and treatment of periodontal diseases are anti-infective in nature (135, 150, 151).
Early attempts to interfere pharmacologically with the inflammatory process in periodontal diseases involved the use of non-steroidal anti-inflammatory drugs (149, 154, 165, 398). These drugs block the activity of cyclooxygenases, preventing the metabolism of arachidonic acid. A series of animal studies in the 1980s demonstrated that non-steroidal anti-inflammatory drugs were capable of slowing the progression of periodontal diseases (154). Human clinical studies followed and demonstrated similar results (398); however, the safety profile of these drugs did not allow for their prolonged continuous use. Drugs that selectively inhibited cyclooxygenase-2 were received with great enthusiasm because they promised to minimize the gastrointestinal adverse effects observed with the use of non-steroidal anti-inflammatory drugs (306). However, the long-term use of these drugs were later associated with the occurrence of congestive heart failure in a significant proportion of subjects (170). The only therapeutic adjunct on the market for periodontal disease therapy that interferes with the host-response is low-dose doxycycline (Periostat®) (116, 117). The mechanism by which this drug is supposed to control periodontal tissue destruction is through its anti-collagenolytic activity, resulting from the inhibition of matrix metalloproteinases (115). However, it has been demonstrated that the use of low-dose doxycycline also results in changes in the proportion of subgingival bacterial species that are resistant to doxycycline, demonstrating its impact on the subgingival microbiota (129). Therefore, the therapeutic effect of this agent cannot be ascribed solely to its modulation of the host response.
Bone sparing drugs such as bisphosphonates have also been tested in the control of periodontal bone loss in several animal studies (287) and in human clinical trials (163, 164). Reports of osteochemonecrosis of the jaw associated with the use of bisphosphonates raises serious questions about the safety profile of this class of drugs (241). Although the prevalence of osteochemonecrosis of the jaw is rather low and the mechanisms involved in its pathogenesis are still not fully understood, there is evidence that they might be associated with infection (234). One of the mechanisms proposed posited that blocking osteoclastic activity would impair the ability of bone tissue to “fight infections”. Bone resorption, including the one observed in periodontal diseases can be interpreted as a mechanism of defense against infectious agents. More recently, agents that can block the activity of receptor activator of nuclear factor kappa B ligand (RANKL) such as anti-RANKL monoclonal antibodies (e.g. denosumab) has also been proposed as an approach to control periodontal bone loss (376).
Because inflammation is part of the minimal causes that lead to periodontal tissue destruction, it is conceivable that periodontal diseases can be controlled by agents that block the inflammatory reaction. Nevertheless, because of the chronic nature of periodontal diseases, susceptible subjects would be required to use this (these) agent(s) for prolonged periods of times. That implies that they would be required to have a rather benign safety profile. Such an approach would be analogous to the use of fluorides to prevent dental caries in the sense that the use of fluorides controls the disease process (i.e. demineralization of enamel) without interfering with its bacterial etiology (i.e. S. mutans). The use of fluorides is a good example of how interfering with pathogenesis, rather than etiology, might lead to a successful strategy for disease control. The analogy is only flawed because the pathogenesis of dental caries involves a biochemical reaction, rather than an immunoinflammatory response. In any case, no anti-inflammatory drug with a safety profile that would afford its long-term use currently exists. Drugs that promote resolution of inflammation might offer a safe alternative to host-response modulation in the near future.
Lesson learned: resolution of inflammation impacts the composition of the adjacent microbiota
Our knowledge of inflammatory pathways has been revolutionized by the recognition that resolution of inflammation is an active mechanism mediated by specific pathways, rather than a “passive decay of proinflammatory signals” (379). Molecules such as lipoxins, aspirin-triggered lipoxins, protectins and resolvins act as agonists of the resolution phase of inflammation and activate the elimination of inflammatory leukocytes and promote tissue healing (171). These agents must be differentiated from the anti-inflammatory drugs discussed above that simply block inflammatory pathways at different points. In contrast, proresolvins promote a return to tissue homeostasis (170). Using a P. gingivalis-induced rabbit model of periodontitis, Hasturk et al. (146) demonstrated that topical applications of resolving E 1 (RvE1) prevented the onset and progression of destructive periodontal disease. The same group later demonstrated that RvE1 was capable of inducing regeneration of periodontal tissues, including new periodontal ligament, cementum and alveolar bone (147). These remarkable results were expanded when daily dietary supplementation with omega-3 polyusaturated fatty acids (the precursors of resolvins and protectins) and low-dose aspirin were used as an adjunct to mechanical periodontal therapy in a human clinical study (91). After 6-months of daily use of 81 mg aspirin and 3g of fish oil (the source of omega-3 polyusaturated fatty acids), the clinical results favored the test group with gains in clinical attachment level and pocket depth reductions comparable to those obtained in studies testing adjunctive systemic antibiotics. The use of this class of host-modulatory drugs promises to revolutionize the way clinicians treat periodontal infections in the near future.
A very interesting outcome of the experiments with the rabbit model was the apparent “disappearance” of P. gingivalis after the application of RvE1 (147). The authors reported that the infection with P. gingivalis altered the composition of the existing microbiota of the animals, resulting in the detection of previous undetected species such as A. actinomycetemcomitans and F. nucleatum and the disappearance of Capnocytophaga curvus, C. rectus, among other changes. Application of RvE1 resulted in a return of the microbiota to the baseline composition together with the elimination of P. gingivalis. Two mechanisms have been proposed to explain these findings: 1) resolvins, which do not have any appreciable direct antimicrobial effect, might have promoted the release of antimicrobial peptides, such as defensins and bactericidal/permeability-increasing protein or 2) the resolution of the inflammation “starved” P. gingivalis because it depends on peptides derived from host-tissue degradation as a source of nutrients (379). According to the author, “Such an explanation would suggest that the magnitude of inflammation generated by the host determines the composition of flora within the biofilm, which is a corollary to the hypothesis that the microbial species present sets the threshold for the inflammatory response” (379). These findings highlight the intimate relationship between microbial community composition and the local environment.
Studies on the inflammatory processes mediated by pro-resolvins are at their early stages and new data continue to expand our knowledge of the wide scope of their activities. The group from Charles Serhan's laboratory recently reported on anti-infective mechanisms of pro-resolving mediators (53). Using a murine model of peritoneal infection induce by E. coli, resolving (Rv) D1 and RvD5 were able to reduce bacterial titers in blood and increase survival. These molecules and protectin D1 (PD1) were also capable of enhancing phagocytosis of E. coli by human neutrophils and macrophages, while RvD5 counter-regulated a panel of pro-inflammatory genes, including nuclear factor-kappaB and tumor necrosis factor-α. Further, the authors demonstrated that RvD1, RvD5 and PD1 in association with ciprofloxacin enhanced host antimicrobial responses. In skin infections, pro-resolvins enhanced vancomycin clearance of S. aureus. The authors concluded that pro-resolving mediators had antiphlogistic actions, enhanced containment of infectious agents and lowered antibiotic doses required for bacterial clearance. Therefore, the effects of resolvins on the prevention of destructive periodontal diseases might also be explained by indirect antimicrobial mechanisms. It is tempting to speculate what would be the clinical effects of the combined use of systemic antibiotics and pro-resolvins in the treatment of periodontal diseases.
Lesson learned: bacteria are not bystanders in their interaction with the host
When one considers host-microbial interactions, much attention has been focused on the potential of this interplay to result in disease; i.e. on the pathogenic potential of this interaction. Still, for the most part, host-microbial interactions do not result in overt disease or tissue destruction. For most bacteria, including pathogens, asymptomatic colonization and/or clearance without blatant disease are the most common results of an encounter with the host. As a result of genetic variability among different strains of the same bacterial species and individuals of the same host species, these interactions might be rather unique, with several possible outcomes, including: immediate clearance, colonization (with or without disease), long-term asymptomatic carriage, invasion, direct damage (e.g. bacterial toxins), and immunopathology. Clearance might happen after other interactions, for instance, as a result of a disease process. In addition, a carrier state may evolve into overt disease. Therefore, disease onset seems to require a fairly specific set of interactions between host and bacterial factors. Even some of the most relevant human pathogens such as S. pneumoniae, Neisseria meningitidis and Mycobacterium tuberculosis cause disease at relatively rare occasions, considering the level of exposure to these microorganisms (279). As we have alluded to above, much attention has been paid to the notion that periodontal tissue destruction results from indirect rather than direct tissue damage induced by bacteria. Immunopathology is a pathogenic mechanism common to many infections; however, the specific immunoinflammatory mechanisms involved in different infectious diseases will be distinct. Considering its polymicrobial nature, it can be anticipated that the host-microbial interactions that occur during periodontal disease initiation are quite variable and rather unique.
It is important to realize that infectious agents are active determinants of the nature of the immune response triggered in the infected host. If one considers, for instance, the T helper cell polarization into T helper cell-1, T helper cell-2, or T regulatory (Treg) subsets, the nature of the antigenic challenge is a major determinant of the nature of T-cell responses (30, 71, 275). Mice immunized with P. gingivalis alone resulted in anti-P. gingivalis IgG1 antibodies (T helper cell-2 response), while prior immunization with F. nucleatum resulted in anti-P. gingivalis IgG2a (T helper cell-1 response) (112). Oral bacterial antigens from Bacteroides fragilis, S. mitis and P. acnes primed human dendritic cells to induce T helper cell-1, T helper cell-2 and Treg cell differentiation, respectively (181). Lymphocytes stimulated with antigens from oral streptococci increased the expression of interleukin-4 (T helper cell-2 cytokine) while stimulation with antigens from Bacteroides species resulted in increased expression of interferon-γ (T helper cell-1 cytokine) (155). Antigens of oral streptococci induced the differentiation of naïve T-cells into T helper cell-2 cells, while antigen preparations of anaerobic bacteria induced T helper cell-1 cells (386). P. gingivalis lipopolysaccharide can also modulate T-cell immunity by downregulating the expression of cytokines such as interferon-γ, interleukin-12 and interleukin-8 (296). Further, A. actinomycetemcomitans is capable of modulating T-cell responses through several mechanisms including induction of T-cell apoptosis (252), expression of superantigens (406) and preferential stimulation of T-cells secreting interleukin-4 and interleukin-10 (407). Therefore, the expression of cytokines that modulate T-cell immunity is influenced by the composition of subgingival biofilms. It is beyond the scope of this manuscript to review all host-microbial interactions that have been reported in periodontal diseases. The lesson learned here is that in host-microbial interactions during periodontal pathogenesis, neither player has a passive role.
In trying to understand the role of inflammatory mediators and the processes that they regulate during periodontal disease pathogenesis, scientists have often opted for reductionist approaches using in vitro systems and/or animal models. Molecules such as cytokines have been dichotomized as either “protective” or “destructive” or “pro-inflammatory” or “anti-inflammatory”. Although the data generated using these approaches have greatly enhanced our knowledge of the patterns of expression of these molecules in periodontal health and disease, it has become apparent that these approaches will no longer suffice for the study of complex host-microbial interactions. The inadequacy of simple scenarios where pro-inflammatory cytokines should be inhibited, while anti-inflammatory cytokines could be used as therapeutics, is illustrated by the realization that their functions might vary depending on the context in which they are studied. For instance, interleukin-6 is a pro-inflammatory cytokine that has been associated with periodontal tissue destruction and has been proposed as a biomarker of periodontal disease progression (111). However, there is evidence that interleukin-6 might act as a potent anti-inflammatory cytokine. In a murine model of periapical lesion, interleukin-6 knockouts and the neutralization of interleukin-6 using antibodies resulted in significantly higher periapical bone resorption (29). Interfering with the cytokine network (or any other class of inflammatory mediator for that matter), without taking into account the consequences to the host-microbial interactions might have disastrous consequences.
The lessons learned from the studies on sepsis might be quite relevant. When anticytokine therapy was used in animal models of sepsis (e.g. anti-tumor necrosis factor-α) not all animal models demonstrated enhanced survival, particularly when cytokines were blocked in live bacterial models compared to models using injection of purified endotoxins (endotoxemia). In fact, these studies demonstrated that an endogenous tumor necrosis factor-α response was essential for the formation of abscesses to isolate the infectious agent (79). This is particularly relevant when drugs that block tumor necrosis factor-α used in the treatment of rheumatoid arthritis, an autoimmune disease, are suggested by some as a potential treatment for periodontal diseases, which are infections in nature (236). There is a growing body of evidence that blocking tumor necrosis factor-α might result in an increased risk for opportunistic infections, including mycobacterial diseases. Further, absence of the interleukin-1 receptor can also result in decreased resistance to Listeria or gram-positive bacteria (79). In his elegant review of the role of cytokines in periodontal disease pathogenesis, Garlet (107) stressed the fallacy of examining cytokines' functions solely under the “protective vs. destructive archetype” and emphasized the need to take into account their role in the “control of infection viewpoint”. In fact, his research team had previously reported a role for tumor necrosis factor-α and interferon-γ in controlling the microbial challenge in periodontal diseases. Using knockout mice and A. actinomycetemcomitans infection to induce periodontal disease, they demonstrated that both cytokines were essential for the control of the experimental infection, as evidenced by an increased bacterial load and elevated acute phase response compared to wild-type controls (108, 109). Here we have another example of how the focus solely on the host response might be counter productive.
As discussed above, shifts in local cytokine levels appear to be guided by the microbiota associated with periodontitis; however, there are very limited data regarding the changes in cytokine levels in relation to the total mass and composition of subgingival biofilms (10, 16, 167). It is also recognized that subgingival biofilms present great variability in their microbial composition (124, 158), neighboring sites on different teeth may differ considerably in levels and proportions of colonizing bacterial species (336). Far less appreciated has been the effect of this microbial diversity on the cytokine milieu released by the adjacent periodontal tissues. Teles et al. (363) examined in periodontally healthy and generalized aggressive periodontitis subjects, in vivo associations among the composition of subgingival biofilms (determined using checkerboard DNA-DNA hybridization) and the levels of 8 cytokines in gingival crevicular fluid (measured using multiplexed bead immunoassay [Luminex]). The microbiotas associated with periodontal health and generalized aggressive periodontitis were characterized using hierarchical cluster analysis of site-specific levels of 40 subgingival species. The results demonstrated that the cytokine profiles associated with the clusters in the periodontally healthy subjects were more homogeneous and presented a similar pattern with interleukin-10 composing over 50% of the total gingival crevicular fluid cytokines. In contrast, the generalized aggressive periodontitis group presented a more diverse pattern of cytokine expression among the microbial clusters (Fig. 19). The authors concluded that different subgingival biofilm profiles are associated with distinct patterns of gingival crevicular fluid cytokine expression.
Fig.19.

Mean counts × 105 of subgingival taxa in the clusters and not-in-cluster group detected in 31 generalized aggressive periodontitis subjects. The counts of 40 subgingival species were measured at each of up to 14 sites in each subject and employed in a cluster analysis using the chord coefficient and an average unweighted linkage sort. Five clusters were formed at > 33% similarity. Four subjects presented at least one site with a microbial profile that did not fit any cluster. The numbers above each panel represent the number of subjects with at least one site with the microbial profile that defined that cluster. The numbers in parentheses represent the total number of sites in each cluster. After the clusters were identified, data were averaged within a subject and then averaged across subjects in each cluster group separately. The numbers above each panel represent the number of subjects with at least one site with the microbial profile that defined that cluster. The numbers in parentheses represent the total number of sites in each cluster. The pie diagrams indicate the mean proportion of each cytokine in the 5 cluster groups and in the not-in-cluster subjects. Significance of differences for each species among cluster groups was sought using the Kruskal-Wallis test; * P<0.05, ** P<0.01, *** P<0.001 and adjusted for 40 comparisons (338). The species were ordered and grouped according to the complexes described by Socransky et al. (337). Significance of differences for the proportion of each cytokine among clusters was also examined using the Kruskal-Wallis test. Printed with permission from Teles et al. (363).
Interestingly, our lack of understanding of the role of specific bacterial species and bacterial communities in host-microbial interactions is not new. In 1963 Bo Krasse had already alerted to this problem writing: “It is obvious that the oral aggregations of microbes are of determinative importance for dental diseases, but their specific role in the dynamic host-microbial relationship is not properly understood” (183). Unfortunately this statement remains truthful and relevant to this day.
New approaches to the study of the etiopathogenesis of periodontal diseases, what the future holds?
A simple lesson learned from the studies on the etiopathogenesis of periodontal diseases is that periodontal infections are complex diseases. This simple statement implies that in order to fully understand the mechanisms that lead to periodontal diseases, scientists in the field will have to use alternative approaches to the ones employed so far. This is not necessarily a new lesson; in a comment about the difficulties in identifying the etiological agents of periodontal infections Socransky wrote: “Unfortunately, at this time it is difficult to even suggest methods to assess the significance of bacterial agents or metabolites in “natural” periodontal diseases” (332). It is not difficult to image that the same statement would apply to the study of the immunoinflammatory response in periodontal diseases. Traditionally, science has taken a reductionist approach, dissecting biological systems into their building blocks and studying them separately. Because individual parts of biological systems (e.g. single genes or proteins) never work in isolation, but rather in integrated networks, reductionist methodologies will never give us a complete picture of how those complex systems work. Because health and disease are the result of the dynamic changes in these integrated networks, to fully understand them, a more holistic “systems biology” approach will be required (105). There is no consensus regarding the definition of systems biology, but it can be described as methods aimed at integrating quantitative data from different outputs with the objective to describe and predict the function of biological systems. The predictive models derived from such methodology should greatly enhance our ability to understand health and disease; they should not only allow for a better description of biological systems, but also help in the study of those systems. In other words, these models should help us discover new properties of the biological systems they attempt to characterize. In the past decade the systems biology paradigm has been favored by many scientists trying to understand the incredible complexity of biological systems and apply this knowledge to clinical practice.
In recent years, several research groups have applied novel systems biology approaches to the study of periodontal diseases which should, in time, provides us with a better understanding of the pathophysiological mechanisms involved in initiation and progression of periodontal diseases (182, 261). For instance, human studies on the transcriptome of gingival tissues in health and disease and at different stages during experimental gingivitis have been conducted (262, 272). Papapanou et al. (272) demonstrated that colonization by T. forsythia and P. gingvalis were associated with the differential expression of thousands of genes in the adjacent gingival tissue, while the host-compatible species A. naeslundii was merely associated with 8 probe sets. The transcriptome of gingival tissues during experimental gingivitis has also been recently characterized (262). The data demonstrated that 131 immune response related genes were significantly up- or down-regulated during induction and/or resolution of gingivitis. These genes corresponded to a relatively small subset (11.9%) of immune response genes present in the gene array, suggesting specificity in the gingival transcriptomic response to plaque accumulation. In addition, new candidate genes and pathways were identified as being altered during experimental gingivitis, including neural processes, epithelial defenses, angiogenesis and would healing. Unbiased metabolomic profiling of gingival crevicular fluid samples from healthy, gingivitis and periodontitis sites demonstrated that the metabolic composition of gingival crevicular fluid was the result of host and bacterial interactions and accurately reflected the disease status of the site (31). The results suggested up-regulation of the inflammatory pathway of purine degradation, depletion of antioxidants, degradation of host cellular components, accumulation of bacterial products, and leakage of host circulation components into disease sites (Fig. 20) (31).
Fig. 20.

Illustration of purine degradation pathway and the levels of inosine, hypoxanthine, xanthine, uric acid, guanosine, and guanine by box plot for site categories (data distribution of the 22 participants in this study) and scatter plot for each individual in the gingival crevicular fluid samples. The metabolites that are up-regulated and down-regulated by the diseases are indicated by up and down block arrows, respectively. For the box plots, the top and bottom of the box represent the 75th and 25th percentiles, respectively. The top and bottom bars (“whiskers”) represent the entire spread of the datapoints for the participants, excluding “extreme” points, which are indicated with squares. The filled triangle indicates the mean value, and the open triangle indicates the median value. P-values < 0.05 are marked with an asterisk. H, healthy sites; G, gingivitis sites; P, periodontitis sites. The analytical variations for the compounds measured were below 15%. Adapted with permission from Barnes et al. (31).
Among the many limitations that techniques of the past have imposed, the study of the microbiota of periodontal health and disease has been “limited” to its composition, while its function has been “ignored”. Periodontal disease initiation and progression might not only be associated with shifts in biofilm composition but also result from changes in the biofilm metabolism and synthesis of virulence factors. Hence, there was a need to develop new tools to study the physiology of these microorganisms in their environment. Newly developed metatranscriptomic approaches have been engineered for characterizing gene expression of entire microbial communities (103); studies using this approach, combined with metagenomics, were recently initiated at the Forsyth Institute (102). Metagenomic and gene expression analysis of entire complex bacterial communities in situ provide the information required to understand the activity and relative importance of the constituents of the pathogenic biofilm during periodontal infections, including uncultivable microorganisms. Metagenomics and metatranscriptomics treat the microbial community and its constituent genes as a whole. In metagenomic analysis, DNA is extracted from the entire community and large-scale sequencing is conducted on the community's genomic material. This approach overcomes bias and other problems associated with PCR amplification (77) and it is not limited to analysis of a pre-determined number of bacteria, or to a specific gene such as 16S rRNA (100).
Despite intense interest in examining gene expression of entire microbial communities, investigations have been limited to following individual genes using quantitative RT-PCR or individual organisms using microarray analysis (106, 144). The key to assess expression analysis in whole microbial communities is the linear amplification of small quantities of RNA extracted from the bacterial assemblages. The final amplified RNA is a direct representation of the relative abundance of mRNA in the original sample. Linear RNA amplification methods have been previously used to study gene expression in eukaryotic tissues but are not generally applicable to microbial mRNA because of the requirement of a poly(A) tail. Wendisch et al. (397) developed a method for the polyadenylation of bacterial messenger RNA, which facilitates preferential isolation of bacterial mRNA from rRNA in crude extracts. The second aspect that facilitates metatranscriptomic analysis of whole bacterial communities is that pyrosequencing techniques now allow for the sequencing of large amounts of DNA, avoiding cloning biases associated with classical techniques. Taking advantage of this new technology, Frias-Lopez et al. (103) developed a method to study expression profiles of whole microbial communities in situ, opening the possibility of studying the physiology of microbes in their environment. This method allows for identifying the species present in a complex microbial community in addition to determining their metabolic activity. We anticipate that this approach will lead us to in situ identification of virulence factors in oral biofilms that lead to disease progression in periodontitis. The ability to characterize gene expression and gene interactions among entire bacterial communities in situ during different stages of periodontal disease progression has the potential to revolutionize the study of these infections.
Figure 21 illustrates the proof-of-principle of this technology after the processing of pooled samples from 74 sites from a periodontally healthy subject (sites sampled had no signs of gingivitis and pocket depth <2 mm) and pooled samples from 27 diseased sites from a subject with untreated chronic periodontitis (sites sampled were deep [>6 mm] bleeding pockets). Phylogenetic assignments of metatranscriptomic reads were done using software for analyzing metagenomes (MEtaGenome ANalyzer [MEGAN]). Illumina sequences from metatranscriptomic analysis were assigned to phylogenetic groups using BLAT results. The figure shows the comparison between reads from healthy and patients with periodontal disease. Numbers of reads were normalized to database size, represented by different sizes of the pies. Reads from healthy patients are in white and from periodontal disease patients in red. Green semicircles represent the statistical significance of the differences after Holm-Bonferroni correction. The taxa identified were clustered according to phyla and genus. Overall, the data were consistent with our current understanding of the pathogenesis of chronic periodontal diseases. Genera that encompass putative periodontal pathogens such as Prophyromonas, Tannerella, Prevotella, Treponema, Campylobacter and Parvimonas presented a higher level of gene expression in samples from the periodontitis subject. Conversely, genera characteristic of commensal species such as Streptococcus, Veillonella, Neisseria, and Actinomyces had a higher level of gene expression in samples from the healthy subject. Further, the phylum Synergistetes that contains only yet uncultivable members and that has been recently associated with periodontal diseases also demonstrated a higher level of gene expression in the samples from the periodontitis patient.
Fig. 21.

Metatranscriptomics from pooled samples from 74 sites from a periodontally healthy subject (no bleeding on probing, no gingival redness and pocket depth <2 mm) and pooled samples from 27 diseased sites from a subject with untreated chronic periodontitis (pocket depth >6 mm with bleeding on probing). Phylogenetic assignments of metatranscriptomic reads were done using software for analyzing metagenomes (MEtaGenome ANalyzer [MEGAN]). Illumina sequences from metatranscriptomic analysis were assigned to phylogenetic groups using BLAT results. Numbers of reads were normalized to database size, represented by different sizes of the pies. Reads from healthy patients are in white and from periodontal disease patients in red. Green semicircles represent the statistical significance of the differences after Holm-Bonferroni correction. The taxa identified were clustered according to phyla and genus.
The computational power of nowadays computers; the development of sophisticated analytical methods and computer modeling; and the development of accessible high-throughput multiplexing technologies for genetic and epigenetic studies, microbiomics, metabolomics, proteomics and transcriptomics analyses, afford the new generation of scientists in the field of periodontal microbiology resources that its pioneers could not even imagine. A brave new world is ahead of us; let's make sure that new lessons continue to be learned.
Fig. 6.
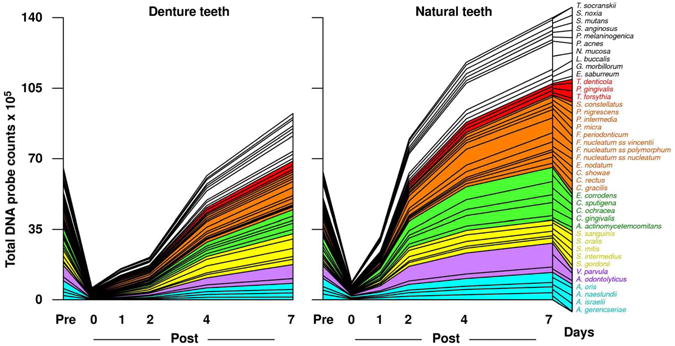
Cumulative mean counts (×105) of 41 bacterial species in samples taken from 55 subjects with natural teeth and 62 subjects with full mouth dentures. Samples were taken prior to professional removal of the dental biofilms, immediately after cleaning, and after 1, 2, 4 and 7 days of re-development. The subjects refrained from oral hygiene procedures for the 7 day test period. Samples were removed from the mesio-buccal aspect of each natural tooth (excluding third molars) and each denture tooth before cleaning and immediately post-cleaning. In addition, samples were obtained from up to 7 teeth in randomly selected quadrants at 1, 2, 4 and 7 days after tooth cleaning. All samples were individually analyzed for their content of 41 taxa using checkerboard DNA-DNA hybridization. Species counts in the samples were averaged within each subject at each time point and then averaged across subjects in the 2 clinical groups. The plots present the cumulative mean values at each time point in each clinical group. The species were ordered and color-coded according to previously described microbial complexes (337). Printed with permission from Teles et al. (360).
Acknowledgments
This work was supported in part by the research grants DE-021127, DE-017400, DE-021565, DE-021742 and DE-021553 from the National Institute of Dental and Craniofacial Research.
References
- 1.Consensus report Periodontal diseases: pathogenesis and microbial factors. Ann Periodontol. 1996;1:926–932. doi: 10.1902/annals.1996.1.1.926. [DOI] [PubMed] [Google Scholar]
- 2.Ann Periodontol; Proceedings of the 1996 World Workshop in Periodontics; Lansdowne, Virginia. July 13-17, 1996; 1996. pp. 1–947. [PubMed] [Google Scholar]
- 3.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abiko Y, Sato T, Mayanagi G, Takahashi N. Profiling of subgingival plaque biofilm microflora from periodontally healthy subjects and from subjects with periodontitis using quantitative real-time PCR. J Periodontal Res. 2010;45:389–395. doi: 10.1111/j.1600-0765.2009.01250.x. [DOI] [PubMed] [Google Scholar]
- 5.Abramson JS, Mills EL. Depression of neutrophil function induced by viruses and its role in secondary microbial infections. Rev Infect Dis. 1988;10:326–341. doi: 10.1093/clinids/10.2.326. [DOI] [PubMed] [Google Scholar]
- 6.Ahn J, Yang L, Paster BJ, Ganly I, Morris L, Pei Z, et al. Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. Plos One. 2011;6:e22788. doi: 10.1371/journal.pone.0022788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Ahmad A, Wunder A, Auschill TM, Follo M, Braun G, Hellwig E, et al. The in vivo dynamics of Streptococcus spp., Actinomyces naeslundii, Fusobacterium nucleatum and Veillonella spp in dental plaque biofilm as analysed by five-colour multiplex fluorescence in situ hybridization. Journal of Medical Microbiology. 2007;56:681–687. doi: 10.1099/jmm.0.47094-0. [DOI] [PubMed] [Google Scholar]
- 8.Albandar JM, Brunelle JA, Kingman A. Destructive periodontal disease in adults 30 years of age and older in the United States, 1988-1994. J Periodontol. 1999;70:13–29. doi: 10.1902/jop.1999.70.1.13. [DOI] [PubMed] [Google Scholar]
- 9.Albandar JM, Khattab R, Monem F, Barbuto SM, Paster BJ. The subgingival microbiota of papillon-lefevre syndrome. J Periodontol. 2012;83:902–908. doi: 10.1902/jop.2011.110450. [DOI] [PubMed] [Google Scholar]
- 10.Albandar JM, Kingman A, Lamster IB. Crevicular fluid level of beta-glucuronidase in relation to clinical periodontal parameters and putative periodontal pathogens in early-onset periodontitis. J Clin Periodontol. 1998;25:630–639. doi: 10.1111/j.1600-051x.1998.tb02499.x. [DOI] [PubMed] [Google Scholar]
- 11.Ali RW, Martin L, Haffajee AD, Socransky SS. Detection of identical ribotypes of Porphyromonas gingivalis in patients residing in the United States, Sudan, Romania and Norway. Oral Microbiol Immunol. 1997;12:106–111. doi: 10.1111/j.1399-302x.1997.tb00625.x. [DOI] [PubMed] [Google Scholar]
- 12.Allison KR, Brynildsen MP, Collins JJ. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amann R, Ludwig W. Ribosomal RNA-targeted nucleic acid probes for studies in microbial ecology. FEMS Microbiol Rev. 2000;24:555–565. doi: 10.1111/j.1574-6976.2000.tb00557.x. [DOI] [PubMed] [Google Scholar]
- 14.Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aparna MS, Yadav S. Biofilms: microbes and disease. Braz J Infect Dis. 2008;12:526–530. doi: 10.1590/s1413-86702008000600016. [DOI] [PubMed] [Google Scholar]
- 16.Apatzidou DA, Riggio MP, Kinane DF. Impact of smoking on the clinical, microbiological and immunological parameters of adult patients with periodontitis. J Clin Periodontol. 2005;32:973–983. doi: 10.1111/j.1600-051X.2005.00788.x. [DOI] [PubMed] [Google Scholar]
- 17.Armitage GC, Dickinson WR, Jenderseck RS, Levine SM, Chambers DW. Relationship between the percentage of subgingival spirochetes and the severity of periodontal disease. J Periodontol. 1982;53:550–556. doi: 10.1902/jop.1982.53.9.550. [DOI] [PubMed] [Google Scholar]
- 18.Arno A, Waerhaug J, Lovdal A, Schei O. Incidence of gingivitis as related to sex, occupation, tobacco consumption, toothbrushing, and age. Oral Surg Oral Med Oral Pathol. 1958;11:587–595. doi: 10.1016/0030-4220(58)90004-5. [DOI] [PubMed] [Google Scholar]
- 19.Ashimoto A, Chen C, Bakker I, Slots J. Polymerase chain reaction detection of 8 putative periodontal pathogens in subgingival plaque of gingivitis and advanced periodontitis lesions. Oral Microbiol Immunol. 1996;11:266–273. doi: 10.1111/j.1399-302x.1996.tb00180.x. [DOI] [PubMed] [Google Scholar]
- 20.Asikainen S, Chen C. Oral ecology and person-to-person transmission of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Periodontol 2000. 1999;20:65–81. doi: 10.1111/j.1600-0757.1999.tb00158.x. [DOI] [PubMed] [Google Scholar]
- 21.Asikainen S, Dogan B, Turgut Z, Paster BJ, Bodur A, Oscarsson J. Specified species in gingival crevicular fluid predict bacterial diversity. Plos One. 2010;5:e13589. doi: 10.1371/journal.pone.0013589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atwood KC, Schneider LK, Ryan FJ. Periodic selection in Escherichia coli. Proc Natl Acad Sci U S A. 1951;37:146–155. doi: 10.1073/pnas.37.3.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Axelsson P, Lindhe J. Effect of controlled oral hygiene procedures on caries and periodontal disease in adults. J Clin Periodontol. 1978;5:133–151. doi: 10.1111/j.1600-051x.1978.tb01914.x. [DOI] [PubMed] [Google Scholar]
- 24.Axelsson P, Lindhe J. Effect of controlled oral hygiene procedures on caries and periodontal disease in adults. Results after 6 years. J Clin Periodontol. 1981;8:239–248. doi: 10.1111/j.1600-051x.1981.tb02035.x. [DOI] [PubMed] [Google Scholar]
- 25.Axelsson P, Lindhe J. Effect of oral hygiene instruction and professional toothcleaning on caries and gingivitis in schoolchildren. Community Dent Oral Epidemiol. 1981;9:251–255. doi: 10.1111/j.1600-0528.1981.tb00340.x. [DOI] [PubMed] [Google Scholar]
- 26.Axelsson P, Lindhe J, Nystrom B. On the prevention of caries and periodontal disease Results of a 15-year longitudinal study in adults. J Clin Periodontol. 1991;18:182–189. doi: 10.1111/j.1600-051x.1991.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 27.Axelsson P, Nystrom B, Lindhe J. The long-term effect of a plaque control program on tooth mortality, caries and periodontal disease in adults. Results after 30 years of maintenance. J Clin Periodontol. 2004;31:749–757. doi: 10.1111/j.1600-051X.2004.00563.x. [DOI] [PubMed] [Google Scholar]
- 28.Badersten A, Nilveus R, Egelberg J. Scores of plaque, bleeding, suppuration and probing depth to predict probing attachment loss 5 years of observation following nonsurgical periodontal therapy. J Clin Periodontol. 1990;17:102–107. doi: 10.1111/j.1600-051x.1990.tb01070.x. [DOI] [PubMed] [Google Scholar]
- 29.Balto K, Sasaki H, Stashenko P. Interleukin-6 deficiency increases inflammatory bone destruction. Infect Immun. 2001;69:744–750. doi: 10.1128/IAI.69.2.744-750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barnard A, Mahon BP, Watkins J, Redhead K, Mills KH. Th1/Th2 cell dichotomy in acquired immunity to Bordetella pertussis: variables in the in vivo priming and in vitro cytokine detection techniques affect the classification of T-cell subsets as Th1, Th2 or Th0. Immunology. 1996;87:372–380. doi: 10.1046/j.1365-2567.1996.497560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnes VM, Teles R, Trivedi HM, Devizio W, Xu T, Mitchell MW, et al. Acceleration of purine degradation by periodontal diseases. J Dent Res. 2009;88:851–855. doi: 10.1177/0022034509341967. [DOI] [PubMed] [Google Scholar]
- 32.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 33.Bayles KW. The biological role of death and lysis in biofilm development. Nat Rev Microbiol. 2007;5:721–726. doi: 10.1038/nrmicro1743. [DOI] [PubMed] [Google Scholar]
- 34.Beikler T, Peters U, Prior K, Ehmke B, Flemmig TF. Sequence variations in rgpA and rgpB of Porphyromonas gingivalis in periodontitis. J Periodontal Res. 2005;40:193–198. doi: 10.1111/j.1600-0765.2005.00783.x. [DOI] [PubMed] [Google Scholar]
- 35.Berezow AB, Darveau RP. Microbial shift and periodontitis. Periodontol 2000. 2011;55:36–47. doi: 10.1111/j.1600-0757.2010.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernard CS, Giraud C, Spagnolo J, Bentzmann Sd. In: Biofilms: the secret story of microbial communities. Locht C, Simonet M, editors. Norfolk: Caister Academic Press; 2012. pp. 129–168. [Google Scholar]
- 37.Bjarnsholt T. In: Introduction to biofilms. Bjarnsholt T, Moser C, Jenses PO, Hoiby N, editors. New York: Springer; 2011. pp. 1–10. [Google Scholar]
- 38.Bogren A, Teles RP, Torresyap G, Haffajee AD, Socransky SS, Wennstrom JL. Clinical and microbiologic changes associated with the combined use of a powered toothbrush and a triclosan/copolymer dentifrice: a 3-year prospective study. J Periodontol. 2007;78:1708–1717. doi: 10.1902/jop.2007.070028. [DOI] [PubMed] [Google Scholar]
- 39.Booth V, Downes J, Van den Berg J, Wade WG. Gram-positive anaerobic bacilli in human periodontal disease. J Periodontal Res. 2004;39:213–220. doi: 10.1111/j.1600-0765.2004.00726.x. [DOI] [PubMed] [Google Scholar]
- 40.Botero JE, Vidal C, Contreras A, Parra B. Comparison of nested polymerase chain reaction (PCR), real-time PCR and viral culture for the detection of cytomegalovirus in subgingival samples. Oral Microbiol Immunol. 2008;23:239–244. doi: 10.1111/j.1399-302X.2007.00418.x. [DOI] [PubMed] [Google Scholar]
- 41.Boutaga K, van Winkelhoff AJ, Vandenbroucke-Grauls CM, Savelkoul PH. The additional value of real-time PCR in the quantitative detection of periodontal pathogens. J Clin Periodontol. 2006;33:427–433. doi: 10.1111/j.1600-051X.2006.00925.x. [DOI] [PubMed] [Google Scholar]
- 42.Brinig MM, Lepp PW, Ouverney CC, Armitage GC, Relman DA. Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microbiol. 2003;69:1687–1694. doi: 10.1128/AEM.69.3.1687-1694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broadbent JM, Thomson WM, Boyens JV, Poulton R. Dental plaque and oral health during the first 32 years of life. J Am Dent Assoc. 2011;142:415–426. doi: 10.14219/jada.archive.2011.0197. [DOI] [PubMed] [Google Scholar]
- 44.Brunner J, Wittink FR, Jonker MJ, de Jong M, Breit TM, Laine ML, et al. The core genome of the anaerobic oral pathogenic bacterium Porphyromonas gingivalis. BMC Microbiol. 2010;10:252. doi: 10.1186/1471-2180-10-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campos MS, Marchini L, Bernardes LA, Paulino LC, Nobrega FG. Biofilm microbial communities of denture stomatitis. Oral Microbiol Immunol. 2008;23:419–424. doi: 10.1111/j.1399-302X.2008.00445.x. [DOI] [PubMed] [Google Scholar]
- 46.Carranza F. In: Microbiology. Carranza F, Shklar G, editors. Carol Stream: Quintessence Publishing Co, Inc; 2003. pp. 80–91. [Google Scholar]
- 47.Cassai E, Galvan M, Trombelli L, Rotola A. HHV-6, HHV-7, HHV-8 in gingival biopsies from chronic adult periodontitis patients. A case-control study. J Clin Periodontol. 2003;30:184–191. doi: 10.1034/j.1600-051x.2003.00220.x. [DOI] [PubMed] [Google Scholar]
- 48.Chalabi M, Rezaie F, Moghim S, Mogharehabed A, Rezaei M, Mehraban B. Periodontopathic bacteria and herpesviruses in chronic periodontitis. Mol Oral Microbiol. 25:236–240. doi: 10.1111/j.2041-1014.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- 49.Chandler DP, Fredrickson JK, Brockman FJ. Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol Ecol. 1997;6:475–482. doi: 10.1046/j.1365-294x.1997.00205.x. [DOI] [PubMed] [Google Scholar]
- 50.Chen CK, Sunday GJ, Zambon JJ, Wilson ME. Restriction endonuclease analysis of Eikenella corrodens. J Clin Microbiol. 1990;28:1265–1270. doi: 10.1128/jcm.28.6.1265-1270.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The human oral microbiome database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010:baq013. doi: 10.1093/database/baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, Zhang M, Zhou C, Kallenbach NR, Ren D. Control of bacterial persister cells by Trp/Arg-containing antimicrobial peptides. Appl Environ Microbiol. 2011;77:4878–4885. doi: 10.1128/AEM.02440-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiang N, Fredman G, Backhed F, Oh SF, Vickery T, Schmidt BA, et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–528. doi: 10.1038/nature11042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cionca N, Giannopoulou C, Ugolotti G, Mombelli A. Amoxicillin and metronidazole as an adjunct to full-mouth scaling and root planing of chronic periodontitis. J Periodontol. 2009;80:364–371. doi: 10.1902/jop.2009.080540. [DOI] [PubMed] [Google Scholar]
- 55.Cionca N, Giannopoulou C, Ugolotti G, Mombelli A. Microbiologic testing and outcomes of full-mouth scaling and root planing with or without amoxicillin/metronidazole in chronic periodontitis. J Periodontol. 2010;81:15–23. doi: 10.1902/jop.2009.090390. [DOI] [PubMed] [Google Scholar]
- 56.Claffey N, Egelberg J. Clinical indicators of probing attachment loss following initial periodontal treatment in advanced periodontitis patients. J Clin Periodontol. 1995;22:690–696. doi: 10.1111/j.1600-051x.1995.tb00828.x. [DOI] [PubMed] [Google Scholar]
- 57.Cohan FM. Bacterial species and speciation. Syst Biol. 2001;50:513–524. doi: 10.1080/10635150118398. [DOI] [PubMed] [Google Scholar]
- 58.Colombo AP, Bennet S, Cotton SL, Goodson JM, Kent R, Haffajee AD, et al. Impact of periodontal therapy on the subgingival microbiota of severe periodontitis: Comparison between good responders and “refractory” subjects by the human oral microbe identification microarray (HOMIM) J Periodontol. 2012 doi: 10.1902/jop.2012.110566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, et al. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol. 2009;80:1421–1432. doi: 10.1902/jop.2009.090185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colombo AP, Teles RP, Torres MC, Souto R, Rosalem WJ, Mendes MC, et al. Subgingival microbiota of Brazilian subjects with untreated chronic periodontitis. J Periodontol. 2002;73:360–369. doi: 10.1902/jop.2002.73.4.360. [DOI] [PubMed] [Google Scholar]
- 61.Contreras A, Nowzari H, Slots J. Herpesviruses in periodontal pocket and gingival tissue specimens. Oral Microbiol Immunol. 2000;15:15–18. doi: 10.1034/j.1399-302x.2000.150103.x. [DOI] [PubMed] [Google Scholar]
- 62.Contreras A, Slots J. Herpesviruses in human periodontal disease. J Periodontal Res. 2000;35:3–16. doi: 10.1034/j.1600-0765.2000.035001003.x. [DOI] [PubMed] [Google Scholar]
- 63.Contreras A, Umeda M, Chen C, Bakker I, Morrison JL, Slots J. Relationship between herpesviruses and adult periodontitis and periodontopathic bacteria. J Periodontol. 1999;70:478–484. doi: 10.1902/jop.1999.70.5.478. [DOI] [PubMed] [Google Scholar]
- 64.Cortelli JR, Aquino DR, Cortelli SC, Fernandes CB, de Carvalho-Filho J, Franco GC, et al. Etiological analysis of initial colonization of periodontal pathogens in oral cavity. J Clin Microbiol. 2008;46:1322–1329. doi: 10.1128/JCM.02051-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Costerton JW, Lewandowski Z, DeBeer D, Caldwell D, Korber D, James G. Biofilms, the customized microniche. J Bacteriol. 1994;176:2137–2142. doi: 10.1128/jb.176.8.2137-2142.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 68.Cugini MA, Haffajee AD, Smith C, Kent RL, Jr, Socransky SS. The effect of scaling and root planing on the clinical and microbiological parameters of periodontal diseases: 12-month results. J Clin Periodontol. 2000;27:30–36. doi: 10.1034/j.1600-051x.2000.027001030.x. [DOI] [PubMed] [Google Scholar]
- 69.Dahlén G, Leonhardt A. A new checkerboard panel for testing bacterial markers in periodontal disease. Oral Microbiol Immunol. 2006;21:6–11. doi: 10.1111/j.1399-302X.2005.00243.x. [DOI] [PubMed] [Google Scholar]
- 70.Dalwai F, Spratt DA, Pratten J. Modeling shifts in microbial populations associated with health or disease. Appl Environ Microbiol. 2006;72:3678–3684. doi: 10.1128/AEM.72.5.3678-3684.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Jong EC, Vieira PL, Kalinski P, Schuitemaker JH, Tanaka Y, Wierenga EA, et al. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse th cell-polarizing signals. J Immunol. 2002;168:1704–1709. doi: 10.4049/jimmunol.168.4.1704. [DOI] [PubMed] [Google Scholar]
- 72.de Lillo A, Ashley FP, Palmer RM, Munson MA, Kyriacou L, Weightman AJ, et al. Novel subgingival bacterial phylotypes detected using multiple universal polymerase chain reaction primer sets. Oral Microbiol Immunol. 2006;21:61–68. doi: 10.1111/j.1399-302X.2005.00255.x. [DOI] [PubMed] [Google Scholar]
- 73.De Souza RF, Nascimento C, Regis RR, Silva-Lovato CH, Paranhos HF. Effects of the domestic use of a disclosing solution on the denture biofilm: a preliminary study. J Oral Rehabil. 2009;36:491–497. doi: 10.1111/j.1365-2842.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 74.del Pozo JL, Patel R. The challenge of treating biofilm-associated bacterial infections. Clin Pharmacol Ther. 2007;82:204–209. doi: 10.1038/sj.clpt.6100247. [DOI] [PubMed] [Google Scholar]
- 75.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dewhirst FE, Tamer MA, Ericson RE, Lau CN, Levanos VA, Boches SK, et al. The diversity of periodontal spirochetes by 16S rRNA analysis. Oral Microbiol Immunol. 2000;15:196–202. doi: 10.1034/j.1399-302x.2000.150308.x. [DOI] [PubMed] [Google Scholar]
- 77.Diaz PI. Microbial diversity and interactions in subgingival biofilm communities. Front Oral Biol. 2012;15:17–40. doi: 10.1159/000329669. [DOI] [PubMed] [Google Scholar]
- 78.Diaz PI, Dupuy AK, Abusleme L, Reese B, Obergfell C, Choquette L, et al. Using high throughput sequencing to explore the biodiversity in oral bacterial communities. Mol Oral Microbiol. 2012;27:182–201. doi: 10.1111/j.2041-1014.2012.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dinarello CA. Anti-cytokine therapeutics and infections. Vaccine. 2003;21(2):S24–34. doi: 10.1016/s0264-410x(03)00196-8. [DOI] [PubMed] [Google Scholar]
- 80.Donachie SP, Foster JS, Brown MV. Culture clash: challenging the dogma of microbial diversity. ISME J. 2007;1:97–99. doi: 10.1038/ismej.2007.22. [DOI] [PubMed] [Google Scholar]
- 81.Downes J, Dewhirst FE, Tanner AC, Wade WG. Alloprevotella rava gen. nov., sp. nov., isolated from the human oral cavity, and reclassification of Prevotella tannerae (Moore, Johnson & Moore, 1994) as Alloprevotella tannerae gen. nov., comb. nov. Int J Syst Evol Microbiol. 2012 doi: 10.1099/ijs.0.041376-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Downes J, Tanner AC, Dewhirst FE, Wade WG. Prevotella saccharolytica sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol. 2010;60:2458–2461. doi: 10.1099/ijs.0.014720-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Downes J, Vartoukian SR, Dewhirst FE, Izard J, Chen T, Yu WH, et al. Pyramidobacter piscolens gen. nov., sp. nov a member of the phylum ‘Synergistetes’ isolated from the human oral cavity. Int J Syst Evol Microbiol. 2009;59:972–980. doi: 10.1099/ijs.0.000364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dumitrescu AL, Abd-El-Aleem S, Morales-Aza B, Donaldson LF. A model of periodontitis in the rat: effect of lipopolysaccharide on bone resorption, osteoclast activity, and local peptidergic innervation. J Clin Periodontol. 2004;31:596–603. doi: 10.1111/j.1600-051X.2004.00528.x. [DOI] [PubMed] [Google Scholar]
- 85.Duran-Pinedo AE, Paster B, Teles R, Frias-Lopez J. Correlation network analysis applied to complex biofilm communities. Plos One. 2011;6:e28438. doi: 10.1371/journal.pone.0028438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dzink JL, Sheenan MT, Socransky SS. Proposal of three subspecies of Fusobacterium nucleatum Knorr 1922: Fusobacterium nucleatum subsp. nucleatum subsp. nov., comb. nov.; Fusobacterium nucleatum subsp. polymorphum subsp. nov., nom. rev., comb. nov.; and Fusobacterium nucleatum subsp. vincentii subsp. nov., nom. rev., comb. nov. Int J Syst Bacteriol. 1990;40:74–78. doi: 10.1099/00207713-40-1-74. [DOI] [PubMed] [Google Scholar]
- 87.Dzink JL, Socransky SS, Haffajee AD. The predominant cultivable microbiota of active and inactive lesions of destructive periodontal diseases. J Clin Periodontol. 1988;15:316–323. doi: 10.1111/j.1600-051x.1988.tb01590.x. [DOI] [PubMed] [Google Scholar]
- 88.Dzink JL, Tanner AC, Haffajee AD, Socransky SS. Gram negative species associated with active destructive periodontal lesions. J Clin Periodontol. 1985;12:648–659. doi: 10.1111/j.1600-051x.1985.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 89.Eckert KA, Kunkel TA. DNA polymerase fidelity and the polymerase chain reaction. PCR Methods Appl. 1991;1:17–24. doi: 10.1101/gr.1.1.17. [DOI] [PubMed] [Google Scholar]
- 90.Ehmke B, Moter A, Beikler T, Milian E, Flemmig TF. Adjunctive antimicrobial therapy of periodontitis: long-term effects on disease progression and oral colonization. J Periodontol. 2005;76:749–759. doi: 10.1902/jop.2005.76.5.749. [DOI] [PubMed] [Google Scholar]
- 91.El-Sharkawy H, Aboelsaad N, Eliwa M, Darweesh M, Alshahat M, Kantarci A, et al. Adjunctive treatment of chronic periodontitis with daily dietary supplementation with omega-3 Fatty acids and low-dose aspirin. J Periodontol. 2010;81:1635–1643. doi: 10.1902/jop.2010.090628. [DOI] [PubMed] [Google Scholar]
- 92.Enersen M, Olsen I, Caugant DA. Genetic diversity of Porphyromonas gingivalis isolates recovered from single “refractory” periodontitis sites. Appl Environ Microbiol. 2008;74:5817–5821. doi: 10.1128/AEM.00225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Enersen M, Olsen I, van Winkelhoff AJ, Caugant DA. Multilocus sequence typing of Porphyromonas gingivalis strains from different geographic origins. J Clin Microbiol. 2006;44:35–41. doi: 10.1128/JCM.44.1.35-41.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Farrelly V, Rainey FA, Stackebrandt E. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl Environ Microbiol. 1995;61:2798–2801. doi: 10.1128/aem.61.7.2798-2801.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Faveri M, Mayer MP, Feres M, de Figueiredo LC, Dewhirst FE, Paster BJ. Microbiological diversity of generalized aggressive periodontitis by 16S rRNA clonal analysis. Oral Microbiol Immunol. 2008;23:112–118. doi: 10.1111/j.1399-302X.2007.00397.x. [DOI] [PubMed] [Google Scholar]
- 96.Feres M. Antibiotics in the treatment of periodontal diseases: microbiological basis and clinical applications. Ann R Australas Coll Dent Surg. 2008;19:37–44. [PubMed] [Google Scholar]
- 97.Filoche S, Wong L, Sissons CH. Oral biofilms: emerging concepts in microbial ecology. J Dent Res. 2010;89:8–18. doi: 10.1177/0022034509351812. [DOI] [PubMed] [Google Scholar]
- 98.Fine DH, Mandel ID. Indicators of periodontal disease activity: an evaluation. J Clin Periodontol. 1986;13:533–546. doi: 10.1111/j.1600-051x.1986.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 99.Fine DH, Markowitz K, Furgang D, Fairlie K, Ferrandiz J, Nasri C, et al. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol. 2007;45:3859–3869. doi: 10.1128/JCM.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol. 2008;24:4–10. doi: 10.1097/MOG.0b013e3282f2b0e8. [DOI] [PubMed] [Google Scholar]
- 101.Fraser C, Alm EJ, Polz MF, Spratt BG, Hanage WP. The bacterial species challenge: making sense of genetic and ecological diversity. Science. 2009;323:741–746. doi: 10.1126/science.1159388. [DOI] [PubMed] [Google Scholar]
- 102.Frias-Lopez J, Duran-Pinedo A. Effect of periodontal pathogens on the metatranscriptome of a healthy multispecies biofilm model. J Bacteriol. 2012;194:2082–2095. doi: 10.1128/JB.06328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, et al. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci U S A. 2008;105:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Friedman MT, Barber PM, Mordan NJ, Newman HN. The “plaque-free zone” in health and disease: a scanning electron microscope study. J Periodontol. 1992;63:890–896. doi: 10.1902/jop.1992.63.11.890. [DOI] [PubMed] [Google Scholar]
- 105.Galitski T. Molecular networks in model systems. Annu Rev Genomics Hum Genet. 2004;5:177–187. doi: 10.1146/annurev.genom.5.061903.180053. [DOI] [PubMed] [Google Scholar]
- 106.Gao H, Yang ZK, Gentry TJ, Wu L, Schadt CW, Zhou J. Microarray-based analysis of microbial community RNAs by whole-community RNA amplification. Appl Environ Microbiol. 2007;73:563–571. doi: 10.1128/AEM.01771-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Garlet GP. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 2010;89:1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 108.Garlet GP, Cardoso CR, Campanelli AP, Ferreira BR, Avila-Campos MJ, Cunha FQ, et al. The dual role of p55 tumour necrosis factor-alpha receptor in Actinobacillus actinomycetemcomitans-induced experimental periodontitis: host protection and tissue destruction. Clin Exp Immunol. 2007;147:128–138. doi: 10.1111/j.1365-2249.2006.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garlet GP, Cardoso CR, Campanelli AP, Garlet TP, Avila-Campos MJ, Cunha FQ, et al. The essential role of IFN-gamma in the control of lethal Aggregatibacter actinomycetemcomitans infection in mice. Microbes Infect. 2008;10:489–496. doi: 10.1016/j.micinf.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 110.Garlet GP, Giozza SP, Silveira EM, Claudino M, Santos SB, Avila-Campos MJ, et al. Association of human T lymphotropic virus 1 amplification of periodontitis severity with altered cytokine expression in response to a standard periodontopathogen infection. Clin Infect Dis. 2010;50:e11–18. doi: 10.1086/649871. [DOI] [PubMed] [Google Scholar]
- 111.Geivelis M, Turner DW, Pederson ED, Lamberts BL. Measurements of interleukin-6 in gingival crevicular fluid from adults with destructive periodontal disease. J Periodontol. 1993;64:980–983. doi: 10.1902/jop.1993.64.10.980. [DOI] [PubMed] [Google Scholar]
- 112.Gemmell E, Bird PS, Ford PJ, Ashman RB, Gosling P, Hu Y, et al. Modulation of the antibody response by Porphyromonas gingivalis and Fusobacterium nucleatum in a mouse model. Oral Microbiol Immunol. 2004;19:247–251. doi: 10.1111/j.1399-302X.2004.00147.x. [DOI] [PubMed] [Google Scholar]
- 113.Genco RJ, Loos BG. The use of genomic DNA fingerprinting in studies of the epidemiology of bacteria in periodontitis. J Clin Periodontol. 1991;18:396–405. doi: 10.1111/j.1600-051x.1991.tb02307.x. [DOI] [PubMed] [Google Scholar]
- 114.Genco RJ, Slots J. Host responses in periodontal diseases. J Dent Res. 1984;63:441–451. doi: 10.1177/00220345840630031601. [DOI] [PubMed] [Google Scholar]
- 115.Golub LM. Reduction with tetracyclines of excessive collagen degradation in periodontal and other diseases. N Y State Dent J. 1990;56:24–26. [PubMed] [Google Scholar]
- 116.Golub LM, Goodson JM, Lee HM, Vidal AM, McNamara TF, Ramamurthy NS. Tetracyclines inhibit tissue collagenases. Effects of ingested low-dose and local delivery systems. J Periodontol. 1985;56:93–97. doi: 10.1902/jop.1985.56.11s.93. [DOI] [PubMed] [Google Scholar]
- 117.Golub LM, McNamara TF, Ryan ME, Kohut B, Blieden T, Payonk G, et al. Adjunctive treatment with subantimicrobial doses of doxycycline: effects on gingival fluid collagenase activity and attachment loss in adult periodontitis. J Clin Periodontol. 2001;28:146–156. doi: 10.1034/j.1600-051x.2001.028002146.x. [DOI] [PubMed] [Google Scholar]
- 118.Goncalves LF, Fermiano D, Feres M, Figueiredo LC, Teles FR, Mayer MP, et al. Levels of Selenomonas species in generalized aggressive periodontitis. J Periodontal Res. 2012 doi: 10.1111/j.1600-0765.2012.01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goodson JM, Haffajee AD, Socransky SS, Kent R, Teles R, Hasturk H, et al. Control of periodontal infections: a randomized controlled trial I. The primary outcome attachment gain and pocket depth reduction at treated sites. J Clin Periodontol. 2012;39:526–536. doi: 10.1111/j.1600-051X.2012.01870.x. [DOI] [PubMed] [Google Scholar]
- 120.Goodson JM, Tanner AC, Haffajee AD, Sornberger GC, Socransky SS. Patterns of progression and regression of advanced destructive periodontal disease. J Clin Periodontol. 1982;9:472–481. doi: 10.1111/j.1600-051x.1982.tb02108.x. [DOI] [PubMed] [Google Scholar]
- 121.Greene JC. Oral hygiene and periodontal disease. Am J Public Health Nations Health. 1963;53:913–922. doi: 10.2105/ajph.53.6.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Greisen K, Loeffelholz M, Purohit A, Leong D. PCR primers and probes for the 16S rRNA gene of most species of pathogenic bacteria, including bacteria found in cerebrospinal fluid. J Clin Microbiol. 1994;32:335–351. doi: 10.1128/jcm.32.2.335-351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grenier G, Gagnon G, Grenier D. Detection of herpetic viruses in gingival crevicular fluid of patients suffering from periodontal diseases: prevalence and effect of treatment. Oral Microbiol Immunol. 2009;24:506–509. doi: 10.1111/j.1399-302X.2009.00542.x. [DOI] [PubMed] [Google Scholar]
- 124.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6:1176–1185. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guerrero A, Griffiths GS, Nibali L, Suvan J, Moles DR, Laurell L, et al. Adjunctive benefits of systemic amoxicillin and metronidazole in non-surgical treatment of generalized aggressive periodontitis: a randomized placebo-controlled clinical trial. J Clin Periodontol. 2005;32:1096–1107. doi: 10.1111/j.1600-051X.2005.00814.x. [DOI] [PubMed] [Google Scholar]
- 126.Haffajee AD, Bogren A, Hasturk H, Feres M, Lopez NJ, Socransky SS. Subgingival microbiota of chronic periodontitis subjects from different geographic locations. J Clin Periodontol. 2004;31:996–1002. doi: 10.1111/j.1600-051X.2004.00597.x. [DOI] [PubMed] [Google Scholar]
- 127.Haffajee AD, Cugini MA, Dibart S, Smith C, Kent RL, Jr, Socransky SS. The effect of SRP on the clinical and microbiological parameters of periodontal diseases. J Clin Periodontol. 1997;24:324–334. doi: 10.1111/j.1600-051x.1997.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 128.Haffajee AD, Cugini MA, Tanner A, Pollack RP, Smith C, Kent RL, Jr, et al. Subgingival microbiota in healthy, well-maintained elder and periodontitis subjects. J Clin Periodontol. 1998;25:346–353. doi: 10.1111/j.1600-051x.1998.tb02454.x. [DOI] [PubMed] [Google Scholar]
- 129.Haffajee AD, Patel M, Socransky SS. Microbiological changes associated with four different periodontal therapies for the treatment of chronic periodontitis. Oral Microbiol Immunol. 2008;23:148–157. doi: 10.1111/j.1399-302X.2007.00403.x. [DOI] [PubMed] [Google Scholar]
- 130.Haffajee AD, Socransky SS. Attachment level changes in destructive periodontal diseases. J Clin Periodontol. 1986;13:461–475. doi: 10.1111/j.1600-051x.1986.tb01491.x. [DOI] [PubMed] [Google Scholar]
- 131.Haffajee AD, Socransky SS. Microbial etiological agents of destructive periodontal diseases. Periodontol 2000. 1994;5:78–111. doi: 10.1111/j.1600-0757.1994.tb00020.x. [DOI] [PubMed] [Google Scholar]
- 132.Haffajee AD, Socransky SS, Dzink JL, Taubman MA, Ebersole JL. Clinical, microbiological and immunological features of subjects with refractory periodontal diseases. J Clin Periodontol. 1988;15:390–398. doi: 10.1111/j.1600-051x.1988.tb01017.x. [DOI] [PubMed] [Google Scholar]
- 133.Haffajee AD, Socransky SS, Goodson JM. Comparison of different data analyses for detecting changes in attachment level. J Clin Periodontol. 1983;10:298–310. doi: 10.1111/j.1600-051x.1983.tb01278.x. [DOI] [PubMed] [Google Scholar]
- 134.Haffajee AD, Socransky SS, Goodson JM. Periodontal disease activity. J Periodontal Res. 1982;17:521–522. doi: 10.1111/j.1600-0765.1982.tb02045.x. [DOI] [PubMed] [Google Scholar]
- 135.Haffajee AD, Socransky SS, Gunsolley JC. Systemic anti-infective periodontal therapy. A systematic review. Ann Periodontol. 2003;8:115–181. doi: 10.1902/annals.2003.8.1.115. [DOI] [PubMed] [Google Scholar]
- 136.Haffajee AD, Socransky SS, Patel MR, Song X. Microbial complexes in supragingival plaque. Oral Microbiol Immunol. 2008;23:196–205. doi: 10.1111/j.1399-302X.2007.00411.x. [DOI] [PubMed] [Google Scholar]
- 137.Haffajee AD, Socransky SS, Smith C, Dibart S. Microbial risk indicators for periodontal attachment loss. J Periodontal Res. 1991;26:293–296. doi: 10.1111/j.1600-0765.1991.tb01662.x. [DOI] [PubMed] [Google Scholar]
- 138.Haffajee AD, Socransky SS, Smith C, Dibart S. Relation of baseline microbial parameters to future periodontal attachment loss. J Clin Periodontol. 1991;18:744–750. doi: 10.1111/j.1600-051x.1991.tb00066.x. [DOI] [PubMed] [Google Scholar]
- 139.Haffajee AD, Teles RP, Patel MR, Song X, Veiga N, Socransky SS. Factors affecting human supragingival biofilm composition. I Plaque mass. J Periodontal Res. 2009;44:511–519. doi: 10.1111/j.1600-0765.2008.01154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Haffajee AD, Teles RP, Patel MR, Song X, Yaskell T, Socransky SS. Factors affecting human supragingival biofilm composition. II. Tooth position. J Periodontal Res. 2009;44:520–528. doi: 10.1111/j.1600-0765.2008.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Haffajee AD, Teles RP, Socransky SS. Association of Eubacterium nodatum and Treponema denticola with human periodontitis lesions. Oral Microbiol Immunol. 2006;21:269–282. doi: 10.1111/j.1399-302X.2006.00287.x. [DOI] [PubMed] [Google Scholar]
- 142.Haffajee AD, Teles RP, Socransky SS. The effect of periodontal therapy on the composition of the subgingival microbiota. Periodontol 2000. 2006;42:219–258. doi: 10.1111/j.1600-0757.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- 143.Haffajee ADSSSFMX-FLA. Plaque microbiology in health and disease. In: Wilson HNNM, editor. Dental plaque revisited: Oral biofilms in health and disease. Vol. 600 Royal College of Physicians, London, UK: Bioline; 1999. [Google Scholar]
- 144.Hamano-Hasegawa K, Morozumi M, Nakayama E, Chiba N, Murayama SY, Takayanagi R, et al. Comprehensive detection of causative pathogens using real-time PCR to diagnose pediatric community-acquired pneumonia. J Infect Chemother. 2008;14:424–432. doi: 10.1007/s10156-008-0648-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Han X, Kawai T, Taubman MA. Interference with immune-cell-mediated bone resorption in periodontal disease. Periodontol 2000. 2007;45:76–94. doi: 10.1111/j.1600-0757.2007.00215.x. [DOI] [PubMed] [Google Scholar]
- 146.Hasturk H, Kantarci A, Ebrahimi N, Andry C, Holick M, Jones VL, et al. Topical H2 antagonist prevents periodontitis in a rabbit model. Infect Immun. 2006;74:2402–2414. doi: 10.1128/IAI.74.4.2402-2414.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. 2007;179:7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 148.Hausmann E, Weinfeld N, Miller WA. Effects of lipopolysaccharides on bone resorption in tissue culture. Calcif Tissue Res. 1972;9:272–282. doi: 10.1007/BF02061967. [DOI] [PubMed] [Google Scholar]
- 149.Heasman PA, Benn DK, Kelly PJ, Seymour RA, Aitken D. The use of topical flurbiprofen as an adjunct to non-surgical management of periodontal disease. J Clin Periodontol. 1993;20:457–464. doi: 10.1111/j.1600-051x.1993.tb00389.x. [DOI] [PubMed] [Google Scholar]
- 150.Heitz-Mayfield LJ. How effective is surgical therapy compared with nonsurgical debridement? Periodontol 2000. 2005;37:72–87. doi: 10.1111/j.1600-0757.2004.03797.x. [DOI] [PubMed] [Google Scholar]
- 151.Herrera D, Sanz M, Jepsen S, Needleman I, Roldan S. A systematic review on the effect of systemic antimicrobials as an adjunct to scaling and root planing in periodontitis patients. J Clin Periodontol. 2002;29(3):136–159. doi: 10.1034/j.1600-051x.29.s3.8.x. discussion 160-132. [DOI] [PubMed] [Google Scholar]
- 152.Hill AB. The Environment and Disease: Association or causation? Proc R Soc Med. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ho W, Eubank T, Leblebicioglu B, Marsh C, Walters J. Azithromycin decreases crevicular fluid volume and mediator content. J Dent Res. 2010;89:831–835. doi: 10.1177/0022034510368650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Howell TH. Blocking periodontal disease progression with anti-inflammatory agents. J Periodontol. 1993;64:828–833. doi: 10.1902/jop.1993.64.8s.828. [DOI] [PubMed] [Google Scholar]
- 155.Hudler P, Gubina M, Ihan Hren N, Seme K, Malovrh T, Gale N, et al. A mouse model of chronic bacterial lesions (a cotton trap) for studying oral bacteria-lymphocyte interactions. Pflugers Arch. 2000;440:R91–93. [PubMed] [Google Scholar]
- 156.Hujoel P. In: Grading evidence with a focus on etiology, surrogates, and clinical devices. Lesaffre E, Feine J, Leroux B, Declerck D, editors. Chichester: John Wiley & Sons Ltd; 2009. pp. 13–26. [Google Scholar]
- 157.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Huttenhower C, Hofmann O. A quick guide to large-scale genomic data mining. PLoS Comput Biol. 2010;6:e1000779. doi: 10.1371/journal.pcbi.1000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hutter G, Schlagenhauf U, Valenza G, Horn M, Burgemeister S, Claus H, et al. Molecular analysis of bacteria in periodontitis: evaluation of clone libraries, novel phylotypes and putative pathogens. Microbiology. 2003;149:67–75. doi: 10.1099/mic.0.25791-0. [DOI] [PubMed] [Google Scholar]
- 160.Imbronito AV, Grande SR, Freitas NM, Okuda O, Lotufo RF, Nunes FD. Detection of Epstein-Barr virus and human cytomegalovirus in blood and oral samples: comparison of three sampling methods. J Oral Sci. 2008;50:25–31. doi: 10.2334/josnusd.50.25. [DOI] [PubMed] [Google Scholar]
- 161.Ismail AI, Morrison EC, Burt BA, Caffesse RG, Kavanagh MT. Natural history of periodontal disease in adults: findings from the Tecumseh Periodontal Disease Study, 1959-87. J Dent Res. 1990;69:430–435. doi: 10.1177/00220345900690020201. [DOI] [PubMed] [Google Scholar]
- 162.Jandik KA, Belanger M, Low SL, Dorn BR, Yang MC, Progulske-Fox A. Invasive differences among Porphyromonas gingivalis strains from healthy and diseased periodontal sites. J Periodontal Res. 2008;43:524–530. doi: 10.1111/j.1600-0765.2007.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jeffcoat MK. Safety of oral bisphosphonates: controlled studies on alveolar bone. Int J Oral Maxillofac Implants. 2006;21:349–353. [PubMed] [Google Scholar]
- 164.Jeffcoat MK, Cizza G, Shih WJ, Genco R, Lombardi A. Efficacy of bisphosphonates for the control of alveolar bone loss in periodontitis. J Int Acad Periodontol. 2007;9:70–76. [PubMed] [Google Scholar]
- 165.Jeffcoat MK, Reddy MS, Haigh S, Buchanan W, Doyle MJ, Meredith MP, et al. A comparison of topical ketorolac, systemic flurbiprofen, and placebo for the inhibition of bone loss in adult periodontitis. J Periodontol. 1995;66:329–338. doi: 10.1902/jop.1995.66.5.329. [DOI] [PubMed] [Google Scholar]
- 166.Jeraldo P, Chia N, Goldenfeld N. On the suitability of short reads of 16S rRNA for phylogeny-based analyses in environmental surveys. Environ Microbiol. 2011;13:3000–3009. doi: 10.1111/j.1462-2920.2011.02577.x. [DOI] [PubMed] [Google Scholar]
- 167.Jin L, Soder B, Corbet EF. Interleukin-8 and granulocyte elastase in gingival crevicular fluid in relation to periodontopathogens in untreated adult periodontitis. J Periodontol. 2000;71:929–939. doi: 10.1902/jop.2000.71.6.929. [DOI] [PubMed] [Google Scholar]
- 168.Kaeberlein T, Lewis K, Epstein SS. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science. 2002;296:1127–1129. doi: 10.1126/science.1070633. [DOI] [PubMed] [Google Scholar]
- 169.Kamma JJ, Contreras A, Slots J. Herpes viruses and periodontopathic bacteria in early-onset periodontitis. J Clin Periodontol. 2001;28:879–885. doi: 10.1034/j.1600-051x.2001.028009879.x. [DOI] [PubMed] [Google Scholar]
- 170.Kantarci A, Hasturk H, Van Dyke TE. Host-mediated resolution of inflammation in periodontal diseases. Periodontol 2000. 2006;40:144–163. doi: 10.1111/j.1600-0757.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- 171.Kantarci A, Van Dyke TE. Lipoxin signaling in neutrophils and their role in periodontal disease. Prostaglandins Leukot Essent Fatty Acids. 2005;73:289–299. doi: 10.1016/j.plefa.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 172.Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, et al. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008;87:1016–1020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- 173.Keyes P. Are periodontal pathoses caused by bacterial infections on cervicoradicular surfaces of teeth? J Dent Res. 1970;2:223–228. [Google Scholar]
- 174.Keyes PH, Rams TE. A rationale for management of periodontal diseases: rapid identification of microbial ‘therapeutic targets’ with phase-contrast microscopy. J Am Dent Assoc. 1983;106:803–812. doi: 10.14219/jada.archive.1983.0436. [DOI] [PubMed] [Google Scholar]
- 175.Kidane D, Carrasco B, Manfredi C, Rothmaier K, Ayora S, Tadesse S, et al. Evidence for different pathways during horizontal gene transfer in competent Bacillus subtilis cells. PLoS Genet. 2009;5:e1000630. doi: 10.1371/journal.pgen.1000630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kigure T, Saito A, Seida K, Yamada S, Ishihara K, Okuda K. Distribution of Porphyromonas gingivalis and Treponema denticola in human subgingival plaque at different periodontal pocket depths examined by immunohistochemical methods. J Periodontal Res. 1995;30:332–341. doi: 10.1111/j.1600-0765.1995.tb01284.x. [DOI] [PubMed] [Google Scholar]
- 177.Koehler A, Karch H, Beikler T, Flemmig TF, Suerbaum S, Schmidt H. Multilocus sequence analysis of Porphyromonas gingivalis indicates frequent recombination. Microbiology. 2003;149:2407–2415. doi: 10.1099/mic.0.26267-0. [DOI] [PubMed] [Google Scholar]
- 178.Kolenbrander PE, Palmer RJ, Jr, Rickard AH, Jakubovics NS, Chalmers NI, Diaz PI. Bacterial interactions and successions during plaque development. Periodontol 2000. 2006;42:47–79. doi: 10.1111/j.1600-0757.2006.00187.x. [DOI] [PubMed] [Google Scholar]
- 179.Konstantinidis A, Sakellari D, Papa A, Antoniadis A. Real-time polymerase chain reaction quantification of Epstein-Barr virus in chronic periodontitis patients. J Periodontal Res. 2005;40:294–298. doi: 10.1111/j.1600-0765.2005.00796.x. [DOI] [PubMed] [Google Scholar]
- 180.Kopczynski ED, Bateson MM, Ward DM. Recognition of chimeric small-subunit ribosomal DNAs composed of genes from uncultivated microorganisms. Appl Environ Microbiol. 1994;60:746–748. doi: 10.1128/aem.60.2.746-748.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kopitar AN, Ihan Hren N, Ihan A. Commensal oral bacteria antigens prime human dendritic cells to induce Th1, Th2 or Treg differentiation. Oral Microbiol Immunol. 2006;21:1–5. doi: 10.1111/j.1399-302X.2005.00237.x. [DOI] [PubMed] [Google Scholar]
- 182.Kornman KS. Mapping the pathogenesis of periodontitis: a new look. J Periodontol. 2008;79:1560–1568. doi: 10.1902/jop.2008.080213. [DOI] [PubMed] [Google Scholar]
- 183.Krasse B. Oral aggregations of microbes. J Dent Res. 1963;2:521–528. doi: 10.1177/00220345630420016201. [DOI] [PubMed] [Google Scholar]
- 184.Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci U S A. 1999;96:14547–14552. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kubar A, Saygun I, Ozdemir A, Yapar M, Slots J. Real-time polymerase chain reaction quantification of human cytomegalovirus and Epstein-Barr virus in periodontal pockets and the adjacent gingiva of periodontitis lesions. J Periodontal Res. 2005;40:97–104. doi: 10.1111/j.1600-0765.2005.00770.x. [DOI] [PubMed] [Google Scholar]
- 186.Kuboniwa M, Inaba H, Amano A. Genotyping to distinguish microbial pathogenicity in periodontitis. Periodontol 2000. 2010;54:136–159. doi: 10.1111/j.1600-0757.2010.00352.x. [DOI] [PubMed] [Google Scholar]
- 187.Kumar PS, Brooker MR, Dowd SE, Camerlengo T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. Plos One. 2011;6:e20956. doi: 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ. New bacterial species associated with chronic periodontitis. J Dent Res. 2003;82:338–344. doi: 10.1177/154405910308200503. [DOI] [PubMed] [Google Scholar]
- 189.Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J Clin Microbiol. 2005;43:3944–3955. doi: 10.1128/JCM.43.8.3944-3955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol. 2006;44:3665–3673. doi: 10.1128/JCM.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kumar PS, Mason MR, Brooker MR, O'Brien K. Pyrosequencing reveals unique microbial signatures associated with healthy and failing dental implants. J Clin Periodontol. 2012;39:425–433. doi: 10.1111/j.1600-051X.2012.01856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 193.Laine ML, Appelmelk BJ, van Winkelhoff AJ. Prevalence and distribution of six capsular serotypes of Porphyromonas gingivalis in periodontitis patients. J Dent Res. 1997;76:1840–1844. doi: 10.1177/00220345970760120601. [DOI] [PubMed] [Google Scholar]
- 194.Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. doi: 10.1128/iai.63.10.3878-3885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lang NP, Cumming BR, Loe H. Toothbrushing frequency as it relates to plaque development and gingival health. J Periodontol. 1973;44:396–405. doi: 10.1902/jop.1973.44.7.396. [DOI] [PubMed] [Google Scholar]
- 197.Lang NP, Joss A, Orsanic T, Gusberti FA, Siegrist BE. Bleeding on probing. A predictor for the progression of periodontal disease? J Clin Periodontol. 1986;13:590–596. doi: 10.1111/j.1600-051x.1986.tb00852.x. [DOI] [PubMed] [Google Scholar]
- 198.Lawrence JG, Retchless AC. The interplay of homologous recombination and horizontal gene transfer in bacterial speciation. Methods Mol Biol. 2009;532:29–53. doi: 10.1007/978-1-60327-853-9_3. [DOI] [PubMed] [Google Scholar]
- 199.Lee A, Ghaname CB, Braun TM, Sugai JV, Teles RP, Loesche WJ, et al. Bacterial and salivary biomarkers predict the gingival inflammatory profile. J Periodontol. 2011 doi: 10.1902/jop.2011.110060. [DOI] [PubMed] [Google Scholar]
- 200.Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol. 2007;5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 201.Lewis K. Riddle of biofilm resistance. Antimicrob Agents Chemother. 2001;45:999–1007. doi: 10.1128/AAC.45.4.999-1007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Leys EJ, Lyons SR, Moeschberger ML, Rumpf RW, Griffen AL. Association of Bacteroides forsythus and a novel Bacteroides phylotype with periodontitis. J Clin Microbiol. 2002;40:821–825. doi: 10.1128/JCM.40.3.821-825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Li J, Helmerhorst EJ, Leone CW, Troxler RF, Yaskell T, Haffajee AD, et al. Identification of early microbial colonizers in human dental biofilm. J Appl Microbiol. 2004;97:1311–1318. doi: 10.1111/j.1365-2672.2004.02420.x. [DOI] [PubMed] [Google Scholar]
- 204.Lin YL, Chang PC, Wang Y, Li M. Identification of novel viral interleukin-10 isoforms of human cytomegalovirus AD169. Virus Res. 2008;131:213–223. doi: 10.1016/j.virusres.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lin YL, Li M. Human cytomegalovirus and Epstein-Barr virus inhibit oral bacteria-induced macrophage activation and phagocytosis. Oral Microbiol Immunol. 2009;24:243–248. doi: 10.1111/j.1399-302X.2009.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lindhe J, Hamp SE, Loe H. Experimental periodontitis in the beagle dog. Int Dent J. 1973;23:432–437. [PubMed] [Google Scholar]
- 207.Listgarten MA. The structure of dental plaque. Periodontol 2000. 1994;5:52–65. doi: 10.1111/j.1600-0757.1994.tb00018.x. [DOI] [PubMed] [Google Scholar]
- 208.Listgarten MA. Structure of the microbial flora associated with periodontal health and disease in man. A light and electron microscopic study. J Periodontol. 1976;47:1–18. doi: 10.1902/jop.1976.47.1.1. [DOI] [PubMed] [Google Scholar]
- 209.Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, Sommer DD, et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. Plos One. 2012;7:e37919. doi: 10.1371/journal.pone.0037919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Loe H. Human research models for the production and prevention of marginal periodontal disease and caries. Annu Meet Am Inst Oral Biol. 1973;30:48–58. [PubMed] [Google Scholar]
- 211.Loe H, Anerud A, Boysen H, Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. J Clin Periodontol. 1986;13:431–445. doi: 10.1111/j.1600-051x.1986.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 212.Loe H, Anerud A, Boysen H, Smith M. The natural history of periodontal disease in man. The rate of periodontal destruction before 40 years of age. J Periodontol. 1978;49:607–620. doi: 10.1902/jop.1978.49.12.607. [DOI] [PubMed] [Google Scholar]
- 213.Loe H, Theilade E, Jensen SB. Experimental gingivitis in man. J Periodontol. 1965;36:177–187. doi: 10.1902/jop.1965.36.3.177. [DOI] [PubMed] [Google Scholar]
- 214.Loenen WA, Bruggeman CA, Wiertz EJ. Immune evasion by human cytomegalovirus: lessons in immunology and cell biology. Semin Immunol. 2001;13:41–49. doi: 10.1006/smim.2001.0294. [DOI] [PubMed] [Google Scholar]
- 215.Loesche WJ, Bretz WA, Kerschensteiner D, Stoll J, Socransky SS, Hujoel P, et al. Development of a diagnostic test for anaerobic periodontal infections based on plaque hydrolysis of benzoyl-DL-arginine-naphthylamide. J Clin Microbiol. 1990;28:1551–1559. doi: 10.1128/jcm.28.7.1551-1559.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Loos BG, Dyer DW. Restriction fragment length polymorphism analysis of the fimbrillin locus, fimA, of Porphyromonas gingivalis. J Dent Res. 1992;71:1173–1181. doi: 10.1177/00220345920710050901. [DOI] [PubMed] [Google Scholar]
- 217.Loos BG, Dyer DW, Whittam TS, Selander RK. Genetic structure of populations of Porphyromonas gingivalis associated with periodontitis and other oral infections. Infect Immun. 1993;61:204–212. doi: 10.1128/iai.61.1.204-212.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Loos BG, Mayrand D, Genco RJ, Dickinson DP. Genetic heterogeneity of Porphyromonas (Bacteroides) gingivalis by genomic DNA fingerprinting. J Dent Res. 1990;69:1488–1493. doi: 10.1177/00220345900690080801. [DOI] [PubMed] [Google Scholar]
- 219.Loos BG, van Winkelhoff AJ, Dunford RG, Genco RJ, de Graaff J, Dickinson DP, et al. A statistical approach to the ecology of Porphyromonas gingivalis. J Dent Res. 1992;71:353–358. doi: 10.1177/00220345920710020201. [DOI] [PubMed] [Google Scholar]
- 220.Lopez NJ, Gamonal JA, Martinez B. Repeated metronidazole and amoxicillin treatment of periodontitis. A follow-up study. J Periodontol. 2000;71:79–89. doi: 10.1902/jop.2000.71.1.79. [DOI] [PubMed] [Google Scholar]
- 221.Lopez NJ, Socransky SS, Da Silva I, Japlit MR, Haffajee AD. Effects of metronidazole plus amoxicillin as the only therapy on the microbiological and clinical parameters of untreated chronic periodontitis. J Clin Periodontol. 2006;33:648–660. doi: 10.1111/j.1600-051X.2006.00957.x. [DOI] [PubMed] [Google Scholar]
- 222.Lopez NJ, Socransky SS, Da Silva I, Japlit MR, Haffajee AD. Subgingival microbiota of chilean patients with chronic periodontitis. J Periodontol. 2004;75:717–725. doi: 10.1902/jop.2004.75.5.717. [DOI] [PubMed] [Google Scholar]
- 223.Lovdal A, Arno A, Schei O, Waerhaug J. Combined effect of subgingival scaling and controlled oral hygiene on the incidence of gingivitis. Acta Odontol Scand. 1961;19:537–555. doi: 10.3109/00016356109043406. [DOI] [PubMed] [Google Scholar]
- 224.Lovdal A, Arno A, Waerhaug J. Incidence of clinical manifestations of periodontal disease in light of oral hygiene and calculus formation. J Am Dent Assoc. 1958;56:21–33. doi: 10.14219/jada.archive.1958.0021. [DOI] [PubMed] [Google Scholar]
- 225.Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Distribution of selected bacterial species on intraoral surfaces. J Clin Periodontol. 2003;30:644–654. doi: 10.1034/j.1600-051x.2003.00376.x. [DOI] [PubMed] [Google Scholar]
- 226.Magnusson I, Lindhe J, Yoneyama T, Liljenberg B. Recolonization of a subgingival microbiota following scaling in deep pockets. J Clin Periodontol. 1984;11:193–207. doi: 10.1111/j.1600-051x.1984.tb01323.x. [DOI] [PubMed] [Google Scholar]
- 227.Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ, et al. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol. 1998;64:795–799. doi: 10.1128/aem.64.2.795-799.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Mark KE, Wald A, Magaret AS, Selke S, Olin L, Huang ML, et al. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. doi: 10.1086/591913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279–294. doi: 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- 230.Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263–271. doi: 10.1177/08959374940080022001. [DOI] [PubMed] [Google Scholar]
- 231.Marsh PD, Devine DA. How is the development of dental biofilms influenced by the host? J Clin Periodontol. 2011;38(11):28–35. doi: 10.1111/j.1600-051X.2010.01673.x. [DOI] [PubMed] [Google Scholar]
- 232.Matarazzo F, Figueiredo LC, Cruz SE, Faveri M, Feres M. Clinical and microbiological benefits of systemic metronidazole and amoxicillin in the treatment of smokers with chronic periodontitis: a randomized placebo-controlled study. J Clin Periodontol. 2008;35:885–896. doi: 10.1111/j.1600-051X.2008.01304.x. [DOI] [PubMed] [Google Scholar]
- 233.Matto J, Saarela M, von Troil-Linden B, Kononen E, Jousimies-Somer H, Torkko H, et al. Distribution and genetic analysis of oral Prevotella intermedia and Prevotella nigrescens. Oral Microbiol Immunol. 1996;11:96–102. doi: 10.1111/j.1399-302x.1996.tb00342.x. [DOI] [PubMed] [Google Scholar]
- 234.Mawardi H, Giro G, Kajiya M, Ohta K, Almazrooa S, Alshwaimi E, et al. A role of oral bacteria in bisphosphonate-induced osteonecrosis of the jaw. J Dent Res. 90:1339–1345. doi: 10.1177/0022034511420430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mayanagi G, Sato T, Shimauchi H, Takahashi N. Detection frequency of periodontitis-associated bacteria by polymerase chain reaction in subgingival and supragingival plaque of periodontitis and healthy subjects. Oral Microbiol Immunol. 2004;19:379–385. doi: 10.1111/j.1399-302x.2004.00172.x. [DOI] [PubMed] [Google Scholar]
- 236.Mayer Y, Balbir-Gurman A, Machtei EE. Anti-tumor necrosis factor-alpha therapy and periodontal parameters in patients with rheumatoid arthritis. J Periodontol. 2009;80:1414–1420. doi: 10.1902/jop.2009.090015. [DOI] [PubMed] [Google Scholar]
- 237.McCracken GI, Preshaw PM, Steen IN, Swan M, deJager M, Heasman PA. Measuring plaque in clinical trials: index or weight? J Clin Periodontol. 2006;33:172–176. doi: 10.1111/j.1600-051X.2006.00877.x. [DOI] [PubMed] [Google Scholar]
- 238.McKillip JL, Jaykus LA, Drake M. rRNA stability in heat-killed and UV-irradiated enterotoxigenic Staphylococcus aureus and Escherichia coli O157:H7. Appl Environ Microbiol. 1998;64:4264–4268. doi: 10.1128/aem.64.11.4264-4268.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Menard C, Mouton C. Clonal diversity of the taxon Porphyromonas gingivalis assessed by random amplified polymorphic DNA fingerprinting. Infect Immun. 1995;63:2522–2531. doi: 10.1128/iai.63.7.2522-2531.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Mestnik MJ, Feres M, Figueiredo LC, Duarte PM, Lira EA, Faveri M. Short-term benefits of the adjunctive use of metronidazole plus amoxicillin in the microbial profile and in the clinical parameters of subjects with generalized aggressive periodontitis. J Clin Periodontol. 2010;37:353–365. doi: 10.1111/j.1600-051X.2010.01538.x. [DOI] [PubMed] [Google Scholar]
- 241.Migliorati CA, Siegel MA, Elting LS. Bisphosphonate-associated osteonecrosis: a long-term complication of bisphosphonate treatment. Lancet Oncol. 2006;7:508–514. doi: 10.1016/S1470-2045(06)70726-4. [DOI] [PubMed] [Google Scholar]
- 242.Miller WD. The microorganisms of the human mouth. The local and general diseases which are caused by them. Philadelphia (PA): SS White. 1890:274–441. [Google Scholar]
- 243.Mogensen TH, Paludan SR. Molecular pathways in virus-induced cytokine production. Microbiol Mol Biol Rev. 2001;65:131–150. doi: 10.1128/MMBR.65.1.131-150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Mombelli A. Microbiological monitoring. J Clin Periodontol. 1996;23:251–257. doi: 10.1111/j.1600-051x.1996.tb02084.x. [DOI] [PubMed] [Google Scholar]
- 245.Mombelli A, Cionca N, Almaghlouth A. Does adjunctive antimicrobial therapy reduce the perceived need for periodontal surgery? Periodontol 2000. 2011;55:205–216. doi: 10.1111/j.1600-0757.2010.00356.x. [DOI] [PubMed] [Google Scholar]
- 246.Moore WE, Moore LV. The bacteria of periodontal diseases. Periodontol 2000. 1994;5:66–77. doi: 10.1111/j.1600-0757.1994.tb00019.x. [DOI] [PubMed] [Google Scholar]
- 247.Mousques T, Listgarten MA, Phillips RW. Effect of scaling and root planing on the composition of the human subgingival microbial flora. J Periodontal Res. 1980;15:144–151. doi: 10.1111/j.1600-0765.1980.tb00268.x. [DOI] [PubMed] [Google Scholar]
- 248.Munson MA, Banerjee A, Watson TF, Wade WG. Molecular analysis of the microflora associated with dental caries. J Clin Microbiol. 2004;42:3023–3029. doi: 10.1128/JCM.42.7.3023-3029.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Munson MA, Pitt-Ford T, Chong B, Weightman A, Wade WG. Molecular and cultural analysis of the microflora associated with endodontic infections. J Dent Res. 2002;81:761–766. doi: 10.1177/0810761. [DOI] [PubMed] [Google Scholar]
- 250.Nachtwey J, Spencer JV. HCMV IL-10 suppresses cytokine expression in monocytes through inhibition of nuclear factor-kappaB. Viral Immunol. 2008;21:477–482. doi: 10.1089/vim.2008.0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Nakagawa I, Amano A, Ohara-Nemoto Y, Endoh N, Morisaki I, Kimura S, et al. Identification of a new variant of fimA gene of Porphyromonas gingivalis and its distribution in adults and disabled populations with periodontitis. J Periodontal Res. 2002;37:425–432. doi: 10.1034/j.1600-0765.2002.01637.x. [DOI] [PubMed] [Google Scholar]
- 252.Nalbant A, Zadeh HH. Actinobacillus actinomycetemcomitans induces apoptosis of T lymphocytes by the Fas and Fas ligand pathway. Oral Microbiol Immunol. 2002;17:277–284. doi: 10.1034/j.1399-302x.2002.170503.x. [DOI] [PubMed] [Google Scholar]
- 253.Newman MG, Socransky SS, Savitt ED, Propas DA, Crawford A. Studies of the microbiology of periodontosis. J Periodontol. 1976;47:373–379. doi: 10.1902/jop.1976.47.7.373. [DOI] [PubMed] [Google Scholar]
- 254.Nichols D, Lewis K, Orjala J, Mo S, Ortenberg R, O'Connor P, et al. Short peptide induces an “uncultivable” microorganism to grow in vitro. Appl Environ Microbiol. 2008;74:4889–4897. doi: 10.1128/AEM.00393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Nocker A, Camper AK. Selective removal of DNA from dead cells of mixed bacterial communities by use of ethidium monoazide. Appl Environ Microbiol. 2006;72:1997–2004. doi: 10.1128/AEM.72.3.1997-2004.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Noiri Y, Ebisu S. Identification of periodontal disease-associated bacteria in the “plaque-free zone”. J Periodontol. 2000;71:1319–1326. doi: 10.1902/jop.2000.71.8.1319. [DOI] [PubMed] [Google Scholar]
- 257.Noiri Y, Li L, Ebisu S. The localization of periodontal-disease-associated bacteria in human periodontal pockets. J Dent Res. 2001;80:1930–1934. doi: 10.1177/00220345010800101301. [DOI] [PubMed] [Google Scholar]
- 258.Noiri Y, Li L, Yoshimura F, Ebisu S. Localization of Porphyromonas gingivalis-carrying fimbriae in situ in human periodontal pockets. J Dent Res. 2004;83:941–945. doi: 10.1177/154405910408301210. [DOI] [PubMed] [Google Scholar]
- 259.Noiri Y, Ozaki K, Nakae H, Matsuo T, Ebisu S. An immunohistochemical study on the localization of Porphyromonas gingivalis, Campylobacter rectus and Actinomyces viscosus in human periodontal pockets. J Periodontal Res. 1997;32:598–607. doi: 10.1111/j.1600-0765.1997.tb00937.x. [DOI] [PubMed] [Google Scholar]
- 260.Nonnenmacher C, Dalpke A, Rochon J, Flores-de-Jacoby L, Mutters R, Heeg K. Real-time polymerase chain reaction for detection and quantification of bacteria in periodontal patients. J Periodontol. 2005;76:1542–1549. doi: 10.1902/jop.2005.76.9.1542. [DOI] [PubMed] [Google Scholar]
- 261.Offenbacher S, Barros SP, Beck JD. Rethinking periodontal inflammation. J Periodontol. 2008;79:1577–1584. doi: 10.1902/jop.2008.080220. [DOI] [PubMed] [Google Scholar]
- 262.Offenbacher S, Barros SP, Paquette DW, Winston JL, Biesbrock AR, Thomason RG, et al. Gingival transcriptome patterns during induction and resolution of experimental gingivitis in humans. J Periodontol. 2009;80:1963–1982. doi: 10.1902/jop.2009.080645. [DOI] [PubMed] [Google Scholar]
- 263.Olson JC, Cuff CF, Lukomski S, Lukomska E, Canizales Y, Wu B, et al. Use of 16S ribosomal RNA gene analyses to characterize the bacterial signature associated with poor oral health in West Virginia. BMC Oral Health. 2011;11:7. doi: 10.1186/1472-6831-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Ouverney CC, Armitage GC, Relman DA. Single-cell enumeration of an uncultivated TM7 subgroup in the human subgingival crevice. Appl Environ Microbiol. 2003;69:6294–6298. doi: 10.1128/AEM.69.10.6294-6298.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Ozmeric N, Preus NR, Olsen I. Genetic diversity of Porphyromonas gingivalis and its possible importance to pathogenicity. Acta Odontol Scand. 2000;58:183–187. doi: 10.1080/000163500429190. [DOI] [PubMed] [Google Scholar]
- 266.Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000. 1997;14:9–11. doi: 10.1111/j.1600-0757.1997.tb00189.x. [DOI] [PubMed] [Google Scholar]
- 267.Page RC, Offenbacher S, Schroeder HE, Seymour GJ, Kornman KS. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontol 2000. 1997;14:216–248. doi: 10.1111/j.1600-0757.1997.tb00199.x. [DOI] [PubMed] [Google Scholar]
- 268.Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976;34:235–249. [PubMed] [Google Scholar]
- 269.Page RC, Sturdivant EC. Noninflammatory destructive periodontal disease (NDPD) Periodontol 2000. 2002;30:24–39. doi: 10.1034/j.1600-0757.2002.03003.x. [DOI] [PubMed] [Google Scholar]
- 270.Papaioannou W, Gizani S, Haffajee AD, Quirynen M, Mamai-Homata E, Papagiannoulis L. The microbiota on different oral surfaces in healthy children. Oral Microbiol Immunol. 2009;24:183–189. doi: 10.1111/j.1399-302X.2008.00493.x. [DOI] [PubMed] [Google Scholar]
- 271.Papapanou PN, Baelum V, Luan WM, Madianos PN, Chen X, Fejerskov O, et al. Subgingival microbiota in adult Chinese: prevalence and relation to periodontal disease progression. J Periodontol. 1997;68:651–666. doi: 10.1902/jop.1997.68.7.651. [DOI] [PubMed] [Google Scholar]
- 272.Papapanou PN, Behle JH, Kebschull M, Celenti R, Wolf DL, Handfield M, et al. Subgingival bacterial colonization profiles correlate with gingival tissue gene expression. BMC Microbiol. 2009;9:221. doi: 10.1186/1471-2180-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Papapanou PN, Teanpaisan R, Obiechina NS, Pithpornchaiyakul W, Pongpaisal S, Pisuithanakan S, et al. Periodontal microbiota and clinical periodontal status in a rural sample in southern Thailand. Eur J Oral Sci. 2002;110:345–352. doi: 10.1034/j.1600-0722.2002.21361.x. [DOI] [PubMed] [Google Scholar]
- 274.Parfrey LW, Knight R. Spatial and temporal variability of the human microbiota. Clin Microbiol Infect. 2012;18(4):8–11. doi: 10.1111/j.1469-0691.2012.03861.x. [DOI] [PubMed] [Google Scholar]
- 275.Pashine A, John B, Rath S, George A, Bal V. Th1 dominance in the immune response to live Salmonella typhimurium requires bacterial invasiveness but not persistence. Int Immunol. 1999;11:481–489. doi: 10.1093/intimm/11.4.481. [DOI] [PubMed] [Google Scholar]
- 276.Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Paster BJ, Dewhirst FE. Molecular microbial diagnosis. Periodontol 2000. 2009;51:38–44. doi: 10.1111/j.1600-0757.2009.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Paster BJ, Olsen I, Aas JA, Dewhirst FE. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006;42:80–87. doi: 10.1111/j.1600-0757.2006.00174.x. [DOI] [PubMed] [Google Scholar]
- 279.Paterson GK, Maskell DJ. In: Genetic determinants of bacterial pathogenesis. Locht C, Simonet M, editors. Norfolk: Caister Academic Press; 2012. pp. 51–68. [Google Scholar]
- 280.Petit MD, van Steenbergen TJ, Scholte LM, van der Velden U, de Graaff J. Epidemiology and transmission of Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans among children and their family members. A report of 4 surveys. J Clin Periodontol. 1993;20:641–650. doi: 10.1111/j.1600-051x.1993.tb00709.x. [DOI] [PubMed] [Google Scholar]
- 281.Polz MF, Cavanaugh CM. Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol. 1998;64:3724–3730. doi: 10.1128/aem.64.10.3724-3730.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol 2000. 2000;24:153–192. doi: 10.1034/j.1600-0757.2000.2240108.x. [DOI] [PubMed] [Google Scholar]
- 283.Quirynen M, Bollen CML. The influence of surface roughness and surface free energy on supragingival and subgingival plaque formation in man - a review of the literature. J Clin Periodontol. 1995;22:1–14. doi: 10.1111/j.1600-051x.1995.tb01765.x. [DOI] [PubMed] [Google Scholar]
- 284.Quirynen M, Marechal M, Busscher HJ, Weerkamp AH, Darius PL, van Steenberghe D. The influence of surface free energy and surface roughness on early plaque formation. An in vivo study in man. J Clin Periodontol. 1990;17:138–144. doi: 10.1111/j.1600-051x.1990.tb01077.x. [DOI] [PubMed] [Google Scholar]
- 285.Rams TE, Keyes PH. A rationale for the management of periodontal diseases: effects of tetracycline on subgingival bacteria. J Am Dent Assoc. 1983;107:37–41. doi: 10.14219/jada.archive.1983.0177. [DOI] [PubMed] [Google Scholar]
- 286.Razumov AS. Direct method for counting bacteria in water. A comparison with the method after Koch. Mikrobiologiya (Moscow) 1932;1:131. [Google Scholar]
- 287.Reddy MS, Weatherford TW, 3rd, Smith CA, West BD, Jeffcoat MK, Jacks TM. Alendronate treatment of naturally-occurring periodontitis in beagle dogs. J Periodontol. 1995;66:211–217. doi: 10.1902/jop.1995.66.3.211. [DOI] [PubMed] [Google Scholar]
- 288.Relman DA. The human microbiome: ecosystem resilience and health. Nutr Rev. 2012;70(1):S2–9. doi: 10.1111/j.1753-4887.2012.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Relman DA. New technologies, human-microbe interactions, and the search for previously unrecognized pathogens. J Infect Dis. 2002;186(2):S254–258. doi: 10.1086/344935. [DOI] [PubMed] [Google Scholar]
- 290.Reysenbach AL, Giver LJ, Wickham GS, Pace NR. Differential amplification of rRNA genes by polymerase chain reaction. Appl Environ Microbiol. 1992;58:3417–3418. doi: 10.1128/aem.58.10.3417-3418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Riley MA, Lizotte-Waniewski M. Population genomics and the bacterial species concept. Methods Mol Biol. 2009;532:367–377. doi: 10.1007/978-1-60327-853-9_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ritz HL. Microbial population shifts in developing human dental plaque. Arch Oral Biol. 1967;12:1561–1568. doi: 10.1016/0003-9969(67)90190-2. [DOI] [PubMed] [Google Scholar]
- 293.Riviere GR, Elliot KS, Adams DF, Simonson LG, Forgas LB, Nilius AM, et al. Relative proportions of pathogen-related oral spirochetes (PROS) and Treponema denticola in supragingival and subgingival plaque from patients with periodontitis. J Periodontol. 1992;63:131–136. doi: 10.1902/jop.1992.63.2.131. [DOI] [PubMed] [Google Scholar]
- 294.Riviere GR, Wagoner MA, Baker-Zander SA, Weisz KS, Adams DF, Simonson L, et al. Identification of spirochetes related to Treponema pallidum in necrotizing ulcerative gingivitis and chronic periodontitis. N Engl J Med. 1991;325:539–543. doi: 10.1056/NEJM199108223250803. [DOI] [PubMed] [Google Scholar]
- 295.Riviere GR, Weisz KS, Simonson LG, Lukehart SA. Pathogen-related spirochetes identified within gingival tissue from patients with acute necrotizing ulcerative gingivitis. Infect Immun. 1991;59:2653–2657. doi: 10.1128/iai.59.8.2653-2657.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Roberts FA, Darveau RP. Beneficial bacteria of the periodontium. Periodontol 2000. 2002;30:40–50. doi: 10.1034/j.1600-0757.2002.03004.x. [DOI] [PubMed] [Google Scholar]
- 297.Rothman K, Greenland S. In: Causation and Causal Inference. Rothman K, Greenland S, editors. Vol. 1998. Philadelphia: Lippincott Williams & Wilkins; pp. 7–28. [Google Scholar]
- 298.Rotola A, Cassai E, Farina R, Caselli E, Gentili V, Lazzarotto T, et al. Human herpesvirus 7, Epstein-Barr virus and human cytomegalovirus in periodontal tissues of periodontally diseased and healthy subjects. J Clin Periodontol. 2008;35:831–837. doi: 10.1111/j.1600-051X.2008.01301.x. [DOI] [PubMed] [Google Scholar]
- 299.Sachdeo A, Haffajee AD, Socransky SS. Biofilms in the edentulous oral cavity. J Prosthodont. 2008;17:348–356. doi: 10.1111/j.1532-849X.2008.00301.x. [DOI] [PubMed] [Google Scholar]
- 300.Saglie FR, Carranza FA, Jr, Newman MG. The presence of bacteria within the oral epithelium in periodontal disease. I. A scanning and transmission electron microscopic study. J Periodontol. 1985;56:618–624. doi: 10.1902/jop.1985.56.10.618. [DOI] [PubMed] [Google Scholar]
- 301.Saglie R, Carranza FA, Jr, Newman MG, Pattison GA. Scanning electron microscopy of the gingival wall of deep periodontal pockets in humans. J Periodontal Res. 1982;17:284–293. doi: 10.1111/j.1600-0765.1982.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 302.Sakamoto M, Takeuchi Y, Umeda M, Ishikawa I, Benno Y. Rapid detection and quantification of five periodontopathic bacteria by real-time PCR. Microbiol Immunol. 2001;45:39–44. doi: 10.1111/j.1348-0421.2001.tb01272.x. [DOI] [PubMed] [Google Scholar]
- 303.Sakamoto M, Umeda M, Ishikawa I, Benno Y. Comparison of the oral bacterial flora in saliva from a healthy subject and two periodontitis patients by sequence analysis of 16S rDNA libraries. Microbiol Immunol. 2000;44:643–652. doi: 10.1111/j.1348-0421.2000.tb02545.x. [DOI] [PubMed] [Google Scholar]
- 304.Salari MH, Kadkhoda Z. Rate of cultivable subgingival periodontopathogenic bacteria in chronic periodontitis. J Oral Sci. 2004;46:157–161. doi: 10.2334/josnusd.46.157. [DOI] [PubMed] [Google Scholar]
- 305.Salvi GE, Franco LM, Braun TM, Lee A, Rutger Persson G, Lang NP, et al. Pro-inflammatory biomarkers during experimental gingivitis in patients with type 1 diabetes mellitus: a proof-of-concept study. J Clin Periodontol. 2010;37:9–16. doi: 10.1111/j.1600-051X.2009.01500.x. [DOI] [PubMed] [Google Scholar]
- 306.Salvi GE, Lang NP. The effects of non-steroidal anti-inflammatory drugs (selective and non-selective) on the treatment of periodontal diseases. Curr Pharm Des. 2005;11:1757–1769. doi: 10.2174/1381612053764878. [DOI] [PubMed] [Google Scholar]
- 307.Santos AL, Siqueira JF, Jr, Rocas IN, Jesus EC, Rosado AS, Tiedje JM. Comparing the bacterial diversity of acute and chronic dental root canal infections. Plos One. 2011;6:e28088. doi: 10.1371/journal.pone.0028088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Sanz M, van Winkelhoff AJ, Herrera D, Dellemijn-Kippuw N, Simon R, Winkel E. Differences in the composition of the subgingival microbiota of two periodontitis populations of different geographical origin. A comparison between Spain and The Netherlands Eur. J Oral Sci. 2000;108:383–392. doi: 10.1034/j.1600-0722.2000.108005383.x. [DOI] [PubMed] [Google Scholar]
- 309.Saygun I, Kubar A, Ozdemir A, Slots J. Periodontitis lesions are a source of salivary cytomegalovirus and Epstein-Barr virus. J Periodontal Res. 2005;40:187–191. doi: 10.1111/j.1600-0765.2005.00790.x. [DOI] [PubMed] [Google Scholar]
- 310.Saygun I, Kubar A, Ozdemir A, Yapar M, Slots J. Herpesviral-bacterial interrelationships in aggressive periodontitis. J Periodontal Res. 2004;39:207–212. doi: 10.1111/j.1600-0765.2004.00728.x. [DOI] [PubMed] [Google Scholar]
- 311.Saygun I, Kubar A, Sahin S, Sener K, Slots J. Quantitative analysis of association between herpesviruses and bacterial pathogens in periodontitis. J Periodontal Res. 2008;43:352–359. doi: 10.1111/j.1600-0765.2007.01043.x. [DOI] [PubMed] [Google Scholar]
- 312.Sbordone L, Ramaglia L, Gulletta E, Iacono V. Recolonization of the subgingival microflora after scaling and root planing in human periodontitis. J Periodontol. 1990;61:579–584. doi: 10.1902/jop.1990.61.9.579. [DOI] [PubMed] [Google Scholar]
- 313.Schatzle M, Loe H, Burgin W, Anerud A, Boysen H, Lang NP. Clinical course of chronic periodontitis. I. Role of gingivitis. J Clin Periodontol. 2003;30:887–901. doi: 10.1034/j.1600-051x.2003.00414.x. [DOI] [PubMed] [Google Scholar]
- 314.Schatzle M, Loe H, Lang NP, Burgin W, Anerud A, Boysen H. The clinical course of chronic periodontitis. J Clin Periodontol. 2004;31:1122–1127. doi: 10.1111/j.1600-051X.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 315.Schei OW J, Lovdal A, Arno A. Alveolar bone loss as related to oral hygiene and age. J Periodontol. 1959;30:7. [Google Scholar]
- 316.Scheu PM, Berghof K, Stahl U. Detection of pathogenic and spoilage microorganisms in food with the polymerase chain reaction. Food Microbiol. 1998;15:13–31. [Google Scholar]
- 317.Schillinger C, Petrich A, Lux R, Riep B, Kikhney J, Friedmann A, et al. Co-localized or randomly distributed? Pair cross correlation of in vivo grown subgingival biofilm bacteria quantified by digital image analysis. Plos One. 2012;7:e37583. doi: 10.1371/journal.pone.0037583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Shaddox LM, Huang H, Lin T, Hou W, Harrison PL, Aukhil I, et al. Microbiological characterization in children with aggressive periodontitis. J Dent Res. 2012 doi: 10.1177/0022034512456039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. Persisters: a distinct physiological state of E. coli. BMC Microbiol. 2006;6:53. doi: 10.1186/1471-2180-6-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Siegrist BE, Brecx MC, Gusberti FA, Joss A, Lang NP. In vivo early human dental plaque formation on different supporting substances. A scanning electron microscopic and bacteriological study. Clin Oral Implants Res. 1991;2:38–46. doi: 10.1034/j.1600-0501.1991.020105.x. [DOI] [PubMed] [Google Scholar]
- 321.Silva MP, Feres M, Sirotto TA, Soares GM, Mendes JA, Faveri M, et al. Clinical and microbiological benefits of metronidazole alone or with amoxicillin as adjuncts in the treatment of chronic periodontitis: a randomized placebo-controlled clinical trial. J Clin Periodontol. 2011;38:828–837. doi: 10.1111/j.1600-051X.2011.01763.x. [DOI] [PubMed] [Google Scholar]
- 322.Silva N, Dutzan N, Hernandez M, Dezerega A, Rivera O, Aguillon JC, et al. Characterization of progressive periodontal lesions in chronic periodontitis patients: levels of chemokines, cytokines, matrix metalloproteinase-13, periodontal pathogens and inflammatory cells. J Clin Periodontol. 2008;35:206–214. doi: 10.1111/j.1600-051X.2007.01190.x. [DOI] [PubMed] [Google Scholar]
- 323.Sizova MV, Hohmann T, Hazen A, Paster BJ, Halem SR, Murphy CM, et al. New approaches for isolation of previously uncultivated oral bacteria. Appl Environ Microbiol. 2012;78:194–203. doi: 10.1128/AEM.06813-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Slots J. Herpesviral-bacterial interactions in periodontal diseases. Periodontol 2000. 2010;52:117–140. doi: 10.1111/j.1600-0757.2009.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Slots J. Herpesviral-bacterial synergy in the pathogenesis of human periodontitis. Curr Opin Infect Dis. 2007;20:278–283. doi: 10.1097/QCO.0b013e3280964da0. [DOI] [PubMed] [Google Scholar]
- 326.Slots J. Herpesviruses, the missing link between gingivitis and periodontitis? J Int Acad Periodontol. 2004;6:113–119. [PubMed] [Google Scholar]
- 327.Slots J. Human viruses in periodontitis. Periodontol 2000. 2010;53:89–110. doi: 10.1111/j.1600-0757.2009.00325.x. [DOI] [PubMed] [Google Scholar]
- 328.Slots J, Contreras A. Herpesviruses: a unifying causative factor in periodontitis? Oral Microbiol Immunol. 2000;15:277–280. doi: 10.1034/j.1399-302x.2000.150501.x. [DOI] [PubMed] [Google Scholar]
- 329.Slots J, Emrich LJ, Genco RJ, Rosling BG. Relationship between some subgingival bacteria and periodontal pocket depth and gain or loss of periodontal attachment after treatment of adult periodontitis. J Clin Periodontol. 1985;12:540–552. doi: 10.1111/j.1600-051x.1985.tb01388.x. [DOI] [PubMed] [Google Scholar]
- 330.Slots J, Saygun I, Sabeti M, Kubar A. Epstein-Barr virus in oral diseases. J Periodontal Res. 2006;41:235–244. doi: 10.1111/j.1600-0765.2006.00865.x. [DOI] [PubMed] [Google Scholar]
- 331.Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 332.Socransky SS. Relationship of bacteria to the etiology of periodontal disease. J Dent Res. 1970;49:203–222. doi: 10.1177/00220345700490020401. [DOI] [PubMed] [Google Scholar]
- 333.Socransky SS, Gibbons RJ, Dale AC, Bortnick L, Rosenthal E, Macdonald JB. The microbiota of the gingival crevice area of man. I. Total microscopic and viable counts and counts of specific organisms. Arch Oral Biol. 1963;8:275–280. doi: 10.1016/0003-9969(63)90019-0. [DOI] [PubMed] [Google Scholar]
- 334.Socransky SS, Haffajee AD. The bacterial etiology of destructive periodontal disease: current concepts. J Periodontol. 1992;63:322–331. doi: 10.1902/jop.1992.63.4s.322. [DOI] [PubMed] [Google Scholar]
- 335.Socransky SS, Haffajee AD. Evidence of bacterial etiology: a historical perspective. Periodontol 2000. 1994;5:7–25. doi: 10.1111/j.1600-0757.1994.tb00016.x. [DOI] [PubMed] [Google Scholar]
- 336.Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000. 2005;38:135–187. doi: 10.1111/j.1600-0757.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- 337.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 338.Socransky SS, Haffajee AD, Smith C, Dibart S. Relation of counts of microbial species to clinical status at the sampled site. J Clin Periodontol. 1991;18:766–775. doi: 10.1111/j.1600-051x.1991.tb00070.x. [DOI] [PubMed] [Google Scholar]
- 339.Socransky SS, Haffajee AD, Smith C, Martin L, Haffajee JA, Uzel NG, et al. Use of checkerboard DNA-DNA hybridization to study complex microbial ecosystems. Oral Microbiol Immunol. 2004;19:352–362. doi: 10.1111/j.1399-302x.2004.00168.x. [DOI] [PubMed] [Google Scholar]
- 340.Socransky SS, Haffajee AD, Smith GL, Dzink JL. Difficulties encountered in the search for the etiologic agents of destructive periodontal diseases. J Clin Periodontol. 1987;14:588–593. doi: 10.1111/j.1600-051x.1987.tb01520.x. [DOI] [PubMed] [Google Scholar]
- 341.Socransky SS, Smith C, Martin L, Paster BJ, Dewhirst FE, Levin AE. “Checkerboard” DNA-DNA hybridization. Biotechniques. 1994;17:788–792. [PubMed] [Google Scholar]
- 342.Sorsa T, Tjaderhane L, Konttinen YT, Lauhio A, Salo T, Lee HM, et al. Matrix metalloproteinases: contribution to pathogenesis, diagnosis and treatment of periodontal inflammation. Ann Med. 2006;38:306–321. doi: 10.1080/07853890600800103. [DOI] [PubMed] [Google Scholar]
- 343.Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- 344.Stashenko P, Dewhirst FE, Peros WJ, Kent RL, Ago JM. Synergistic interactions between interleukin 1, tumor necrosis factor, and lymphotoxin in bone resorption. J Immunol. 1987;138:1464–1468. [PubMed] [Google Scholar]
- 345.Stashenko P, Dewhirst FE, Rooney ML, Desjardins LA, Heeley JD. Interleukin-1 beta is a potent inhibitor of bone formation in vitro. J Bone Miner Res. 1987;2:559–565. doi: 10.1002/jbmr.5650020612. [DOI] [PubMed] [Google Scholar]
- 346.Stern J, Shai E, Zaks B, Halabi A, Houri-Haddad Y, Shapira L, et al. Reduced expression of gamma interferon in serum and marked lymphoid depletion induced by Porphyromonas gingivalis increase murine morbidity and mortality due to cytomegalovirus infection. Infect Immun. 2004;72:5791–5798. doi: 10.1128/IAI.72.10.5791-5798.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Sugano N, Ikeda K, Oshikawa M, Idesawa M, Tanaka H, Sato S, et al. Relationship between Porphyromonas gingivalis, Epstein-Barr virus infection and reactivation in periodontitis. J Oral Sci. 2004;46:203–206. doi: 10.2334/josnusd.46.203. [DOI] [PubMed] [Google Scholar]
- 348.Sumi Y, Kagami H, Ohtsuka Y, Kakinoki Y, Haruguchi Y, Miyamoto H. High correlation between the bacterial species in denture plaque and pharyngeal microflora. Gerodontology. 2003;20:84–87. doi: 10.1111/j.1741-2358.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- 349.Sunde PT, Olsen I, Enersen M, Beiske K, Grinde B. Human cytomegalovirus and Epstein-Barr virus in apical and marginal periodontitis: a role in pathology? J Med Virol. 2008;80:1007–1011. doi: 10.1002/jmv.21180. [DOI] [PubMed] [Google Scholar]
- 350.Sunde PT, Olsen I, Enersen M, Grinde B. Patient with severe periodontitis and subgingival Epstein-Barr virus treated with antiviral therapy. J Clin Virol. 2008;42:176–178. doi: 10.1016/j.jcv.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 351.Suomi JD, Greene JC, Vermillion JR, Doyle J, Chang JJ, Leatherwood EC. The effect of controlled oral hygiene procedures on the progression of periddontal disease in adults: results after third and final year. J Periodontol. 1971;42:152–160. doi: 10.1902/jop.1971.42.3.152. [DOI] [PubMed] [Google Scholar]
- 352.Suzuki MT, Giovannoni SJ. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol. 1996;62:625–630. doi: 10.1128/aem.62.2.625-630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Tanner AC, Dzink JL, Socransky SS, Des Roches CL. Diagnosis of periodontal disease using rapid identification of “activity-related” gram-negative species. J Periodontal Res. 1987;22:207–208. doi: 10.1111/j.1600-0765.1987.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 354.Tanner AC, Kent R, Jr, Kanasi E, Lu SC, Paster BJ, Sonis ST, et al. Clinical characteristics and microbiota of progressing slight chronic periodontitis in adults. J Clin Periodontol. 2007;34:917–930. doi: 10.1111/j.1600-051X.2007.01126.x. [DOI] [PubMed] [Google Scholar]
- 355.Tanner AC, Sonis AL, Lif Holgerson P, Starr JR, Nunez Y, Kressirer CA, et al. White-spot lesions and gingivitis microbiotas in orthodontic patients. J Dent Res. 2012;91:853–858. doi: 10.1177/0022034512455031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Teanpaisan R, Douglas CW, Eley AR, Walsh TF. Clonality of Porphyromonas gingivalis, Prevotella intermedia and Prevotella nigrescens isolated from periodontally diseased and healthy sites. J Periodontal Res. 1996;31:423–432. doi: 10.1111/j.1600-0765.1996.tb00511.x. [DOI] [PubMed] [Google Scholar]
- 357.Teles F, Haffajee AD, Socransky SS. Multiple displacement amplification as an aid in checkerboard DNA-DNA hybridization. Oral Microbiol Immunol. 2007;22:118–125. doi: 10.1111/j.1399-302X.2007.00333.x. [DOI] [PubMed] [Google Scholar]
- 358.Teles FR, Haffajee AD, Socransky SS. The reproducibility of curet sampling of subgingival biofilms. J Periodontol. 2008;79:705–713. doi: 10.1902/jop.2008.070424. [DOI] [PubMed] [Google Scholar]
- 359.Teles FR, Teles RP, Martin L, Socransky SS, Haffajee AD. Relationships among interleukin-6, tumor necrosis factor-alpha, adipokines, vitamin D, and chronic periodontitis. J Periodontol. 2012;83:1183–1191. doi: 10.1902/jop.2011.110346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Teles FR, Teles RP, Sachdeo A, Uzel NG, Song XQ, Torresyap G, et al. Comparison of microbial changes in early redeveloping biofilms on natural teeth and dentures. J Periodontol. 2012;83:1139–1148. doi: 10.1902/jop.2012.110506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Teles FR, Teles RP, Siegelin Y, Paster B, Haffajee AD, Socransky SS. RNA-oligonucleotide quantification technique (ROQT) for the enumeration of uncultivated bacterial species in subgingival biofilms. Mol Oral Microbiol. 2011;26:127–139. doi: 10.1111/j.2041-1014.2010.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Teles R, Sakellari D, Teles F, Konstantinidis A, Kent R, Socransky S, et al. Relationships among gingival crevicular fluid biomarkers, clinical parameters of periodontal disease, and the subgingival microbiota. J Periodontol. 81:89–98. doi: 10.1902/jop.2009.090397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Teles RP, Gursky LC, Faveri M, Rosa EA, Teles FR, Feres M, et al. Relationships between subgingival microbiota and GCF biomarkers in generalized aggressive periodontitis. J Clin Periodontol. 2010;37:313–323. doi: 10.1111/j.1600-051X.2010.01534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Teles RP, Haffajee AD, Socransky SS. Microbiological goals of periodontal therapy. Periodontol 2000. 2006;42:180–218. doi: 10.1111/j.1600-0757.2006.00192.x. [DOI] [PubMed] [Google Scholar]
- 365.Teles RP, Patel M, Socransky SS, Haffajee AD. Disease progression in periodontally healthy and maintenance subjects. J Periodontol. 2008;79:784–794. doi: 10.1902/jop.2008.070485. [DOI] [PubMed] [Google Scholar]
- 366.Teughels W, Van Assche N, Sliepen I, Quirynen M. Effect of material characteristics and/or surface topography on biofilm development. Clinical Oral Implants Research. 2006;17:68–81. doi: 10.1111/j.1600-0501.2006.01353.x. [DOI] [PubMed] [Google Scholar]
- 367.Theilade E, Budtz-Jorgensen E, Theilade J. Predominant cultivable microflora of plaque on removable dentures in patients with healthy oral mucosa. Arch Oral Biol. 1983;28:675–680. doi: 10.1016/0003-9969(83)90101-2. [DOI] [PubMed] [Google Scholar]
- 368.Theilade E, Wright WH, Jensen SB, Loe H. Experimental gingivitis in man. II. A longitudinal clinical and bacteriological investigation. J Periodontal Res. 1966;1:1–13. doi: 10.1111/j.1600-0765.1966.tb01842.x. [DOI] [PubMed] [Google Scholar]
- 369.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 2009;5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Tribble GD, Rigney TW, Dao DH, Wong CT, Kerr JE, Taylor BE, et al. Natural competence is a major mechanism for horizontal DNA transfer in the oral pathogen. Porphyromonas gingivalis MBio. 2012;3 doi: 10.1128/mBio.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Tsalikis LE, Kaklamanos EG, Kavadia-Tsatala S, Chasapopoulou E, Pidonia-Manika I. Association of gingival crevicular fluid and serum intracytoplasmic enzyme levels in periodontally healthy homozygous (major) beta-thalassemia patients. J Clin Periodontol. 2004;31:356–363. doi: 10.1111/j.1600-051X.2004.00485.x. [DOI] [PubMed] [Google Scholar]
- 372.Tuite-McDonnell M, Griffen AL, Moeschberger ML, Dalton RE, Fuerst PA, Leys EJ. Concordance of Porphyromonas gingivalis colonization in families. J Clin Microbiol. 1997;35:455–461. doi: 10.1128/jcm.35.2.455-461.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Uzel NG, Teles FR, Teles RP, Song XQ, Torresyap G, Socransky SS, et al. Microbial shifts during dental biofilm re-development in the absence of oral hygiene in periodontal health and disease. J Clin Periodontol. 2011;38:612–620. doi: 10.1111/j.1600-051X.2011.01730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Valenza G, Veihelmann S, Peplies J, Tichy D, Roldan-Pareja Mdel C, Schlagenhauf U, et al. Microbial changes in periodontitis successfully treated by mechanical plaque removal and systemic amoxicillin and metronidazole. Int J Med Microbiol. 2009;299:427–438. doi: 10.1016/j.ijmm.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 375.Valm AM, Mark Welch JL, Rieken CW, Hasegawa Y, Sogin ML, Oldenbourg R, et al. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci U S A. 2011;108:4152–4157. doi: 10.1073/pnas.1101134108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Valverde P. Pharmacotherapies to manage bone loss-associated diseases: a quest for the perfect benefit-to-risk ratio. Curr Med Chem. 2008;15:284–304. doi: 10.2174/092986708783497274. [DOI] [PubMed] [Google Scholar]
- 377.van der Velden U, Abbas F, Armand S, de Graaff J, Timmerman MF, van der Weijden GA, et al. The effect of sibling relationship on the periodontal condition. J Clin Periodontol. 1993;20:683–690. doi: 10.1111/j.1600-051x.1993.tb00716.x. [DOI] [PubMed] [Google Scholar]
- 378.Van der Weijden GA, Timmerman MF, Reijerse E, Wolffe GN, van Winkelhoff AJ, van der Velden U. The prevalence of A. actinomycetemcomitans, P. gingivalis and P. intermedia in selected subjects with periodontitis. J Clin Periodontol. 1994;21:583–588. doi: 10.1111/j.1600-051x.1994.tb00747.x. [DOI] [PubMed] [Google Scholar]
- 379.Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol. 2008;79:1601–1608. doi: 10.1902/jop.2008.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.van Steenbergen TJ, Petit MD, Scholte LH, van der Velden U, de Graaff J. Transmission of Porphyromonas gingivalis between spouses. J Clin Periodontol. 1993;20:340–345. doi: 10.1111/j.1600-051x.1993.tb00370.x. [DOI] [PubMed] [Google Scholar]
- 381.van Steenbergen TJ, van Winkelhoff AJ, de Graaff J. Pathogenic synergy: mixed infections in the oral cavity. Antonie Van Leeuwenhoek. 1984;50:789–798. doi: 10.1007/BF02386241. [DOI] [PubMed] [Google Scholar]
- 382.van Winkelhoff AJ, Herrera D, Oteo A, Sanz M. Antimicrobial profiles of periodontal pathogens isolated from periodontitis patients in The Netherlands and Spain. J Clin Periodontol. 2005;32:893–898. doi: 10.1111/j.1600-051X.2005.00782.x. [DOI] [PubMed] [Google Scholar]
- 383.van Winkelhoff AJ, Loos BG, van der Reijden WA, van der Velden U. Porphyromonas gingivalis, Bacteroides forsythus and other putative periodontal pathogens in subjects with and without periodontal destruction. J Clin Periodontol. 2002;29:1023–1028. doi: 10.1034/j.1600-051x.2002.291107.x. [DOI] [PubMed] [Google Scholar]
- 384.van Winkelhoff AJ, Rijnsburger MC, van der Velden U. Clonal stability of Porphyromonas gingivalis in untreated periodontitis. J Clin Periodontol. 2008;35:674–679. doi: 10.1111/j.1600-051X.2008.01285.x. [DOI] [PubMed] [Google Scholar]
- 385.van Winkelhoff AJ, Rodenburg JP, Goene RJ, Abbas F, Winkel EG, de Graaff J. Metronidazole plus amoxycillin in the treatment of Actinobacillus actinomycetemcomitans associated periodontitis. J Clin Periodontol. 1989;16:128–131. doi: 10.1111/j.1600-051x.1989.tb01626.x. [DOI] [PubMed] [Google Scholar]
- 386.Vardjan N, Kopitar AN, Ihan Hren N, Malovrh T, Wraber B, Ihan A. Immune response in lymphocyte cultures stimulated by oral bacteria preparations. Pflugers Arch. 2000;440:R67–69. [PubMed] [Google Scholar]
- 387.Vartoukian SR, Downes J, Palmer RM, Wade WG. Fretibacterium fastidiosum gen. nov., sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol. 2012 doi: 10.1099/ijs.0.041038-0. [DOI] [PubMed] [Google Scholar]
- 388.Vartoukian SR, Palmer RM, Wade WG. Cultivation of a Synergistetes strain representing a previously uncultivated lineage. Environ Microbiol. 12:916–928. doi: 10.1111/j.1462-2920.2009.02135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Vartoukian SR, Palmer RM, Wade WG. Diversity and morphology of members of the phylum “synergistetes” in periodontal health and disease. Appl Environ Microbiol. 2009;75:3777–3786. doi: 10.1128/AEM.02763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Vartoukian SR, Palmer RM, Wade WG. Strategies for culture of ‘unculturable’ bacteria. FEMS Microbiol Lett. 309:1–7. doi: 10.1111/j.1574-6968.2010.02000.x. [DOI] [PubMed] [Google Scholar]
- 391.Vianna ME, Horz HP, Gomes BP, Conrads G. Microarrays complement culture methods for identification of bacteria in endodontic infections. Oral Microbiol Immunol. 2005;20:253–258. doi: 10.1111/j.1399-302X.2005.00221.x. [DOI] [PubMed] [Google Scholar]
- 392.von Wintzingerode F, Gobel UB, Stackebrandt E. Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev. 1997;21:213–229. doi: 10.1111/j.1574-6976.1997.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 393.Wade WG. Has the use of molecular methods for the characterization of the human oral microbiome changed our understanding of the role of bacteria in the pathogenesis of periodontal disease? J Clin Periodontol. 2011;38(11):7–16. doi: 10.1111/j.1600-051X.2010.01679.x. [DOI] [PubMed] [Google Scholar]
- 394.Wadelius M, Chen LY, Downes K, Ghori J, Hunt S, Eriksson N, et al. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenomics J. 2005;5:262–270. doi: 10.1038/sj.tpj.6500313. [DOI] [PubMed] [Google Scholar]
- 395.Waters JC. Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol. 2009;185:1135–1148. doi: 10.1083/jcb.200903097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Weinberg A, Belton CM, Park Y, Lamont RJ. Role of fimbriae in Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1997;65:313–316. doi: 10.1128/iai.65.1.313-316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Wendisch VF, Zimmer DP, Khodursky A, Peter B, Cozzarelli N, Kustu S. Isolation of Escherichia coli mRNA and comparison of expression using mRNA and total RNA on DNA microarrays. Anal Biochem. 2001;290:205–213. doi: 10.1006/abio.2000.4982. [DOI] [PubMed] [Google Scholar]
- 398.Williams RC, Jeffcoat MK, Howell TH, Rolla A, Stubbs D, Teoh KW, et al. Altering the progression of human alveolar bone loss with the non-steroidal anti-inflammatory drug flurbiprofen. J Periodontol. 1989;60:485–490. doi: 10.1902/jop.1989.60.9.485. [DOI] [PubMed] [Google Scholar]
- 399.Winkel EG, van Winkelhoff AJ, Timmerman MF, van der Velden U, van der Weijden GA. Amoxicillin plus metronidazole in the treatment of adult periodontitis patients. A double-blind placebo-controlled study. J Clin Periodontol. 2001;28:296–305. doi: 10.1034/j.1600-051x.2001.028004296.x. [DOI] [PubMed] [Google Scholar]
- 400.Wu JY, Jiang XT, Jiang YX, Lu SY, Zou F, Zhou HW. Effects of polymerase, template dilution and cycle number on PCR based 16 S rRNA diversity analysis using the deep sequencing method. BMC Microbiol. 2010;10:255. doi: 10.1186/1471-2180-10-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Wu YM, Yan J, Ojcius DM, Chen LL, Gu ZY, Pan JP. Correlation between infections with different genotypes of human cytomegalovirus and Epstein-Barr virus in subgingival samples and periodontal status of patients. J Clin Microbiol. 2007;45:3665–3670. doi: 10.1128/JCM.00374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Wyss C, Choi BK, Schupbach P, Moter A, Guggenheim B, Gobel UB. Treponema lecithinolyticum sp. nov., a small saccharolytic spirochaete with phospholipase A and C activities associated with periodontal diseases. Int J Syst Bacteriol. 1999;49 Pt 4:1329–1339. doi: 10.1099/00207713-49-4-1329. [DOI] [PubMed] [Google Scholar]
- 403.Wyss C, Moter A, Choi BK, Dewhirst FE, Xue Y, Schupbach P, et al. Treponema putidum sp. nov., a medium-sized proteolytic spirochaete isolated from lesions of human periodontitis and acute necrotizing ulcerative gingivitis. Int J Syst Evol Microbiol. 2004;54:1117–1122. doi: 10.1099/ijs.0.02806-0. [DOI] [PubMed] [Google Scholar]
- 404.Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol. 2000;27:648–657. doi: 10.1034/j.1600-051x.2000.027009648.x. [DOI] [PubMed] [Google Scholar]
- 405.Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Microbial composition of supra- and subgingival plaque in subjects with adult periodontitis. J Clin Periodontol. 2000;27:722–732. doi: 10.1034/j.1600-051x.2000.027010722.x. [DOI] [PubMed] [Google Scholar]
- 406.Zadeh HH, Nalbant A, Park K. Large-scale early in vitro response to Actinobacillus actinomycetemcomitans suggests superantigenic activation of T-cells. J Dent Res. 2001;80:356–362. doi: 10.1177/00220345010800011101. [DOI] [PubMed] [Google Scholar]
- 407.Zadeh HH, Tanavoli S, Haines DD, Kreutzer DL. Despite large-scale T cell activation, only a minor subset of T cells responding in vitro to Actinobacillus actinomycetemcomitans differentiate into effector T cells. J Periodontal Res. 2000;35:127–136. doi: 10.1034/j.1600-0765.2000.035003127.x. [DOI] [PubMed] [Google Scholar]
- 408.Zambon JJ, Sunday GJ, Smutko JS. Molecular genetic analysis of Actinobacillus actinomycetemcomitans epidemiology. J Periodontol. 1990;61:75–80. doi: 10.1902/jop.1990.61.2.75. [DOI] [PubMed] [Google Scholar]
- 409.Zappa U, Reinking-Zappa M, Graf H, Espeland M. Cell populations and episodic periodontal attachment loss in humans. J Clin Periodontol. 1991;18:508–515. doi: 10.1111/j.1600-051x.1991.tb00082.x. [DOI] [PubMed] [Google Scholar]
- 410.Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Zijnge V, van Leeuwen MBM, Degener JE, Abbas F, Thurnheer T, Gmur R, et al. Oral biofilm architecture on natural teeth. Plos One. 5 doi: 10.1371/journal.pone.0009321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Zou S, Zhou J. Relationship between the activity of leukocyte chemokines in gingival crevicular fluid and periodontitis. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 1997;19:442–446. [PubMed] [Google Scholar]
- 413.Zuger J, Luthi-Schaller H, Gmur R. Uncultivated Tannerella BU045 and BU063 are slim segmented filamentous rods of high prevalence but low abundance in inflammatory disease-associated dental plaques. Microbiology. 2007;153:3809–3816. doi: 10.1099/mic.0.2007/010926-0. [DOI] [PubMed] [Google Scholar]