

Abstract
Professional APCs, such as dendritic cells, are routinely used in vitro for the generation of cytotoxic T lymphocytes specific for tumor antigens. In addition to dendritic cells, CD40-activated B cells and variant K562 leukemic cells can be readily transfected with nucleic acids for in vitro and in vivo antigen presentation. However, the expression of immunoproteasome components in dendritic cells may preclude display of tumor antigens such as Mart1/MelanA. Here, we use three target epitopes, two derived from tumor antigens [Mart126–34 (M26) and Cyp1B1239–247 (Cyp239)] and one derived from the Influenza A viral antigen [FluM158–66 (FluM58)], to demonstrate that CD40-activated B cells, like dendritic cells, have a limited capability to process certain tumor antigens. In contrast, the K562 HLA-A*0201 transfectant efficiently processes and presents M26 and Cyp239 as well as the influenza FluM58 epitopes to T cells. These results demonstrate that the choice of target APC for gene transfer of tumor antigens may be limited by the relative efficacy of proteasome components to process certain tumor epitopes. Importantly, K562 can be exploited as an artificial APC, efficient in processing both M26 and Cyp239 epitopes and presumably, by extension, other relevant tumor antigens.
Keywords: Vaccination, Tumor Immunity, Antigen Processing, Dendritic Cells
Introduction
The development of effective tumor immunotherapy requires identification of target antigens and the MHC-restricted antigenic epitopes that are naturally processed by tumor cells. Expression of immunoproteasome components by APCs, such as EBV-transformed lymphoblastoid cell line (LCL) and dendritic cells, is thought to limit autoimmunity to self antigens. This results in poor antigen presentation of certain self-antigenic epitopes, such as the Mart126–34 epitope[1], which can be reversed by siRNA inhibition of the immunoproteasome[2]. Tumor cells also have mechanisms to impair immunoproteasome induction, which may function to aid tumor escape via alteration of HLA-dependent peptide display[3, 4] To identify tumor-specific epitopes, it is essential to develop model systems that present antigenic epitopes processed by the constitutive proteasome functioning in the majority of tumor cells, rather than by the immunoproteasome restricted to professional APCs. In addition, identifying optimal APCs for the immune monitoring of whole antigen-based vaccines will facilitate the identification of novel tumor antigenic epitopes.
While dendritic cells are efficient APCs, their low numbers and laborious isolation from human blood have led immunotherapists to search for other sources of cells able to display tumor antigens for immune stimulation. Two alternative APCs in development are autologous CD40-activated B (CD40-B) cells and human erythroleukemia cell line K562. CD40-B cells are a renewable resource that are readily produced from small quantities of peripheral blood and efficiently present peptide antigens to T cells[5, 6]. CD40-B cells are more rapidly generated than autologous EBV-LCL. An additional advantage over the LCL is that they do not express EBV antigens, yet express high levels of costimulatory molecules and HLA class I and class II molecules. CD40-B cells are readily transfected with nucleic acid or retrovirus[7, 8], and when pulsed with tumor lysates can stimulate CD4+ cells[9]. This has led to their preclinical development as APCs [10, 11].
In addition, the leukemic K562 cell line is currently being tested in multiple clinical trials as a bystander cell line for GM-CSF secretion and are in early phase clinical development as artificial APCs[12, 13]. HLA-transfected K562 cells have been shown to efficiently present peptides[14] and minigene epitopes[15] derived from tumor antigens to T cells in vitro. With the development of whole antigen delivery by nucleic acid transfection, a systematic assessment of the relative antigen processing capability of these APCs for different antigenic epitopes is critical for the development of cellular immunotherapeutics.
Here, we demonstrate that CD40-B cells, like dendritic cells, manifest impaired antigen processing of Mart1 protein, resulting in the loss of the Mart126–35 epitope. This is associated with high levels of expression of the immunoproteasome subunits LMP2 and LMP7 in CD40-B cells. To determine if this impaired processing extends to other tumor antigens, we assessed the processing and presentation of the Cyp239 epitope of cytochrome P450 1B1 (Cyp1B1), which is currently in early-phase DNA vaccine clinical trials [16, 17] and found it defective as well. In contrast, K562 cells retain the constitutive proteasome and are highly efficient at antigen presentation of both the Mart1 and the Cyp239 tumor antigenic epitopes. The influenza M1 epitope, which is presented well in the presence of the immunoproteasome subunit LMP7[18], was processed efficiently by all three APCs (CD40-B cells, dendritic cells, and K562 cells). The impact of these findings on APC selection and epitope recognition will be discussed.
Materials and Methods
Generation of antigen presenting cells
Peripheral blood mononuclear cells were obtained with written informed consent from healthy blood donors under Institutional Review Board approval at the Dana-Farber Cancer Institute. For the generation of dendritic cells, CD14 positive cells were isolated from leukapheresis material by immunomagnetic enrichment (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s guidelines. Purified CD14 cells were cultured in IMDM (Invitrogen, Carlsbad, CA) supplemented with 10% human AB serum (Cellgro, Herndon, VA), 2 mM glutamine (Cellgro), 50 μg/ml human transferrin (Roche, Basel, Switzerland), 5 μg/ml human insulin (Sigma-Aldrich, St. Louis, MO), and 15 μg/ml gentamycin (Invitrogen) in the presence of 50 ng/ml GM-CSF (Genzyme, Cambridge, MA) and 20 ng/ml IL-4 (R&D Systems, Minneapolis, MN). On days three and six, 50% fresh medium with cytokines was added. On day seven, cells were matured for 48 hours with either 10 ng/ml interferon-γ (Pierce, Rockford, IL, Figures 3,4) or 1 μg/ml prostaglandin E2 (Sigma-Aldrich), 10 ng/ml IL-1β (BD Bioscience, San Jose, CA), 1000 U/ml IL-6 (BD Bioscience) and 10 ng/ml TNFα (Genzyme) (Figure 5). CD40-B cells were isolated from peripheral blood by activation on CD40L-expressing irradiated feeder cells in the presence of IL-4 (R&D Systems) and cyclosporin A (Novartis, Basel, Switzerland) as described[5]. K562 cells were obtained from ATCC and nucleofected (Amaxa Inc, Gaithersburg, MD) with the full-length HLA-A*0201 gene. The Mart1-positive melanoma cell lines K008 (HLA-A2−) and K029 (HLA-A2+) were obtained from Dr. Glenn Dranoff (Dana-Farber Cancer Institute). The Mart1-positive melanoma cell line C32TG (HLA-A2+), and the Mart1-negative breast cancer cell lines MCF-7 (HLA-A2+) and SKBR-3 (HLA-A2−) were obtained from ATCC. The TAP-deficient cell line T2 was obtained from Dr. Peter Cresswell (Yale University School of Medicine).
Figure 3. The Mart1 M26 epitope is poorly presented by CD40-B cells and dendritic cells, but efficiently presented by K562 cells.
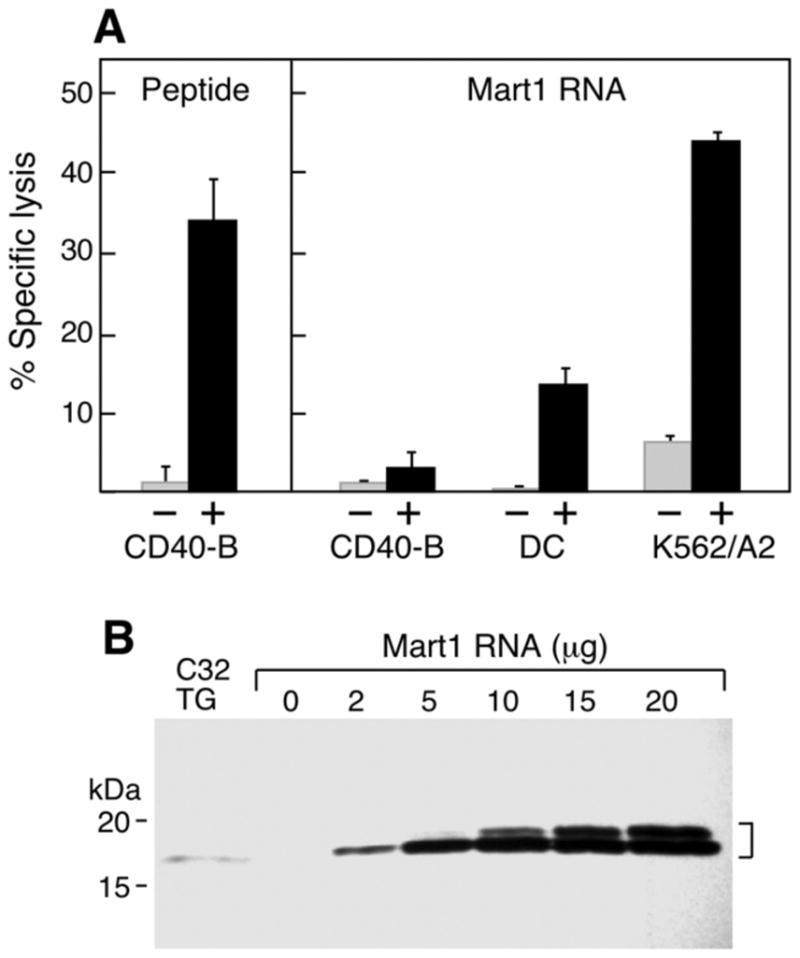
A. In comparison to K562/A2 cells, CD40-B cells and dendritic cells (DC) expressing Mart1 antigen are poorly lysed by the Mart1 M26-specific T cell clone. Europium-labeled HLA-A2+ target cells (5,000 target cells/well) were co-cultured with the Mart1 M26 specific T cell clone at an E:T of 1:1, and cytolysis measured by standard europium release assay. CD40-B targets pulsed with M26 peptide (black) but not control peptide (at 2 μg/ml, left panel, grey) were efficiently lysed. In contrast, target CD40-B cells nucleofected with full-length Mart1 RNA (black) or control RNA (20 μg, right panel, grey) were poorly lysed by the Mart1 M26 T cell clone. In contrast, Mart1 RNA-nucleofected dendritic cells were moderately lysed by the T cells, while K562/A2 cells were efficiently lysed. Standard deviation<2.7%, DC vs. K562A2 p=0.0004. B. Mart1 protein is strongly expressed as a doublet after RNA transfer of full-length Mart1 into CD40-B cells, shown by immunoblot using a Mart1-specific monoclonal antibody. The melanoma cell line C32TG endogenously expressing Mart1 was used as a positive control.
Figure 4. CD40-B cells efficiently process FluM1 antigen but not Mart1 M26 or Cyp239 antigen.

Target cells (25,000 cells/well) were coincubated with the FluM1 (left column), Mart1 M26 (center column), or Cyp239 (right column) specific T cell clones (500 cells/well) in triplicate and γ-interferon secretion was determined by ELISPOT. CD40-B cells pulsed with specific peptide (FluM1, M26, or Cyp239), but not control peptide, specifically stimulated all three T cell clones. CD40B cells and dendritic cells (DC) nucleofected with 20 μg of RNA encoding the specific full-length antigen for FluM1 (left) but not control RNA stimulated the FluM1 clone (left, middle). In contrast, CD40B cells or dendritic cells (DC) nucleofected with Mart1 RNA (center), or Cyp1B1 RNA (right) poorly stimulated the T cell clones. In comparison, RNA encoding all three antigens were nucleofected into K562/A2 cells and were efficiently presented to the T cell clones (bottom row).
Figure 5. Specific processing of the Cyp239 epitope by K562/A2 cells by cytolysis.

(A), γ-interferon ELISPOT (B), and γ-interferon ELISA (C). A. The Cyp239 T cell clone was cocultured with tumor cell targets (5,000 cells/well), and cytolytic activity was measured by standard europium release assay at the varying E:T ratios shown. Dendritic cells (DC) and K562/A2 cells were nucleofected with 20 μg of RNA encoding Cyp1B1 antigen or the control CML66 tumor antigen. Only K562/A2/Cyp1B1+ cells are efficiently lysed by the T cell clone. The positive control (T2 cells pulsed with Cyp239) and negative control targets (T2 cells pulsed with the Her2 369 peptide) are shown. B. The target cells from A (25,000 cells/well) were cocultured with the Cyp239 T cell clone (500 cells/well) and γ-interferon secretion was determined by ELISPOT assay. Only Cyp1B1-transfected K562/A2 cells and Cyp239 peptide-pulsed cells efficiently stimulated γ-interferon secretion. C. The target cells from A and B were titrated as shown for their ability to stimulate the Cyp239 T cell clone (10,000 cells/well), and γ-interferon secretion was measured by ELISA assay.
Plasmids, peptides, and RNA synthesis
Peptides encoding the Mart126–35 epitope (M26, EAAGIGILTV), the Cyp1B1239–247 epitope (Cyp239, SLVDVMPWL), the influenza M158–66 epitope (FluM1, GILGFVFTL), and the control Her2369–377 epitope (H369, KIFGSLAFL), were synthesized (New England Peptide, Gardner, MA). The full-length HLA-A*0201 gene under control of the CMV promoter was a kind gift of Dr. Gordon Freeman (Dana-Farber Cancer Institute). The M1 cDNA was a kind gift of Dr. Matthew Albert (Institut Pasteur), and was subcloned into pCDNA3.1CT GFP topo (Invitrogen) with a stop codon so that GFP was not expressed. The pOBT7-Mart1 cDNA and the Cyp1B1 cDNA were obtained from ATCC. The CML66 expression plasmid was generated in our laboratory from a CML expression library[19]. For RNA production, endotoxin-free plasmids were linearized with the restriction enzyme Ssp1 (NE Biolabs, Beverly, MA) and in vitro transcribed using the mMessage mMachine and in vitro polyadenylation systems (Ambion, Foster City, CA). The size and quality of in vitro generated RNA was confirmed using an Agilent 2100 Bioanalyzer (Palo Alto, CA).
Nucleofection of RNA into target cells
Polyadenylated RNA (2–20 μg) was nucleofected (Amaxa gmbh) into target cells (2 × 106 cells) in 100 μl of PBS/10%HEPES buffer. The programs used were Q-004, T-016, and U-010 for CD40-B cells, K562 cells, and dendritic cells, respectively. The efficiency (>90%) of nucleofection and cell viability (80–90%) were first determined using mRNA encoding eGFP (Clontech, Mountain View, CA). For dendritic cell nucleofection, immature dendritic cells were nucleofected and incubated overnight in GM-CSF, IL-4, and maturation cytokines prior to T cell assays.
Generation of Mart126-35-specific, Cyp239-specific, and Influenza M1-specific T-cell clones
CD40-B cells generated from peripheral blood mononuclear cells (PBMCs) of HLA-A2+ healthy donors were used to stimulate autologous CD8+ T-cells, purified with anti-CD8 bead (Miltenyi Biotec), as described[5]. CD8+ T-cells (2 × 106 cells/well in 24 well plate) were cultured with irradiated, 10 μg/ml M26-, Cyp239-, or FluM1-peptide pulsed CD40 B-cells in RPMI1640 (Invitrogen) supplemented with 10% human AB serum (Cellgro), 2 mM glutamine (Cellgro), 10 mM HEPES (Cellgro), and 15 μg/ml gentamycin (Invitrogen) in the presence of 10 ng/ml of IL-7 (Endogen Inc., Woburn, MA) on day 0 and 20–50 IU/ml of IL-2 (Chiron Corp., Emeryville, CA) on days 1 and 4. Restimulation of T cells was performed with irradiated, 2 μg/ml peptide pulsed CD40 B-cells every week. Cells were cloned by limiting dilution using irradiated allogeneic PBMCs and EBV-LCL feeder cells in the presence of phytohemagglutinin (Sigma-Aldrich) and 100 IU/ml IL-2 (Chiron Corp). Specificity of the T cell clones was determined by testing with M26-specific, Cyp239-specific, and FluM1-specific multimers.
Immune Assays
Europium cytolytic assays were performed with europium-labeled target cells (PerkinElmer, Wellesley MA)[20]. Target cells (5,000 cells/well in 96 well plate) were labelled for 30 minutes with europium, washed extensively, and coincubated with different ratios of T cells to target cells in triplicate for 2 hours, and specific europium release was measured. ELISA was performed with varying numbers (1562–50,000 cells/well) of APCs coincubated with T cell clones (10,000 cells/well) in triplicate in 96 round-bottom plates for 24 hours. Supernatants were harvested and the secretion of interferon-γ was measured (Endogen, Rockford, IL). ELISPOT was performed using peptide-pulsed target cells (25,000 cells/well) coincubated with the T cell clones (500 cells/well) in triplicate in ELISPOT plates (Millipore, Billerica, MA) for 24 hours. Interferon-γ secretion was detected using capture and detection antibodies as directed (Mabtech AB, Nacka Strand, Sweden) and imaged using an ImmunoSpot Series Analyzer (Cellular Technology, Ltd, Cleveland, OH). The proteasome inhibitors lactacystin (Santa Cruz, Santa Cruz CA) and MG-132 (Biovision, Mountain View CA) were used at 10 μM.
MHC class I multimer analysis
Fluorescent HLA-A2 tetramers were synthesized in association with FluM1 peptide or Cyp239 peptide and β2-microglobulin as previously described[21]. The Mart1/MelanA pentamer was obtained from Proimmune (Oxford, UK) and used as directed. The T cell clones were incubated with the specific tetramer or pentamer and anti-CD8 monoclonal antibody (Beckman-Coulter, Miami, FL) and analysed by flow cytometry (Beckman-Coulter FC500). Data were compensated and analyzed with WinList 3.0 (Verity Software House, Inc., Topsham, ME).
Immunoblotting
Tumor cells were washed in PBS, lysed and separated on 10% SDS-PAGE. Proteins were transferred to PVDF membrane (Millipore), and the presence of Mart1 protein was assessed using anti-Mart1 mAb (sc-20032; Santa Cruz Biotechnology, Inc, Santa Cruz, CA). Immunoblots for the proteasome subunits β1 and β1i (LMP2) were performed using the antibodies PW9255 and PW8840 (Affiniti Research Products, Exeter, UK). Immunoproteasomes were induced in K562 cells by incubation for 48 hours with 10 ng/ml interferon-γ (Pierce, Rockford, IL).
Quantitative real-time RT-PCR
Total RNA was isolated by using the RNeasy Mini kit (Qiagen, Valencia, CA), and RNA integrity was checked with an Agilent 2100 Bioanalyzer and quantified spectrophotometrically. Two microgram RNA was reverse transcribed into first-strand cDNA, and quantitative real-time PCR was performed with the 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA), according to the manufacturer’s guidelines. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) copy number was measured in each cDNA sample as an internal control. The sequences of primers and hybridization probes were as follows: LMP2 forward 5′-CGACAGCCTTTTGCCATTG-3′, LMP2 reverse 5′-ATGCTGCATCCACATAACCATAGA-3′, LMP2 probe 5′-FAM-CTCCGGCAGCACCT-MGBNFQ-3′; LMP7 forward 5′-CCAAGGAATGCAGGCTGTACTAT-3′, LMP7 reverse 5′-GAGGCTGCCGACACTGAAA-3′, LMP7 probe 5′-FAM-TGCGAAATGGAGAACGT-MGBNFQ-3′; LMP10 forward 5′-ACCCACAGAGCCCGTGAAG-3′, LMP10 reverse 5′-TCAGGACAGCTGTGGTTCCA-3′, LMP10 probe 5′-FAM-CCGCTACCACTTTGT-MGBNFQ-3′; GAPDH forward 5′-ATGGAAATCCCATCACCATCTT-3′, GAPDH reverse 5′-CGCCCCACTTGATTTTGG-3′, GAPDH probe 5′-FAM-CAGGAGCGAGATCC-MGBNFQ-3′.
Proteasomal cleavage prediction
To determine if existing algorithms for constitutive and immunoproteasomal cleavage sites could predict the differential processing of these epitopes, we performed a bioinformatics analysis for proteasomal cleavage prediction using the NetChop3.0[22] and PaProc II[23] algorithms. NetChop3.0 predicts only constitutive proteasomal cleavage sites; and a threshold of 0.8 was selected for the analysis.
Statistical Analysis
All assays were performed in triplicate. Where indicated, values are plotted as mean ± SD. Values were compared using an unpaired t-test. Cytotoxicity, ELISPOT, and ELISA assays are representative of 3–5 independent assays. Quantitation of signal intensities of immunoblots were performed using ImageJ[24].
Results
Generation of FluM1, M26, and Cyp239 peptide-specific T cell clones
To directly compare the antigen processing and presentation of an immunoproteasome-preferential antigen (FluM1), an immunoproteasome-sensitive antigen (M26), and an antigen (Cyp239) of unknown proteosomal sensitivity, HLA-A2-restriced T cell clones were generated to all three epitopes. PBMCs from HLA-A2+ healthy blood donors were stimulated with peptide-pulsed CD40-B cells as described[5]. CD8+ cytotoxic T cells were cloned by limiting dilution, and the high avidity T cell clones for each peptide were selected for further analysis. Antigenic specificity was confirmed by tetramer or pentamer flow cytometry (Figure 1A). By europium cytolytic assay, all three T cell clones were confirmed as cytolytic to peptide-pulsed HLA-A2+ T2 targets (Figure 1B), but not to unpulsed targets (Figure 1B, background) or to control peptides (data not shown). The FluM1.F7 and Cyp239.128 clones had higher avidity, reaching half-maximal cytolysis at 1–10 ng/ml, compared with the Mart1 M26.M14 clone at 10–100 ng/ml (Figure 1B). All three T cell clones secreted interferon-γ upon stimulation with peptide-pulsed CD40-B cells, as measured by ELISA (data not shown).
Figure 1. The FluM1, Mart1 M26, and Cyp239 T cell clones are peptide-specific and cytolytic.

A. Detection of FluM1, Mart1 M26, and Cyp239 T cell clones by flow cytometry using antigen-specific tetramer (left, right) and pentamer (middle). B. All three T cell clones are cytolytic to peptide-pulsed HLA-A2+ CD40-B cells (5,000 target cells/well) at an E:T ratio of 10:1. Specific peptides were titrated in the concentrations shown. Background is unpulsed target cells.
The M26 T cell clone recognizes naturally-processed antigen on tumor cells
We confirmed that the Mart1 M26-specific T cell clone would specifically recognize naturally-processed antigen on tumor cell lines. The M26.M14 clone was cocultured with tumor cell targets and cytolysis of the tumor targets was measured (Figure 2). The positive control cells (T2 cells pulsed with Mart1 M26 peptide) were efficiently lysed. The M26.M14 T cell clone efficiently lysed the melanoma cells K029 (HLA-A2+ Mart1+) and C32TG (HLA-A2+ Mart1+). The melanoma cells K008 (HLA-A2− Mart1+) and the breast cancer cell lines MCF-7 (HLA-A2+ Mart1−) and SKBR-3 (HLA-A2− Mart1−) were not lysed.
Figure 2. The Mart1 T cell clone recognizes naturally-processed antigen on tumor cells.

The Mart1 M26 T cell clone was cocultured with tumor cell targets (5,000 target cells/well), and cytolytic activity was measured by standard europium release assay at the varying E:T ratios shown. The Mart1 T cell clone efficiently lyses HLA-A2+ melanoma cells, including HLA-A2+ Mart1+ cells (melanoma cells K029 and C32TG), but not HLA A2− Mart1+ cells (K008), HLA-A2+ Mart1− cells (MCF-7), nor HLA-A2− Mart1− cells (SKBR-3). The positive control (T2 cells pulsed with Mart1 peptide M26) and negative control targets (T2 cells pulsed with FluM1 peptide) are shown.
The M26 epitope is presented poorly by CD40-B cells and dendritic cells, but efficiently presented by K562 cells
It has been shown that the Mart1 M26 epitope is internally cleaved and destroyed by the immunoproteasome, limiting efficient antigen presentation by dendritic cells[1, 25]. Although the M26 epitope can be processed and presented by CD40-B cells[8], we predicted that CD40-B cells would also be relatively inefficient at presentation of this epitope due to internal cleavage of the epitope by the immunoproteasome. To assess the relative efficiency of antigen processing and presentation, RNA transfer into dendritic cells, CD40-B cells and K562 cells was optimized. The optimization of gene transfer using an Amaxa nucleofector was first performed using eGFP-encoding RNA, with greater than 90% eGFP+ cells and viabilities seen at 24–96 hours after nucleofection (data not shown). The high quality of the in vitro-generated RNA was confirmed using an Agilent bioanalyzer (data not shown). To determine the relative efficiency of Mart1 M26 antigen presentation, full-length RNA was transfected into all three APCs, and specific lysis by the Mart1 M26.M14 T cell clone was measured (Figure 3A). While K562/A2 cells are efficiently lysed, both dendritic cells and CD40-B cells show relatively impaired (but not completely absent) presentation of the M26 epitope to the T cells.
Expression of Mart1 antigen after RNA transfer in CD40-B cells
The impaired antigen presentation of CD40-B cells in Figure 3A could be due to inefficient protein expression or antigen processing. To confirm that RNA transfer into CD40-B cells results in protein overexpression, full-length Mart1-encoding RNA was nucleofected into CD40-B cells at varying concentrations. The cells were cultured for 24 hours to allow for protein expression, then the Mart1 concentration was assessed by immunoblot using a Mart1 specific monoclonal antibody (Figure 3B). The Mart1+ melanoma cell line C32TG was used as a positive control. Strong expression of Mart1 antigen was observed at all RNA concentrations, with a larger protein band (doublet) observed at higher concentrations of RNA. Wild-type Mart1 has been observed as a doublet in tumor cells by Western blot[26]. For the antigen presentation studies, 20 μg of Mart1 RNA was used. All cell lines strongly express the MelanA and Cyp1B1 antigens after transfection of 20 ug of RNA (data not shown). When RNA levels were titrated to levels resulting in equal expression of MelanA antigen, the same ratios of antigen presentation of Mart1 antigen were observed (K562>DC>CD40B cells), Supplemental Figure 1).
Differential antigen processing of CD40-B cells, dendritic cells, and K562 cells
To confirm that the poor presentation of Mart1 M26 epitope by CD40-B cells is antigen-specific and not just differential sensitivity of target cells to cytolysis, FluM1 RNA was transferred into the APCs, with highly efficient M1 antigen presentation observed in all three APCs as measured by γ-interferon ELISPOT assay (Figure 4, left column), but weaker presentation by K562/A2 cells compared with CD40-B and DC (Table 1A), as expected for an immunoproteasome-dependent epitope[18]. As little as 2 μg of M1 RNA was efficiently processed, and increased quantities of M1 RNA did not improve recognition by T cells (data not shown). This is consistent with the prediction that 2 μg of antigen-specific RNA potentially encodes 10 million antigenic epitopes/cell, far in excess of the measured MHC class I density on CD40-B cells (up to 107,000 antigenic binding sites per cell)[5].
Table 1A.
Quantitation of ELISPOT data: average number of spots (standard deviation)
| APC | RNA | FluM1 | p-value | Mart1 | p-value | Cyp239 | p-value |
|---|---|---|---|---|---|---|---|
| CD40-B | No peptide | 4.0 (1.0) | 0 (0) | 5.3 (1.5) | |||
| Specific peptide | 205 (15.6) | p=0.002 | 86.3 (32.3) | p=0.044 | 195 (3.2) | p<0.0001 | |
| CD40-B | control | 5.7 (4.0) | 0.3 (0.6) | 7.7 (1.5) | |||
| specific | 222 (29.1) | p=0.005 | 1.0 (1.7) | p=0.581 | 6.7 (3.2) | p=0.661 | |
| DC | control | 0.7 (0.6) | 3.0 (1.7) | 14.0 (2.0) | |||
| specific | 312 (31.3) | p=0.003 | 11.7 (4.0) | p=0.049 | 39 (4.4) | p=0.004 | |
| K562/A2 | control | 13.3 (5.9) | 7.7 (3.5) | 8.0 (1.7) | |||
| specific | 95.3 (17.0) | p=0.008 | 121 (22.5) | p=0.011 | 344 (5.1) | p<0.001 |
In comparison, both RNA-transfected CD40-B cells and dendritic cells did not efficiently present the nucleofected Mart1 antigen (Figure 4, middle column), although both dendritic cells and CD40-B cells did induce weak γ-interferon secretion. We also found that the tumor antigenic epitope Cyp239 was poorly processed by both dendritic cells and CD40B cells (Figure 4, right column). To determine if K562 cells stably expressing HLA-A2 (K562/A2) could process and present these antigens, K562/A2 cells were nucleofected with RNA encoding the full-length antigens (Figure 4, bottom 2 rows). All three antigenic epitopes were efficiently presented by K562/A2 cells, as measured by γ-interferon ELISPOT assay. Numerical data from Figure 4 is shown in Table 1A. K562/A2 cells more efficiently presented the Mart1 epitope derived from RNA compared with DC’s (p=0.011) and CD40-B cells (p=0.011). Similarly, K562/A2 cells more efficiently presented Cyp239 epitope derived from RNA compared with DC’s (p<0.001) and CD40-B cells (p<0.001). To confirm proteasomal dependence, presentation of the Mart1 epitope was markedly inhibited in DC’s in the presence of the proteasomal inhibitors lactacystin (mean 74 vs. 39, p=0.01) or MG-132 (mean 74 vs. 15, p=0.003) (Supplemental Figure 2). Similarly, inhibition was observed in K562 cells by lactacystin (mean 314 vs 44, p=0.001) and with MG-132 (mean 314 vs. 28, p=0.0002).
Confirmation of selective antigen presentation of Cyp239 by K562 cells
To confirm that the Cyp239 epitope is efficiently processed and presented by K562/A2 cells but not by CD40-B cells or dendritic cells under different maturation conditions, both specificity by cytolysis (Figure 5A) and interferon-γ secretion by ELISPOT (Figure 5B) were performed. In Figures 3 and 4, dendritic cells were matured with IFN-γ. In Figure 5, dendritic cells were matured with a combination of prostaglandin E2, IL-1β, IL-6, and TNFα. Full-length RNA encoding Cyp1B1 or the control antigen CML66 were nucleofected into dendritic cells and K562/A2 cells, and the antigens were presented to the Cyp239 epitope-specific T cell clone Cyp239.128. Peptide-pulsed T2 cells were used as controls. Figures 5A and 5B show that K562/A2 cells efficiently process the nucleofected Cyp1B1 RNA. Dendritic cells weakly process this epitope, as revealed by cytolytic activity and IFN-γ production from Cyp239.128 T cell clone (DC vs. K562/A2, p<0.0001, data shown in Table 1B). Similarly, the SV40 immortalized human fibroblast cell line GM847 (HLA-A2+cyp1B1−) is poorly lysed by Cyp239 specific T cell clones (<10% at E:T 30:1), but the DNA transfectant GM847/Cyp1B1 is lysed at >80% (data not shown). Figure 5C shows stimulatory cell titrations for the induction of IFNγ by this same clone. Dendritic cells are only weakly able to stimulate IFN-γ at the highest APC density. The maturation conditions of the dendritic cells did not impact processing of Cyp239.
Table 1B.
Quantitation of ELISPOT data for CYP239 T cell clone: average number of spots (standard deviation)
| APC | Antigen | Cyp1B1 | p-value |
|---|---|---|---|
| T2 | H369 peptide | 2.7 (2.9) | |
| Cyp239 peptide | 200.7 (13.7) | p=0.001 | |
| DC | CML66 RNA | 2 (1) | |
| CYP1B1 RNA | 18 (5.6) | p=0.035 | |
| K562/A2 | CML66 RNA | 2.3 (3.2) | |
| CYP1B1 RNA | 164.7 (8.3) | p=0.0002 |
Comparison of constitutive and immunoproteasome expression in CD40-B cells, dendritic cells, and K562 cells
Our observation that the Mart1 M26 and Cyp239 epitopes are poorly presented by dendritic cells, and that K562 cells efficiently present both of these epitopes, suggested that differential expression of immunoproteasomes in these APCs could be the primary factor regulating processing of these antigens. We performed quantitative real-time RT-PCR of three immunoproteasomal subunits, LMP10 (MECL-1,β2i), LMP2 (β1i), and LMP7 (β5i), in all three APCs (Figure 6A). The expression of all of these subunits was normalized to expression in K562 cells, which are low in expression of all three subunits[15]. Both CD40B cells and dendritic cells express markedly increased quantities of LMP2 and LMP7. Furthermore, K562 cells can be readily induced by interferon-γ to express these subunits.
Figure 6. Immunoproteasomal subunits LMP2 (β1i) and LMP7 are poorly expressed in K562 cells in comparison to CD40B cells and dendritic cells.

A. Total RNA derived from different APCs were used in quantitative real-time RT-PCR to measure relative expression of the proteasomal subunits LMP10 (left), LMP2 (center) and LMP7 (right). All RNA expression levels are normalized to K562. Only mature dendritic cells and K562 cells treated with interferon-γ showed upregulation of LMP10. The immunoproteasomal subunits LMP2 and LMP7 are strongly expressed in both dendritic cells (DC) and CD40B cells relative to K562, which can upregulate both subunits after treatment with interferon-γ. LCL, EBV-transformed lymphoblastoid cell line; iDC, immature dendritic cells; mDC, mature dendritic cells. Standard error<0.09. B. Immunoblot of DC, CD40B cells, and K562 cells, showing expression of the constitutive proteasome subunits (β1, β2, and β5) in all cells, and poor expression of the immunoproteasomal components (LMP2, LMP10, and LMP7) in K562 cells. C. Rapid induction of the immunoproteasomal components (LMP2, LMP10) and later induction of LMP7 in K562 cells after 1–5 days (D1–D5) of treatment with interferon-γ.
The differential expression of LMP2, LMP10, and LMP7 was confirmed by western blot in Figure 6B. K562 cellsin contrast to DC and CD40B cells, poorly express the immunoproteasome components LMP2, LMP10, and LMP7. All three immunoproteasomal components are induced with interferon-γ treatment (Figure 6C). Induction of the immunoproteasome in K562 cells with 5 days of interferon-γ treatment resulted in a 34% decrease in Mart1 antigen presentation as measured by interferon-γ ELISPOT (mean 187 vs 124, p=0.12, data not shown).
The function of the proteasome is to cleave proteins into smaller peptides and is thought to be primarily responsible for generating the C terminus of MHC class I binding peptides [27, 28]. The immunoproteasome is thought to preferentially cleave after hydrophobic and bulky amino acids such as leucine and phenylalanine, while the constitutive proteasome favors cleavage after negatively charged amino acids, such as glutamic and aspartic acids[29]. As seen in Table 2, NetChop3.0 accurately predicted the C-terminal cleavage sites for all three epitopes, but predicted for internal cleavage sites for both FluM1 and Cyp239. In contrast, PaProc II predicted no C-terminal cleavage for Cyp239, and also predicted internal cleavage sites for all three epitopes. Therefore, neither the NetChop3.0 nor the PaProc II algorithms predict for the observed differential antigen processing observed here.
Table 2.
Proteasomal processing predictions
| EPITOPE | SEQUENCE | Proteasome | NetChop3.0 | PaProC II |
|---|---|---|---|---|
| FluM1_58 | GILGFVFTL | Constitutive Immuno | /LTK/GILGF/VFTL/ | L/TK/GILGF/VF/T/L/ LT/KGIL/G/FVF/TL/ |
| Mart1_M26 | EAAGIGILTV | Constitutive Immuno | AE/EAAGIGILTV/ | A/E/E/AA/G/I/GIL/TV/ AEEAAGI/GIL/TV/ |
| Cyp239 | SLVDVMPWL | Constitutive Immuno | AGSL/VDVMPWL/ | AGS/LV/DVM/PWL… AG/SLVD/VM/PWL… |
Discussion
Peptide-based cancer immunotherapies have resulted in limited clinical responses with measured objective response rates of 2.9% reported across multiple tumor types[30]. Despite advances in the cell manufacturing of APCs, such as dendritic cells, the delivery of single MHC class I-binding peptides has resulted in measurable immune responses but limited therapeutic utility. This has led to the development of nucleic acid transfer of tumor antigens into APCs for immunotherapy, and to identify multiple MHC class I and class-II restricted epitopes for future vaccine development. While dendritic cells have been the prototype APC for clinical trials[31], their use for antigen delivery (in vitro or in vivo) has been limited by multiple factors, including costs of production, reproducibility, and the requirement of leukapheresis to achieve sufficient cell numbers for either APC production or vaccine monitoring. As alternative APCs, CD40-B cells[5] and modified K562 cells are being developed for clinical use[13].
The differential antigen processing by constitutive proteasomes versus immunoproteasomes has been demonstrated for Mart1 and other tumor antigens[1, 25]. Our data suggests that the Cyp239 epitope is also differentially processed by the immunoproteasome. Dendritic cells (both immature and mature) and EBV-transformed B cells express LMP7, while normal B lymphocytes do not[25]. This dramatically impacts in vivo T-cell responses to Mart1 in A2-transgenic mice[32]. The addition of interferon-γ upregulates the proteasome subunits β1i (LMP2), β2i (LMP7) and β 5i (MECL1, LMP10), which results in proteasomes with higher chymotrypsin-like activity [33]. This causes internal cleavage and destruction of certain antigenic peptides[29, 34]. Proteasomal prediction algorithms do not predict for the observed differences in presentation of Mart1 or Cyp239, in agreement with the observation that proteasome cleavage predictions are less reliable than predictions of peptide binding to MHC molecules or to TAP transporter[35, 36]. This likely reflects quantitative, rather than qualitative, differences in cleavage site usage between standard and immunoproteasomes.
Here, we exploit the differential antigenic processing of the Mart1 M26 and the Cyp239 epitopes, in comparison to the influenza M158–66 epitope, to demonstrate that both CD40-B cells and dendritic cells have a limited ability to process the full-length Mart1 and Cyp1B1 antigens for presentation to T cells. It has been shown that RNA-transfected CD40-B cells may be used for stimulation of primary Mart1-specific T cells[8], but this may reflect other factors beyond peptide/MHC density, such as efficiency of costimulation. Here we demonstrate that, as T cell targets, antigen processing and presentation of the Mart1 epitope by CD40-B cells is relatively poor, in comparison to K562 cells. This may limit the usefulness of RNA transfected CD40-B cells for immune monitoring of tumor immunotherapy, although they are highly effective at presentation of peptide epitopes.
The leukemic cell line K562 has been used for years as a target of NK activity in vitro due to its limited expression of endogenous MHC molecules. HLA-transfectants of the K562 cell line, further modified to express the costimulatory molecules B7.1 and CD83, are also highly efficient at presenting peptide antigenic epitopes for in vitro T cell stimulation[13, 15]. Since HLA-transfectants of K562 may still be sensitive to NK-mediated cell lysis[37] or induce immunity to minor histocompatibility antigens, the in vivo use of these cells may induce in situ IFN-γ and alter antigen processing. Here, we have demonstrated that HLA-A*0201 transfected K562 cells process the constitutive proteasome-dependent epitopes, Mart126–34 and the Cyp239 epitope from cytochrome P450 1B1. K562 cells, which are readily cultured and easily transfected, provide a rapid and reliable system for presenting epitopes that are processed via the constitutive proteasome. As potential artificial APC, K562 cells have not been demonstrated to prime T-cells, which requires fully matured APC expressing high levels of co-stimulatory molecules including CD80, CD86 and CD40. These studies have focused on the use of K562 cells for stimulating memory T cell responses. Engineered expression of these or other co-stimulatory molecules may allow for priming of naïve T cells as well.
Acknowledgments
We thank Dr. Lee Nadler for providing valuable scientific reagents. This study was supported by a research grant from the deBeaumont Foundation and the National Cancer Institute/Avon Foundation (5 P30 CA006516-44, KSA), R21 CA115043-2 (CW) and the Cancer Vaccine Center at the Dana-Farber Cancer Institute.
Footnotes
Contribution: K.S.A. designed and analyzed experiments, collected and reviewed data, and prepared the manuscript; W.Z., M.S., D.D., and Y-J.K. designed and performed experiments; T.S., C.W., and V.B. designed and analyzed experiments, and E.R. reviewed data and prepared the manuscript.
The authors declare no competing financial interests.
References
- 1.Van den Eynde BJ, Morel S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr Opin Immunol. 2001 Apr;13(2):147–53. doi: 10.1016/s0952-7915(00)00197-7. [DOI] [PubMed] [Google Scholar]
- 2.Dannull J, Lesher DT, Holzknecht R, Qi W, Hanna G, Seigler H, et al. Immunoproteasome down-modulation enhances the ability of dendritic cells to stimulate antitumor immunity. Blood. 2007 Dec 15;110(13):4341–50. doi: 10.1182/blood-2007-04-083188. [DOI] [PubMed] [Google Scholar]
- 3.Meidenbauer N, Zippelius A, Pittet MJ, Laumer M, Vogl S, Heymann J, et al. High frequency of functionally active Melan-a-specific T cells in a patient with progressive immunoproteasome-deficient melanoma. Cancer Res. 2004 Sep 1;64(17):6319–26. doi: 10.1158/0008-5472.CAN-04-1341. [DOI] [PubMed] [Google Scholar]
- 4.Heink S, Fricke B, Ludwig D, Kloetzel PM, Kruger E. Tumor cell lines expressing the proteasome subunit isoform LMP7E1 exhibit immunoproteasome deficiency. Cancer Res. 2006 Jan 15;66(2):649–52. doi: 10.1158/0008-5472.CAN-05-2872. [DOI] [PubMed] [Google Scholar]
- 5.von Bergwelt-Baildon MS, Vonderheide RH, Maecker B, Hirano N, Anderson KS, Butler MO, et al. Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: potential for clinical application. Blood. 2002 May 1;99(9):3319–25. doi: 10.1182/blood.v99.9.3319. [DOI] [PubMed] [Google Scholar]
- 6.Schultze JL, Grabbe S, von Bergwelt-Baildon MS. DCs and CD40-activated B cells: current and future avenues to cellular cancer immunotherapy. Trends Immunol. 2004 Dec;25(12):659–64. doi: 10.1016/j.it.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Kondo E, Topp MS, Kiem HP, Obata Y, Morishima Y, Kuzushima K, et al. Efficient generation of antigen-specific cytotoxic T cells using retrovirally transduced CD40-activated B cells. J Immunol. 2002 Aug 15;169(4):2164–71. doi: 10.4049/jimmunol.169.4.2164. [DOI] [PubMed] [Google Scholar]
- 8.Coughlin CM, Vance BA, Grupp SA, Vonderheide RH. RNA-transfected CD40-activated B cells induce functional T-cell responses against viral and tumor antigen targets: implications for pediatric immunotherapy. Blood. 2004 Mar 15;103(6):2046–54. doi: 10.1182/blood-2003-07-2379. [DOI] [PubMed] [Google Scholar]
- 9.Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, Hwu P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003 Jun 1;63(11):2836–43. [PubMed] [Google Scholar]
- 10.von Bergwelt-Baildon M, Maecker B, Schultze J, Gribben JG. CD40 activation: potential for specific immunotherapy in B-CLL. Ann Oncol. 2004 Jun;15(6):853–7. doi: 10.1093/annonc/mdh213. [DOI] [PubMed] [Google Scholar]
- 11.von Bergwelt-Baildon M, Schultze JL, Maecker B, Menezes I, Nadler LM Correspondence re RLapointe et al. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003;63:2836–43. [PubMed] [Google Scholar]; Cancer Res. 2004 Jun 1;64(11):4055–6. doi: 10.1158/0008-5472.CAN-03-3606. author reply 6–7. [DOI] [PubMed] [Google Scholar]
- 12.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007 May;15(5):981–8. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler MO, Lee JS, Ansen S, Neuberg D, Hodi FS, Murray AP, et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin Cancer Res. 2007 Mar 15;13(6):1857–67. doi: 10.1158/1078-0432.CCR-06-1905. [DOI] [PubMed] [Google Scholar]
- 14.Hirano N, Butler MO, Xia Z, Ansen S, von Bergwelt-Baildon MS, Neuberg D, et al. Engagement of CD83 ligand induces prolonged expansion of CD8+ T cells and preferential enrichment for antigen specificity. Blood. 2006 Feb 15;107(4):1528–36. doi: 10.1182/blood-2005-05-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirano N, Butler MO, Xia Z, Berezovskaya A, Murray AP, Ansen S, et al. Efficient presentation of naturally processed HLA class I peptides by artificial antigen-presenting cells for the generation of effective antitumor responses. Clin Cancer Res. 2006 May 15;12(10):2967–75. doi: 10.1158/1078-0432.CCR-05-2791. [DOI] [PubMed] [Google Scholar]
- 16.Maecker B, Sherr DH, Vonderheide RH, von Bergwelt-Baildon MS, Hirano N, Anderson KS, et al. The shared tumor-associated antigen cytochrome P450 1B1 is recognized by specific cytotoxic T cells. Blood. 2003 Nov 1;102(9):3287–94. doi: 10.1182/blood-2003-05-1374. [DOI] [PubMed] [Google Scholar]
- 17.Gribben JG, Ryan DP, Boyajian R, Urban RG, Hedley ML, Beach K, et al. Unexpected association between induction of immunity to the universal tumor antigen CYP1B1 and response to next therapy. Clin Cancer Res. 2005 Jun 15;11(12):4430–6. doi: 10.1158/1078-0432.CCR-04-2111. [DOI] [PubMed] [Google Scholar]
- 18.Gileadi U, Moins-Teisserenc HT, Correa I, Booth BL, Jr, Dunbar PR, Sewell AK, et al. Generation of an immunodominant CTL epitope is affected by proteasome subunit composition and stability of the antigenic protein. J Immunol. 1999 Dec 1;163(11):6045–52. [PubMed] [Google Scholar]
- 19.Yang XF, Wu CJ, McLaughlin S, Chillemi A, Wang KS, Canning C, et al. CML66, a broadly immunogenic tumor antigen, elicits a humoral immune response associated with remission of chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2001 Jun 19;98(13):7492–7. doi: 10.1073/pnas.131590998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blomberg K, Granberg C, Hemmila I, Lovgren T. Europium-labelled target cells in an assay of natural killer cell activity. II. A novel non-radioactive method based on time-resolved fluorescence. significance and specificity of the method. J Immunol Methods. 1986 Aug 21;92(1):117–23. doi: 10.1016/0022-1759(86)90511-9. [DOI] [PubMed] [Google Scholar]
- 21.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996 Oct 4;274(5284):94–6. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005 Apr;57(1–2):33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 23.Schuler MM, Nastke MD, Stevanovikc S. SYFPEITHI: database for searching and T-cell epitope prediction. Methods Mol Biol. 2007;409:75–93. doi: 10.1007/978-1-60327-118-9_5. [DOI] [PubMed] [Google Scholar]
- 24.Rasband WS. ImageJ. 1997–2009 http://rsbinfonihgov/ij/
- 25.Morel S, Levy F, Burlet-Schiltz O, Brasseur F, Probst-Kepper M, Peitrequin AL, et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity. 2000 Jan;12(1):107–17. doi: 10.1016/s1074-7613(00)80163-6. [DOI] [PubMed] [Google Scholar]
- 26.De Maziere AM, Muehlethaler K, van Donselaar E, Salvi S, Davoust J, Cerottini JC, et al. The melanocytic protein Melan-A/MART-1 has a subcellular localization distinct from typical melanosomal proteins. Traffic. 2002 Sep;3(9):678–93. doi: 10.1034/j.1600-0854.2002.30909.x. [DOI] [PubMed] [Google Scholar]
- 27.Niedermann G, King G, Butz S, Birsner U, Grimm R, Shabanowitz J, et al. The proteolytic fragments generated by vertebrate proteasomes: structural relationships to major histocompatibility complex class I binding peptides. Proc Natl Acad Sci U S A. 1996 Aug 6;93(16):8572–7. doi: 10.1073/pnas.93.16.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craiu A, Akopian T, Goldberg A, Rock KL. Two distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptide. Proc Natl Acad Sci U S A. 1997 Sep 30;94(20):10850–5. doi: 10.1073/pnas.94.20.10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toes RE, Nussbaum AK, Degermann S, Schirle M, Emmerich NP, Kraft M, et al. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J Exp Med. 2001 Jul 2;194(1):1–12. doi: 10.1084/jem.194.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004 Sep;10(9):909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005 Apr;5(4):296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 32.Chapatte L, Ayyoub M, Morel S, Peitrequin AL, Levy N, Servis C, et al. Processing of tumor-associated antigen by the proteasomes of dendritic cells controls in vivo T-cell responses. Cancer Res. 2006 May 15;66(10):5461–8. doi: 10.1158/0008-5472.CAN-05-4310. [DOI] [PubMed] [Google Scholar]
- 33.Chapiro J, Claverol S, Piette F, Ma W, Stroobant V, Guillaume B, et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J Immunol. 2006 Jan 15;176(2):1053–61. doi: 10.4049/jimmunol.176.2.1053. [DOI] [PubMed] [Google Scholar]
- 34.Basler M, Moebius J, Elenich L, Groettrup M, Monaco JJ. An altered T cell repertoire in MECL-1-deficient mice. J Immunol. 2006 Jun 1;176(11):6665–72. doi: 10.4049/jimmunol.176.11.6665. [DOI] [PubMed] [Google Scholar]
- 35.Peters B. Modeling the MHC-I pathway. Berlin, Germany: Humboldt University; 2003. [Google Scholar]
- 36.Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008;9:8. doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maziarz RT, Mentzer SJ, Burakoff SJ, Faller DV. Distinct effects of interferon-gamma and MHC class I surface antigen levels on resistance of the K562 tumor cell line to natural killer-mediated lysis. Cell Immunol. 1990 Oct 15;130(2):329–38. doi: 10.1016/0008-8749(90)90276-w. [DOI] [PubMed] [Google Scholar]