

Abstract
Pituitary adenylate cyclase-activating polypeptide (PACAP) is a pleiotropic neuropeptide expressed in the brain, where it may act as a neuromodulator or neurotransmitter contributing to different behavioral processes and stress responses. PACAP is highly expressed in the amygdala, a subcortical brain area involved in both innate and learned fear, suggesting a role for PACAP-mediated signaling in fear-related behaviors. It remains unknown, however, whether and how PACAP affects neuronal and synaptic functions in the amygdala. In this study, we focused on neurons in the lateral division of the central nucleus (CeL), where PACAP-positive presynaptic terminals were predominantly found within the amygdala. In our experiments on rat brain slices, exogenous application of PACAP did not affect either resting membrane potential or membrane excitability of CeL neurons. PACAP enhanced, however, excitatory synaptic transmission in projections from the basolateral nucleus (BLA) to the CeL, while inhibitory transmission in the same pathway was unaffected. PACAP-induced potentiation of glutamatergic synaptic responses persisted after the washout of PACAP and was blocked by the VPAC1 receptor antagonist, suggesting that VPAC1 receptors might mediate synaptic effects of PACAP in the CeL. Moreover, potentiation of synaptic transmission by PACAP was dependent on postsynaptic activation of protein kinase A and calcium/calmodulin-dependent protein kinase II, as well as synaptic targeting of GluR1 subunit-containing AMPA receptors. Thus, PACAP may upregulate excitatory neurotransmission in the BLA-CeL pathway postsynaptically, consistent with the known roles of PACAP in control of fear-related behaviors.
INTRODUCTION
Pituitary adenylate cyclase-activating polypeptide (PACAP) is an excitatory neuropeptide, originally isolated from hypothalamic tissue, which was found to increase the production of cAMP in mammalian anterior pituitary cells (Miyata et al., 1989). Subsequent studies revealed that PACAP is expressed in various areas throughout the brain (Köves et al., 1991; Basille et al., 2000), where it serves neurotrophic, neuromodulatory and neurotransmitting functions (Masuo et al., 1993; Stella and Magistretti, 1996; Villalba et. al., 1997; Roberto et al., 2001; Nielsen et al., 2004; reviewed by Vaudry et al., 2009), binding to three different G protein-coupled receptors with similar affinity. One receptor, PAC1, is specific to actions of PACAP, while two other receptors, VPAC1 and VPAC2, are shared by PACAP and vasoactive intestinal peptide (VIP) (Ishihara et al., 1992; Pisegna and Wank, 1993). As a neuromodulator, PACAP may control the release of hormones and regulate neurotransmission and plasticity by activating different signaling molecules, including adenylate cyclase, phospholipase C, mitogen-activated protein kinases (MAPK) and increasing intracellular Ca2+ concentration (Hashimoto et al., 1993; Sreedharan et al., 1994; Macdonald et al., 2005; Vaudry et al., 2009; Yang et al., 2010). At the behavioral level, PACAP was shown to regulate many pivotal functions, such as food intake, psychomotor activity, stress responses, learning and memory (Hashimoto et al., 2001; Otto et al., 2001a; Hashimoto et al., 2006; Hammack et al., 2009).
Notably, previous studies provided several lines of evidence for the involvement of PACAP in regulation of fear-related behavioral processes. Thus, PACAP-containing fibers and PACAP receptors are densely expressed in the amygdala, a subcortical brain area essential for innate and learned fear (Piggins et al., 1996; Shioda et al., 1997; Kozicz and Arimura, 2002; Joo et al., 2004; Shumyatsky et al., 2005; Pape and Pare, 2010). Consistent with this expression pattern, direct infusions of PACAP into the central nucleus of the amygdala had an anxiogenic effect, resulting in a shift from an active to passive mode of responding to an aversive stimulus (Legradi et al., 2007). Moreover, genetically modified mice lacking either PACAP or type I PACAP receptor (PAC1) exhibited reduced anxiety levels (Hashimoto et al., 2001; Otto et al., 2001b), suggesting that endogenous PACAP may regulate innate fear. Finally, it was demonstrated recently that PACAP blood level may predict symptoms of posttraumatic stress disorder in human female subjects exhibiting abnormally enhanced fear responses (Ressler et al., 2011). These previous findings suggest a possibility that PACAP may regulate anxiety-like behaviors by modulating neuronal and synaptic activity in the components of innate fear circuitry, such as the amygdala, yet very little is known about the effects of PACAP in the amygdala at the cellular and molecular level. Here, we show that PACAP-containing fibers are abundant in the lateral division of the central nucleus of the amygdala (CeL), and that PACAP facilitates neurotransmission at excitatory glutamatergic projections from the BLA to the CeL, a projection known to be implicated in responses to innately aversive stimuli (Tye et a, 2011), through postsynaptically expressed synaptic strengthening.
MATERIALS AND METHODS
PACAP immunohistochemistry
Sprague-Dawley rats (350–375 g) of either sex were sacrificed by an overdose of sodium pentobarbitol (130 mg/kg) and perfused intracardially with 0.9 % saline (200 ml) followed by 2 % paraformaldehyde, 0.05 % glutaraldehyde, and 0.2 % picric acid in 0.1 M PBS (500 ml). After the fixation, the brains were stored for 4 days in a 30 % sucrose/0.1 M PBS (pH 7.4) solution at 4 °C and subsequently, 20 μm frozen sections were cut through the amygdala. Every third section through the rostro-caudal extent of the amygdala was then collected in glass vials for immunoprocessing using the avidin-biotin-peroxidase (ABC) method (Hsu et al., 1981). All incubations were performed on a rocker (low setting) at room temperature. Briefly, the sections were incubated in 0.6 % hydrogen peroxide in 0.1 M PBS (pH 7.4) for 30 min to eliminate endogenous peroxidase activity. To reduce non-specific binding, the sections were preincubated in the blocking solution (2 % normal donkey serum, 1% bovine serum albumin, 0.3 % Triton X-100 in 0.1 M PBS, pH 7.4) for 2 hr followed by incubation for 24 h with the rabbit polyclonal primary antibody against PACAP38 (1:1000; Bachem Americas Inc., Torrance, CA). The sections were washed with PBS and incubated for 1 h with donkey anti-rabbit biotinylated secondary antibody (1:400; minimum species cross-reactivity; Jackson ImmunoResearch Laboratories, West Grove, PA). The sections were washed with PBS and incubated for 30 min with the avidin-biotin complex (Vector Laboratories, Burlingame, CA). The sections were then incubated for 5 min in 3,3′-diaminobenzidine (DAB)/H2O2 (Sigma Fast; Sigma, St Louis, MO) as a chromagen for visualization of PACAP38 reaction-product. Sections were mounted on gelatin-coated slides and coverslipped with Permount (Fisher Scientific, Unionville, Ontario).
Fluorescent immunohistochemistry
Alternate sections through the amygdala were cut to determine whether PACAP was localized in amygdala principal cells. Briefly, sections were preincubated in the blocking solution for 2 h followed by incubation for 24 h with rabbit polyclonal primary antibody against PACAP38. Sections were washed with PBS and incubated for 1.5 h with Alexa Fluor 488-conjugated donkey anti-rabbit secondary antibody (Molecular Probes). Sections were then incubated in NeuroTrace 640/660 deep-red fluorescent Nissl stain (1:200; Molecular Probes) in 0.1 M PBS for 20 min. Sections were mounted under low light on gelatin-coated slides using Gel Mount (Sigma, St. Louis, MO).
Light (DAB-processed section) microscopy of PACAP-positive fibers in the amygdala sections was performed using Zeiss Axioskop 2 microscope. Still frame images were captured with a digital camera (AxioCam, Zeiss) interfaced with a Macintosh G4 computer. PACAP innervation of Nissl-labeled neurons in the amygdala was further imaged using a Leica TSC NT laser scanning confocal microscope (Leica Microsystems Inc., Bannockburn, IL).
Cloning of the Rat VPAC1R cDNA and generation of the riboprobes for in situ hybridization
To clone the VPAC1R cDNA, total RNA was isolated from the rat brain using total RNA purification kit (Qiagen). One microgram of total RNA was reverse-transcribed using oligo(dT) 16 (Applied Biosystems). Double-stranded cDNA was amplified by PCR. The pairs of primers designed to amplify the VPAC1R were as follows: F: GCCCCCATCCTCCTCTCCATC; R: TCCGCCTGCACCTCACCATTG. The PCR-amplified product was cloned into pCRII-TOPO vector (Invitrogen). The construct was checked by restriction analysis and sequencing. The VPAC1R antisense riboprobes were generated from the plasmid linearized with Hind III and transcribed with T7 RNA polymerase. The sense riboprobes were synthesized after linearization with Xba I and transcription with SP6 RNA polymerase. Riboprobes were transcribed in the presence of DIG-UTP (Roche).
Coronal sections from fresh-frozen rat brains were cut at 20 μm thickness on the cryostat (Leica) and hybridized according to the published protocol with modifications (Schaeren-Wiemers and Gerfin-Moser, 1993). Briefly, the sections were fixed with 4 % paraformaldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.4) for 10 min, rinsed in PBS treated with diethylpyrocarbonate (DEPC), and treated with acetic anhydride in 0.1 M triethanolamine/distil water for 10 minutes at room temperature. Next, the sections were washed in PBS-DEPC solution, and incubated in prehybridization buffer [50 % formamide, 5 X standard saline citrate (SSC), 5 X Denhardt’s, 250 μg/ml E.coli tRNA and 500 μg/ml herring sperm DNA] for 1 hr at room temperature. Hybridization was performed overnight at 45 °C in prehybridization buffer. After hybridization, the sections were washed in 5 X SSC at 45 °C. After high stringency washing (three times in 0.2 X SSC at 45 °C followed by 0.2 X SSC at room temperature and B1 [0.1M Tris-HCl, pH 7.5, 0.15M NaCl]) the sections were processed for immunological detection of DIG-labeled probes with alkaline-phosphatase (AP)-conjugated anti-DIG antibody (Roche) overnight at 4 °C. The AP reaction was visualized by NBT/BCIP (Promega).
Electrophysiological recordings
Brains were dissected quickly from 30- to 34-day old Sprague-Dawley rats of either sex after decapitation, and chilled in ice-cold Ca2+-free artificial cerebrospinal fluid (ACSF) containing 125 mM NaCl, 2.5 mM KCl, 3.5 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose and equilibrated with 95 % O2 and 5 % CO2. Coronal brain slices containing the amygdala (300 μm thick) were prepared with a vibratome. Slices were recovered in the standard Ca2+-containing ACSF (125 mM NaCl, 2.5 mM KCl, 2.5 mM CaCl2, 1 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose and equilibrated with 95 % O2 and 5 % CO2, pH 7.3 – 7.4) for at least an hour at room temperature (22–25°C), and used for experiments up to 5 hours after preparation. Slices were placed in the recording chamber and perfused continuously with the standard ACSF (~1 mL per minute). Picrotoxin (PTX, 50 μM) was added to the ACSF to inhibit fast GABAergic transmission unless indicated otherwise. Whole-cell recordings were obtained from neurons in the lateral division of the CeA under visual guidance (infrared differential interference contrast optics) with an EPC-9 amplifier and Pulse v8.54 software (Heka Elektronik) as described earlier (Shin et al., 2006; Cho et al., 2012). The patch electrodes (3–5 MΩ resistance) contained 150 mM K-gluconate, 5 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 0.2 EGTA, 4 mM MgATP, and 1.2 mM NaGTP (adjusted to pH 7.25 with KOH, 280–290 mOsm). In the experiments where the EPSCAMPAR/EPSCNMDAR ratio was determined or NMDAR EPSCs were recorded, 140 mM Cs-methanesulfonate was substituted for K-gluconate. Synaptic responses were filtered at 1 kHz and digitized at 2.5 kHz. Input and series resistances were monitored throughout the experiment, and the experiment was terminated when series resistance changed by more than 20 %. Series resistance was not compensated. EPSCs or EPSPs in the CeL neurons were evoked by a bipolar stimulation electrode with 115 μm spacing (FHC Inc.), that was placed within the BLA approximately 250 μm laterally to the border between the BLA and the CeA, with the square current pulses (50–250 μA, 100-μs duration) using Master-8 Pulse Stimulator (AMPI). Stimulation intensity was adjusted in individual experiments to evoke the EPSCs of 50–150 pA amplitude. Membrane potential was held constant at −70 mV throughout the experiments in voltage-clamp mode unless indicated otherwise. Baseline EPSCs or EPSPs were evoked by stimulation of the BLA every 20 seconds. To minimize the effects of washout in whole-cell recordings, an application of PACAP started within 10 min after the establishment of whole-cell configuration. Offline data analysis was performed using Clampfit 9 program (Molecular Devices).
Pharmacological reagents
Reagents were prepared as stock solutions in water (GDP-β-S, Rp-cAMPS, AIP, and Pep1-TGL), ethanol (picrotoxin) or DMSO (Xestospongin-C and chelerythrine) at 200 to 2000-fold concentrations, and stored at −20 °C. Stock solutions of peptides (PACAP38, PACAP6-38, and PG 97-269) were prepared in the standard ACSF at the concentration of 0.25 mM, and stored at −20 °C. Reagents were purchased from Sigma Aldrich (picrotoxin, GDP-β-S, and BAPTA), Enzo Life Sciences (Xestospongin-C) and Tocris Bioscience (chelerythrine, Rp-cAMPS, and AIP). PACAP38 was purchased from Bachem or Tocris, PACAP6-38 from Bachem or American Peptide Company, PG 97-269 from Phoenix Pharmaceuticals, and pep1-TGL from Tocris.
Data Analysis
The baseline EPSC or EPSP amplitude was calculated by averaging peak amplitudes of responses recorded for 5 minutes just before the application of PACAP. Peak amplitudes of EPSCs or EPSPs were normalized to the baseline EPSC or EPSP (% baseline). Data are presented as means ± the standard error of the mean (SEM). In most experiments, peak amplitudes of EPSCs recorded 45–50 minutes after the onset of PACAP application, were averaged and compared with the baseline EPSC. As appropriate, a two-tailed Student’s t test (paired or unpaired) was used for statistical analyses. Statistical analyses were performed with Minitab15 software (Minitab), and p < 0.05 was considered to be statistically significant.
RESULTS
PACAP-immunoreactive fibers densely innervate the CeL
Consistent with previous reports describing immunohistochemical localization of PACAP in the brain (Piggins et al., 1996; Kozicz and Arimura, 2002), we found that PACAP immunopositive fibers innervate densely the central nucleus of the amygdala (CeA) (Fig. 1 A,B), whereas PACAP immunoreactivity was not detected in lateral or basolateral nuclei of the amygdala. Specifically, PACAP immunopositive fibers were found predominantly within the lateral division of the central nucleus (CeL). Notably, PACAP immunopositive fibers formed “collar-like” contacts with neurons in the CeL (Fig. 1 C), suggesting that PACAPergic fibers may establish synaptic connections with these cells. These findings support the notion that endogenously-released PACAP may affect neuronal functions in the CeL.
Figure 1.

PACAP- positive nerve fibers innervate the lateral division of the CeA. A, A low magnification image of coronal section through the amygdala showing PACAP staining (DAB chromagen; brown) in the CeA. LA and BLA, lateral and basolateral nuclei of the amygdala. CeL and CeM, lateral and medial divisions of the CeA, respectively. B, A high magnification image of PACAP staining in the CeL shown in A. C, Fluorescent Nissl staining (NeuroTrace) of neurons in the CeL combined with immunohistochemistry for PACAP. Arrowheads indicate “collar-like” contacts by PACAP-containing fibers (labeled green) on proximal dendrites of CeL neurons with fluorescent Nissl staining that is labeled blue. Scale bars: 500 μm (A), 100 μm (B), 10 μm (C).
PACAP potentiates excitatory synaptic transmission in the BLA-CeL projections
As PACAP-containing fibers were found predominantly within the CeL, we first examined whether PACAP may affect membrane excitability of CeL neurons. In current-clamp mode, CeL neurons fired action potentials in response to depolarizing current pulses exhibiting spike-frequency adaptation (Fig. 2 A). The exogenous application of PACAP38 (5 nM, 10 min), a biologically active and evolutionarily conserved form of endogenously-released PACAP (Ishihara et al., 1992; Hashimoto et al., 1993; Lutz et al., 1993; Hashimoto et al., 2001), had no effect on the number of spikes in recorded cells indicating that firing properties of CeL neurons were unaffected under these conditions (Fig. 2 A; n = 6, two-way ANOVA, p = 0.98).
Figure 2.

PACAP potentiates excitatory transmission in the BLA to the CeL pathway. A, Summary plots of the number of action potentials in CeL neurons evoked by current injections of increasing intensity before (open circles) and after 10-min application of PACAP38 (5 nM; filled circles; n = 6). Traces on top represent responses of CeL neurons to depolarizing current injection (100 pA, 1 s) recorded in current-clamp mode under control conditions and after application of PACAP38. B, Representation of the slice preparation, showing locations of stimulation (Stim) and recording (Rec) electrodes. Synaptic responses were evoked by stimulation of the BLA and recorded from neurons in the CeL. C, Summary graphs demonstrating the time course of EPSP (upper graph, filled circles; n = 10) and IPSP (lower graph, open circles; n = 5) amplitude changes produced by PACAP38 application (red horizontal bars). Insets show the average of 10 biphasic synaptic responses in a CeL neuron before (black trace) and after (red trace) PACAP38 application. The EPSP/IPSP sequences were recorded at a slightly depolarized membrane potential (−55 mV) to increase the driving force for Cl− currents underlying GABAA receptor-mediated IPSPs. D, Summary of the EPSP (n = 10) and IPSP (n = 5) amplitude changes 35–40 minutes after the start of PACAP application for the experiments shown in C. The larger filled circles represent mean values. E, Upper graph, the time course of EPSP amplitude changes in the presence of 50 μM PTX before and after PACAP38 application (red horizontal bar, n = 7). EPSPs were recorded in CeL neurons in current-clamp mode at −70 mV. Lower graph shows the time course of changes in the resting membrane potential, ΔVm, relative to the pre-PACAP value. Insets show averaged BLA-CeL EPSPs recorded before (black trace) and after (red trace) PACAP38 application. Error bars indicate SEM.
Recent studies, utilizing optogenetics techniques, indicate that the BLA-CeL projections may contribute to the expression of innate fear (Tye et a, 2011; Knobloch et al., 2012). To explore whether PACAP, which is known to produce anxiety-like behaviors (Legradi et al., 2007; Hammack et al., 2009), may modulate neurotransmission in the components of innate fear circuitry, we examined the effects of PACAP on synaptic transmission in the pathway from the BLA to the CeL, densely innervated by PACAP-positive terminals (Fig. 1). Synaptic responses in the CeL neurons were induced by stimulating the BLA (Fig. 2 B), which, in the absence of the GABA receptor A (GABAAR) antagonist PTX, evoked biphasic postsynaptic potentials in a fraction of the recorded neurons (5 out of 10 neurons tested exhibited biphasic responses) under current-clamp conditions at the membrane potential of −55 mV (Fig. 2 C). The biphasic responses consisted of the initial excitatory postsynaptic potential (EPSP) which was followed by the PTX-sensitive GABAAR-mediated inhibitory postsynaptic potential (IPSP; Fig. 2 C,E). The latter was likely reflecting feed-forward inhibition of CeL neurons by intercalated cells (Royer et al., 1999; Amano et al., 2010). The AMPA receptor-mediated EPSP, as evidenced by its sensitivity to the specific AMPAR antagonist NBQX (10 μM; not shown), had a short and constant synaptic delay indicating that the EPSP was monosynaptic in nature (Tsvetkov et al., 2002). After recording stable baseline responses for 5 minutes, we perfused slices with the ACSF containing 5 nM PACAP38 for 10 minutes. The peak amplitude of the EPSP was increased to 131 % ± 9 % of its baseline value 35–40 minutes after the start of PACAP38 application (Fig. 2 C, top graph and Fig. 2 D; n = 10, paired t test, p < 0.01 versus baseline), whereas the amplitude of IPSP, observed in the EPSP/IPSP sequences, was unaffected by PACAP38, remaining at 92 % ± 10 % of the baseline value (Fig. 2 C, lower graph and Fig. 2 D; n = 5, p = 0.49). Notably, the EPSPs remained potentiated even after the washout of PACAP38 for >30 minutes. We also examined the action of PACAP38 on isolated BLA-CeL EPSPs in the presence of 50 μM PTX, blocking disynaptic GABAAR-mediated IPSPs, at resting membrane potential (approximately −70 mV). In these experiments, the duration of exogenous PACAP application was increased to 50 min. The magnitude of PACAP38-induced-potentiation under these conditions did not differ from that when PACAP38 was applied for a much shorter period of time. Thus, the EPSP amplitude was 148 % ± 16 % of the baseline amplitude after 30 min washout of PACAP38 which was preceded by 50 min-long PACAP38 application (Fig. 2 E; n = 7, paired t test, p < 0.05 versus baseline; not significantly different from the peak EPSP amplitude following 10 min-long application of PACAP38 followed by 30 min washout, t test, p = 0.38). The observed synaptic enhancements were not associated with changes in the resting membrane potential in the course of prolonged application of PACAP38 (Fig. 2 E, lower graph). These findings indicate that PACAP may induce selective and lasting potentiation of neurotransmission at excitatory glutamatergic synapses in the BLA-CeL projections.
PACAP-induced potentiation of excitatory transmission at the BLA-CeL synapses is postsynaptically expressed
PACAP could facilitate excitatory synaptic transmission either by increasing the probability of release from presynaptic BLA terminals or enhancing the responsiveness of postsynaptic CeL neurons to the unchanged amount of glutamate (Regehr and Stevens, 2001). To distinguish between these alternative possibilities, we recorded excitatory postsynaptic currents (EPSCs) in CeL neurons under voltage-clamp conditions at a holding potential of −70 mV in the presence of 50 μM PTX in external medium. As in the above-described experiments, synaptic responses were evoked by stimulation electrodes placed in the BLA. Similar to the effects of PACAP38 on the EPSP (Fig. 2 C,E), addition of PACAP38 to the ACSF produced a progressive increase in the peak amplitude of the EPSC (Fig. 3 A). The magnitude of the EPSC remained elevated to 130 % ± 6 % of its baseline value even after the washout of PACAP38 for 35–40 min (Fig. 3 A,B; n = 12, paired t test, p < 0.001 versus baseline).
Figure 3.

Potentiation of synaptic transmission in the BLA-CeL projections by PACAP implicates postsynaptic expression mechanisms. A, Summary graphs demonstrating the time course of EPSC (filled symbols, n = 12) and paired-pulse ratio (PPR, open symbols, n = 12) magnitude changes produced by PACAP38 application (red horizontal bar). The EPSCs were recorded at −70 mV in voltage-clamp mode. Insets show averaged paired BLA-CeL EPSCs (50 ms interpulse interval) obtained before (black trace) and after (red trace) PACAP38 application. B, Summary of EPSC amplitude changes induced by PACAP38 for the experiments shown in A (n =12). C, Summary of PPR magnitude changes for the experiments shown in A (n = 12). D, Examples of EPSCs (average of 10–15 traces) recorded at −70 mV or +50 mV before (black traces) and after PACAP38 application (red traces) in the presence of PTX (50 μM). The amplitude of AMPA receptor-mediated EPSC (EPSCAMPAR) was determined at −70 mV (at a black dot), while the amplitude of NMDA receptor EPSC (EPSCNMDAR) was determined at +50 mV and measured 50 ms after the stimulus (at a vertical dotted line). E, Summary plot demonstrating the time course of EPSCAMPAR magnitude changes in CeL neurons affected by PACAP38 application (red horizontal bar, n = 9). F, Values of the EPSCAMPAR/EPSCNMDAR ratio calculated before and after application of PACAP38 (n = 9) for the same recordings as in E. G, Examples of NMDAR EPSCs recorded in CeL neurons at +30 mV before (black trace) and after (red trace) PACAP38 application. Recordings were performed in the presence of PTX (50 μM) and NBQX (10 μM). H, Time course of EPSCNMDAR amplitude changes in response to PACAP application (red horizontal bar, n = 7). I, The EPSCNMDAR amplitude values before and after PACAP38 application (n = 7) for the same recordings as in H. Error bars indicate SEM.
To explore the synaptic site of PACAP38-induced potentiation, we first tested the effects of exogenous PACAP38 on paired-pulse ratio (PPR, 50 ms interval), which is inversely related to the presynaptic release probability, and, therefore, changes in PPR reflect presynaptic modifications (Zucker and Regehr, 2002). We found that potentiation of synaptic transmission following PACAP38 application was not associated with changes in the PPR magnitude (Fig. 3 A,C; PPR magnitude was 1.28 ± 0.05 and 1.20 ± 0.07 before and after PACAP38 application, respectively; n = 12, paired t test, p = 0.21), indicating that PACAP38 had no effect on the probability of release at the BLA-CeL synapses. To examine further the expression mechanism of PACAP-induced potentiation, we obtained and compared the EPSCAMPAR/EPSCNMDAR ratio, as a measure of changes in the postsynaptic sensitivity to glutamate, before and after PACAP38 application. The NMDA receptor-mediated component of the EPSC was determined at +50 mV, to relieve the voltage-dependent block of the NMDAR channels by external Mg2+, and measured 50 ms after the stimulus, while the peak amplitude of the AMPA receptor EPSC was determined at −70 mV (Fig. 3 D). The EPSCAMPAR was significantly potentiated by 10-min application of 5 nM PACAP38 (Fig. 3 E; n = 9, paired t test, p < 0.05 versus baseline). However, the amplitude of EPSCNMDAR did not change significantly in these neurons after the addition of PACAP38 (n = 9, EPSCNMDAR was 32 ± 4 pA and 37 ± 5 pA before and after PACAP38 application, respectively; paired t test, p = 0.31). Consistent with these results, the EPSCAMPAR/EPSCNMDAR ratio increased significantly after PACAP38 application (Fig. 3 F; n = 9, paired t test, p < 0.05), supporting the notion that the application of PACAP led to a selective increase in the EPSCAMPAR amplitude, while the EPSCNMDAR remained unchanged.
We also tested the effect of PACAP on isolated NMDAR EPSCs, performing recordings from CeL neurons at +30 mV in the presence of the AMPA receptor antagonist NBQX (10 μM) and PTX (50 μM). We found that PACAP (5 nM) had no effect on the amplitude of NMDAR EPSCs over the course of recordings (Fig. 3 G–I), with the amplitude remaining at 98% ± 8 % of the baseline value (Fig. 3 G–I; n = 7, paired t test, p = 0.85 versus baseline) 25–30 min after the start of PACAP application. Together, these findings demonstrate that PACAP-induced potentiation at the BLA-CeL synapses is associated with increased postsynaptic responsiveness of CeL neurons to glutamate released from the BLA terminals, and, thus is postsynaptically expressed.
Potentiating effect of PACAP at the BLA-CeL synapses is mediated by activation of VPAC1 receptors
PACAP exerts its physiological functions through binding to two different receptor subtypes: type I receptor (PAC1), which is a specific receptor for endogenous PACAP, and type II receptors, VPAC1 and VPAC2, which could be activated by both PACAP and VIP (Ishihara et al., 1992; Hashimoto et al., 1993; Lutz et al., 1993). To identify the receptor subtype mediating the observed potentiation at the BLA-CeL synapses, we first tested the effects of PACAP38 (5 nM) in the presence of PACAP6-38 (250 nM), known to block both PAC1 and VPAC2 receptors (Robberecht et al., 1992; Fizanne et al., 2004), but not VPAC1 receptors when used in this concentration (Dickinson et al., 1997). Under these conditions, PACAP38 application still resulted in potentiation of EPSCs in CeL neurons to 120 % ± 7 % of the baseline value (Fig. 4 A; n = 11, paired t test, p < 0.05 versus baseline). There was no significant difference in PACAP-induced potentiation of the EPSC amplitude between control (as shown in Fig. 3 A,B) and PACAP6-38-treated slices (unpaired t test, p = 0.31). On the other hand, a selective VPAC1 receptor antagonist, PG 97-269 (250 nM) (Gourlet et al., 1997), prevented potentiation at the BLA-CeL synapses by PACAP38, as the EPSC amplitude remained at 105 % ± 4 % of its baseline value (Fig. 4 B,C; n = 10, paired t test, p = 0.18 versus baseline). Importantly, PG 97-269 has a very low affinity for PAC1 and VPAC2 receptors in a concentration used (Gourlet et al., 1997; Dickson et al., 2006).
Figure 4.

Activation of VPAC1 receptors mediates PACAP-induced potentiation at the BLA-CeL synapses. A, Left, examples of BLA-CeL EPSCs recorded before (black trace) and after (red trace) application of PACAP38 (red horizontal bar) at −70 mV in the presence of PACAP6-38 (250 nM) in the external medium. Right, summary graph demonstrating the time course of EPSC amplitude changes produced by PACAP38 (red horizontal bar) in the presence of PACAP6-38 (n = 11). The period of antagonist application is shown by a blue horizontal bar. B, Same as in A but the experiments were performed in the presence of PG 97-269 (250 nM, blue bar) in the external solution (n = 10). C, Summary of EPSC amplitude changes produced by PACAP 38 application in the presence of PACAP6-38 (n = 11) or PG 97-269 (n = 10). Error bars indicate SEM. D and E, RNA in situ hybridization with anti-sense (D) and sense probes (E) demonstrates expression of VPACR1 in the rat brain (purple). Low magnification images in the left column show the general pattern of VPAC1R expression in coronal brain sections. Higher magnification images in the middle and right columns demonstrate VPAC1R expression in the lateral (LA), basolateral (BLA) and central nuclei (lateral division, CeL; medial division, CeM) of the amygdala. Dotted lines indicate borders of the different amygdaloid nuclei. Scale bars: 400 μm (middle), and 200 μm (right).
We also analyzed the expression pattern of the VPAC1R gene in the amygdala by mRNA in situ hybridization (Fig. 4 D,E). VPAC1R mRNA was present in different amygdaloid nuclei, including the CeL (Fig. 4 D), which is densely innervated by the PACAP-containing fibers (Fig. 1 A,B), thus confirming previous reports (reviewed in Vaudry et al., 2009). These results suggest that PACAP38-induced potentiation in projections from the BLA to the CeL could be mediated by VPAC1 receptors.
Signal transduction mechanisms underlying PACAP-induced synaptic enhancements in the CeL
The findings that PACAP38-induced potentiation of the BLA-CeL EPSC is postynaptically expressed and mediated by VPAC1 receptor, which is G protein-coupled (Vaudry et al., 2009), suggest that the potentiating action of PACAP38 may occur via activation of the G protein-mediated signaling pathways in postsynaptic CeL neurons. Consistent with this notion, we found that loading postsynaptic CeL neurons with a non-phosphorylatable analog of GDP, GDP-β-S (1 mM), which inhibits the function of G proteins, prevented potentiation of the BLA-CeL EPSCs by PACAP38, with the EPSC amplitude remaining at 99 % ± 5 % of the baseline EPSC value (Fig. 5A–C; n = 9, paired t test, p = 0.91 versus baseline). However, the EPSC amplitude was potentiated to 129 % ± 9 % of its baseline value (n = 6, paired t test, p < 0.05) following PACAP38 application when the recording pipette solution contained GTP. This indicates that activation of postsynaptic G protein-coupled signaling mechanisms is implicated in PACAP38-induced potentiation of glutamatergic synaptic transmission in the BLA-CeL projections.
Figure 5.

G protein-coupled signaling and rise in intracellular Ca2+ concentration are required for PACAP-induced potentiation. A, Examples of EPSCs recorded in CeL neurons before (black traces) and after PACAP38 application (red traces) when pipette solution contained either GTP (upper traces) or GDP-β-S (1 mM; lower traces). EPSCs were evoked by stimulation of the BLA every 20 seconds and recorded at −70 mV. B, Time course of EPSC amplitude changes produced by PACAP38 (red horizontal bar) in the presence of GTP (n = 6, open circles) or GDP-β-S (n = 9, filled circles) in recording pipette solution. C, Summary of the EPSP amplitude changes after PACAP38 application for the experiments shown in B (n = 6 and n = 9 for the experiments with GTP or GDP-β-S, respectively). D, Examples of EPSCs recorded before (black traces) and after PACAP38 application (red traces) under control conditions (0.2 mM EGTA in pipette solution, upper traces) and in the presence of 10 mM BAPTA in pipette solution (lower traces). E, Time course of EPSC amplitude changes produced by PACAP38 (red horizontal bar) in CeL neurons loaded with either 0.2 mM EGTA (n = 6, open circles) or 10 mM BAPTA (n = 8, filled circles). F, Summary of the EPSP amplitude changes after PACAP38 application for the experiments shown in E (n = 6 for EGTA, n = 8 for BAPTA). G, EPSCs recorded before (black trace) and after PACAP38 application (red trace) when CeL neurons were loaded with 2.5 μM Xestospongin-C. H, Time course of EPSC amplitude changes produced by PACAP in CeL neurons either loaded with Xestospongin-C (n = 6, filled circles) or under control conditions (n = 7, open circles). Error bars indicate SEM.
Binding of PACAP to VPAC1 receptor results in activation of Gαs protein leading to the increased production of the cAMP and stimulation of the cAMP/PKA signaling pathway (Sreedharan et al, 1994; Dickson et al, 2006; Yang et al., 2010). VPAC1 receptor was also shown to be coupled to Gαq protein stimulating the inositol 1,4,5-trisphosphate (InsP3)/Ca2+-dependent intracellular mechanisms (Langer and Robberecht, 2005; Dickson and Finlayson, 2009). Thus, it has been demonstrated previously in heterologous expression systems that activation of VPAC1 receptors may lead to the increase in intracellular Ca2+ concentration (Sreedharan et al., 1994). To determine whether the potentiating action of PACAP38 on synaptic transmission in the amygdala may depend on the rise of Ca2+ concentration in postsynaptic CeL neurons, we included a high concentration of the fast Ca2+ chelator BAPTA (10 mM) in the recording pipette solution. Under these conditions, PACAP38-induced potentiation was suppressed, with the EPSC amplitude remaining at 99 % ± 3 % of the baseline value 35–40 min after the end of PACAP38 application (Fig. 5D–F; n = 8, paired t test, p = 0.78 versus baseline). PACAP38 retained, however, its ability to potentiate the BLA-CeL EPSCs when the recording pipette solution contained 0.2 mM EGTA, a low affinity Ca2+ chelator not preventing increases in intracellular Ca2+ when used in a given concentration. The EPSC was potentiated by PACAP38 in these experiments to 136 % ± 12% of the baseline amplitude (n = 6, paired t test, p < 0.05). Therefore, an increase in postsynaptic Ca2+ concentration is required for potentiation of synaptic transmission at the BLA-CeL synapses by PACAP38.
Moreover, we investigated the source of postsynaptic Ca2+ necessary for the potentiating effect of PACAP38. As VPAC1 receptor is Gαq-coupled and may activate phospholipase C (Langer and Robberecht, 2005), the effects of PACAP on synaptic transmission in the amygdala could be mediated by release of Ca2+ from the internal Ca2+ stores. Consistent with this notion, potentiation of the BLA-CeL EPSCs by PACAP38 was blocked when Xestospongin-C (2.5 μM), an inhibitor of InsP3 receptors (Gafni et al., 1997), was included in the recording pipette solution (Fig. 5 G,H). Under these experimental conditions, the EPSC amplitude was 96 % ± 8 % of its baseline value following PACAP38 application (n = 6, paired t test, p = 0.61 versus baseline), whereas the EPSC was potentiated by PACAP38 to 149 % ± 9 % of the baseline amplitude (n = 7, p < 0.01) in the absence of Xestospongin-C in pipette solution. This indicates that Ca2+ release from the InsP3-sensitive Ca2+ stores may contribute to the PACAP38-induced potentiation at synaptic inputs to CeL neurons formed by projections originating in the BLA.
To investigate further the signaling mechanisms lying downstream of the VPAC1 receptor activation, we dialyzed recorded CeL neurons with pipette solution containing specific protein kinase inhibitors. First, we examined whether the potentiating effect of PACAP on the BLA-CeL EPSCs depended on activation of protein kinase A (PKA) by loading CeL neurons with Rp-cAMPS (100 μM), a competitive inhibitor of the cAMP-induced activation of PKA (Van Haastert et al., 1984). The intracellular introduction of Rp-cAMPS resulted in a complete block of synaptic potentiation by PACAP38, with the EPSC amplitude remaining at 106 % ± 6 % of the baseline value (Fig. 6 A,D; n = 7, paired t test, p = 0.38), suggesting that activation of the cAMP/PKA pathways might be required for the PACAP effect on synaptic transmission.
Figure 6.

Postsynaptic mechanism of PACAP38-induced synaptic potentiation in CeL neurons. A, Left, examples of EPSCs recorded in CeL neurons before (black trace) and after PACAP38 application (red trace) when pipette solution contained the cAMP antagonist Rp-cAMPS (100 μM). Right, time course of EPSC amplitude changes produced by PACAP38 (red horizontal bar) in CeL neurons loaded with Rp-cAMPS (n = 7). B, Effects of PACAP38 were tested in the presence of PKC inhibitor chelerythrine (10 μM in external solution (n = 7). C, Effects of PACAP38 were tested in CeL neurons loaded with CamKII inhibitor autocamtide-2-related inhibitory peptide (AIP, 5 μM; n = 6). D, Summary of the EPSP amplitude changes after PACAP38 application for the experiments shown in A–C (n = 7 for the experiments with Rp-cAMPS, n = 7 for chelerythrine, n = 6 for AIP). E, Left, examples of EPSCs recorded in CeL neurons before (black traces) and after PACAP38 application (red traces) under control conditions and when pipette solution contained Pep1-TGL (200 μM). Loading postsynaptic CeL neurons with Pep1-TGL blocked synaptic potentiation by PACAP38. This indicates that synaptic targeting of GluR1 subunit-containing AMPA receptors was necessary for PACAP-induced potentiation in CeL neurons. Right, time course of EPSC amplitude changes produced by PACAP38 (red horizontal bar) in CeL neurons. F, Summary of the EPSP amplitude changes produced by PACAP38 application under control conditions and in the presence of pep1-TGL in pipette solution (control, n = 5; Pep1-TGL, n = 9). Error bars indicate SEM.
As VPAC1 receptor is coupled to the Gαq protein, which is capable of activating phospholipase C (Langer and Robberecht, 2005), and our results indicate that the PACAP-induced potentiation at the BLA-CeL synapses depended on the rise in postsynaptic Ca2+ concentration, we next asked whether activation of protein kinase C (PKC) by postsynaptic Ca2+ may contribute to the observed effects of PACAP on synaptic function in CeL neurons. We found, however, that an addition of chelerythrine (10 μM), a specific cell-permeable PKC antagonist (Herbert et al, 1990), to the external solution did not suppress the ability of PACAP38 to potentiate synaptic transmission in the BLA-CeL pathway. Thus, the EPSC amplitude was potentiated by PACAP38 to 130 % ± 8 % of the baseline value (Fig. 6 B,D; n = 7, paired t test, p < 0.05), which was not significantly different from potentiation under control conditions (as shown in Fig. 3 A,B; t test, p = 0.99 between experimental groups). This finding indicates that PACAP-induced synaptic enhancements in the CeL do not require activation of PKC. On the other hand, the dialysis of CeL neurons with intrapipette solution containing autocamtide-2-related inhibitory peptide (AIP, 5 μM), which blocks Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Ishida et al., 1995), suppressed PACAP38-induced potentiation. The amplitude of the BLA-CeL EPSC was 103 % ± 3 % of the baseline value 35–40 min after the end of PACAP38 application (Fig. 6 C,D; n = 6, paired t test, p = 0.41), showing that activation of postsynaptic CaMKII kinase might be required for the effect of PACAP38 on synaptic function. Thus, both cAMP/PKA pathway and CaMKII-dependent signaling in postsynaptic CeL neurons are implicated in potentiation of neurotransmission in the BLA-CeL pathway by PACAP38.
Enhanced synaptic targeting of GluR1 subunit-containing AMPA receptors may underlie PACAP-induced potentiation
The involvement of postsynaptic signaling mechanisms in synaptic effects of PACAP, as well as postsynaptic expression of PACAP38-induced potentiation (see Fig. 3 A,C), suggested to us that postsynaptic trafficking of GluR1 subunit-containing AMPA receptor to the sites of synaptic transmission could contribute to PACAP-induced synaptic strengthening (Hayashi et al., 2000). To explore this possibility, we loaded postsynaptic CeL neurons with Pep1-TGL (200 μM), a synthetic peptide which corresponds to the C-terminus of the AMPA receptor GluR1 subunit and inhibits the delivery of GluR1-containing AMPA receptors to the postsynaptic sites by disrupting the interaction of GluR1 C-terminus with the synapse-associated protein SAP97 (Lin et al., 2010; Polepalli et al., 2010; Hayashi et al., 2000; Shi et al., 2001; Yang et al., 2008). This experimental intervention resulted in a suppression of PACAP38-induced potentiation, with the EPSC amplitude remaining at 107 % ± 5 % of the baseline value (Fig. 6 E,F; n = 9, paired t test, p = 0.18), whereas in the absence of Pep1-TGL in pipette solution the BLA-CeL EPSC was potentiated to 141 % ± 12 % of the baseline amplitude (n = 5, p < 0.05 versus baseline; the difference between two experimental groups was highly significant, t test, p < 0.05). Thus, PACAP38 could possibly enhance postsynaptic responsiveness to glutamate in CeL neurons by promoting synaptic targeting of GluR1 subunit-containing AMPA receptors.
Furthermore, we examined whether the induction of long-term potentiation (LTP) at the BLA-CeL synapses could be affected by PACAP administration. LTP was readily induced in the studied pathway with the pairing protocol (80 pulses/2 Hz at a holding potential of +30 mV) under control conditions (50 μM PTX, no PACAP in the external solution). LTP-inducing stimulation resulted in potentiation of the BLA-CeL EPSCs to 141 % ± 9 % of the baseline amplitude value 35–40 min after the induction (Fig. 7A–C; n = 7, paired t test, p < 0.01 versus baseline). We found that PPR was unaltered following the induction of LTP (n = 7; paired t test, p = 0.24), suggesting that LTP under these conditions was expressed postsynaptically. Next, we pretreated slices with 5 nM PACAP for >10 min, and then performed whole-cell patch-clamp recordings from CeL neurons in the continuous presence of PACAP. After the baseline recording (which was of the same duration as in the experiments in control external solution), we attempted to induce LTP by delivering the above-described pairing protocol. Under these conditions, the amplitude of the BLA-CeL EPSC remained at 108 % ± 9 % of the baseline value (n = 8, paired t test, p = 0.39 versus baseline; significantly different from control LTP, unpaired t test, p < 0.05), suggesting that LTP in the studied pathway might be largely occluded by the prior PACAP-induced potentiation. Taken together with the finding that PACAP-induced potentiation also did not affect the amplitude of PPR (Fig. 3 A–C), these results suggest that PACAP-induced potentiation and LTP induced by electrical stimulation may share common postsynaptic mechanisms (e.g., protein kinase pathways and/or synaptic targeting of GluR1-containing AMPA receptors).
Figure 7.

Pairing-induced LTP in the BLA-CeL pathway is occluded in slices treated with PACAP. A, A diagram illustrating a pairing protocol which was used for the induction of LTP (top). Below, EPSCs recorded in CeL neurons before (black traces) and 35–40 minutes after (red traces) the delivery of LTP-inducing stimulation either under control conditions (Pairing) or in the presence of 5 nM PACAP (PACAP + pairing). Recordings were performed in the presence of 50 μM PTX. B, Top, summary graph of LTP experiments performed in control external solution (n = 7). LTP was induced at the red arrow. Bottom, summary graph of LTP experiments performed on PACAP38-treated slices (n = 8). Red horizontal bar indicates the duration of PACAP38 application (5 nM), which started >10 min prior to the establishing of whole-cell configuration. C, Summary plot of the EPSC amplitude changes after the delivery of pairing protocol with (PACAP + pairing) or without PACAP pretreatment (Pairing). Error bars indicate SEM.
PACAP increases firing output of CeL neurons in response to synaptic activation of inputs from the BLA
Finally, we explored the role of synaptic enhancements in the BLA-CeL pathway, produced by PACAP38, in regulation of neuronal functions in the CeL. For this, we tested the effect of PACAP on the number of extracellular spikes evoked in individual CeL neurons by presynaptic stimuli of constant intensity delivered to the BLA (Riccio et al., 2009). The estimates of spiking output of CeL neurons were obtained in a cell-attached recording configuration (Fig. 8 A) to minimize the effects of prolonged whole-cell recordings on the intracellular milieu which could possibly affect functional properties of the recorded cells. We found that synaptically-induced spike firing in CeL neurons, expressed as the average number of spikes triggered by a single presynaptic stimulus, was significantly facilitated by the application of PACAP38 (Fig. 8 B,C; n = 6, paired t test, p < 0.01 versus pre-PACAP baseline; the estimates of the effect of PACAP on neuronal firing were obtained after its 40 min washout). These findings indicate that PACAP-induced potentiation of synaptic transmission at the BLA-CeL synapses might be functionally relevant, as it is translated into the enhanced firing output of CeL neurons in response to afferent signals arising from the BLA.
Figure 8.

PACAP38 increases synaptically-driven firing output of CeL neurons during activation of the BLA. A, Left, superimposed extracellular spikes evoked in a CeL neuron by stimulation of the BLA and recorded in a cell-attached patch configuration (left, as in Riccio et al., 2009). Right, recordings from the same neuron under current-clamp conditions after a whole-cell recording configuration was established. In given examples, ten traces in cell-attached configuration and three traces in whole-cell current-clamp mode were superimposed. B, Left, examples of responses in the CeL neuron in the course of BLA stimulation (once every 10 min) by electrical pulses of the constant intensity under control conditions. Right, spike firing output of the same CeL neuron 40 min after the start of PACAP38 application (5 nM, 10 min). C, Time course of changes in firing output of CeL neurons produced by PACAP38 application (red horizontal bar) (n = 6). Firing output was quantified as the average number of spikes per stimulus (the total number of spikes divided by the number of stimuli), which was then normalized to its baseline pre-PACAP value (marked with a horizontal dotted line). Error bars indicate SEM.
DISCUSSION
Our results demonstrate that PACAP potentiates glutamatergic synaptic transmission in projections from the BLA to the CeL via activation of Gαs- and Gαq-coupled postsynaptic VPAC1 receptors. This potentiation was associated with increased postsynaptic responsiveness of CeL neurons to glutamate, without significant changes in presynaptic function, through the intracellular mechanism which implicated both cAMP/PKA and CaMKII-dependent signaling, thus resulting in increased targeting of GluR1 subunit-containing AMPA receptors to the sites of synaptic transmission (Fig. 9; Hayashi et al., 2000).
Figure 9.
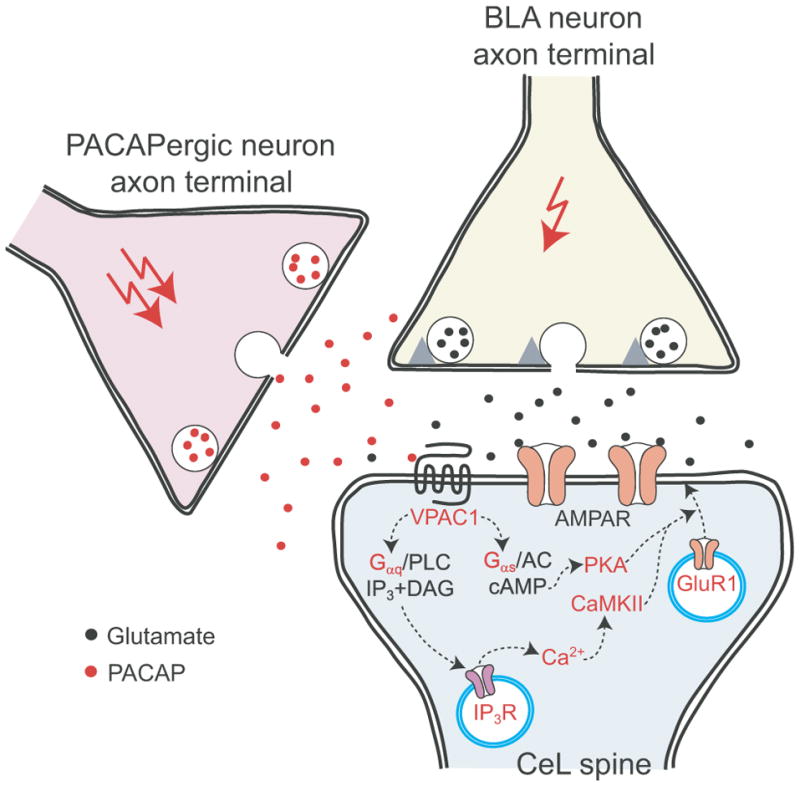
A mechanism for postsynaptically-expressed PACAP-induced synaptic enhancements in the BLA-CeL projections. PACAP binding to Gαq and Gαs-coupled VAPC1 receptors resulted in release of Ca2+ from InsP3-sensitive stores with subsequent activation of CaMKII kinase and activation of cAMP/PKA signaling pathway, respectively. This was associated with increased targeting of GluR1 subunit-containing AMPA receptors to the sites of synaptic transmission, thus enhancing the efficacy of glutamatergic synaptic transmission in the BLA-CeL projections.
PACAP-containing neurons in the dorsal vagal complex (DVC) of the brainstem are believed to contribute to ascending innervation of the extended amygdala (Kozicz et al., 1998; Das et al., 2007). PACAP-containing fibers and VPAC1 receptors are highly expressed in the CeL, suggesting that PACAP/VPAC1 pathway might be strategically positioned for modulation of neuronal functions in the CeL under conditions associated with endogenous release of PACAP (e.g., acute and chronic stress or anxiety-provoking situations). Notably, PAC1 receptor, a specific receptor for PACAP, is also expressed in the CeL (Joo et al., 2004). Moreover, it has been demonstrated previously that PACAP binds to these two receptors, PAC1 and VPAC1, with similar affinity (Hashimoto et al., 1993; Ishida et al., 1995). As the potentiating effect of PACAP on synaptic transmission was mediated exclusively by VPAC1 receptors, it is plausible that specific characteristics of the surface distribution and density of different PACAP receptor subtypes in CeL neurons could explain the receptor selectivity of PACAP-induced synaptic potentiation. Additionally, neurons in the CeL receive both excitatory and inhibitory inputs in the course of BLA activation. The GABAA receptor-mediated feed-forward inhibition of CeL neurons is likely provided by GABAergic intercalated cells, residing in small neuronal clusters between the BLA and the central nucleus (Royer et al., 1999). Our observation that the actions of PACAP were specific to glutamatergic synapses on CeL neurons suggests that the ability of PACAP to affect synaptic function might be synapse and cell type specific. As PACAP shares VPAC1 and VPAC2 receptors with VIP (Kd ~ 3 nM), a neuropeptide with 68 % sequence homology with PACAP which is also expressed in the CeL (Loren et al., 1979; Roberts et al., 1982; Gray et al., 1984; Ishihara et al., 1992), it might be interesting to examine in future studies whether VIP could also affect excitatory synaptic transmission in the BLA to the CeL pathway, possibly acting in concert with PACAP and thus contributing to the amygdala-based behaviors.
Only a handful of earlier studies have addressed the mechanisms of modulatory actions of PACAP at central synapses, mostly in the hippocampus, while synaptic effects of PACAP in the amygdala have not been previously explored. In the hippocampus, the described effects of PACAP on glutamatergic synaptic transmission are somewhat contradictory with some studies reporting that PACAP was inducing long-lasting potentiation at Schaffer collateral-CA1 synapses (Roberto and Brunelli, 2000; Cunha-Reis et al., 2005), whereas other investigators showed that PACAP could depress synaptic responses in the same pathway (Ciranna and Cavallaro, 2003). This experimental divergence could possibly be attributed to the differences in recording conditions and the dosing of PACAP (Roberto et al., 2001). Notably, in a low concentration, PACAP38 (1 nM) was shown previously to increase NMDAR-mediated synaptic currents, both at the Schaffer collateral-CA1 synapses in hippocampal slices and isolated CA1 neurons, via activation of PAC1 receptors, and, subsequently, Gαq and PKC (MacDonald et al., 2005). This differs from our findings in the amygdala, where the potentiating effect of PACAP38 was mediated by a different receptor subtype, VPAC1, and limited to the AMPA receptor-mediated synaptic responses, emphasizing the role of regional variations in the expression of receptor subtypes activated by PACAP and their coupling to distinct signaling pathways.
The observation that PACAP-induced strengthening of glutamatergic synaptic transmission in the BLA-CeL projections could be translated into increased spiking output of neurons in the CeL in response to the BLA-driven synaptic activation is consistent with the demonstrated roles of PACAP in regulation of innate fear responses (Otto et al., 2001b; Legradi et al., 2007). Neuromodulatory functions of PACAP in the CeL appear to be specific to its actions on synaptic transmission, as the membrane properties of CeL neurons were unaffected by this neuropeptide in our experiments. While the neural substrates of innate fear are diffuse and not completely characterized (Shumyatsky et al., 2005; Riccio et al., 2009), it is clear that perturbancies in the function of individual components of innate fear circuitry would affect the behavioral outcome.
Recently, several groups independently provided detailed descriptions of microcircuits in the amygdala and their functional roles in connection to the regulation of learned and innate fear responses (Herry et al., 2008; Ciocci et al., 2010; Haubensak et al., 2010; Tye et al., 2011; Knobloch et al., 2012). Notably, selective activation of the BLA to the CeL pathway was shown to produce an acute anxiolytic effect whereas inhibition of this pathway increased anxiety (Tye et al., 2011), suggesting that CeL neurons may function as an inhibitory gate between the BLA and the medial division of the central nucleus of the amygdala (CeM). Then, if CeL neurons simply inhibit CeM in response to BLA activation, endogenous release of PACAP, which was found to potentiate excitatory transmission in the BLA to the CeL pathway, would be expected to exert anxiolytic effect in vivo. However, functional roles of the CeL in control of information processing within the amygdala might be more complex. Thus, recent studies, combining in vivo electrophysiological recordings and optogenetics, demonstrated that the CeL may contain two different subpopulations of GABAergic neurons (CeLon/PKC-δ− and CeLoff/PKC-δ+ neurons) (Haubensak et al., 2010; Ciocchi et al., 2010). CeLon neurons, which receive inputs from the BLA, display increased firing during presentation of conditioned stimulus (CS). The CeL may disinhibit the CeM through inhibition of CeLoff neurons by activated CeLon neurons. Although CeLon neurons also send direct (monosynaptic) inhibitory projection to the CeM, disinhibitory CeLon-CeLoff-CeM pathway seems to predominate over inhibitory CeLon-CeM pathway (Ciocchi et al., 2010; Haubensak et al., 2010). Based on this model, PACAP would facilitate information flow from the BLA to the CeM if it potentiates excitatory transmission from the BLA to the CeLon neurons. Disinhibiting the CeM through decreased activation of CeLoff neurons would promote fear responses. Our current findings, however, are insufficient to support this model directly, as neurons in the CeL were chosen for recordings randomly. Thus, it may be interesting to explore in future studies whether PACAPergic axon fibers innervate specific populations of CeL neurons (CeLon versus CeLoff neurons). Double immunohistochemistry for PACAP and PKCδ could address this question directly. It would also be important to examine whether PACAP could affect neurotransmission in inputs to CeLon/PKC-δ− or CeLoff/PKC-δ+ neurons differentially.
While the CeL may function as a relay station, either inhibitory or disinhibitory, between the BLA and the CeM, neurons in the CeL also project to the bed nucleus of the stria terminalis (BNST; Sun et al., 1991). The BNST and CeA, acting in concert with other brain areas (e.g., the paraventricular nucleus of the hypothalamus), promote anxiety-related behaviors (Hammack et al., 2009). Therefore, modulation of neurotransmission in the BLA-CeL pathway by PACAP, released endogenously in response to stressful or anxiety-provoking events, may change the activity of both the BNST and CeA, thus affecting anxiety levels. To understand more fully the role of PACAP in regulation of fear responses, it might be necessary to characterize its neuronal and synaptic functions in the components of fear circuitry receiving projections from the amygdala, including the BNST.
Acknowledgments
This work was supported by National Institutes of Health grants MH090464 (to V.Y.B.) and MH080328 (to G.P.S.), and Whitehall Foundation (to G.P.S. and V.Y.B.). We thank Yan Li for comments on the manuscript.
References
- Amano T, Unal CT, Pare D. Synaptic correlates of fear extinction in the amygdala. Nat Neurosci. 2010;13:489–494. doi: 10.1038/nn.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basille M, Vaudry D, Coulouarn Y, Jegou S, Lihrmann I, Fournier A, Vaudry H, Gonzalez B. Comparative distribution of pituitary adenylate cyclase-activating polypeptide (PACAP) binding sites and PACAP receptor mRNAs in the rat brain during development. J Comp Neurol. 2000;425:495–509. [PubMed] [Google Scholar]
- Cho JH, Bayazitov IT, Meloni EG, Myers KM, Carlezon WA, Zakharenko SS, Bolshakov VY. Coactivation of thalamic and cortical pathways induces input timing-dependent plasticity in amygdala. Nature Neurosci. 2012;15:113–122. doi: 10.1038/nn.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, Muller C, Luthi A. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- Ciranna L, Cavallaro S. Opposing effects by pituitary adenylate cyclase-activating polypeptide and vasoactive intestinal peptide on hippocampal synaptic transmission. Exp Neurol. 2003;184:778–784. doi: 10.1016/S0014-4886(03)00300-5. [DOI] [PubMed] [Google Scholar]
- Cunha-Reis D, Ribeiro JA, Sebastiao AM. VIP enhances synaptic transmission to hippocampal CA1 pyramidal cells through activation of both VPAC1 and VPAC2 receptors. Brain Res. 2005;1049:52–60. doi: 10.1016/j.brainres.2005.04.077. [DOI] [PubMed] [Google Scholar]
- Das M, Vihlen CS, Legradi G. Hypothalamic and brainstem sources of pituitary adenylate cyclase-activating polypeptide nerve fibers innervating the hypothalamic paraventricular nucleus of the rat. Journal of Comparative Neurology. 2007;500:761–776. doi: 10.1002/cne.21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson T, Fleetwood-Walker SM, Mitchell R, Lutz EM. Evidence for roles of vasoactive intestinal polypeptide (VIP) and pituitary adenylate cyclase activating polypeptide (PACAP) receptors in modulating the responses of rat dorsal horn neurons to sensory inputs. Neuropeptides. 1997;31:175–185. doi: 10.1016/s0143-4179(97)90087-1. [DOI] [PubMed] [Google Scholar]
- Dickson L, Aramori I, McCulloch J, Sharkey J, Finlayson K. A systematic comparison of intracellular cyclic AMP and calcium signalling highlights complexities in human VPAC/PAC receptor pharmacology. Neuropharmacology. 2006;51:1086–1098. doi: 10.1016/j.neuropharm.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Dickson L, Finlayson K. VPAC and PAC receptors: From ligands to function. Pharmacol Ther. 2009;121:294–316. doi: 10.1016/j.pharmthera.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Fizanne L, Sigaudo-Roussel D, Saumeet JL, Fromy B. Evidence for the involvement of VPAC1 and VPAC2 receptors in pressure-induced vasodilatation in rodents. J Physiol. 2004;554:519–528. doi: 10.1113/jphysiol.2003.053835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- Gourlet P, De Neef P, Cnudde J, Waelbroeck M, Robberecht P. In vitro properties of a high affinity selective antagonist of the VIP1 receptor. Peptides. 1997;18:1555–1560. doi: 10.1016/s0196-9781(97)00230-1. [DOI] [PubMed] [Google Scholar]
- Gray TS, Cassell MD, Nilaver G, Zimmerman EA, Williams TH. The distribution and ultrastructure of VIP-immunoreactivity in the central nucleus of the rat amygdala. Neuroscience. 1984;11:399–408. doi: 10.1016/0306-4522(84)90032-0. [DOI] [PubMed] [Google Scholar]
- Hammack SE, Roman CW, Lezak KR, Kocho-Shellenberg M, Grimmig B, Falls WA, Braas K, May V. Roles for pituitary adenylate cyclase-activating peptide (PACAP) expression and signaling in the bed nucleus of the stria terminalis (BNST) in mediating the behavioral consequences of chronic stress. J Mol Neurosci. 2009;42:327–340. doi: 10.1007/s12031-010-9364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Ishihara T, Shigemoto R, Mori K, Nagata S. Molecular cloning and tissue distribution of a receptor for pituitary adenylate cyclase-activating polypeptide. Neuron. 1993;11:333–342. doi: 10.1016/0896-6273(93)90188-w. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Shintani N, Baba A. New insights into the central PACApergic system from the phenotypes in PACAP- and PACAP receptor-knockout mice. Ann NY Acad Sci. 2006;1070:75–89. doi: 10.1196/annals.1317.038. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Shintani N, Tanaka K, Mori W, Hirose M, Matsuda T, Sakaue M, Miyazaki J, Niwa H, Tashiro F, Yamamoto K, Koga K, Tomimoto S, Kunugi A, Suetake S, Baba A. Altered psychomotor behaviors in mice lacking pituitary adenylate cyclase-activating polypeptide (PACAP) Proc Natl Acad Sci U S A. 2001;98:13355–13360. doi: 10.1073/pnas.231094498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong HW, Deisseroth K, Callaway EM, Fanselow MS, Luthi A, Anderson DJ. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Shigemoto R, Mori K, Takahashi K, Nagata S. Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron. 1992;8:811–819. doi: 10.1016/0896-6273(92)90101-i. [DOI] [PubMed] [Google Scholar]
- Joo KM, Chung YH, Kim MK, Nam RH, Lee BL, Lee KH, Cha CI. Distribution of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide receptors (VPAC1, VPAC2, and PAC1 receptor) in the rat brain. J Comp Neurol. 2004;476:388–413. doi: 10.1002/cne.20231. [DOI] [PubMed] [Google Scholar]
- Knobloch HS, Charlet A, Hoffmann LC, Eliava M, Khrulev S, Cetin AH, Osten P, Schwarz MK, Seeburg PH, Stoop R, Grinevich V. Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron. 2012;73:553–566. doi: 10.1016/j.neuron.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Köves K, Arimura A, Görcs TG, Somogyvári-Vigh A. Comparative distribution of immunoreactive pituitary adenylate cyclase activating polypeptide and vasoactive intestinal polypeptide in rat forebrain. Neuroendocrinology. 1991;54:159–169. doi: 10.1159/000125864. [DOI] [PubMed] [Google Scholar]
- Kozicz T, Vigh S, Arimura A. The source of origin of PACAP- and VIP-immunoreactive fibers in the laterodorsal division of the bed nucleus of the stria terminalis in the rat. Brain Research. 1998;810:211–219. doi: 10.1016/s0006-8993(98)00692-1. [DOI] [PubMed] [Google Scholar]
- Kozicz T, Arimura A. Dopamine- and cyclic AMP-regulated phosphoprotein-immunoreactive neurons activated by acute stress are innervated by fiber terminals immunopositive for pituitary adenylate cyclase-activating polypeptide in the extended amygdala in the rat. Regul Pept. 2002;109:63–70. doi: 10.1016/s0167-0115(02)00188-x. [DOI] [PubMed] [Google Scholar]
- Langer I, Robberecht P. Mutations in the carboxy-terminus of the third intracellular loop of the human recombinant VPAC1 receptor impair VIP-stimulated [Ca2+]i increase but not adenylate cyclase stimulation. Cell Signal. 2005;17:17–24. doi: 10.1016/j.cellsig.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Legradi G, Das M, Giunta B, Hirani K, Mitchell EA, Diamond DM. Microinfusion of pituitary adenylate cyclase-activating polypeptide into the central nucleus of amygdala of the rat produces a shift from an active to passive mode of coping in the shock-probe fear/defensive burying test. Neural Plast. 2007;2007:79102. doi: 10.1155/2007/79102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Lujan R, Watanabe M, Frerking M, Maylie J, Adelman JP. Coupled activity-dependent trafficking of synaptic SK2 channels and AMPA receptors. J Neurosci. 2010;30:11726–11734. doi: 10.1523/JNEUROSCI.1411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loren I, Emson PC, Fahrenkrug J, Bjorklund A, Alumets J, Hakanson R, Sundler F. Distribution of vasoactive intestinal polypeptide in the rat and mouse brain. Neuroscience. 1979;4:1953–1976. doi: 10.1016/0306-4522(79)90068-x. [DOI] [PubMed] [Google Scholar]
- Lutz EM, Sheward WJ, West KM, Morrow JA, Fink G, Harmar AJ. The VIP2 receptor: molecular characterisation of a cDNA encoding a novel receptor for vasoactive intestinal peptide. FEBS Lett. 1993;334:3–8. doi: 10.1016/0014-5793(93)81668-p. [DOI] [PubMed] [Google Scholar]
- Macdonald DS, Weerapura M, Beazely MA, Martin L, Czerwinski W, Roder JC, Orser BA, MacDonald JF. Modulation of NMDA receptors by pituitary adenylate cyclase activating peptide in CA1 neurons requires G alpha q, protein kinase C, and activation of Src. J Neurosci. 2005;25:11374–84. doi: 10.1523/JNEUROSCI.3871-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuo Y, Matsumoto Y, Tokito F, Tsuda M, Fujino M. Effects of vasoactive intestinal polypeptide (VIP) and pituitary adenylate cyclase activating polypeptide (PACAP) on the spontaneous release of acethylcholine from hippocampus by brain microdilaysis. Brain Res. 1993;611:207–215. doi: 10.1016/0006-8993(93)90504-g. [DOI] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164:567–574. doi: 10.1016/0006-291x(89)91757-9. [DOI] [PubMed] [Google Scholar]
- Nielsen KM, Chaverra M, Hapner SJ, Nelson BR, Todd V, Zigmond RE, Lefcort F. PACAP promotes sensory neuron differentiation: blockade by neurotrophic factors. Mol Cell Neurosci. 2004;25:629–641. doi: 10.1016/j.mcn.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Otto C, Kovalchuk Y, Wolfer DP, Gass P, Martin M, Zuschratter W, Grone HJ, Kellendonk C, Tronche F, Maldonado R, Lipp HP, Konnerth A, Schutz G. Impairment of mossy fiber long-term potentiation and associative learning in pituitary adenylate cyclase activating polypeptide type I receptor-deficient mice. J Neurosci. 2001a;21:5520–5527. doi: 10.1523/JNEUROSCI.21-15-05520.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C, Martin M, Wolfer DP, Lipp HP, Maldonado R, Schutz G. Altered emotional behavior in PACAP-type-I-receptor-deficient mice. Brain Res Mol Brain Res. 2001b;92:78–84. doi: 10.1016/s0169-328x(01)00153-x. [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–63. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggins HD, Stamp JA, Burns J, Rusak B, Semba K. Distribution of pituitary adenylate cyclase activating polypeptide (PACAP) immunoreactivity in the hypothalamus and extended amygdala of the rat. J Comp Neurol. 1996;376:278–294. doi: 10.1002/(SICI)1096-9861(19961209)376:2<278::AID-CNE9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Pisegna JR, Wank SA. Molecular cloning and functional expression of the pituitary adenylate cyclase-activating polypeptide type I receptor. Proc Natl Acad Sci U S A. 1993;90:6345–6349. doi: 10.1073/pnas.90.13.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polepalli JS, Sullivan RK, Yanagawa Y, Sah P. A specific class of interneuron mediates inhibitory plasticity in the lateral amygdala. J Neurosci. 2010;30:14619–14629. doi: 10.1523/JNEUROSCI.3252-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Steven CF. Physiology of synaptic transmission and short-term plasticity. In: Cowan, Südhof, Stevens, editors. Synapses. Baltimore and London: Johns Hopkins University Press; 2001. pp. 135–175. [Google Scholar]
- Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, Norrholm SD, Kilaru V, Smith AK, Myers AJ, Ramirez M, Engel A, Hammack SE, Toufexis D, Braas KM, Binder EB. May V Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Li Y, Moon J, Kim KS, Smith KS, Rudolph U, Gapon S, Yao GL, Tsvetkov E, Rodig SJ, Van’t Veer A, Meloni EG, Carlezon WA, Bolshakov VY, Clapham DE. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137:761–772. doi: 10.1016/j.cell.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht P, Gourlet P, De Neef P, Woussen-Colle MC, Vandermeers-Piret MC, Vandermeers A, Christophe J. Structural requirements for the occupancy of pituitary adenylate-cyclase-activating-peptide (PACAP) receptors and adenylate cyclase activation in human neuroblastoma NB-OK-1 cell membranes. Discovery of PACAP(6-38) as a potent antagonist. Eur J Biochem. 1992;207:239–246. doi: 10.1111/j.1432-1033.1992.tb17043.x. [DOI] [PubMed] [Google Scholar]
- Roberto M, Brunelli M. PACAP-38 enhances excitatory synaptic transmission in the rat hippocampal CA1 region. Learn Mem. 2000;7:303–311. doi: 10.1101/lm.34200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Scuri R, Brunelli M. Differential effects of PACAP-38 on synaptic responses in rat hippocampal CA1 region. Learn Mem. 2001;8:265–271. doi: 10.1101/lm.40501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GW, Woodhams PL, Polak JM, Crow TJ. Distribution of neuropeptides in the limbic system of the rat: the amygdaloid complex. Neuroscience. 1982;7:99–131. doi: 10.1016/0306-4522(82)90156-7. [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Pare D. An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci. 1999;19:10575–10583. doi: 10.1523/JNEUROSCI.19-23-10575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Shioda S, Shuto Y, Somogyvari-Vigh A, Legradi G, Onda H, Coy DH, Nakajo S, Arimura A. Localization and gene expression of the receptor for pituitary adenylate cyclase-activating polypeptide in the rat brain. Neurosci Res. 1997;28:345–354. doi: 10.1016/s0168-0102(97)00065-5. [DOI] [PubMed] [Google Scholar]
- Shin RM, Tsvetkov E, Bolshakov VY. Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron. 2006;52:883–896. doi: 10.1016/j.neuron.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumyatsky GP, Malleret G, Shin RM, Takizawa S, Tully K, Tsvetkov E, Zakharenko SS, Joseph J, Vronskaya S, Yin DQ, Schubart UK, Kandel ER, Bolshakov VY. Stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell. 2005;123:697–709. doi: 10.1016/j.cell.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Sreedharan SP, Patel DR, Xia M, Ichikawa S, Goetzl EJ. Human vasoactive intestinal peptide1 receptors expressed by stable transfectants couple to two distinct signaling pathways. Biochem Biophys Res Commun. 1994;203:141–148. doi: 10.1006/bbrc.1994.2160. [DOI] [PubMed] [Google Scholar]
- Stella N, Magistretti PJ. Vasoactive intestinal polypeptide (VIP) and pituitary adenylate cyclase activating polypeptide (PACAP) potentiate the glutamate-evoked release of arachidonic acid from mouse cortical neurons. J Biol Chem. 1996;271:23705–23710. doi: 10.1074/jbc.271.39.23705. [DOI] [PubMed] [Google Scholar]
- Sun N, Roberts L, Cassell MD. Rat central amygdaloid nucleus projections to the bed nucleus of the stria terminalis. Brain Res Bull. 1991;27:651–662. doi: 10.1016/0361-9230(91)90041-h. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron. 2002;34:289–300. doi: 10.1016/s0896-6273(02)00645-1. [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Haastert PJ, Van Driel R, Jastorff B, Baraniak J, Stec WJ, De Wit RJ. Competitive cAMP antagonists for cAMP-receptor proteins. J Biol Chem. 1984;259:10020–10024. [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev. 2009;61:283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L. Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci. 1997;17:83–90. doi: 10.1523/JNEUROSCI.17-01-00083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Lei G, Jackson MF, Macdonald JF. The involvement of PACAP/VIP system in the synaptic transmission in the hippocampus. J Mol Neurosci. 2010;42:319–326. doi: 10.1007/s12031-010-9372-7. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, Zhou Q. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc Natl Acad Sci U S A. 2008;105:11388–11393. doi: 10.1073/pnas.0802978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]