

Abstract
CYP51 (sterol 14α-demethylase) is a cytochrome P450 enzyme essential for sterol biosynthesis and the primary target for clinical and agricultural antifungal azoles. The azoles that are currently in clinical use for systemic fungal infections represent modifications of two basic scaffolds, ketoconazole and fluconazole, all of them being selected based on their antiparasitic activity in cellular experiments. By studying direct inhibition of CYP51 activity across phylogeny including human pathogens Trypanosoma brucei, Trypanosoma cruzi and Leishmania infantum, we identified three novel protozoa-specific inhibitory scaffolds, their inhibitory potency correlating well with antiprotozoan activity. VNI scaffold (carboxamide containing β-phenyl-imidazoles) is the most promising among them: killing T. cruzi amastigotes at low nanomolar concentration, it is also easy to synthesize and nontoxic. Oral administration of VNI (up to 400 mg/kg) neither leads to mortality nor reveals significant side effects up to 48 h post treatment using an experimental mouse model of acute toxicity. Trypanosomatidae CYP51 crystal structures determined in the ligand-free state and complexed with several azole inhibitors as well as a substrate analog revealed high rigidity of the CYP51 substrate binding cavity, which must be essential for the enzyme strict substrate specificity and functional conservation. Explaining profound potency of the VNI inhibitory scaffold, the structures also outline guidelines for its further development. First steps of the VNI scaffold optimization have been undertaken; the results presented here support the notion that CYP51 structure-based rational design of more efficient, pathogen-specific inhibitors represents a highly promising direction.
Keywords: Sterol 14α-demethylase, CYP51, Inhibition, Crystal structure
1. Introduction
1.1. Human infections with Trypanosomatidae
Human infections with eukaryotic pathogens represent one of the most severe global health problems. While fungal diseases receive significant attention from pharmaceutical companies, the pathologies caused by protozoan parasites are considered not profitable and remain highly ignored (Chaudhary and Roos, 2005). Chagas disease, sleeping sickness and leishmaniasis are the three most neglected tropical diseases with the major impact among the poorest (www.who.int/neglected_diseases/en/). The etiologic agents are unicellular organisms from the order Kinetoplastida, family Trypanosomatidae: genera Trypanosoma and Leishmania. Trypanosoma cruzi causes Chagas disease, endemic in 18 countries in South and Central America (www.who.int/neglected_diseases/diseases/chagas/en). In this area, 8–10 million people are infected, ~50,000 die each year and >100 millions are at risk. The disease is transmitted by kissing bugs (Triatomine); other ways of infection include contaminated food, blood, organs and from mother to child. In mammals, Trypanosoma cruzi cycles as non-replicative bloodstream trypomastigotes and replicative intracellular amastigotes. The severity of the acute stage varies from non-symptomatic to fatal, depending on the parasite strain, burden and host genetics. Chronic stage manifests ~10–20 years later, primarily affecting the heart and/or gastrointestinal tract. Sleeping sickness is caused by Trypanosoma brucei and transmitted by tsetse fly (Glossina) (www.who.int/trypanosomiasis_african/en). This parasite is extracellular, the bloodstream form eventually crossing the blood-brain barrier and invading cerebrospinal fluid. Sleeping sickness is endemic in 36 African countries, with 300,000 new cases and ~30,000 deaths per year, >60 million people are at risk. Leishmania is transmitted by sand fly (Plebotomine) (www.who.int/leishmaniasis/en). In mammals these are obligatory intracellular parasites, multiplying within macrophages. Depending on the Leishmania species, at least four different forms of the disease can be distinguished. Visceral leishmaniasis (Leishmania donovani, Leishmania infantum, Leishmania chagasi), also known as kala-azar, or black fever, is the most severe form affecting visceral tissues, primarily spleen, liver and bone marrow. Leishmaniasis is endemic in 88 countries in Africa, America, Asia and Europe, with 12 million people infected, 1–2 million new cases and ~60,000 deaths occurring each year. Without treatment, visceral leishmaniasis, sleeping sickness and chronic symptomatic Chagas disease are fatal. Due to human migrations and HIV-coinfections these diseases are now spreading worldwide and becoming a real challenge in North America, Europe and Asia. There are no vaccines (Cavalli and Bolognesi, 2009), and a handful of drugs that have limited efficacy display severe side effects and high levels of resistance (Lepesheva and Waterman, 2011a; Soeiro et al., 2009). Safer and more efficient drugs are desperately needed (Clayton, 2010; Leslie, 2011; Paniz Mondolfi et al., 2011).
1.2. Sterol biosynthesis and the CYP51 reaction
Sterols can be regarded as markers of eukaryotic cells (Rohmer et al., 1979). They are essential membrane components and also serve as regulatory (hormonal) molecules required for cell growth and development (Nes, 2011). Beginning with acetyl-CoA condensation, sterol biosynthesis proceeds to squalene 2,3-epoxide, which in non-photosynthetic organisms is then converted into lanosterol, the first cyclized sterol precursor of the pathway (Fig. 1). The three-step reaction shown in the black rectangle is catalyzed by a cytochrome P450 (CYP) monooxygenase, CYP51, also known as sterol 14α-demethylase (EC 1.14.13.70) (Lepesheva and Waterman, 2007). As all cytochrome P450 enzymes, CYP51 is a cysteine-coordinated hemoprotein, which cleaves molecular oxygen with the formation of one molecule of water and insertion of the other oxygen atom into the substrate. In order to fulfill its catalytic function and remove the 14α-methyl group from the sterol ring, CYP51 must repeat one P450 catalytic cycle three times, with the substrate molecule remaining properly oriented within the enzyme active site for the three sequential monooxygenation steps. Being an efficient target for clinical and agricultural antifungals because of its essentiality (Vanden Bossche, 1988), CYP51 is potentially an excellent subject for further drug development. As a cytochrome P450 it “senses” ligands spectrally, providing investigators with basic information on the nature and binding affinity of potential new inhibitors (Fig. 2).
Fig. 1.

Schematic representation of sterol biosynthesis. Trypanosomatidae CYP51 substrates, CYP51 reaction and the major end products of the pathway are shown.
Fig. 2.

P450 spectral responses to ligand binding. (a) In the ferric (Fe3+) state CYP51 contains a water molecule (red sphere) coordinated to the heme iron (orange sphere); low-spin hexacoordinated state, the Soret band maximum is at 417 nm. (b) When the iron is reduced (Fe2+) and binds CO, the Soret band maximum shifts to ~450 nm. (c) When the substrate (eburicol is shown in light blue) binds to the enzyme, it displaces water from the iron coordination sphere; Fe3+ becomes pentacoordinated high-spin, the Soret band maximum shifts to the left (394 nm), producing type 1 spectral response. (d) When a ligand stronger than water directly coordinates to the heme iron (carbon atoms of fluconazole are colored in pink) the Soret band maximum shifts further to the right, producing type 2 spectral response. (e) Spectral responses of T. cruzi CYP51 to two VNI derivatives, VNI-T and FF-VNI-T, and the corresponding titration curves. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
1.3. Protozoa-specific CYP51 inhibitory scaffolds
Using this approach as a starting point, we tested protozoan, fungal and human CYP51s against commercial antifungals, experimental azole derivatives, substrate analogs and also conducted optical high throughput screening followed by web-search for structurally similar compounds. As a result, novel CYP51 inhibitory scaffolds have been identified, our current work being focused on three of them (Fig. 3). VNI and VNF represent the most potent inhibitory scaffold (Lepesheva et al., 2007): at the standard CYP51 reaction conditions they completely inhibit activity of the T. cruzi and T. brucei CYP51 orthologs at 1:1 molar ratio to the enzyme and cannot be replaced in the CYP51 active center by substrate over time yet do not inhibit the human ortholog (Lepesheva et al., 2008). Other important advantages of this scaffold include low general cytotoxicity (EC50 (HL60) >50 μM), which in comparison with cellular effect on T. cruzi amastigotes (EC50 = 1.2 nM for VNI) (Lepesheva et al., 2011) produces selectivity index human/T. cruzi >50,000. Also, we found that, contrary to the antifungal drugs posaconazole and fluconazole, they do not induce T. cruzi CYP51 gene expression, which suggests lower propensity to cause resistance (Lepesheva et al., 2010a). Curative effect of VNI in the murine model of Chagas disease has been confirmed (Villalta et al., submitted for publication) and its testing in the model of visceral leishmaniasis is currently in progress. It is noteworthy that VNI slows down parasitemia in the mouse model of T. brucei infection (Lepesheva et al., 2010b), where endogenously produced T. brucei sterols are required only for regulatory functions (Nes et al., 2012); this finding advocates for a possibility of VNI use against T. brucei in combination with the traditional anti-sleeping sickness drugs.
Fig. 3.

CYP51 inhibitors. (a) Antifungal drugs, posaconazole and ravuconazole have entered clinical trials for Chagas disease. (b) Three novel experimental CYP51 inhibitory scaffolds. Indometacin amid derivatives (INDO-pyridine scaffold) are interesting because of their potential dual action in vivo: as non-azole heme binding CYP51 inhibitors and as COX2 inhibitors (anti-inflammatory effect) (Konkle et al., 2009). 14α-Methylenecyclopropyl-24.25-dihydrolanosterol (MCP) acts as a T. cruzi CYP51 selective suicide substrate, which is the first example of an effective mechanism-based CYP51 inhibitor (Hargrove et al., 2012). Carboxamide containing β-phenyl imidazoles (VNI and VNF) are the most potent inhibitors of protozoan CYP51s.
1.4. Rigidity of the binding cavity as the most characteristic CYP51 structural feature
By the time when the first eukaryotic CYP51 structures were resolved (Lepesheva et al., 2010b), it was quite generally accepted that high flexibility represents the major characteristic feature of the cytochrome P450 structural fold (Poulos, 2003; Scott et al., 2003; Williams et al., 2004; Ekroos and Sjögren, 2006), which ultimately meant that P450 structures are unlikely to be reasonably helpful in directing rational design of new P450-targeting inhibitors (Williams et al., 2004). However, we found that, unlike CYPs with expressed substrate promiscuity, CYP51s have a highly rigid substrate binding cavity. By determining X-ray structures of CYP51s from three protozoan pathogens (T. brucei, T. cruzi and L. infantum), in ligand-free state [PDB ID: 3g1q] and complexed with different azole inhibitors ((Lepesheva et al., 2010a,b; Hargrove et al., 2011; Buckner et al., in press), and substrate analog MCP (Hargrove et al., 2012) we observed that CYP51s do not reveal significant conformational changes, either upon ligand binding, or across species (Lepesheva and Waterman, 2011b). This led us to the hypothesis that structural rigidity of the CYP51 binding cavity (1) underlies the enzyme ability to preserve strict catalytic conservation at very low sequence identity across phylogeny and (2) should allow prediction of scaffold modifications that would potentially create a better match of an inhibitor to the enzyme active site topology. Interestingly, although VNI and VNF are structurally very similar (Fig. 3), they bind to CYP51 in the opposite orientation, the inhibitory pharmacophore (Fig. 4) revealing the empty volume that extends beyond each of the bound inhibitors. First steps for the CYP51 structure-based VNI scaffold modification have been undertaken and the results are presented below.
Fig. 4.

CYP51 structure-based inhibitory pharmacophore. VNI (cyan, PDB ID: 3gw9), VNF (salmon, PDB ID: 3ksw), MCP (green, PDB ID: 3p99) and a tipifarnib derivative JKF (blue, PDB ID: 3tik) co-crystallized within the CYP51 binding cavity. The heme is shown in yellow, the secondary structural elements that represent cytochrome P450 substrate recognition sites (SRS) 1 (helix B′), 4 (helix I), 5 (β 1–4) and 6 (β 4-hairpin) are depicted as ribbons. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. Materials and methods
The proteins (T. cruzi, T. brucei, L. infantum, Candida albicans and human CYP51s and rat and T. brucei cytochrome P450 reductase (CPR)) were expressed in Escherichia coli and purified as previously described (Lepesheva et al., 2004, 2006; Hargrove et al., 2011). P450 concentration was determined from reduced CO-binding spectra using the coefficient of molar extinction of Δε450– 490 = 91−1 cm−1 (Omura and Sato, 1964).
2.1. Synthetic scheme and purity analysis of VNI and derivatives
The racemic mixture of VNI and its S-enantiomer was synthesized by PyBOP amide coupling reaction between 1-(2,4-dichlorophenyl)- 2-(1H-imidazol-1-yl)ethanamine 2 and 4-(5-phenyl-1,3, 4-oxadiazol-2-yl)benzoic acid 4. The compound 2 (Chevreuil et al., 2006) was prepared from commercially available 1-(2,4-dichlorophenyl)- 2-(1H-imidazol-1-yl)ethanol 1 in three steps starting with mesylation with methanesulfonyl chloride in the presence of triethylamine to afford mesylated compound which was treated with sodium azide, followed by reduction with lithium aluminum hydride to provide compound 2 in 52% overall yield. Synthesis of 4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzoic acid 4 commenced from benzohydrazide 3 with methyl 4-(chlorocarbonyl)benzoate to afford methyl 4-(2-benzoylhydrazinecarbonyl)benzoate. Oxadiazole cyclization with POCl3 followed by hydrolysis with NaOH afforded benzoic acid 4 in 90%.
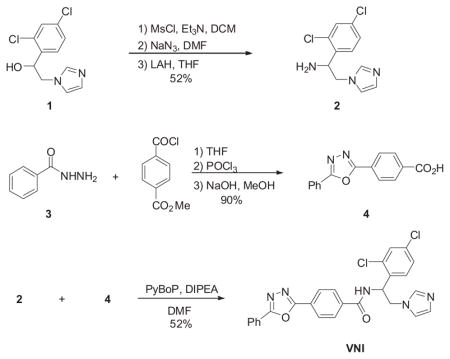
The other VNI derivatives were synthesized using similar method and their S-enantiomers were separated by Supercritical Fluid Chromatography (SFC).
2.1.1. VNI
1H NMR (DMSO, 400 MHz) δ (ppm) 9.39 (d, J = 8.0 Hz, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.16 (dd, J = 1.6, 8.0 Hz, 2H), 8.04 (d, J = 8.4, 2H), 7.76–7.59 (m, 5H), 7.51 (dd, J = 2.4, 8.4 Hz, 1H), 7.19 (s, 1H), 6.86 (s, 1H), 5.78–5.62 (m, 1H), 4.40 (dd, J = 10.0, 14.4 Hz, 1H), 4.31 (dd, J = 5.2, 14.4 Hz, 1H); LCMS, single peak, 0.89 min, m/e, 504.2 (M+1); Chiral resolution, CHIRALPAK IB (4.6 × 250 mm) 50% isocratic with 0.1 DEA.
2.1.2. VNI-triazole
1H NMR (CDCl3, 400 MHz) δ (ppm) 8.55 (d, J = 7.2 Hz, 1H), 8.27 (d, J = 8.4 Hz, 2H), 8.16 (dd, J = 2.0, 8.0 Hz, 2H), 8.11 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.87 (s, 1H, NH), 7.62–7.52 (m, 3H), 7.45 (d, J = 2.0 Hz, 1H), 7.07 (dd, J = 2.0, 8.4 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 6.02–5.93 (m, 1H), 4.75 (dd, J = 4.8, 14.4 Hz, 1H), 4.71 (dd, J = 3.6, 14.4 Hz, 1H); LCMS, single peak, 0.80 min, m/e, 506.1 (M+1); Chiral resolution, CHIRALPAK IB (4.6 × 250 mm) 55% isocratic with 0.1 DEA.
2.1.3. Difluoro-β-phenyl-VNI-triazole
1H NMR (DMSO, 400 MHz) δ (ppm) 9.29 (d, J = 8.4 Hz, 1H), 8.48 (s, 1H), 8.23 (d, J = 8.4 Hz, 2H), 8.15 (dd, J = 1.6, 8.0 Hz, 2H), 8.00 (d, J = 8.4 Hz, 2H), 7.96 (s, 1H), 7.70–7.60 (m, 4H), 7.25 (dt, J = 2.4, 10.8 Hz, 1H), 7.15 (dt, J = 2.4, 8.4 Hz, 1H), 5.85–5.72 (m, 1H), 4.70 (dd, J = 9.2, 13.6 Hz, 1H), 4.55 (dd, J = 5.6, 13.6 Hz, 1H); LCMS, single peak, 0.72 min, m/e, 473.2 (M+1); Chiral resolution, CHIRALPAK IB (4.6 × 250mm) 50% isocratic with 0.1 DEA.
2.1.4. VNI/VNF combination
1H NMR (DMSO, 400 MHz) δ (ppm) 8.24 (d, J = 8.4 Hz, 2H), 8.16 (d, J = 6.8 Hz, 2H), 7.96 (d, J = 8.4 Hz, 2H), 7.68–7.50 (m, 7H), 7.45 (d, J = 8.4 Hz, 1H), 7.42 (d, J = 12.0 Hz, 1H), 7.22–7.10 (m, 3H), 6.96 (s, 1H), 5.84 (t, J = 7.6 Hz, 1H), 4.63–4.45 (m, 2H); LCMS, single peak, 0.75 min, m/e, 548.2 (M+1); Chiral resolution, CHIRALPAK IB (4.6 × 250 mm) 55% isocratic with 0.1 DEA.
2.2. CYP51 spectral titration
The titrations of CYP51s with VNI and its derivatives were carried out in 2 ml tandem cuvettes at P450 concentrations of ~2 μM in the wavelength range 350–450 nm in a 50 mM potassium phosphate buffer, pH 7.4, containing 200 mM NaCl and 0.1 mM EDTA using a Shimadzu UV-2401PC spectrophotometer. The inhibitors were added to the sample cuvette in the concentration range 0.15–5.0 μM from a 0.2 mM stock solution in DMSO. The apparent dissociation constants (Kd) were calculated by plotting the absorbance changes in the difference spectra (ΔA) upon titration against free ligand concentration and fitting the data to a rectangular hyperbola in Sigma Plot Statistics as described previously (Lepesheva et al., 2007). The concentrations of free ligand were calculated using the equation:
where [L] and [E] are the concentrations of the ligands (VNI derivatives) and the enzyme used for the titration, respectively, ΔA is the difference in the absorption observed at given ligand concentrations and ΔAmax is the maximal difference in the absorption that has been achieved upon titration.
2.3. Reconstitution of CYP51 activity in vitro
The CYP51 reaction conditions were modified from those previously described (Lepesheva et al., 2007) in order to increase the excess of the substrate over the enzyme. In the standard reaction mixture we use 1 μM CYP51 concentration, and therefore the maximal achievable enzyme/substrate ratio is 1/50 (due to low solubility of the CYP51 substrates). The modified reaction mixture contained 0.1 μM CYP51, 2 μM CPR and 50 μM radiolabeled sterol substrates, eburicol for T. cruzi, norlanosterol for T. brucei and L. infantum and lanosterol for C. albicans and human CYP51 orthologs. The inhibitors were added from 0.1 mM DMSO stocks to final concentration 0.2 μM. The reaction was initiated by the addition of 100 μM NADPH, the mixture was incubated for 1 h at 37°C. The reaction was stopped by extraction of the sterols with ethyl acetate. Reaction products were dried, dissolved in methanol, and analyzed by a reverse phase HPLC system (Waters) equipped with a β-RAM detector (INUS Systems Inc.) using a Nova Pak C18 column and linear gradient water:acetonitrile:methanol (1.0:4.5:4.5) (solvent A) and methanol (solvent B) from 0% to 100% B for 30 min at a flow rate of 1 ml/min. Though at these conditions the CYP51 turnover became about 40-fold slower, we were able to see some substrate conversion at 2/1 molar ratio VNI/CYP51 after 1 h reaction. This increase in the sensitivity of the assay was used to monitor influence of chemical modifications on the inhibitory potencies of the VNI derivatives. The experiments were performed in triplicates and presented as the mean ± standard deviation.
2.4. Cell culture and growth inhibition assay using 5-bromodeoxyuridine incorporation
NIH/3T3 mouse embryonic fibroblasts were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle Medium (DMEM) containing 10% fetal bovine serum and 1% penicillin–streptomycin (Life Technologies, Gaithersburg, MD) at 37 °C and in 5% CO2. NIH/3T3 cells were plated in 96-well plates (5000 cells per well), incubated for 24 h and treated with vehicle (DMSO) or VNI derivatives for 24 h. Incorporation of 5-bromodeoxyuridine (BrdU) was determined in each well with the use of an enzyme-linked immunosorbent assay (ELISA)-based 5-bromo-2′-deoxy-uridine (BrdU) Labeling and Detection Kit III (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s instructions. Briefly, after treatment, cells were labeled with BrdU for 16 h, fixed, washed and incubated with monoclonal anti-BrdU antibody conjugated with peroxidase for 90 min at room temperature. Then cells were washed and incubated with substrate solution (TMB, tetramethyl-benzidine) for 5 min at room temperature. Reaction was stopped by addition of 1M H2SO4, and optical density (OD) was measured immediately at 450 nm using SpectraMAX 190 microplate reader (Molecular Devices). BrdU incorporation was quantified by a change of OD. Samples were analyzed in quadruplicates.
The cell growth inhibition effect was evaluated as percent of proliferating cells (cells treated with VNI derivatives vs cells treated with DMSO; mean ± SEM). Since this method measures synthesis of cellular DNA, i.e. the rate of cell growth and proliferation, it is considered more sensitive in evaluation of cytotoxic effects compared to cellular viability assays such as based on cell staining with MTT or trypan blue. Therefore, it is more suitable to analyze compounds with low cytotoxicity and/or without cytolytic activity.
2.5. Inhibitory effect on CYP3A4
The effect of VNI and its derivatives on the activity of human CYP3A4 was compared with that of ketoconazole using a BD Biosciences Screening Kit according to the manufacturer’s instructions. Fluorescence was measured using fluorometer Molecular Devices SpectraMax M5. Samples were analyzed in triplicates.
2.6. Acute mice toxicity study
Female Swiss Webster mice (25–26 g) were obtained from the Fundação Oswaldo Cruz (CECAL-FIOCRUZ) animal facilities (Rio de Janeiro, Brazil). Mice were housed at maximum 8 per cage and kept in a conventional room at 20–24 °C under a 12/12 h light/dark cycle. The animals were provided with sterilized water and chow ad libitum and all procedures were carried out in accordance with the guidelines established by the FIOCRUZ Committee of Ethics for the Use of Animals (CEUA 0028/09). In order to determine the NOAEL (No Observed Adverse Effect Level), acute toxicity studies were performed as previously reported (Silva et al., 2008). Briefly, on day 1, female mice were treated with VNI per oral (p.o.) at doses ranging from 25 to 400 mg/kg. Then, mice were inspected up to 48 h for toxic and sub-toxic symptoms according to OECD guidelines (Organization for Economic Co-operation and Development) (Batista et al., 2010). After this period, the NOAEL (no observable adverse effect level) values were determined, the animals were euthanized and their blood and organs collected for hematological and biochemical measurements performed through the Program for Technological Development in Tools for Health (PDTIS-FIOCRUZ). Gross pathology and organ weight were also evaluated as reported (Silva et al., 2012). The data are representative of three independent experiments and statistical analysis was performed individually for each assay using a variance (ANOVA) program with the level of statistical significance set at p ≤ 0.05.
3. Results and discussion
VNI ((R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide) is a highly potent inhibitor of protozoan CYP51s and represents a promising drug candidate. We found that it has good oral bioavailability (>30%): at a single dosage of 25 mg/kg its concentration in the mouse plasma reaches 40 μM within 2 h and remains above 20 μM for 8 h (Lepesheva and Hargrove, unpublished). Preliminary acute toxicity studies demonstrate that the VNI dosages up to 400 mg/kg do not cause significant toxicity as no detectable side effect (including no changes in characteristic mice behavior, motility, neurological conditions, skin and fur profile) could be noticed throughout the entire period of analysis (48 h). The measurement of mice body weight also did not reveal significant alterations (Fig. 5a, p > 0.1). The gross pathology did not show alterations in color and topological aspects of the studied organs (spleen, lung, heart, liver and kidneys) as compared to those mice that received vehicle (data not shown). Regarding organ weight analysis (spleen, kidney, lung, heart and liver), only a minor decrease (about 10%) in heart weight was detected when the 25 and 400 mg/kg doses (p = 0.04 and 0.035, respectively) were administrated (Fig. 5b). Also, a small variation in liver size (increase <10%; p = 0.05) was detected using 200 mg/kg VNI. Biochemical analysis of blood from VNI treated mice did not demonstrate major differences as compared to the animals treated with vehicle only (Table 1). Also, the analysis of alanine aminotransferase (ALT) measurement did not suggest hepatic damage since no significant variations were found among VNI treated and DMSO-treated mice (Table 1). Altogether these data show that VNI is well tolerated since it presents NOAEL values, in female mice, at 400 mg/kg via p.o., thus suggesting low toxicity.
Fig. 5.

Acute toxicity of VNI using Swiss female mice. Single doses were administered on day 1 (starting at 25 mg/kg up to 400 mg/kg – p.o.) and then mice were inspected up to 48 h for toxic and sub-toxic symptoms. (a) Mean ± SD values (n = 3) of three independent assays showing the percentage (%) of animal body weight variation (0–48 h posttreatment). (b) Mean ± SD values (n = 3) of three independent assays showing ratio values of organ weight/body weight measurements after 48 h of VNI administration; p < 0.05.
Table 1.
Biochemical analysis of blood samples from control and VNI-treated female mice (n = 3).
| VNI (mg/kg) | 0 | 25 | 50 | 100 | 200 | 400 |
|---|---|---|---|---|---|---|
| Red blood cells (million cells/mcL) | 8.73 ± 0.8 | 8.19 ± 0.7 | 8.59 ± 0.6 | 8.76 ± 0.8 | 8.69 ± 0.7 | 7.97 ± 0.7 |
| Hemoglobin (gm/dL) | 15 ± 1.4 | 13.7 ± 1.1 | 14.6 ± 1.2 | 14.2 ± 1.3 | 14.1 ± 1.1 | 13.4 ± 1.2 |
| Hematocrit (%) | 49.7 ± 4.1 | 45.8 ± 3.7 | 48.4 ± 3.6 | 49.0 ± 4.1 | 48.8 ± 3.9 | 45.6 ± 4.0 |
| Average red blood cell size (MCV – femtoliter) | 56.9 ± 4.9 | 55.9 ± 4.7 | 56.3 ± 5.0 | 55.9 ± 4.3 | 56.2 ± 4.5 | 57.2 ± 4.7 |
| Hemoglobin amount per red blood cell (MCH-pg/cell) | 17.2 ± 1.4 | 16.7 ± 1.5 | 17.0 ± 1.4 | 16.2 ± 1.5 | 16.2 ± 1.3 | 16.8 ± 1.3 |
| Amount of hemoglobin relative to the size of the cell (hemoglobin concentration) per red blood cell (MCHC-gm/dL) | 30.2 ± 2.7 | 29.9 ± 2.5 | 30.2 ± 3.0 | 29.0 ± 2.4 | 28.9 ± 2.4 | 29.4 ± 2.6 |
| White blood cell (WBC-cells/mcL) | 7.9 ± 0.7 | 9.6 ± 0.8 | 12.8 ± 1.1 | 13.3 ± 1.1 | 12.2 ± 1.0 | 9.4 ± 0.7 |
| Platelets | 1483 ± 118 | 1587 ± 127 | 1652 ± 139 | 1574 ± 126 | 1398 ± 110 | 1173 ± 95 |
| Naa | 156 ± 13 | 153 ± 13 | 152 ± 10 | 154 ± 11 | 152 ± 12 | 153 ± 11 |
| Ka | 9.5 ± 0.8 | 9.3 ± 0.7 | 7.9 ± 0.6 | 7.7 ± 0.6 | 8.0 ± 0.7 | 7.4 ± 0.7 |
| Cla | nd | 91 ± 7 | 92 ± 9 | 92 ± 8 | nd | nd |
| Pa | 13.7 ± 1.0 | 13.8 ± 1.1 | 12.8 ± 1.0 | 14.1 ± 1.4 | 13.2 ± 1.2 | 13.3 ± 0.9 |
| Glucosea | 199 ± 13 | 251 ± 19 | 211 ± 17 | 228 ± 15 | 262 ± 21 | 234 ± 18 |
| Alanine aminotransferase (ALT)a | 107 ± 7 | nd | 81 ± 6 | 78 ± 5 | 75 ± 6 | 82 ± 7 |
Reference values (CECAL/Fiocruz): Na (124–174), K (4.6–8), Cl (92–120), P (6.1–10.1), glucose (90–192) and ALT (up to 132).
However, VNI inhibits L. infantum CYP51 slightly weaker (Table 2) than the two trypanosomal orthologs (perhaps because of the larger volume of the L. infantum CYP51 substrate binding cavity (Hargrove et al., 2011)). Also it would be potentially desirable to further increase the drug lifetime in plasma while weakening its inhibitory effect on CYP3A4 (the major drug metabolizing cytochrome P450 in the human body).
Table 2.
VNI and its derivatives as CYP51 inhibitors.
| Effect | Compound
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VNI | S-enantiomer | FF-VNI | VNI-T | S-enantiomer | FF-VNI-T | S-enantiomer | VNI/VNF | S-enantiomer | Ketoconazole | |
| Binding to T. cruzi CYP51, Kd (μM) | 0.09 ± 0.01 | 0.12 ± 0.01 | 0.02 ± 0.006 | 0.15 ± 0.012 | 0.55 ± 0.04 | 0.07 ± 0.01 | 0.17 ± 0.02 | 0.04 ± 0.007 | 0.10 ± 0.01 | 0.24 ± 0.02 |
| CYP51 inhibition | ||||||||||
| •Pathogens (activity at I/E/Sa = 2/1/500, 60′ reaction, %) | ||||||||||
| T. cruzi | 7 ± 0.5 | 17 ± 1.5 | 9 ± 0.7 | 8 ± 0.6 | 70 ± 4.3 | 90 ± 6.7 | 85 ± 6.4 | 0 | 26 ± 1.7 | 38 ± 2.8 |
| T. brucei | 2 ± 0.1 | 65 ± 5.8 | 11 ± 109 | 90 ± 6.4 | 56 ± 3.8 | 93 ± 8.0 | 83 ± 6.6 | 0 | 74 ± 5.3 | 54 ± 3.9 |
| L. infantum | 12 ± 1.1 | 96 ± 6.3 | 58 ± 4.9 | 17 ± 1.2 | 97 ± 7.1 | 68 ± 5.1 | 95 ± 7.4 | 2 ± 0.2 | 32 ± 2.2 | 24 ± 1.6 |
| C. albicans | 40 ± 3.4 | nt | 68 ± 5.5 | 100 ± 6.9 | 75 ± 5.7 | 100 ± 7.7 | 95 ± 6.9 | 30 ± 2.4 | 90 ± 6.7 | 9 ± 0. |
| •Human (inhibitor/enzyme molar ratio for 50% inhibition, 60′ reaction) | >200 | >200 | >200 | >200 | >200 | >200 | >200 | 15 ± 0.9 | >200 | 80 ± 5.9 |
| Mammalian cell growth: % of viable 3T3 cells at 50 μMb | 76.8 ± 2.1 | 77.1 ± 2.6 | 65.9 ± 3.1 | 98.1 ± 1.8 | 97.9 ± 1.9 | 101.0 ± 1.4 | 107.2 ± 1.7 | 77.2 ± 6.3 | 74.6 ± 5.4 | 0 |
| LogP (ChemDraw) | 4.6 | 4.6 | 3.7 | 4.9 | 4.9 | 4.1 | 4.1 | 5.4 | 5.4 | 3.5 |
| CYP3A4 inhibition (IC50, nM) | 405 ± 23 | 290 ± 19 | 180 ± 14 | 2910 ± 227 | 2370 ± 152 | nt | nt | 3580 ± 271 | 2040 ± 16 | 8 ± 0.7 |
![]()
Chiral atom (VNI and all other structures shown are R-enantiomers).
Molar ratio inhibitor/enzyme/substrate; experimental details can be found in Section 2.
The concentration range was limited by the compound solubility in DMSO (5 mMfor the imidazole and 20 mMfor the triazole derivatives), maximal DMSO concentration in the cell cultures not exceeding 1%.
The initial VNI modifications described here were undertaken (1) to study whether the changes that improved the properties of commercial antifungal azoles (Chen and Sorrell, 2007) (β-phenyl ring fluorination and replacement of the imidazole ring with the 1,2,4-triazole ring, see ketoconazole vs posaconazole and ravuconazole in Fig. 3) will be favorable for the VNI scaffold as well; and (2) to test whether filling the CYP51 substrate binding cavity through VNI/VNF combination in one chemical structure such as adding an additional aromatic ring to the β-phenyl rind (i.e. Table 2, VNI/VNF) will further increase the inhibitory potency.
Variations in the heme-coordinating heterocyclic ring (imidazole vs triazole) were pursued to establish the significance of the basicity of the iron-coordinating atom (imidazole > triazole) (El Ghomari et al., 1997) and possible influence on the drug toxicity (Zonios and Bennett, 2008), since it is generally believed that clinical triazole antifungal drugs are less toxic and more selective than imidazoles (Chen and Sorrell, 2007). Replacement of the Cl atoms in the VNI β-phenyl ring by fluorines was mainly aimed to examine whether this substitution might ultimately be used to further enhance the compound pharmacokinetics (PK), particularly metabolic stability in vivo (Zonios and Bennett, 2008; Hu et al., 2010) without affecting CYP51 inhibition, since we already know that minimal alterations in this ring composition are often crucial for the inhibitory potency, sometimes assigning additional selectivity to this scaffold (Lepesheva et al., 2007). Fluorine is now extensively used in drug discovery and is increasingly found in drugs entering the marketplace (Maddaford, 2012). It is quite well established that the high electronegativity of fluorine allows for manipulation of drug properties including reduction of pKa, modification of lipophilisity, improving metabolic stability and duration of action (Hagmann, 2008; Hu et al., 2010), enhancing potency, reducing the potential for covalent protein binding and idiosyncratic toxicity, attenuation of biliary clearance (Uetrecht, 2007), etc. Except for FF-VNI, both R and S-enantiomers were tested to monitor influence of stereochemistry on the compound properties. Ketoconazole was used as a control (Table 2).
Interestingly, all the derivatives displayed high apparent binding affinity (shown in Table 2 for T. cruzi CYP51). Examples of experimental titration curves for VNI-T (stronger inhibitor, Kd = 0.15 μM) and FF-VNI-T (weaker inhibitor, Kd = 0.07 μM) are presented in Fig. 2e. The data support our previous finding that coordination of the N atom to the heme iron which is reflected in the spectral responses, being useful for primary identification of ligand binding, does not necessarily correlate with the inhibitory potency of the compound in the reconstituted enzyme reaction, as some of the ligands can be later replaced by the substrate in the enzyme active center (Lepesheva et al., 2007).
3.1. VNI-triazole (VNI-T)
Significant decrease in the cellular growth inhibition was the most obvious advantage of the heme coordinating ring modification (98% of proliferating cells for 50 μM VNI-T vs. 77% for 50 μM VNI, Table 2). However, no increase in the inhibitory potency was observed. While T. cruzi and L. infantum CYP51s presented about the same values as those obtained with VNI, the T. brucei ortholog inhibition with VNI-T was much weaker. Quite unexpectedly, T. brucei CYP51 was more strongly inhibited by the S-enantiomer, which affected the other protozoan CYP51s relatively weaker than the S-enantiomer of VNI. Thus, the basicity of the heterocyclic nitrogen does not seem to be crucial for CYP51 inhibition, at least for the VNI scaffold. This is in agreement with the hypothesis that it must be the interaction of the nonligated portion of the azole molecule with the protein moiety which defines the compound inhibitory potency (Correia and Ortiz de Montellano, 2005). Overall, the data suggest that VNI-T is worth testing as a drug candidate for Chagas disease and leishmaniasis, particularly because of potentially greater safety.
3.2. Difluoro-β-phenyl-VNI (FF-VNI)
Although in the CYP51 structures the β-phenyl rings of posaconazole and VNI are positioned similarly (Lepesheva et al., 2010a), substitution of the two Cl atoms to fluorines in the VNI scaffold did not display any advantages. Only for the T. cruzi enzyme the potency of FF-VNI was comparable to that of VNI, while the other orthologs were inhibited weaker. Also, our preliminary studies of FF-VNI PK did not reveal any improvement (not shown), which implies that the primary site(s) of in vivo metabolism in the VNI scaffold must be different from that of the posaconazole/ravuconazole scaffolds and needs to be identified prior to further introduction of the fluorine atoms.
3.3. Difluoro-β-phenyl-VNI-triazole (FF-VNI-T)
The finding that the β-phenyl ring is not the right place for the replacement of Cl to F in VNI was further confirmed upon testing of FF-VNI-T. The compound was found to be a much weaker inhibitor for all pathogenic CYP51s, though the fact of the decrease in toxicity as a result of the triazole ring presence was confirmed (Table 2).
3.4. VNI/VNF combination (VNI/VNF)
The compound which was designed based on the structures of CYP51 complexes with VNI [3gw9] and VNF [3ksw] with the purpose to fill the majority of the CYP51 substrate binding cavity was indeed found to be the most potent inhibitor of all three protozoan CYP51s. Its effect on C. albicans CYP51 (structure not yet determined) remains relatively weaker than the effect of ketoconazole. VNI/VNF also inhibits human CYP51 stronger than the other VNI derivatives (Table 2). It remains to be clarified (and is a good opportunity to do so) whether the observed decrease in selectivity might have any harmful effect in mammals in vivo, though we anticipate that replacement of the imidazole ring in VNI/VNF with the triazole ring should increase the inhibitor selectivity while further decreasing its toxicity and inhibitory effect on CYP3A4. The S-form of this compound, similar to the S-enantiomer of VNI, displays a weaker inhibitory potency towards all three protozoan CYP51s.
To conclude, the VNI scaffold is highly promising for the development of novel drugs for human infections with Trypanosomatidae. CYP51 structure can successfully be used to guide further, rational development of this scaffold in order to produce highly selective, pathogen-specific drugs.
Acknowledgments
This work was supported by NIH GM 067871 (G.I.L., M.R.W.) and Vanderbilt Institute of Chemical Biology Pilot Project grant 2011 (G.I.L.). The present study was also supported by Fiocruz and by Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ – Programa de Pesquisa para o Sistema Único de Saúde (PPSUS), APQ1 and Pensa-Rio, Conselho Nacional Desenvolvimento científico e Tecnológico (CNPq), PDTIS/Fiocruz. The authors thank the Program for Technological Development in Tools for Health-PDTIS-FIOCRUZ for use of their facilities.
References
- Batista DG, Batista MM, Oliveira GM, Amaral PB, Lannes-Vieira J, Britto CC, Junqueira A, Lima MM, Romanha AJ, Sales PA, Junior, Stephens CE, Boykin DW, Soeiro MD. Arylimidamide DB766: a potential chemotherapeutic candidate for chagas disease treatment. Antimicrob Agents Chemother. 2010;54:2940–2952. doi: 10.1128/AAC.01617-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner FS, Bahia MT, Suryadevara PK, White KL, Shackleford DM, Chennamaneni NK, Hulverson MA, Laydbak JU, Chatelain E, Scandale I, Verlinde CL, Charman SA, Lepesheva GI, Gelb MH. Pharmacological characterization, structural studies, and in vivo activity of anti-chagas disease lead compounds derived from tipifarnib. Antimicrob Agents Chemother. doi: 10.1128/AAC.06244-11. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli A, Bolognesi ML. Neglected tropical diseases: multi-target-directed ligands in the search for novel lead candidates against Trypanosoma and Leishmania. J Med Chem. 2009;52:7339–7359. doi: 10.1021/jm9004835. [DOI] [PubMed] [Google Scholar]
- Chaudhary K, Roos DS. Protozoan genomics for drug discovery. Nat Biotech. 2005;23:1089–1091. doi: 10.1038/nbt0905-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Sorrell TC. Antifungal agents. Med J Aust. 2007;187:404–409. doi: 10.5694/j.1326-5377.2007.tb01313.x. [DOI] [PubMed] [Google Scholar]
- Chevreuil F, Landreau A, Seraphin D, Larcher G, Bouchara JP, Richomme P. Synthesis and antifungal activity of new thienyl and aryl conazoles. J Enzyme Inhib Med Chem. 2006;3:293–303. doi: 10.1080/14756360600700640. [DOI] [PubMed] [Google Scholar]
- Clayton J. Chagas disease: pushing through the pipeline. Nature. 2010;465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- Correia MA, Ortiz de Montellano PR. Inhibition of cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Plenum Publishing Corp; New York: 2005. pp. 246–322. [Google Scholar]
- da Silva CF, Batista MM, da Batista DG, de Souza EM, da Silva PB, de Oliveira GM, Meuser AS, Shareef AR, Boykin DW, de Soeiro MN. In vitro and in vivo studies of the trypanocidal activity of a diarylthiophene diamidine against Trypanosoma cruzi. Antimicrob Agents Chemother. 2008;52:3307–3314. doi: 10.1128/AAC.00038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva CF, da Batista DG, Oliveira GM, de Souza EM, Hammer ER, da Silva PB, Daliry A, Araujo JS, Britto C, Rodrigues AC, Liu Z, Farahat AA, Kumar A, Boykin DW, de Soeiro MN. In vitro and in vivo investigation of the efficacy of arylimidamide DB1831 and its mesylated salt form – DB1965 – against Trypanosoma cruzi infection. PLoS One. 2012;7:e30356. doi: 10.1371/journal.pone.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekroos M, Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghomari MJ, Mokhlisse R, Laurence C, Le Questel JY, Berthelot M. Basicity of azoles: complexes of diiodine with imidazoles, pyrazoles and triazoles. J Phys Org Chem. 1997;10:669–674. [Google Scholar]
- Hagmann WK. The many roles for fluorine in medicinal chemistry. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- Hargrove TY, Wawrzak Z, Liu J, Nes WD, Waterman MR, Lepesheva GI. Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (cyp51) from Leishmania infantum. J Biol Chem. 2011;286:26838–26848. doi: 10.1074/jbc.M111.237099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove TY, Wawrzak Z, Liu J, Waterman MR, Nes WD, Lepesheva GI. Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl- Δ7-24, 25-dihydrolanosterol. J Lipid Res. 2012;53:311–320. doi: 10.1194/jlr.M021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Negri M, Olgen S, Hartmann RW. The role of fluorine substitution in biphenyl methylene imidazole-type cyp17 inhibitors for the treatment of prostate carcinoma. Chem Med Chem. 2010;5:899–910. doi: 10.1002/cmdc.201000065. [DOI] [PubMed] [Google Scholar]
- Konkle ME, Hargrove TY, Kleshchenko YY, von Kries JP, Ridenour W, Uddin MJ, Caprioli RM, Marnett LJ, Nes WD, Villalta F, Waterman MR, Lepesheva GI. Indomethacin amides as a novel molecular scaffold for targeting Trypanosoma cruzi sterol 14 alpha-demethylase. J Med Chem. 2009;52:2846–2853. doi: 10.1021/jm801643b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Nes WD, Zhou W, Hill GC, Waterman MR. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry. 2004;43:10789–10799. doi: 10.1021/bi048967t. [DOI] [PubMed] [Google Scholar]
- Lepesheva GI, Zaitseva NG, Nes WD, Zhou W, Arase M, Liu J, Hill GC, Waterman MR. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B′ helix defines substrate preferences of sterol 14alpha-demethylase. J Biol Chem. 2006;281:3577–3585. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, Nes WD, Hill GC, Villalta F, Waterman MR. Sterol 14 alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem Bio. 2007;14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Waterman MR. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta. 2007;1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G, Hargrove T, Kleshchenko Y, Nes W, Villalta F, Waterman M. CYP51: a major drug target in the cytochrome p450 superfamily. Lipids. 2008;43:1117–1125. doi: 10.1007/s11745-008-3225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Hargrove TY, Anderson S, Kleshchenko Y, Furtak V, Wawrzak Z, Villalta F, Waterman MR. Structural insights into inhibition of sterol 14 alpha-demethylase in the human pathogen Trypanosoma cruzi. J Biol Chem. 2010a;285:25582–25590. doi: 10.1074/jbc.M110.133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Park HW, Hargrove TY, Vanhollebeke B, Wawrzak Z, Harp JM, Sundaramoorthy M, Nes WD, Pays E, Chaudhuri M, Villalta F, Waterman MR. Crystal structures of Trypanosoma brucei sterol 14 alpha-demethylase and implications for selective treatment of human infections. J Biol Chem. 2010b;285:1773–1780. doi: 10.1074/jbc.M109.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Villalta F, Waterman MR. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51) Adv Parasitol. 2011;75:65–87. doi: 10.1016/B978-0-12-385863-4.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Waterman MR. Sterol 14alpha-Demethylase (CYP51) as a therapeutic target for human Trypanosomiasis and Leishmaniasis. Curr Top Med Chem. 2011a;11:2060–2071. doi: 10.2174/156802611796575902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva GI, Waterman MR. Structural basis for conservation in the CYP51 family. Biochim Biophys Acta. 2011b;1814:88–93. doi: 10.1016/j.bbapap.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie M. Drug developers finally take aim at a neglected disease. Science. 2011;333:933–935. doi: 10.1126/science.333.6045.933. [DOI] [PubMed] [Google Scholar]
- Maddaford SP. A medicinal chemistry perspective on structure-based drug design and development methods. Mol Biol. 2012;841:351–381. doi: 10.1007/978-1-61779-520-6_15. [DOI] [PubMed] [Google Scholar]
- Nes WD. Biosynthesis of Cholesterol and Other Sterols. Chem Reviews. 2011;111:6423–6451. doi: 10.1021/cr200021m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nes CR, Singha SU, Liu J, Ganapathy K, Villalta F, Waterman MR, Lepesheva GI, Chaudhuri M, Nes WD. Novel Sterol Metabolic Network of Trypanosoma brucei Procyclic and Bloodstream Forms. Biochem J. 2012;443:267–277. doi: 10.1042/BJ20111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I evidence for its hemoprotein nature. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- Paniz Mondolfi AE, Stavropoulos C, Gelanew T, Loucas E, Perez Alvarez AM, Benaim G, Polsky B, Schoenian G, Sordillo EM. Successful treatment of old world cutaneous Leishmaniasis caused by Leishmania infantum with posaconazole. Antimicrob Agents Chemother. 2011;55:1774–1776. doi: 10.1128/AAC.01498-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos TL. Cytochrome P450 flexibility. Proc Natl Acad Sci USA. 2003;100:13121–13122. doi: 10.1073/pnas.2336095100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmer M, Bouvier P, Ourisson G. Molecular evolution of biomembranes: structural equivalents and phylogenetic precursors of sterols. Proc Natl Acad Sci USA. 1979;76:847–851. doi: 10.1073/pnas.76.2.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeiro M, de N, Dantas AP, Daliry A, Silva CF, Batista DG, de Souza EM, Oliveira GM, Salomão K, Batista MM, Pacheco MG, Silva PB, Santa-Rita RM, Barreto RF, Boykin DW, Castro SL. Experimental chemotherapy for chagas disease: 15 years of research contributions from in vivo and in vitro studies. Mem Inst Oswaldo Cruz. 2009;104 (Suppl 1):301–310. doi: 10.1590/s0074-02762009000900040. [DOI] [PubMed] [Google Scholar]
- Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. An open conformation of mammalian cytochrome P450 2B4 at 1.6 Å resolution. Proc Natl Acad Sci USA. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht J. Idiosyncratic drug reactions: current understanding. Annu Rev Pharmacol Toxicol. 2007;47:513–539. doi: 10.1146/annurev.pharmtox.47.120505.105150. [DOI] [PubMed] [Google Scholar]
- Vanden Bossche H, editor. Mode of Action of Pyridine, Pyrimidine and Azole Antifungals. Ellis Horwood; Chichester: 1988. pp. 79–119. [Google Scholar]
- Villalta F, Dobish M, Nde P, Kleshchenko Y, Hargrove T, Johnson C, Waterman M, Johnston J, Lepesheva G. VNI cures Chagas disease. doi: 10.1093/infdis/jit042. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Vinković DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome p450 3a4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- Zonios DI, Bennett JE. Update on azole antifungals. Semin Respir Crit Care Med. 2008;29:198–210. doi: 10.1055/s-2008-1063858. [DOI] [PubMed] [Google Scholar]