

Abstract
Calcium (Ca2+) influx is required for the activation and function of all cells in the immune system. It is mediated mainly by store-operated Ca2+ entry (SOCE) through Ca2+ release-activated Ca2+ (CRAC) channels located in the plasma membrane. CRAC channels are composed of ORAI proteins that form the channel pore and are activated by stromal interaction molecules (STIM) 1 and 2. Located in the membrane of the endoplasmic reticulum, STIM1 and STIM2 have the dual function of sensing the intraluminal Ca2+ concentration in the ER and to activate CRAC channels. A decrease in the ER's Ca2+ concentration induces STIM multimerization and translocation into puncta close to the plasma membrane where they bind to and activate ORAI channels. Since the identification of ORAI and STIM genes as the principal mediators of CRAC channel function, substantial advances have been achieved in understanding the molecular regulation and physiological role of CRAC channels in cells of the immune system and other organs. In this review, we discuss the mechanisms that regulate CRAC channel function and SOCE, the role of recently identified proteins and mechanisms that modulate the activation of ORAI/STIM proteins and the consequences of CRAC channel dysregulation for lymphocyte function and immunity.
Keywords: T cells, Lymphocytes, Calcium, CRAC channels, STIM1, ORAI1
Introduction
Lymphocyte function is critically dependent on the regulation of intracellular Ca2+ levels, [Ca2+]i. An increase in [Ca2+]i can originate from two sources: (1) the emptying of intracellular Ca2+ stores, mainly the endoplasmic reticulum (ER), and (2) the influx of extracellular Ca2+ through Ca2+ channels in the plasma membrane. The release of Ca2+ from ER stores follows the generation of second messengers such as inositol 1,4,5-trisphosphate (InsP3) following engagement of the T cell receptor (TCR, Fig. 1) and B cell receptor (BCR) by antigen. Given the small volume of the ER in lymphocytes, store depletion does not result in a sustained rise in [Ca2+]i, which is required for many lymphocyte functions. However, Ca2+ store depletion and the subsequent reduction of ER Ca2+ levels, [Ca2+]ER, is the trigger mechanism for Ca2+ influx through Ca2+ channels in the plasma membrane, ultimately resulting in a large and sustained increase in [Ca2+]i. The hypothesis that the filling state of intracellular Ca2+ stores controls the activity of Ca2+ channels in the plasma membrane was first postulated by J. Putney [1]. Originally termed capacitative Ca2+ entry, this pathway now is commonly referred to as store-operated Ca2+ entry (SOCE).
Fig. 1.

Store-operated Ca2+ entry (SOCE) in T cells. Antigen binding to the T cell receptor (TCR) results in the activation of protein tyrosine kinases Lck and ZAP-70, phosphorylation of signaling molecules LAT and SLP-76, and the activation of phospholipase C (PLC) γ. PLCγ catalyzes the hydrolysis of PIP2 into the second messengers InsP3 and DAG. InsP3 gates InsP3 receptors in the membrane of the ER resulting in the (active) release of Ca2+ from ER Ca2+ stores. Passive depletion of ER Ca2+ stores can be achieved by the inhibition of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump by thapsigargin (TG). The reduction in [Ca2+]ER resulting from active or passive store depletion is sensed by STIM1 and its homologue STIM2, resulting in their oligomerization and translocation to ER-plasma membrane (PM) junctions. There STIM1 and STIM2 bind to and open CRAC channels formed by ORAI proteins located in the plasma membrane, resulting in store-operated Ca2+ entry (SOCE). SOCE is sustained by Ca2+ uptake into mitochondria that are located close to the plasma membrane, thereby preventing Ca2+-dependent inactivation of CRAC channel function. DAG diacylglycerol, InsP3 inositol 1,4,5-trisphosphate, LAT linker of activated T cells, Lck lymphocyte-specific protein tyrosine kinase, PIP2 phosphatidylinositol 4,5-bisphosphate, SHP-76 SH2 domain containing leukocyte protein of 76 kDa, ZAP-70 Zeta-chain-associated protein kinase 70
The molecular identity of the channels that mediate SOCE remained elusive for many years, due in part to the difficulty of measuring the electrical activity of the Ca2+ channels responsible for SOCE in lymphocytes by patch clamp electrophysiology. From indirect measurements of Ca2+ influx, it was clear that whole-cell Ca2+ currents had to be very small. There was some evidence of Ca2+ currents in mast cells and T cells before 1990 [2, 3], but a break-through was achieved when the first bona fide store-operated channel was discovered in mast cells and T cells [4, 5]. This current was named calcium release-activated calcium (CRAC) current [4]. It is present in most, if not all, immune cell types but also in many other electrically non-excitable cells and even in some electrically excitable cell types such as myocytes [6]. In lymphocytes, the activation of CRAC channels can be triggered by active (i.e., TCR or BCR engagement) or passive (i.e., inhibition of ER store refilling) depletion of Ca2+ stores (Fig. 1). Despite the importance of CRAC channels for lymphocyte function, it took another 14 years to discover their molecular nature. In this review, we will discuss the substantial advances that have been made in recent years in deciphering the molecular mechanisms of CRAC channel regulation and their role in lymphocyte function.
ORAI and STIM family proteins are essential CRAC channel components and mediate SOCE
ORAI proteins constitute the CRAC channel pore
Several ion channels were proposed to mediate SOCE in lymphocytes since the discovery of CRAC channel currents in mast cells and T cells [4, 5], including several members of the transient receptor protein (TRP) family of ion channels [7]. In 2006, however, we and others discovered a new class of calcium channels that comprises ORAI1, ORAI2, and ORAI3 (alternatively termed CRACM1–3). ORAI proteins, in particular ORAI1, encode the CRAC channel and are responsible for SOCE in immune cells. Drosophila orai (dOrai) and human ORAI1 genes were identified in genome-wide screens for regulators of Ca2+ influx and the nuclear factor of activated T cells (NFAT), and by linkage analysis in human patients whose T cells lack functional CRAC channels and SOCE resulting in immunodeficiency [8–10]. The mutated gene in patients mapped to a ~4 Mb region on chromosome 12q24 [8], which harbored the hypothetical gene FLJ14466, the human ortholog of the Drosophila gene Olf186-F. The latter was identified as a candidate gene regulating NFAT activation and SOCE by three independent RNAi screens [8–10].
ORAI proteins are structurally unrelated to other calcium channels and contain four-transmembrane domains, a cytoplasmic N- and C-terminus, and two extracellular as well as one intracellular loops (Fig. 2a). Subsequent structure–function analyses by several groups have shown that ORAI1 is the pore-forming subunit of the CRAC channel [11–15]. The first transmembrane domain (TM1) lines the channel pore [11–15] and contains a glutamate (E106) residue that forms a high-affinity Ca2+ binding site that confers the high Ca2+ selectivity to the CRAC channel (Fig. 2a). A mutation of E106 to alanine (A) or glutamine (Q) abolishes CRAC currents and mutation to aspartate renders the channel non-selective [12–14, 16]. E106 is highly conserved in other ORAI homologues and mutating E81 at the same position in TM1 of ORAI3 abolished SOCE [17]. The analysis of other residues in TM1 of ORAI1 indicate that the CRAC channel pore is narrow [11, 15], potentially explaining the CRAC channel's small unitary conductance and therefore small currents observed in lymphocytes.
Fig. 2.
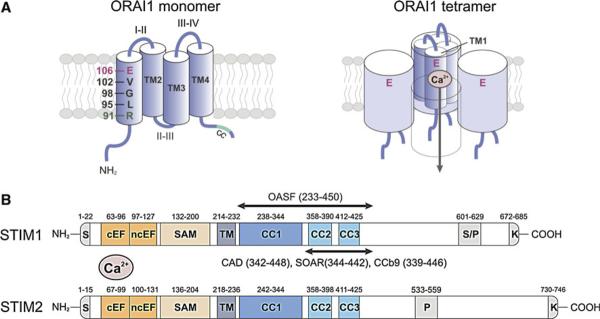
Molecular structure of ORAI1 and STIM1. a ORAI1 is a tetraspanning plasma membrane protein that contains intracellular N-and C-termini (NT, CT), two extracellular loops (I–II, III–IV), and an intracellular loop (II–III; left panel) [8–10]. A coiled-coil (CC) domain in the CT of ORAI1 mediates STIM1 binding and is required for CRAC channel activation [54]. ORAI1 is the pore-forming subunit of the CRAC channel [12–14]. The first transmembrane domain (TM1) of ORAI1 contains several residues that line the channel pore and mediate important CRAC channel functions. Glutamate (E) 106 provides a high-affinity Ca2+ binding site in the channel lumen and is responsible for the high Ca2+ selectivity of the CRAC channel [12–14]. V102 and R91 in the middle and at the cytoplasmic end of TM1, respectively, were proposed to form the channel gate that regulates Ca2+ permeation through the CRAC channel [19, 21]. G98 may function as a molecular hinge that allows the channel to open and close [19]. Functional CRAC channels are formed by assembly of four ORAI1 subunits (right panel) [22–24]. Four TM1 domains are thought to line the channel pore [11, 15], each contributing one E106 residue to form the Ca2+ binding site and Ca2+ selectivity filter [12–14, 16]. In this figure, the fourth ORAI1 subunit is removed to reveal the Ca2+ permeation pathway. b STIM1 and STIM2 are single-pass membrane proteins whose N- and C-termini are located in the lumen of the ER and the cytoplasm, respectively [39]. The NT of STIM1 contains a canonical EF-hand (cEF) that is paired with a noncanonical EF-hand (ncEF) [42]. The cEF hand domain is required for sensing the ER Ca2+ concentration and mutations of acidic residues within it result in impaired Ca2+ binding and constitutive CRAC channel activation independently of ER Ca2+ store depletion [32, 35, 43]. The CT of STIM1 and STIM2 contains two strong (CC1, CC2) and one weak (CC3) coiled-coil domains, which mediate STIM oligomerization upon store depletion. CC2 and CC3 are part of a minimal CRAC channel activation domain (CAD, also called STIM ORAI activating region (SOAR) or coiled-coil domain b9 (CCb9)), which directly binds to ORAI1 to activate CRAC channels [49, 51, 52]. The longer ORAI1-activating short fragment (OASF) also activates CRAC channels [53]. The lysine-rich region at the very CT is required for the recruitment of STIM1 to the plasma membrane (PM) by interacting with negatively charged phospholipids in the PM [59]. CC coiled-coil, EFh EF-hand, K lysine-rich, SAM sterile alpha motif, S signal peptide, S/P serine/proline-rich, TM transmembrane
Sequence analysis in cells from CRAC-deficient patients revealed that they are homozygous for a Arg-to-Trp mis-sense mutation (R91 W) in ORAI1 [8]. R91 is localized at the cytoplasmic end of the first transmembrane domain of ORAI1 and its mutation to Trp interferes with CRAC channel function despite normal ORAI1 expression (Fig. 2a). Hydrophobic substitutions at position 91 impair CRAC channels function, presumably by nserting TM1 of ORAI1 more rigidly in the plasma membrane and interfering with CRAC channel opening [18]. R91 was shown to be a pore-lining residue of the CRAC channel [11] and has been suggested to be part of the gate at the inner mouth of the CRAC channel [19]. Mutation of R91 may therefore directly interfere with ion conduction through the CRAC channel pore. The blocking effect of the R91W mutation could be reversed when combined with mutations of G98 in the middle of TM1, which was therefore proposed to be a gating hinge that allows the CRAC channel to open [19]. Another study identified V102, a residue located in the extracellular region of the pore, as the CRAC channel gate because several mutations at V102 produce constitutively open CRAC channels [20, 21]. A more detailed understanding of the gating mechanism and architecture of the CRAC channel will have to await solving the crystal structure of ORAI proteins. A widely accepted model proposes that functional CRAC channels are formed by assembly of four ORAI1 subunits, whose TM1 domains form the CRAC channel pore [22–24]. A more detailed discussion of the tetrameric CRAC channel structure and the regulation of ion conduction by the CRAC channel can be found in these reviews [20, 25].
Like ORAI1, its homologues ORAI2 and ORAI3 function as Ca2+ channels when overexpressed in a variety of cell types [26, 27]. Their channel properties are similar to those of ORAI1 including regulation by store-depletion and exquisitely high Ca2+ selectivity, but there are also significant differences regarding their inactivation properties and response to pharmacological inhibitors [27]. The physiological roles of ORAI2 and ORAI3 in lymphocytes remain unknown. In human T cells, ORAI1 is the predominant ORAI family member that mediates CRAC channel function and SOCE as emphasized by the complete lack of CRAC channel currents and SOCE in T cells of patients with loss-of-function mutations in ORAI1 [28]. In naive murine T cells, ORAI2 or ORAI3 may contribute to SOCE as naive CD4+ and CD8+ T cells from Orai1−/−mice and Orai1R93W/R93W mice (knock-in mice expressing a non-functional ORAI1-R93W protein that is equivalent to the human ORAI1–R91W mutant) displayed residual SOCE [29–31]. Future studies will have to elucidate the contribution of ORAI2 and ORAI3 to SOCE in lymphocytes and other cell types.
STIM proteins activate CRAC channels
Equally intangible as the nature of CRAC channels themselves was their store-operated activation mechanism. This problem was solved with the identification of stromal interaction molecules (STIM) 1 and STIM2 as essential regulators of SOCE [32–34]. Both proteins are highly conserved single-pass transmembrane proteins that are localized predominantly in the membrane of the ER, although a minor fraction is found in the plasma membrane (PM) [35, 36]. Before its role in SOCE was identified, STIM1 had been implicated in the survival of pre-B cells [37] and as a tumor suppressor linked to the pathogenesis of rhabdomyosarcoma [38]. Early genetic and molecular studies conducted by Dziadek and coworkers identified STIM2 and Drosophila dStim as homologues of STIM1 that display a conserved gene and protein domain structure, suggesting that both genes are derived from one ancestral gene (Fig. 2b) [39]. These studies showed that STIM1 and STIM2 form homo- and heteromultimers and that both proteins are posttranslationally modified by phosphorylation and N-linked glycosylation [36, 39, 40]. The function of STIM proteins, however, remained elusive. In 2005, two groups independently identified STIM1, STIM2, and dStim proteins as regulators of SOCE in HeLa cells and Drosophila S2 cells, respectively, using RNAi screens focusing on signal transduction and ion channel-associated genes [32, 33]. These studies revealed that mammalian STIM1 and dStim are essential for activation of SOCE. Furthermore, they showed that STIM proteins are localized predominantly in the ER where they sense the [Ca2+]ER with the help of an ER-lumenal EF-hand Ca2+ binding domain [32, 33]. Upon Ca2+ store depletion and dissociation of Ca2+ from the EF-hand domain, STIM proteins accumulate in puncta localized at ER-PM junctions where they bind to and open ORAI1 CRAC channels resulting in SOCE. In the next chapter, we will discuss in detail how STIM proteins control the activation and inactivation of ORAI channels.
Mechanisms of ORAI1-CRAC channel activation by STIM proteins
Most studies have investigated the role of STIM1 in CRAC channel activation. In this chapter, we will therefore mainly focus on STIM1, but discuss some relevant differences between STIM1 and STIM2.
Sensing [Ca2+]ER and the conformation of the N-terminus of STIM1
The N-terminus (NT) of STIM proteins contains a pair of canonical and non-canonical EF-hand Ca2+ binding motifs and an adjacent sterile alpha motif (SAM; Fig. 2b) [41, 42]. Several aspartate and glutamate residues within the EF-hand coordinate Ca2+ binding by the N-terminus of STIM1. Mutation of either D76, D84, or E87 residues mimics the Ca2+ dissociated state of the STIM1-NT and results in STIM1 activation and constitutive CRAC channel function in the absence of store depletion [32, 34, 35, 43]. Intriguingly, mutation of D84 in the EFh domain of STIM1 in mice resulted in constitutive Ca2+ influx in T cells but T cell development and function were normal [43] as will be discussed in “Consequences of CRAC channel dysregulation on lymphocyte function” further below.
In the Ca2+-bound state, the EF-SAM domain exists in a closed configuration that is facilitated by hydrophobic interactions between both domains (Fig. 3) [42]. Upon ER store depletion, Ca2+ dissociates from the EF-hand domain resulting in the destabilization of the EF-SAM structure [42]. The latter leads to the unfolding of the STIM1-NT and the oligomerization of neighboring STIM molecules that is facilitated by interactions of their SAM domains [42, 44]. The kinetics with which the NT of STIM1 unfolds upon urea-induced denaturation was reported to be > threefold faster when compared to STIM2 [41]. This difference resulted in the much faster (by ~70-fold) conversion of STIM1 from a monomeric to an oligomeric state compared to STIM2. These findings were explained by the unstructured and oligomeric nature of the Ca2+ free STIM1 EF-SAM domain, whereas the Ca2+ free NT of STIM2 retains a well-folded structure that is only partially different from its Ca2+-bound form [41]. STIM1 and STIM2 also differ significantly with regard to their activation thresholds, i.e. the decrease in [Ca2+]ER that leads to their activation. Half-maximal STIM1 (and CRAC channel) activation are achieved when [Ca2+]ER falls below ~ 200 μM [60], whereas STIM2 responds to small decreases in [Ca2+]ER (from the estimated resting concentration of ~800 μM) [45]. It has therefore been suggested that STIM2 activates SOCE upon small fluctuations in [Ca2+]ER and thus maintains the filling of ER stores and basal [Ca2+]i in resting cells [45]. By contrast, STIM1 is responsible for SOCE in response to cellular stimulation through, for instance, antigen receptors on lymphocytes. These differences in the functional properties of the NT of STIM1 and STIM2 account (together with their different expression levels) for the distinct roles of STIM1 and STIM2 in SOCE in lymphocytes. In the mast cell line RBL-1, it was recently shown that stimulation of tyrosine kinase-coupled FCεRI receptors can couple to both, STIM1 and STIM2, whereas cysteinyl leukotriene type I receptors exclusively operate through STIM1 [46]. Whether distinct antigen receptors on lymphocytes also differentially activate STIM1 and STIM2 is presently unknown.
Fig. 3.

Conformational changes of STIM1 structure during CRAC channel activation. In non-stimulated cells, ER Ca2+ stores are filled and Ca2+ is bound to the cEFh domain in the NT of STIM1. Upon depletion of Ca2+ from ER stores, the [Ca2+]ER drops and Ca2+ dissociates from the EFh, resulting in a conformational change of the STIM1-NT. Structural data support a model in which the Ca2+-free EFh unfolds from the adjacent SAM domain [42], thereby allowing the interaction of SAM domains in neighboring STIM1 molecules and their oligomerization. The STIM1-CT and its CC domains are critical for STIM1 oligomerization and ORAI1 binding. The structure of the STIM1-CT and the conformational changes that occur within it following store depletion are incompletely defined. a In one model, the STIM1-CT is present in a folded, inactive conformation in cells with replete ER stores. CC2 and CC3 (which encompass the majority of the CAD/CCb9/SOAR domain) were proposed to interact with CC1 (which was shown to inhibit the ability of the STIM1-CT to activate CRAC channels) through hydrophobic interactions and hydrogen bonds between acidic (CC1: 302–322) and basic (CC2: 382–386) residues (indicated by minus and plus symbols) [58, 63, 64, 66]. Upon store depletion, the STIM1-CT unfolds, allowing STIM1 oligomerization. b An alternative model proposed by Yang et al. [68] is based on the crystal structure of the human CAD domain and the CC1-CAD domains of C. elegans. The CAD domain, which contains two long alpha helices aligned in an antiparallel manner, exists as a dimer in solution. Dimerization is stabilized by hydrophobic interactions, hydrogen bonds, and stacking interactions between residues located at the N- and C-terminal ends of each CAD domain (for details see text). The CAD dimer forms a V-shaped structure that exposes basic Lys and Arg residues (aa 382–387; indicated by plus symbols), which may mediate binding to ORAI1 as reported earlier [63, 66]. CAD function is restrained by an inhibitory helix at the C-terminal end of CC1 (light blue box) that interacts with CAD in several locations [68]. STIM1 activation is mediated by release of the inhibitory helix from CAD. In contrast to the model shown in a, the structure of the CAD dimer remains unchanged after store depletion. For clarity, only one of the dimer-forming CAD domains is shown in color. CC coiled-coil, CT C-terminus, EFh EF-hand, K lysine-rich, NT N terminus, SAM sterile alpha motif, S/P serine/proline-rich, TM transmembrane
STIM1 puncta formation and ORAI1 interaction
Upon store depletion, STIM proteins translocate within the ER membrane into large clusters, called puncta, that are in close proximity to the plasma membrane [32]. STIM1 puncta formation occurs within ~1 min following store depletion and precedes the opening of CRAC channels [48]. STIM1 puncta formation occurs independently of ORAI1 [49, 50], whereas the recruitment of ORAI1 into puncta is dependent on STIM1 [49]. Luik et al. [47] showed that puncta are the sites of highly localized Ca2+ influx, indicating that these structures form critical signaling microdomains at ER–PM junctions.
CRAC channel activation requires the binding of STIM1 to ORAI1, and several studies have defined the molecular domains in both proteins that mediate this activation. The C-terminus (CT) of STIM1 contains three coiled-coil (CC) domains (Fig. 2b). The more C-terminal second and third CC domains are part of a CRAC channel activation domain (CAD) that is ~110 amino acids (aa) in size and required for ORAI1 activation as reported independently by several groups. Alternative names for CAD (aa 342–448) [49] present in the literature are STIM1-ORAI activating region (SOAR, aa 344–442) [51] and coiled-coil domain b9 (CCb9, aa 339–444) [52]. When expressed as a soluble fragment, STIM1-CAD is localized at the plasma membrane and bound to ORAI1. CAD, and the larger ORAI1-activating small fragment (OASF, aa 233–450), are sufficient to activate CRAC channels in the absence of store depletion [49, 51–53]. An important binding site for STIM1-CAD is the CC domain in the CT of ORAI1. Mutation of hydrophobic residues L273 and L276 within heptad repeats interfere with the structure of the coiled-coil domain in ORAI1-CT and abolish STIM1 binding and CRAC channel activation [54, 55]. In addition, STIM1-CAD also weakly binds to the N-terminus (aa 48–91) of ORAI1 in solution [49, 56], and interactions of STIM1 with both the C- and N-terminus of ORAI1 are thought to mediate CRAC channel activation.
Oligomerization of STIM1 is required for SOCE
For STIM1 to efficiently activate CRAC channels, it has to undergo activation-induced oligomerization. At rest, i.e., in cells with replete ER Ca2+ stores, STIM1 is already oligomerized to some degree based on the observation of resting FRET between STIM1 molecules, their migration as dimers in crosslinking experiments, and the fact that STIM1 molecules can be co-immunoprecipitated in non-stimulated cells [23, 40, 57]. STIM1 interactions in resting cells depend on the cytoplasmic CC regions within STIM1 as shown by co-IP and FRET experiments [57, 58]. Upon store depletion, STIM1 undergoes formation of higher-order oligomers that precede the establishment of puncta and activation of SOCE [54, 59]. Oligomerization of STIM1 mediates CRAC channel activation in part by increasing the avidity of a lysine-rich region at the STIM1-CT for negatively charged plasma membrane phospholipids (PIP2, PIP3), thereby promoting STIM1 translocation to the PM. Deletion of the lysine-rich region (ΔK) impairs STIM1 recruitment to the PM, but does not inhibit store depletion-induced STIM1 oligomerization [59]. The importance of STIM1 oligomerization for the effective opening of CRAC channels was shown by replacing the NT of STIM1 with a rapamycin-inducible oligomerization system. Luik et al. [60] demonstrated that oligomerization of STIM1 driven by N-terminal homotypic binding triggers STIM1 translocation, puncta formation, and activation of CRAC channels, indicating that STIM1 oligomerization is a critical switch in the activation of SOCE following store depletion. The formation of higher-order STIM1 oligomers following store depletion depends on protein domains in the N- and C-termini of STIM1 [41, 42, 57, 58]. The N-termini of STIM1 proteins oligomerize via their EFh-SAM domain following Ca2+ dissociation from the EF-hand and unfolding of the EFh-SAM structure [42, 43]. Deletion of the SAM domain from the NT of STIM1 abolishes SOCE and prevents the formation of STIM1 puncta [57]. In addition, the CT of STIM1 is essential for the formation of higher-order STIM1 complexes. While a STIM1 molecule truncated after the CAD was able to oligomerize upon store depletion, further truncation including the CAD abolished activation-induced oligomerization [58], indicating that CAD—in addition to mediating ORAI1 binding—is required for stabilizing STIM1 oligomers. Recent studies have addressed how many STIM1 molecules are required to activate ORAI1 channels. Using different experimental approaches, Hoover et al. [61] and Li et al. [62] showed that maximal CRAC channel activation is achieved at a STIM1:ORAI1 molar ratio of 2:1. Based on the model that active ORAI1 channels are composed of four subunits [22–24], these findings suggest that eight STIM1 molecules bind to one ORAI1 channel tetramer.
Conformational changes in the STIM1 C-terminus regulate its function
Several recent studies have shed light on the structure of the STIM1-CT and how it regulates CRAC channel activation. They suggest that in resting cells STIM1-CT exists in a closed configuration that is maintained by intramolecular interactions between charged and hydrophobic residues in CC domains [63, 64] (Fig. 3a). Korzeniowski et al. [63] showed that the CC1 domain of STIM1-CT exerts an inhibitory effect on Ca2+ influx and identified an acidic domain (aa 302–322) that was proposed to interact with four lysine residues (aa 382–386) in the CAD domain. The charged interactions between these domains were suggested to keep STIM1 in an inactive state and prevent ORAI1 activation. Upon store depletion, the closed confirmation of STIM1-CT was proposed to give way to an extended conformation that allows the basic STIM1-CT domain (aa 382–386) to interact with an acidic domain in the CT of ORAI1 (aa 272–291) that shows significant sequence homology with the acidic domain in STIM1-CC1 [63]. Thus, the acidic and basic domains in STIM1-CT were proposed to act as an electrostatic clamp that controls the intramolecular conformation of STIM1, thereby regulating its ability to activate ORAI1. In addition, several hydrophobic residues in the CT of STIM1 were shown to control its intramolecular conformation. Muik et al. [64] designed a FRET-based STIM1 reporter that encompasses part of the STIM1 C-terminus (aa 233–474) and that exhibits robust intramolecular FRET. Mutation of several hydrophobic residues in heptad repeats of CC1 (L248, L251), CC2 (L373, A376), and CC3 (L416, L423) that were predicted to interfere with the coiled-coil structure of CC1, CC2, and CC3 resulted in a significant reduction of FRET as well as constitutive localization of STIM1 mutants (L251S and L416S/L423S) into puncta and CRAC channel activation [64]. Collectively, these studies demonstrate that intra- and intermolecular interactions between charged and hydrophobic residues in CC domains within STIM1-CT control its conformation, oligomerization, and ability to bind to ORAI1.
Important insight into the structure of the STIM1-CT was recently provided by Yang et al. [65] who crystallized human STIM1-CAD (aa 345–444) and the entire CC region (aa 212–410) of C. elegans STIM1 (corresponding to aa 233–465 of hSTIM1). When CAD was mutated (L374M, V419A, C437T) to obtain crystals that diffracted to high resolution, it preferentially formed dimers without affecting its ability to colocalize with ORAI1 and mediate SOCE [65]. Importantly, the structural studies show that CAD consists of two long (α1, α4) and two short (α2, α3) alpha helices that are arranged in an “R” shape resulting in an antiparallel alignment of α1 (NT of CAD) and α4 (CT of CAD). In its most stable configuration, CAD forms a dimer in the shape of the letter “V” (Fig. 3b) [65]. The dimer is stabilized by interactions of hydrophobic residues in the NT (L347, L350, L351) and CT (I433, L436) of the two interacting CADs. Additional interactions are provided by hydrogen bonds (R429, T354) and stacking interactions (Y361). Mutation of all three hydrophobic residues abolished the binding of CAD to ORAI1 and the ability of full-length STIM1 to activate SOCE [65], indicating that CRAC channel activation is dependent on STIM1 dimer formation. Structural data by Yang et al. [65] further suggest that binding of STIM1 to ORAI1 is mediated by a polybasic motif (K384–386) in CAD. These lysines are situated in the open part of the V-shaped CAD dimer, providing a peg-like structure that may facilitate CAD binding to ORAI1 (Fig. 3b). These lysines had been suggested before to mediate STIM1 binding to ORAI1 [63, 66]. Their location in the open part of the CAD dimer [65], however, would be inconsistent with their interaction with acidic residues in CC1 (aa 302–322) to keep the CT of STIM1 in a closed, inactive configuration, as was proposed earlier [63, 67]. It is important to note that the structure of the STIM1 dimer as revealed by Yang et al. most likely represents the “resting state” of STIM1 in cells with replete Ca2+ stores; the conformation of the active STIM1 and CAD domain awaits additional structural analyses.
In contrast to CAD, overexpression of the entire STIM1-CT (aa 238–685) results in only a small increase in basal cytosolic Ca2+ [49, 51–53, 63], suggesting that the CAD domain in the context of the entire STIM1-CT is subject to intra- or intermolecular inhibition. Kawasaki et al. [52] showed that while expression of CAD/CCb9 (aa 339–444) resulted in its colocalization with ORAI1 at the PM and constitutive Ca2+ influx, a slightly longer CCb7 fragment (aa 339–485) was located largely in the cytoplasm and was a poor activator of constitutive Ca2+ influx. Direct injection of a synthetic polypeptide encompassing aa 445–475 present in CCb7 (but not CCb9) into Jurkat T cells significantly attenuated CRAC channel function, suggesting that STIM1445–475 curtails the CRAC channel-activating function of CAD. Furthermore, several groups have proposed that STIM1-CT is kept in a closed, inactive configuration through intramolecular interactions between CAD and CC1 as discussed above [63, 64]. Consistent with these findings, the structural studies by Yang et al. [68] defined an inhibitory alpha helix at the C-terminal end of CC1 (corresponding to aa 310–337 in hSTIM1) that interacts with the basal region of the CAD dimer. The authors postulate that the inhibitory alpha helix keeps the CT of STIM1 in a closed configuration and thereby prevents STIM1 multimerization and CRAC channel activation, consistent with the observation that its deletion results in constitutive colocalization of STIM1 with ORAI1 and Ca2+ influx [68]. This idea is consistent with E322 in CC1 being part of an acidic domain that keeps STIM1 in a closed configuration through interaction with a basic domain in the middle of CAD (aa 382–386) [63]. By contrast, the structural data also suggest that the inhibitory helix (aa 310–337) interacts with residues at the N- and C-terminal ends of CAD [68], but not the basic domain (aa 382–386) located in its middle, as previously suggested [63]. An important difference between the models discussed above is whether the structure of the CAD domain (required for CRAC channel activation) is fixed, as suggested by Yang et al. [68] or undergoes a conformational change [63, 64] upon store depletion and STIM1 oligomerization. Important insight into this question will come from solving the crystal structure of STIM1 in an active, oligomerized state.
Despite the unresolved questions discussed above, the following model of STIM1-mediated CRAC channel activation emerges from these studies: In resting cells with replete ER Ca2+ stores, STIM1 exists as a dimer that is stabilized by interactions of CAD and CC1 domains [58, 68]. Upon store depletion and dissociation of Ca2+ from EFh domains in the N terminus of STIM1, the neighboring EFh and SAM domains unfold, allowing the SAM domains of adjacent STIM1 molecules to interact and promote oligomerization. The formation of stable higher-order oligomers of STIM1 requires intermolecular interactions between coiled-coil domains in the CT of STIM1, which become possible after the release of inhibitory intra- and intermolecular interactions between CC1 and CAD domains [63, 64, 66, 68]. Conformational changes in the C-terminus of STIM1 that have yet to be defined promote the translocation of STIM1 to the PM, where the K-rich region at the very CT of STIM1 forms high avidity interactions with acidic phospholipids in the PM. These interactions facilitate STIM1 binding via its CAD domain to the C- and N-termini of ORAI1. Optimal CRAC channel activation is achieved when eight STIM1 molecules bind to one tetrameric ORAI1 channel [61, 62].
Differences between STIM1 and STIM2
The mature STIM1 (685 aa) and STIM2 (746 aa) proteins show 53 % amino acid identity and possess a similar domain architecture, but diverge significantly in their C-terminus [39]. In addition to the conserved EFh, SAM, and CC domains already discussed, both proteins contain lysine-rich tails (5 Lys in STIM1, 7 Lys in STIM2) at their very C-termini, but whereas STIM1 contains a serine- and proline-rich region (aa 600–629), STIM2 possesses a pro-line- and histidine-rich region (aa 533–559). The contributions of these regions to the differential functions of STIM1 and STIM2 are not fully understood. Although STIM1 and STIM2 are ubiquitously expressed in many tissues and cell types [39], expression levels of STIM1 in murine lymphocytes are much higher compared to those of STIM2. STIM1 protein is readily detectable in naive and in vitro-differentiated CD4+ T cells, whereas very little STIM2 is found in naive T cells [69]. STIM2 levels increase after T cell stimulation in vitro whereas levels of STIM1 remain constant. These differences in expression—together with the distinct activation kinetics and thresholds of STIM1 and STIM2 activation discussed further above [41, 45]—may explain why genetic deletion of STIM1 in human and mouse T cells severely impairs SOCE whereas deletion of STIM2 in murine T cells does not affect SOCE levels and CRAC currents, at least in the first minutes following T cell stimulation. STIM2 deletion does, however, abolish sustained Ca2+ influx and activation of the Ca2+ regulated transcription factor NFAT when measured 0.5–6 h after stimulation [69]. Accordingly, expression of cytokine genes by STIM2-deficient T cells is significantly impaired [69–71], resulting in attenuated inflammation in animal models of autoimmune CNS inflammation [70, 71]. Therefore, both STIM1 and STIM2 are required for SOCE and activation of murine T cells although their quantitative contributions differ.
Regulation of CRAC channel inactivation
The tight regulation of CRAC channel function and SOCE in cells of the immune system is important to prevent aberrant immune responses with potentially deleterious consequences. In this chapter, we will discuss the molecular mechanisms that mediate CRAC channel inactivation (Fig. 4).
Fig. 4.
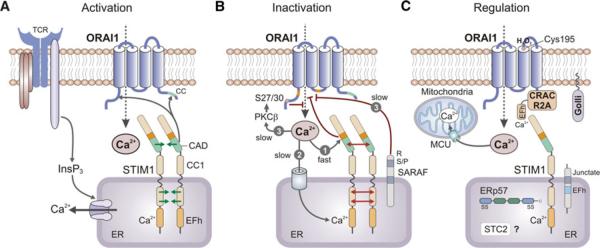
Mechanisms of CRAC channel activation, inactivation, and regulation. a CRAC channel activation. Depletion of Ca2+ from ER stores results in dissociation of Ca2+ from the EF-hand (EFh) in the N-terminus (NT) of STIM1 (and STIM2, not shown), unfolding of the STIM1-NT consisting of EFh and adjacent SAM domains and oligomerization of STIM1 molecules. Oligomerization is mediated by the interaction of multiple protein domains within STIM1 including EFh-SAM, CC1, and CAD domains. The CRAC channel activation domain (CAD, alternatively called SOAR or CCb9) in the C-terminus of STIM1 is sufficient for binding to ORAI1 and CRAC channel activation. b Fast and slow CRAC channel inactivation is mediated by distinct mechanisms. (1) Fast Ca2+-dependent CRAC channel inactivation (CDI) occurs over tens of milliseconds and is mediated by acidic residues in the STIM1-CT immediately adjacent to CAD, a calmodulin-binding domain (aa 70–87) in the N-terminus of ORAI1 and a NVHNL polypeptide in the II–III loop of ORAI1 (all indicated in orange). (2) Slow store-dependent inactivation of CRAC channels occurs over tens of seconds and is mediated by Ca2+ re-uptake into the ER via SERCA pumps, resulting in Ca2+ re-binding to the EFh in STIM1 and STIM1 de-oligomerization. (3) Slow store-independent CRAC channel inactivation also occurs over tens of seconds and is regulated by several mechanisms, including interaction of STIM1 with SARAF and potentially PKCβ-mediated phosphorylation of ORAI1 on N-terminal serine residues S27 and S30 (SF, unpublished). c Regulation of CRAC channel function by several modulatory proteins. CRACR2A forms a ternary complex with ORAI1 and STIM1 to promote puncta formation and CRAC channel activation. Junctate is a single-pass transmembrane protein in the ER that facilitates recruitment of STIM1 (and ORAI1) to ER-PM junctions. Golli is a cytoplasmic protein that is recruited to the PM in a myristoylation-dependent manner and inhibits CRAC channel function. Stanniocalcin 2 (STC2) and the ER-resident oxidoreductase ERp57 bind to STIM1 and inhibit SOCE, but the underlying mechanisms are not well understood. ORAI1 function can be directly modulated by reactive oxygen species (ROS) via redox regulation of a reactive cysteine (C195) in its III–IV loop. Finally, Ca2+ uptake into mitochondria located close the plasma membrane via the mitochondrial uniporter (MCU) prevents Ca2+-dependent CRAC channel inactivation by curtailing a rise in local Ca2+ concentrations. CAD CRAC channel activation domain, CC coiled-coil, EFh EF-hand, SAM sterile-alpha motif, SARAF SOCE-associated regulatory factor, SERCA sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
Fast calcium-dependent inactivation
A biophysical hallmark of CRAC channels is their fast Ca2+-dependent inactivation (CDI). This is observed when CRAC channel currents (ICRAC) are analyzed by patch-clamp electrophysiology following depletion of ER Ca2+ stores. CDI occurs within tens of milliseconds following the peak of ICRAC under conditions where voltage-clamped cells are hyperpolarized (i.e., their membrane potential is artificially held at very negative membrane potentials) [72–74]. Fast CDI can be diminished by chelating intracellular Ca2+ with BAPTA, a fast Ca2+ chelator, but not EGTA, a slow Ca2+ chelator [72, 74]. At the time these observations were made, they suggested that fast CDI results from Ca2+ effects on molecules in close proximity to the site of Ca2+ influx or the CRAC channel itself [73, 74]. After the discovery of ORAI1 and STIM1, the mechanisms underlying fast CDI have been revisited, largely confirming the initial predictions. In cells ectopically expressing ORAI1 and STIM1, fast CDI is observed only when full-length STIM1 or the entire STIM1-CT are expressed, but not with CAD alone, indicating that fast CDI is mediated by a domain in the distal part of STIM1-CT (aa 448–685) [49]. Indeed, a string of acidic residues in STIM1 immediately adjacent to CAD (475-DDVDDMDEE-483) is required for fast CDI and mutation of several D/E residues resulted in loss of fast CDI [75–77] (Fig. 4b). By contrast, overall CRAC channel currents were not altered. It is unlikely that the negative charges in this CRAC modulatory region (CMD) serve as Ca2+ binding sites and directly mediate CDI because the effects of mutating D/E residues on Ca2+ binding and CDI correlated poorly [77].
In addition to STIM1, CDI is mediated by domains within ORAI1 itself. Mullins et al. [77] identified a calmodulin-binding domain in the N-terminus of ORAI1 (aa 70–87) that is critical for CDI and mutation of A73, W76, or Y80 within this domain to Glu abolished calmodulin-binding and fast CDI, suggesting that calmodulin-binding to the NT of ORAI1 is required for CDI (Fig. 4b). In addition, five amino acids (NVHNL) in the intracellular loop of ORAI1 (linking TM domains II and III) were shown to be critical for fast CDI [78]. The authors of this study propose that inactivation of ICRAC is due to direct block of the ion permeation pathway by hydrophobic residues within the NVHNL peptide. Lee et al. [76] showed that fast CDI in ORAI2 and ORAI3 is dependent on three conserved Glu residues in the CT of ORAI2 (E233, E235, E236) and ORAI3 (E281, E283, E284). In this study, fast CDI was more pronounced in ORAI2 and ORAI3 channels compared to ORAI1 [27], and it was suggested that this may be due to the presence of additional glutamate residues (E235 in ORAI2, E283 in ORAI3) compared to ORAI1 [76]. Reconciling these findings, Frischauf et al. [79] argued that a complex interplay between all three cytosolic domains –(NT, CT and II–III loop) of ORAI channels are required—together with STIM1—for fast CDI. Further studies are required to elucidate the structural mechanisms by which STIM1 and ORAI1 mediate CDI. Importantly, it needs to be determined which physiological role fast CDI plays, if any, in modulating Ca2+ signals, lymphocyte function and ultimately immune responses.
Slow inactivation of CRAC channels by ER store-refilling
Inactivation of CRAC channels also occurs on another time scale. CRAC channels slowly inactivate within tens of seconds after their activation. This process is controlled by several mechanisms [80, 81]. One involves the refilling of ER Ca2+ stores following Ca2+ influx via CRAC channels and Ca2+ uptake via sarco/endoplasmic reticulum ATPase (SERCA) pumps (Fig. 4b). Upon refilling of ER stores, Ca2+ binds to the EF-hand domain in the N-terminus of STIM1 leading to de-oligomerization of STIM1, dissolution of puncta and inactivation of ICRAC [59]. Interestingly, although STIM1 oligomerization is entirely dependent on [Ca2+]ER levels, Malli et al. [82] and Shen et al. [83] found that an increase in [Ca2+]ER is required but not sufficient by itself to induce STIM1 de-oligomerization. A localized increase in cytosolic Ca2+ levels was also required for the dissociation of STIM1 puncta and their diffusion away from the plasma membrane. The mechanism by which cytosolic Ca2+ regulates STIM1 de-oligomerization is unknown. Additional mechanisms of slow CRAC channel inactivation that involve posttranslational modification of ORAI/STIM molecules and additional regulatory proteins and mechanisms will be discussed in the following chapters.
Regulation of SOCE by posttranslational modifications of ORAI/STIM proteins and other mechanisms
ORAI1 and STIM1 phosphorylation
In addition to fast CDI and sequestration of Ca2+ in the ER, the activity of CRAC channels is regulated by posttranslational modification of ORAI1 and STIM1 proteins. ATP-dependent phosphorylation leads to the slow inactivation of CRAC currents over tens of seconds. This slow inactivation was enhanced by phorbol esters and prevented by staurosporine and bisindolylmaleimide, indicating that inactivation is mediated by a protein kinase C (PKC)-dependent mechanism [81]. Kawasaki et al. [84] showed that ORAI1 is a direct target of phosphorylation by the Ca2+-dependent PKC isoform PKCβ following Ca2+ influx induced by passive store depletion. PKCβ predominantly phosphorylates the N-terminus of ORAI1 and in particular S27 and S30, but not other intracellular Ser/Thr residues of ORAI1 (Fig. 4b). The phosphorylation of S27 and S30 suppresses SOCE and cells transfected with non-phosphorylatable ORAI1-S27A/S30A mutants showed enhanced SOCE when compared to cells expressing wild-type ORAI1 [84]. In addition, the slow inactivation of CRAC currents is reduced in cells expressing mutant ORAI1-S27/30A compared to wild-type ORAI1 when cells were dialyzed with ATP or non-hydrolysable ATP-γS through the patch-clamp pipette (S.F. unpublished observations). The phosphorylation of ORAI1 on S27 and S30 may therefore represent a Ca2+-regulated, but STIM1-independent negative feedback mechanism for the slow inactivation of CRAC channels. The physiological significance of this mechanism for T cell function remains to be established.
The phosphorylation of STIM1, like that of ORAI1, has been linked to the regulation of CRAC channel function and SOCE during mitosis and meiosis. Smyth et al. [85] observed that SOCE was greatly suppressed in mitotic cells despite normal expression levels of STIM1. ORAI1 expression was slightly decreased in mitotic cells, but its overexpression did not rescue SOCE. Instead, the phosphorylation of two serine residues (486 and 668) in the C-terminus of STIM1 by cyclin-dependent kinase 1 (CDK1) was found to suppress SOCE because expression of STIM1 molecules that were truncated at position 482 or contained mutated S486 and S668 residues rescued SOCE during mitosis [85]. Cells expressing a C-terminally truncated form of STIM1 lacking S486 and S668 proliferate more slowly than cells expressing wild-type STIM1 and a larger percentage of cells are blocked in the G2/M phase of the cell cycle, potentially due to their inability to suppress SOCE during mitosis. Using a Xenopus oocyte model, Machaca and colleagues demonstrated that SOCE is also inhibited during meiosis due to impaired STIM1 puncta formation and enhanced internalization of ORAI1 into an intracellular vesicular compartment [86]. While STIM1 was found to be phosphorylated during meiosis, presumably due to the kinase function of the maturation promoting factor (MPF, a heterodimer containing cyclin B and cdc42), mutation of putative MPF phosphorylation sites had no effect on STIM1 clustering or CRAC channel function [86]. In contrast to its inhibitory effects on SOCE during mitosis and potentially during meiosis, STIM1 phosphorylation was also shown to activate SOCE. STIM1 is phosphorylated by the extracellular-signal-regulated kinases (ERK) 1/2 on serines S575, S608 and S621 located in the STIM1-CT. ERK1/2 phosphorylation is required for the ability of STIM1 to bind to ORAI1 and activate SOCE as mutation of all three serine residues impaired SOCE [87]. While puncta formation by mutant STIM1 remains intact, its colocalization with ORAI1 is inhibited, suggesting that STIM1 phosphorylation on S575/S608/S621 may regulate its ability to bind to ORAI1.
Collectively, the studies discussed above indicate that CRAC channel function is controlled by the phosphorylation status of ORAI1 and STIM1 proteins. The mechanisms underlying this phosphorylation-dependent regulation remain to be elucidated. Given the reported role of interactions between acidic and basic residues in controlling intra- and intermolecular STIM1 interactions and STIM1-ORAI1 binding (see “Mechanisms of ORAI1-CRAC channel activation by STIM proteins” above), it is tempting to speculate that the phosphorylation of serine/threonine residues in STIM1 and ORAI1 provides a means to modulate the strength of these interactions and thereby SOCE. At the cellular level, the physiological relevance of ORAI1 and STIM1 phosphorylation for lymphocyte function during immune responses is not known, but could be a mechanism to regulate T and B cell function through modulation of SOCE.
ORAI1 ubiquitination
The expression of many cell membrane proteins is regulated by their ubiquitination through E3 ubiquitin-protein ligases, which monoubiquitinate and thereby target them for internalization and lysosomal destruction. ORAI1 protein expression is regulated by the ubiquitin ligase Nedd4-2. Overexpression of Nedd4-2 decreased ORAI1 protein expression and SOCE, while interfering with Nedd4-2 expression by siRNA increased ORAI1 levels [88, 89]. The effects of Nedd4-2 on ORAI1 and SOCE were reversed by expression of the serum- and glucocorticoid-inducible kinase 1 (SGK1), a known inhibitor of Nedd4-2 function. Consistent with these findings, mast cells from sgk1−/− mice had reduced ORAI1 expression levels and SOCE. Besides ORAI1, STIM1 is a target for ubiquitination [90]. STIM1 was found to be ubiquitinated and STIM1 levels at the surface of hippocampal neurons increased when cells were treated with proteasome inhibitors. Conversely, overexpression of the E3 ligase POSH (plenty of SH3′s) resulted in decreased surface STIM1 expression. The ubiquitination sites in ORAI1 and STIM1 molecules are unknown and it remains to be investigated whether ORAI1/STIM1 ubiquitination is important for the regulation of SOCE in lymphocytes and immune responses.
Differential redox sensitivity of ORAI channels and its functional implications
ORAI1 and ORAI3 were shown to be differentially regulated by reactive oxygen species (ROS) in human T lymphocytes [91]. ORAI1, but not ORAI3, channels are inhibited by oxidation through H2O2 with an IC50 of ~40 μM. The differential redox sensitivity of ORAI1 and ORAI3 channels depended on a reactive cysteine (C195) localized in the second extracellular loop of ORAI1, but not ORAI3 (Fig. 4c). Thus ORAI channels differ not only in their inactivation properties, ion selectivity, and responsiveness to 2-APB [27], but also their redox sensitivity [91]. Interestingly, naive T cells become progressively less redox-sensitive upon differentiation into effector T cells, an effect that correlated with increased ORAI3 expression [91]. These findings suggest that differential expression of ORAI isoforms is a mechanism to fine tune the redox sensitivity and thereby potentially the function and survival of T cells, for instance under oxidizing conditions in inflamed tissues. Mitochondria produce ROS including H2O2 and their localization close to the immunological synapse (IS) between T cells and antigen-presenting cells (see “Spatial regulation of CRAC channel function at the immunological synapse” below) may influence the activity ORAI1 and ORAI3 by regulating the redox environment in which CRAC channels operate. In the future, the interplay between mitochondrial Ca2+ and redox homeostasis and its potential role for signal transduction at the IS needs to be investigated with tools that allow the subcellular analysis of ROS signaling.
Maintaining CRAC channel activity and SOCE by mitochondrial Ca2+ uptake
Ca2+ sequestration by mitochondria located in close proximity to the PM is an important mechanism to regulate the activity of CRAC channels and thereby the duration and amplitude of Ca2+ influx (Fig. 4c). Mitochondrial Ca2+ uptake prevents the formation of microdomains with high intracellular Ca2+ concentrations close to the mouth of the CRAC channel and thereby Ca2+-dependent ORAI1/CRAC channel inactivation. This was first shown by inhibiting mitochondrial Ca2+ uptake with uncouplers such as carbonyl cyanide m-chlorophenylhydrazone (CCCP) or antimycin A1, which interfered with CRAC channel currents and sustained Ca2+ influx in T cells and mast cells [92–95]. In agreement with the role of mitochondria in buffering Ca2+ entering the cell through CRAC channels, mitochondria were shown to localize close to the PM in T cells following stimulation [96] and to be enriched at the immunological synapse (IS), which is formed between T cells and antigen-presenting cells [97, 98]. Consistent with the localization of overexpressed and endogeneous ORAI1 channels at the IS [97, 99, 100], local Ca2+ domains in close proximity to the IS were modulated by mitochondria [97], thereby preventing slow Ca2+-dependent inactivation of CRAC channels and maintaining SOCE and NFAT translocation to the nucleus. Mitochondria are well suited for this task as they use a high capacity Ca2+ uptake mechanism in the form of the mitochondrial Ca2+ uniporter (MCU) [101, 102]. By contrast, the SERCA-dependent Ca2+ uptake into the ER is slow and ill-suited to prevent CRAC channel inactivation; on the contrary, it inhibits SOCE by promoting the binding of Ca2+ to the EFh of STIM1 and dissolution of STIM1 puncta. Mitochondria ensure that ORAI-CRAC channels localized at the IS are active over long periods of time in contrast to ORAICRAC channels elsewhere in the PM, which are inactivated by increases in [Ca2+]i [98].
Mitochondrial relocalization to the IS itself is highly Ca2+-dependent and requires the actin cytoskeleton [98, 103] as well as motor-assisted transport along microtubules [96]. In neurons, mitochondrial movement depends on kinesin-1, which interacts with the mitochondrial protein Miro [104]. Ca2+ binding to the EFh domains of Miro results in arrest of mitochondrial motility as Ca2+ bound Miro directly interacts with the motor domain of Kinesin-1, thereby preventing motor-microtubule interactions and mitochondrial movement [104]. Whether similar mechanisms are at work in lymphocytes remains to be investigated, but it is tempting to speculate that mitochondria are arrested at sites of high Ca2+ concentrations near ORAI1-CRAC channels at the IS to sustain SOCE and Ca2+-dependent signaling processes and gene expression in T cells [69, 97, 105, 106].
Additional molecules regulating CRAC channel function
STIM1 and ORAI1 are generally considered to be necessary and sufficient to mediate CRAC channel function and SOCE in lymphocytes and other cell types. Genetic deletion of ORAI1 or STIM1 in human and mouse lymphocytes abolishes SOCE or severely impairs it [8, 28, 29, 69, 107, 108]. Conversely, co-expression of ORAI1 and STIM1 in the same cell, but not either protein alone, results in 10–100-fold amplification of CRAC current amplitudes [109, 110], indicating that ORAI1 and STIM1 are sufficient for CRAC channel function and are rate-limiting factors. To prove that STIM1-ORAI1 interaction is sufficient to gate ORAI1 channels in the absence of other proteins, Zhou et al. [56] co-expressed STIM1 and ORAI1 in the yeast Saccharomyces cerevisiae, which lacks endogenous SOCE activity and expresses no STIM or ORAI homologues. Their experiments elegantly showed that STIM1 fragments containing CAD are sufficient to activate ORAI1 and Ca2+ flux in membrane vesicles from S. cerevisiae. While these studies argued that additional molecules are not required for ORAI1 CRAC channel activation, they do not exclude that other proteins may bind to and modulate the function of ORAI1/STIM1 complexes. Indeed, a number of proteins have been identified that either promote or inhibit CRAC channel function (Fig. 4c).
Activators of CRAC channel function
Gwack and coworkers identified a novel family of EF-hand containing proteins, CRACR2A and B, which directly bind to ORAI1 and STIM1 and function as positive regulators of SOCE (Fig. 4c) [111]. At low intracellular Ca2+ concentrations, CRACR2A forms a ternary complex with ORAI1 and STIM1 to promote puncta formation and thereby SOCE. It dissociates from ORAI1/STIM1 puncta upon increases in [Ca2+]i mediated by Ca2+ binding to its EF-hand domain. Deletion of CRACR2A impaired SOCE, whereas expression of EF-hand mutants that mimic the Ca2+-free state of CRACR2A resulted in spontaneous STIM1 puncta formation and enhanced SOCE. In the model proposed by the authors, CRACR2A temporarily stabilizes ORAI1-STIM1 complexes after store-depletion and before [Ca2+]i increases due to SOCE. In search of other ORAI/STIM-interacting molecules, the same group recently identified junctate, a single-pass transmembrane protein that is an alternative splice isoform of junctin and aspartyl β-hydroxylase [112] (Fig. 4c). Junctate is known to be present in triad/dyad junctions in skeletal muscle, where it plays a role in coupling voltage-gated Ca2+ channels to ryanodine receptors in the SR [113]. In Jurkat T cells with replete Ca2+ stores, junctate is localized at ER-PM junctions. Upon store depletion, it colocalizes with STIM1 in puncta and can be co-immunoprecipitated with STIM1 (as well as ORAI1) [112]. Mutation of an EF-hand domain in the ER lumenal part of junctate results in SOCE due to clustering of STIM1 that is independent of store depletion. These data suggest that junctate facilitates recruitment of STIM1 (and ORAI1) to ER-PM junctions via protein interactions in the ER lumen. Another protein recently identified to bind to ORAI1 and STIM1 in Jurkat T cells is a partner of STIM1 (POST), a ten-transmembrane spanning protein of unknown function [114]. While POST relocates to the plasma membrane together with STIM1 upon ER store depletion, it does not directly regulate SOCE as its knock-down by siRNA or its overexpression did not have significant effects on SOCE in T cells. However, POST mediates the binding of STIM1 to plasma membrane Ca2+ ATPases (PMCA) and attenuates their activity. PMCAs pump Ca2+ out of the cytoplasm into the extracellular space, and by inhibiting PMCAs, POST prevents Ca2+ clearance from the cytosol and increases [Ca2+]i independent of SOCE.
A protein that may directly regulate ORAI1 function and Ca2+ influx independent of STIM1 is the secretory pathway Ca2+-ATPase (SPCA2), which is known to regulate Ca2+ sequestration in the Golgi apparatus [115]. Overexpression of SPCA2 in HEK293 cells caused NFAT activation and increased Ca2+ influx, which was suggested to be mediated by direct binding of SPCA2 to the CT of ORAI1 in a STIM1- and store-independent manner. Interestingly, SPCA2 is upregulated in breast cancer cells and suppression of SPCA2 moderately attenuates basal Ca2+ levels and tumorigenicity [115], suggesting that constitutive Ca2+ influx via ORAI1 and SPCA2 may promote tumor growth. Whether SPCA2 can regulate other Ca2+ channels besides ORAI1 and if it plays a role in non-malignant cells including lymphocytes is presently unclear.
Proteins inhibiting CRAC channel function
Several proteins have been identified that negatively regulate CRAC channel function and SOCE (Fig. 4c). One of them is Golli, the product of alternative transcription from the myelin basic protein (MBP) gene, which is expressed throughout the immune system, including T cells [116, 117]. Golli is a cytoplasmic protein but its inhibitory effect on SOCE depended on myristoylation and recruitment to the plasma membrane. The mechanism underlying Golli's effect on SOCE is unknown, but it was suggested that Golli binds to the CT of STIM1 and that Golli, STIM1, and ORAI1 colocalize at the plasma membrane [118]. T cells from Golli−/− mice showed enhanced SOCE in response to anti-CD3 stimulation and ICRAC following thapsigargin treatment, resulting in enhanced T cell proliferation in vitro. Another negative regulator of SOCE is stanniocalcin 2 (STC2), and fibroblasts lacking this protein showed enhanced SOCE [119]. Stanniocalcin 2 directly binds to STIM1 and colocalizes with it in the ER. Although its expression is upregulated in response to ER stress consistent with its potential role in the regulation of SOCE [120], stanniocalcin 2 is best characterized as a secreted glyco-protein. Its role in the ER and the mechanisms by which it inhibits STIM1 function and SOCE remain to be defined. The oxidoreductase ERp57 is localized in the ER, where it functions not only as a component of the peptide-loading complex of MHC class I molecules [121, 122] but is also involved in the modulation of SERCA function [123]. ERp57 was identified by Biacore analysis as a STIM1 binding protein. Genetic deletion of ERp57 moderately enhanced SOCE [124], suggesting that ERp57 inhibits SOCE by interfering with STIM1 function. Mutation of a pair of cysteine residues in the NT of STIM1 (C49, C56) partially reduces the binding of ERp57 to STIM1 (as measured by FRET) but profoundly inhibits STIM1 puncta formation and SOCE [124]. The mechanisms by which ERp57 modulates STIM1 function remain to be elucidated, but the finding by Hawkins et al. [125] that STIM1-C56 is modified by S-glutathionylation in response to oxidative stress resulting in store-independent, constitutive Ca2+ entry suggests that ERp57 may be involved in the redox regulation of SOCE.
Another ER protein that inhibits SOCE by binding to STIM1 is the SOCE-associated regulatory factor (SARAF, Fig. 4c) [126]. SARAF is a single-pass transmembrane protein that, like STIM1 and STIM2, is localized in the membrane of the ER and that is highly expressed in immune and neuronal tissues [126]. Its overexpression reduced basal cytosolic Ca2+ levels, SOCE and CRAC currents, whereas RNAi-mediated depletion had the opposite effect. In cells with replete ER Ca2+ stores, SARAF colocalized with STIM1 and prevented its recruitment into puncta and the activation of Ca2+ influx. Upon store depletion, SARAF was recruited into puncta together with STIM1 in a process that requires the cytoplasmic C-terminus of SARAF. Overexpression of SARAF did not reduce the peak amplitude of ICRAC but resulted in a more pronounced slow CRAC channel inactivation [126]. Inactivation was dependent on Ca2+ influx and correlated with enhanced dissociation of STIM1 oligomers. Collectively, these findings suggest that SARAF prevents STIM1 and STIM2 activation in resting cells and promotes STIM1 deoligomerization following Ca2+ influx, thereby limiting increases in [Ca2+]i. Further studies are required to determine if SARAF detects changes in cytoplasmic and ER Ca2+ levels directly or through its interaction with STIM1. It also remains to be analyzed if SARAF can directly bind to ORAI1 and whether it interacts with other proteins that regulate CRAC channel function described above. Taken together, the fact that several proteins have been identified that inhibit CRAC channel function emphasizes the importance of limiting SOCE to avoid potentially deleterious effects on cellular function and survival.
Spatial regulation of CRAC channel function at the immunological synapse
Spatial rearrangement of signaling molecules and organelles serves as a mechanism to regulate cellular activation. A good example of this principle in the immune system is the polarization that occurs during formation of an immunological synapse (IS) between a T cell and an antigen-presenting cell (APC), which is accompanied by enrichment and depletion of proteins and organelles at the IS [127, 128]. Several studies showed that ORAI1 and STIM1 proteins are recruited to the IS between T cells and APCs [97, 99, 100]. In one study, ORAI1 and STIM1 ectopically expressed in Jurkat T cells and primary human T cells accumulated at the IS formed with superantigen-pulsed Raji B cells or dendritic cells, resulting in localized Ca2+ influx at the IS [100]. Endogenous ORAI1 accumulation at the Jurkat T–Raji B cell interface was observed by Quintana et al. [97]. Another study also reported that ORAI1 and STIM1 may colocalize in clusters in the vicinity of the IS, but furthermore showed that both molecules are located together in cap-like structures at the distal pole of Jurkat T cells away from the TCR [99]. These ORAI1/STIM1-containing caps dynamically rearranged when T cells interacted with superantigen-loaded Raji B cells, resulting in the recruitment of ORAI1 and STIM1 to the existing or newly formed IS at least in a fraction of cells.
It is not understood how ORAI1 and STIM1 are recruited to the IS. Considering that STIM1 puncta form before CRAC channel activation [47], it appears reasonable to assume that STIM1 puncta may form at the IS prior to ORAI1 channel activation. This favors a model where STIM1 anchors ORAI1 at the IS to provide a physical basis for the local activation of Ca2+ influx. Ca2+ influx itself does not appear to be important for ORAI1/STIM1 accumulation at the IS, since expression of a nonconducting mutant of ORAI1 (E106A) that abolishes Ca2+ influx does not interfere with the localization of STIM1 and ORAI1 at the IS [100]. CRAC channel activation in other cell types requires the formation of ER-PM junctions in which both membranes are juxtaposed at close range (10–25 nm) [48] to facilitate interactions between ORAI1 and STIM1 in separate membrane compartments. Indeed, Orci et al. [129] showed by ultrastructural analysis of HeLa cells that STIM1-rich subdomains of the ER form cortical ER domains in close contact with the PM upon depletion of Ca2+ from the ER. Recruitment of STIM1 to the IS may therefore require the formation of ER-PM junctions near the IS, but this has not yet been investigated. It is conceivable that the ER is dynamically recruited to the IS during T cell polarization, potentially with the help of proteins such as junctate as discussed above [112].
Localized Ca2+ influx at the IS is sustained by the recruitment of mitochondria to the IS [97, 98] as mitochondrial Ca2+ uptake was shown to prevent the Ca2+-dependent inactivation of CRAC channels as discussed in “Regulation of SOCE by posttranslational modification and other mechanisms” above [96]. Intriguingly, although both mitochondria and ORAI1 were shown to be recruited to the IS, they did not directly colocalize [97]. This is expected as ORAI1-STIM1 interactions require the formation of tight ER-PM junctions [48], which exclude mitochondria. It is likely, however, that mitochondria are positioned directly outside the ER-PM junctions to take up Ca2+ that fluxes into this narrow space after CRAC channel opening. Thus, in addition to the enrichment of ORAI1 and STIM1 at the IS, the recruitment and retention of mitochondria close to the IS is essential to sustain Ca2+ influx and T cell activation, especially gene transcription that requires prolonged Ca2+ influx.
Localized Ca2+ influx at the IS may furthermore be of physiological importance in cytotoxic CD8+ T cells and NK cells. Formation of a lytic IS between CD8+ T cells and their targets, for instance tumor or virus-infected cells, ensures antigen-specific killing and minimizes unspecific effects on bystander cells [130, 131]. Since secretion of cytolytic granules containing perforin and granzymes by cytotoxic lymphocytes requires Ca2+ influx [132–135], localized SOCE at the IS mediated by polarized ORAI/STIM molecules may facilitate the spatially restricted release of cytolytic molecules.
Consequences of CRAC channel dysregulation on lymphocyte function
Given the many mechanisms that exist to control CRAC channel function, it is reasonable to assume that SOCE and [Ca2+]i have to be tightly regulated to avoid the potentially deleterious consequences of abnormal CRAC channel activity and SOCE on cell function. In the following, we will briefly discuss the outcome of CRAC channel dysregulation on lymphocyte function and adaptive immune responses.
Decreased CRAC channel function
The effect of decreased CRAC channel function on lymphocyte-mediated immune responses is perhaps best highlighted by patients with mutations in ORAI1 or STIM1 genes that abolish ICRAC and Ca2+ influx [8, 28, 108, 136, 137]. The mutations and clinical phenotypes in these patients have been described in greater detail elsewhere [137, 138]. Briefly, the dominant clinical phenotype observed in patients with CRAC channelopathy is immunodeficiency with recurrent and life-threatening bacterial, fungal, and viral infections in early infancy [136, 139]. The spectrum of infections and their typical onset is similar to the immunophenotype observed in patients with severe combined immunodeficiency (SCID). However, in contrast to SCID patients, patients with mutations in STIM1 or ORAI1 have normal lymphocyte numbers and a normal TCR repertoire suggesting that lymphocyte development is not dependent on CRAC channel function and SOCE [140].
Despite normal development, the function of SOCE-deficient lymphocytes is strongly impaired. Lymphocytes from patients with mutations in STIM1 or ORAI1 have a significantly reduced proliferative response to TCR and mitogen stimulation and fail to produce a variety of effector cytokines such as IL-2 and IFNγ [28, 108, 141, 142]. Furthermore, the cytotoxic function of NK cells is significantly impaired in the absence of SOCE [134]. The immunodeficiency in patients with CRAC channelopathy is recapitulated in mice with targeted deletion of Orai and Stim genes. The production of effector cytokines by CD4+ T helper cells such as Th1, Th2, and Th17 cells is impaired in the absence of functional STIM1, STIM2, or ORAI1 [30, 70, 107]. Consistent with decreased T cell function in vitro, ORAI1- and STIM1-deficient mice displayed blunted or abolished T cell-dependent immune responses in vivo. ORAI1-deficient mice tolerated allogeneic skin grafts significantly longer than wild-type control mice and failed to mount Th2-mediated contact hypersensitivity responses [30]. ORAI1- and STIM1-deficient mice were protected from developing autoimmune inflammatory bowel disease (IBD) in a murine model of colitis following adoptive transfer of naive CD4+ CD45RB+ T cells [30]. Furthermore, SOCE is required for the induction of T cell-mediated autoimmune CNS inflammation as mice with T cell-specific or complete deletion of STIM1 failed to develop experimental autoimmune encephalomyelitis (EAE), whereas STIM2-deficient mice were partially protected from EAE [70, 71]. Protection is due to impaired function of Th17 cells that play an important role in the pathology of EAE and multiple sclerosis. In contrast to T cells, the function of B cells seems to be largely preserved in STIM1 and STIM1/STIM2-deficient mice as T-dependent and T-independent antibody responses following immunization of mice was intact [107, 138, 143]. Likewise, no defect in B cell development has been observed in SOCE-deficient patients or mice.
CRAC channel function is required for the development of CD4+ CD25+ Foxp3+ regulatory T (Treg) cells, the numbers of which were moderately reduced in patients with mutations in STIM1 [108, 144] and severely decreased in mice with combined, T cell-specific deletion of Stim1 and Stim2 genes [69]. Reduced Treg numbers are a likely explanation for the autoimmunity observed in the patients, which is characterized by autoimmune hemolytic anemia and thrombocytopenia as well as lymphadenopathy and hepatosplenomegaly [108, 136, 141]. A similar phenotype is observed in STIM1/STIM2-deficient mice, which develop pulmonary, intestinal and salivary gland inflammation due to failed Treg-mediated suppression of leukocyte infiltration and activation in these organs [69, 145]. The function of Treg cells depends on SOCE as deletion of both Stim1 and Stim2 genes in mice abolished Treg-mediated suppression of lymphocyte proliferation in vitro. By contrast, lack of STIM1 or functional ORAI1 in Orai1KI/KI mice only moderately affected Treg function (and had no effect on Treg development). Since small residual SOCE is present in ORAI1- and STIM1-deficient T cells, these findings suggest that Treg cells require very little Ca2+ influx [30, 69, 107]. On the other hand, proinflammatory Th17 cells are more dependent on CRAC channels as even moderately reduced SOCE in STIM2-deficient mice significantly impaired their function [70]. The molecular basis for these distinct SOCE requirements in different T cell lineages remains to be investigated. A more detailed review of the role of CRAC channels in different immune cells including lymphocytes can be found in [146, 147].
Increased CRAC channel function
The number of mechanisms and proteins identified to curtail CRAC channel function discussed above suggest that SOCE needs to be tightly controlled. However, the pathophysiological outcome of elevated CRAC channel activity on lymphocytes and other cell types has been explored only in a couple of studies. One of the best examples of how constitutive CRAC channel activation affects cell function is provided by mice that carry an activating D84G mutation in the EFh domain of STIM1, which were generated by random N-ethyl-N-nitrosourea (ENU) mutagenesis [43]. Mice homozygous for the mutation were embryonically lethal around E13.5 with severe hemorrhages, most likely due to vascular defects or platelet dysfunction. By contrast, heterozygous mice were viable, and T cells from these mice displayed elevated resting [Ca2+]i, but normal SOCE in response to TCR stimulation [43]. Importantly, T cell development, proliferation, and cytotoxic function were normal in Stim1D84G/+ mutant mice. These mice did, however, develop splenomegaly, thrombocytopenia, and a bleeding disorder, which has been attributed to a decreased life span of platelets. Surprisingly perhaps, no other phenotypes such as autoimmunity have been reported in these mice despite constitutive CRAC channel activation.
Feng et al. [117] examined the effects of enhanced SOCE in lymphocytes by studying T cells from mice lacking Golli, a negative regulator of SOCE. T cells from Golli−/− mice had normal resting [Ca2+]i, but increased SOCE and ICRAC following TCR stimulation or passive store depletion, respectively. Golli−/− mice had normal T cell development in the thymus but showed increased numbers of CD3+ T cells in peripheral lymphoid organs and an increased CD4+:CD8+ ratio [117]. This phenotype was attributed to significantly increased T cell proliferation and IL-2 production upon TCR stimulation. Based on the enhanced activation of Golli-deficient T cells observed in vitro, one might have predicted exacerbated T cell-mediated immune responses in vivo, but the opposite was the case. When Golli−/− mice were immunized with MOG peptide, they displayed significantly reduced severity of EAE compare to wild-type littermates. T cells isolated from MOG-immunized Golli−/− mice proliferated poorly when restimulated with MOG in vitro, suggesting that they had become unresponsive to repeated TCR stimulation. This observation is consistent with the model proposed by Macian et al. [133] that Ca2+ signaling in the absence of costimulation causes T cell anergy through activation of NFAT alone, whereas productive T cell responses require activation of NFAT together with other transcription factors such as AP-1 [132]. It will be important to study in more detail how increased SOCE causes T cell anergy, and if, conversely, impaired SOCE results in loss of immunological tolerance and autoimmunity by interfering with the induction of T cell anergy. The effects of deleting other CRAC channel inhibitory proteins such as stanniocalcin 2, ERp57, or SARAF on lymphocyte function and immune responses have not been studied. Likewise, it remains to be elucidated if the mechanisms that result in fast or slow CRAC channel inactivation discussed further above play a role in controlling lymphocyte function.
Conclusions and outlook
CRAC channels and SOCE are present in most, if not all, immune cells, including lymphocytes. It is therefore not surprising that many mechanisms exist to control SOCE and thereby immune responses. Since the crucial discoveries of ORAI1 and STIM1 as the elementary units of store-operated Ca2+ entry [8–10, 32, 33, 47] that mediate CRAC channel function [56], several additional modulators of this pathway have been identified that either promote or restrain SOCE. Future studies will need to clarify the precise molecular mechanisms—including structural information—by which ORAI and STIM proteins regulate CRAC channel function and to identify other proteins that modulate SOCE. Even before the discovery of ORAI/STIM molecules, careful electrophysiological analyses had described processes such as fast and slow CRAC channel inactivation that play potentially important roles in shaping Ca2+ signals and lymphocyte functions. We now possess a better molecular understanding of CRAC channel inactivation and can, for the first time, test if interfering with these processes affects cell function and immune responses. More importantly yet, we are now able to systematically test the role of CRAC channels themselves in lymphocyte function and immunity by gene targeting in animal models or the study of human patients with mutations in CRAC channel genes. Some of the questions to address with regard to the role of CRAC channels in immunity will be which ORAI and STIM homologues mediate leukocyte function, whether distinct requirements for CRAC channel function exist in different lymphocyte lineages and how modulation of SOCE influences immune responses. Answering these questions will be critical to determine whether CRAC channel inhibition might be a safe and effective therapeutic approach for the treatment of autoimmunity, inflammation, and other forms of immune dysregulation in the future.
Acknowledgments
We thank the members of our labs and M. Prakriya for a critical reading of the manuscript and many helpful suggestions. This work was funded by NIH grant AI066128 to S.F., a postdoctoral fellowship by the National Multiple Sclerosis Society to P.S. and grants from the Deutsche Forschungsgemeinschaft SFB 894 (project A1), IRTG 1830, and GRK 1326 to M.H.
References
- 1.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 2.Penner R, Matthews G, Neher E. Regulation of calcium influx by second messengers in rat mast cells. Nature. 1988;334:499–504. doi: 10.1038/334499a0. [DOI] [PubMed] [Google Scholar]
- 3.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 5.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 7.Clapham DE, Runnels LW, Strubing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 8.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 9.Vig M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang SL, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNally BA, Yamashita M, Engh A, Prakriya M. Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci USA. 2009;106:22516–22521. doi: 10.1073/pnas.0909574106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prakriya M, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 13.Vig M, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeromin AV, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y, Ramachandran S, Oh-Hora M, Rao A, Hogan PG. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci USA. 2010;107:4896–4901. doi: 10.1073/pnas.1001169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita M, Navarro-Borelly L, McNally BA, Prakriya M. Orai1 mutations alter ion permeation and Ca2+-dependent inactivation of CRAC channels: evidence for coupling of permeation and gating. J Gen Physiol. 2007;130:525–540. doi: 10.1085/jgp.200709872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gwack Y, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 18.Derler I, et al. Increased hydrophobicity at the N-terminus/membrane interface impairs gating of the SCID-related ORAI1 mutant. J Biol Chem. 2009;23:15903–15915. doi: 10.1074/jbc.M808312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang SL, et al. Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc Natl Acad Sci USA. 2011;108:17838–17843. doi: 10.1073/pnas.1114821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNally BA, Prakriya M. Permeation, selectivity, and gating in store-operated CRAC channels. J Physiol. 2012;590:4179–4191. doi: 10.1113/jphysiol.2012.233098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNally BA, Somasundaram A, Yamashita M, Prakriya M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482:241–245. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penna A, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji W, et al. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA. 2008;105:13668–13673. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–17556. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 27.Lis A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCarl CA, et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124:1311–1318. e1317. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gwack Y, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–5222. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarl CA, et al. Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allo-graft rejection. J Immunol. 2010;185:5845–5858. doi: 10.4049/jimmunol.1001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vig M, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+ - store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang SL, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spassova MA, et al. STIM1 has a plasma membrane role in the activation of store-operated Ca(2+) channels. Proc Natl Acad Sci USA. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manji SS, et al. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000;1481:147–155. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- 37.Oritani K, Kincade PW. Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 1996;134:771–782. doi: 10.1083/jcb.134.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997;57:4493–4497. [PubMed] [Google Scholar]
- 39.Williams RT, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–685. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams RT, et al. Stromal interaction molecule 1 (STIM1), a transmembrane protein with growth suppressor activity, contains an extracellular SAM domain modified by N-linked glycosylation. Biochim Biophys Acta. 2002;1596:131–137. doi: 10.1016/s0167-4838(02)00211-x. [DOI] [PubMed] [Google Scholar]
- 41.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–732. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 42.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–122. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Grosse J, et al. An EF-hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J Clin Invest. 2007;117:3540–3550. doi: 10.1172/JCI32312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 45.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kar P, Bakowski D, Di Capite J, Nelson C, Parekh AB. Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc Natl Acad Sci USA. 2012;109:6969–6974. doi: 10.1073/pnas.1201204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–813. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu P, et al. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem Biophys Res Commun. 2006;350:969–976. doi: 10.1016/j.bbrc.2006.09.134. [DOI] [PubMed] [Google Scholar]
- 51.Yuan JP, et al. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C-terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muik M, et al. A cytosolic homomerization and a modulatory domain within STIM1 C-terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284:8421–8426. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muik M, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 55.Navarro-Borelly L, et al. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Y, et al. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010;17:112–116. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baba Y, et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2006;103:16704–16709. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Covington ED, Wu MM, Lewis RS. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol Biol Cell. 2010;21:1897–1907. doi: 10.1091/mbc.E10-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2011;108:13299–13304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Z, et al. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011;21:305–315. doi: 10.1038/cr.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korzeniowski MK, Manjarres IM, Varnai P, Balla T. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci Signal. 2010;3:ra82. doi: 10.1126/scisignal.2001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muik M, et al. STIM1 couples to ORAI1 via an intra-molecular transition into an extended conformation. EMBO J. 2011;30:1678–1689. doi: 10.1038/emboj.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang X, Jin H, Cai X, Li S, Shen Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2012;109:5657–5662. doi: 10.1073/pnas.1118947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calloway N, Holowka D, Baird B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry. 2010;49:1067–1071. doi: 10.1021/bi901936q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Deng X, Gill DL. Calcium signaling by STIM and Orai: intimate coupling details revealed. Sci Signal. 2010;3:e42. doi: 10.1126/scisignal.3148pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang X, Jin H, Cai X, Li S, Shen Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2012;109:5657–5662. doi: 10.1073/pnas.1118947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh-Hora M, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma J, McCarl CA, Khalil S, Luthy K, Feske S. T-cell specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur J Immunol. 2010;40:3028–3042. doi: 10.1002/eji.201040614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuhmann MK, et al. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol. 2010;184:1536–1542. doi: 10.4049/jimmunol.0902161. [DOI] [PubMed] [Google Scholar]
- 72.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell Calcium. 2003;33:311–321. doi: 10.1016/s0143-4160(03)00045-9. [DOI] [PubMed] [Google Scholar]
- 74.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Derler I, et al. A Ca2(+)release-activated Ca2(+) (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2(+)-dependent inactivation of ORAI1 channels. J Biol Chem. 2009;284:24933–24938. doi: 10.1074/jbc.C109.024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee KP, et al. Molecular determinants of fast Ca2+ - dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci USA. 2009;106:14687–14692. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mullins FM, Park CY, Dolmetsch RE, Lewis RS. STIM1 and calmodulin interact with Orai1 to induce Ca2+ -dependent inactivation of CRAC channels. Proc Natl Acad Sci USA. 2009;106:15495–15500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Srikanth S, Jung HJ, Ribalet B, Gwack Y. The intracellular loop of Orai1 plays a central role in fast inactivation of Ca2+ release-activated Ca2+ channels. J Biol Chem. 2010;285:5066–5075. doi: 10.1074/jbc.M109.072736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frischauf I, et al. Cooperativeness of Orai cytosolic domains tunes subtype-specific gating. J Biol Chem. 2011;286:8577–8584. doi: 10.1074/jbc.M110.187179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J Biol Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- 81.Parekh AB, Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proc Natl Acad Sci USA. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malli R, Naghdi S, Romanin C, Graier WF. Cytosolic Ca2+ prevents the subplasmalemmal clustering of STIM1: an intrinsic mechanism to avoid Ca2+ overload. J Cell Sci. 2008;121:3133–3139. doi: 10.1242/jcs.034496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shen WW, Frieden M, Demaurex N. Local cytosolic Ca2+ elevations are required for STIM1 de-oligomerization and termination of store-operated Ca2+ entry. J Biol Chem. 2011;286:36448–36459. doi: 10.1074/jbc.M111.269415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kawasaki T, Ueyama T, Lange I, Feske S, Saito N. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J Biol Chem. 2010;285:25720–25730. doi: 10.1074/jbc.M109.022996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smyth JT, et al. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol. 2009;11:1465–1472. doi: 10.1038/ncb1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu F, Sun L, Machaca K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc Natl Acad Sci USA. 2009;106:17401–17406. doi: 10.1073/pnas.0904651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pozo-Guisado E, et al. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J Cell Sci. 2010;123:3084–3093. doi: 10.1242/jcs.067215. [DOI] [PubMed] [Google Scholar]
- 88.Eylenstein A, et al. Stimulation of Ca2+ -channel Orai1/STIM1 by serum- and glucocorticoid-inducible kinase 1 (SGK1) Faseb J. 2011;25:2012–2021. doi: 10.1096/fj.10-178210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lang F, Eylenstein A, Shumilina E. Regulation of Orai1/STIM1 by the kinases SGK1 and AMPK. Cell Calcium. 2012 doi: 10.1016/j.ceca.2012.05.005. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 90.Keil JM, Shen Z, Briggs SP, Patrick GN. Regulation of STIM1 and SOCE by the ubiquitin-proteasome system (UPS) PLoS ONE. 2010;5:e13465. doi: 10.1371/journal.pone.0013465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bogeski I, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3:ra24. doi: 10.1126/scisignal.2000672. [DOI] [PubMed] [Google Scholar]
- 92.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoth M, Button DC, Lewis RS. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci USA. 2000;97:10607–10612. doi: 10.1073/pnas.180143997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gilabert JA, Parekh AB. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC) EMBO J. 2000;19:6401–6407. doi: 10.1093/emboj/19.23.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Glitsch MD, Bakowski D, Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 2002;21:6744–6754. doi: 10.1093/emboj/cdf675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Quintana A, et al. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem. 2006;281:40302–40309. doi: 10.1074/jbc.M607896200. [DOI] [PubMed] [Google Scholar]
- 97.Quintana A, et al. Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J. 2011;30:3895–3912. doi: 10.1038/emboj.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Quintana A, et al. T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci USA. 2007;104:14418–14423. doi: 10.1073/pnas.0703126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barr VA, et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol Biol Cell. 2008;19:2802–2817. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lioudyno MI, et al. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci USA. 2008;105:2011–2016. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baughman JM, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria positioning controls local calcium influx in T cells. J Immunol. 2010;184:184–190. doi: 10.4049/jimmunol.0902872. [DOI] [PubMed] [Google Scholar]
- 104.Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 106.Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–324. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- 107.Beyersdorf N, et al. STIM1-independent T cell development and effector function in vivo. J Immunol. 2009;182:3390–3397. doi: 10.4049/jimmunol.0802888. [DOI] [PubMed] [Google Scholar]
- 108.Picard C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peinelt C, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Soboloff J, et al. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 111.Srikanth S, et al. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;12:436–446. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Srikanth S, et al. Junctate is a Ca2+-sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2012;109:8682–8687. doi: 10.1073/pnas.1200667109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu Rev Biochem. 2011;80:973–1000. doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Krapivinsky G, Krapivinsky L, Stotz SC, Manasian Y, Clapham DE. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc Natl Acad Sci USA. 2011;108:19234–19239. doi: 10.1073/pnas.1117231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feng M, et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell. 2010;143:84–98. doi: 10.1016/j.cell.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feng JM. Minireview: expression and function of Golli protein in immune system. Neurochem Res. 2007;32:273–278. doi: 10.1007/s11064-006-9164-1. [DOI] [PubMed] [Google Scholar]
- 117.Feng JM, et al. Golli protein negatively regulates store depletion-induced calcium influx in T cells. Immunity. 2006;24:717–727. doi: 10.1016/j.immuni.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 118.Walsh CM, Doherty MK, Tepikin AV, Burgoyne RD. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem J. 2010;430:453–460. doi: 10.1042/BJ20100650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zeiger W, et al. Stanniocalcin 2 is a negative modulator of store-operated calcium entry. Mol Cell Biol. 2011;31:3710–3722. doi: 10.1128/MCB.05140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ito D, et al. Characterization of stanniocalcin 2, a novel target of the mammalian unfolded protein response with cyto-protective properties. Mol Cell Biol. 2004;24:9456–9469. doi: 10.1128/MCB.24.21.9456-9469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 122.Wearsch PA, Cresswell P. Selective loading of high-affinity peptides onto major histocompatibility complex class I molecules by the tapasin-ERp57 heterodimer. Nat Immunol. 2007;8:873–881. doi: 10.1038/ni1485. [DOI] [PubMed] [Google Scholar]
- 123.Li Y, Camacho P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J Cell Biol. 2004;164:35–46. doi: 10.1083/jcb.200307010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Prins D, Groenendyk J, Touret N, Michalak M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011;12:1182–1188. doi: 10.1038/embor.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hawkins BJ, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190:391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Palty R, Raveh A, Kaminsky I, Meller R, Reuveny E. SARAF inactivates the store-operated calcium entry machinery to prevent excess calcium refilling. Cell. 2012;149:425–438. doi: 10.1016/j.cell.2012.01.055. [DOI] [PubMed] [Google Scholar]
- 127.Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol. 2000;1:23–29. doi: 10.1038/76877. [DOI] [PubMed] [Google Scholar]
- 128.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 129.Orci L, et al. From the cover: STIM1-induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci USA. 2009;106:19358–19362. doi: 10.1073/pnas.0911280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. T cells use two directionally distinct pathways for cytokine secretion. Nat Immunol. 2006;7:247–255. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- 131.Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- 132.Heissmeyer V, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 133.Macian F, et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 134.Maul-Pavicic A, et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci USA. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pores-Fernando AT, Zweifach A. Calcium influx and signaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol Rev. 2009;231:160–173. doi: 10.1111/j.1600-065X.2009.00809.x. [DOI] [PubMed] [Google Scholar]
- 136.Feske S. CRAC channelopathies. Pflugers Arch. 2010;460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin Immunol. 2010;135:169–182. doi: 10.1016/j.clim.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Feske S. Immunodeficiency due to defects in store-operated calcium entry. Ann N Y Acad Sci. 2011;1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Feske S. Immunodeficiency due to defects in store-operated calcium entry. Ann N Y Acad Sci. 2011;1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Oh-hora M. Calcium signaling in the development and function of T-lineage cells. Immunol Rev. 2009;231:210–224. doi: 10.1111/j.1600-065X.2009.00819.x. [DOI] [PubMed] [Google Scholar]
- 141.Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Le Deist F, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–1062. [PubMed] [Google Scholar]
- 143.Matsumoto M, et al. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity. 2011;34:703–714. doi: 10.1016/j.immuni.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 144.Fuchs S, et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol. 2012;188:1523–1533. doi: 10.4049/jimmunol.1102507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng KT, Alevizos I, Liu X, Swaim WD, Yin H, Feske S, Oh-Hora M, Ambudkar IS. STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjogren's syndrome. Proc Natl Acad Sci USA. 2012;109:14544–14549. doi: 10.1073/pnas.1207354109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shaw PJ, Feske S. Physiological and pathophysiological functions of SOCE in the immune system. Frontiers Biosci. 2011;4:2253–2268. doi: 10.2741/540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J Physiol. 2012;590:4157–4167. doi: 10.1113/jphysiol.2012.233221. [DOI] [PMC free article] [PubMed] [Google Scholar]