

Abstract
Bovine Viral Diarrhea Virus (BVDV) is a positive sense, single-stranded RNA virus which exhibits two biotypes in standard cell culture systems. The cytopathic strains of this virus (cpBVDV) induce dramatic cytoplasmic vacuolization in cell cultures, while infection with the non-cytopathic (NCP-BVDV) strains produces no overt changes in the host cells. Our results show that extensive cytoplasmic vacuolization is the earliest morphological change in response to cpBVDV infection in MDBK cells. Cells with extensive vacuolization showed no co-existing chromatin condensation, caspase activation, or loss of membrane integrity. In addition, the caspase inhibitor (zVAD-fmk), although improving cell viability of infected cells from 6.7±2.2% to 18.8±2.2%, did not prevent vacuolization. On the ultrastructural level, the virus-induced cytoplasmic vacuoles are single membrane structures containing organelles and cellular debris, which appear capable of fusing with other vacuoles and engulfing surrounding cytoplasmic materials. LysoTracker Red which marks lysosomes did not stain the virus-induced cytoplasmic vacuoles. In addition, this lysosomal dye could be observed in the cytoplasm of vacuolized cells, suggesting a lysosomal abnormality. Our data demonstrate that cpBVDV induced a novel cell death pathway in MDBK cells that is primarily associated with lysosomal dysfunction and the formation of phagocytic cytoplasmic vacuoles, and this mode of cell death is different from apoptosis and necrosis.
INTRODUCTION
Bovine Viral Diarrhea Virus (BVDV) is a positive sense, single-stranded RNA virus that belongs to the Flaviviridae family (Collett et al., 1988). BVDV is known to cause severe lesions in the gastrointestinal tract and death in affected animals (Bolin et al., 1985, Brownlie et al., 1984). Two biotypes, cytopathic (cpBVDV) and noncytopathic (ncpBVDV), can be isolated from infected animals exhibiting the classical mucosal disease syndrome(Brownlie, 1991). However, only cpBVDV induces cytopathology in sensitive cell types, such as Madin-Darby bovine kidney cells (MDBK).
cpBVDV induces dramatic cytoplasmic vacuolization in infected cell cultures and in tissues of infected calves (Bielefeldt Ohmann & Bloch, 1982), commonly referred to as cytopathic effects (CPE) of cpBVDV . To date, the nature of this vacuolization during BVDV infection is not understood. Recently, it was suggested that cpBVDV-induced apoptosis is responsible for the virus-associated CPE (Grummer et al., 1998, Zhang et al., 1996). However, the morphology of apoptotic cell death is generally not associated with cytoplasmic vacuoles. Furthermore, although inhibitors of apoptosis, including zVAD-fmk, blocked apoptosis in cpBVDV-infected cells and significantly improved cell survival, they failed to achieve suppression of cytoplasmic vacuolization (Bendfeldt et al., 2003, Grummer et al., 2002a).
In this paper we report that cpBVDV induced rapid and extensive vacuolization in MDBK cells, which is morphologically different from apoptosis and necrosis. This cellular response was characterized by electron microscopy, which shows the presence of enlarged single membrane vacuoles containing organelles and cellular debris, capable of fusing with other vacuoles and engulfing surrounding cytoplasmic materials.
EXPERIMENTAL PROCEDURES
1) Chemicals
All cell culture supplies and fluorescent probes were obtained from Invitrogen (Carlsbad, CA). Unless specified, all other reagents were supplied by Sigma Aldrich (St. Louis, MO).
2) Cells and Viruses
MDBK cells (ATCC-CCL22) or fetal bovine kidney cells (FBK) derived from primary tissue, were grown in Dulbecco's Modified Eagle's Minimum Essential Medium (DMEM) containing 4.5 g of glucose, and 10% horse serum in cell culture incubator (5% CO2, 37°C). Cells were infected with plaque-purified cpBVDV strain NADL in cell culture medium, the ncpBVDV strain NY-1 or mock-infected with cell-culture medium alone. The titer of BVDV used in our studies was sufficient to generate an input MOI of 3−5 which can completely destroy MDBK cell monolayers and kill 90% of cells within 72 hours of infection or for ncpNY-1 produce a positive fluorescent stain in 100% of the infected cells.
After initial incubation for 1 h in cell a culture incubator (5% CO2, 37°C), the culture medium was changed to a fresh virus-free medium. For some experiments, either wortmannin (PI3-kinase inhibitor) or zVAD-fmk (pan-caspase inhibitor) was added in required final concentration and then incubated for the next 48 or 72 hours. zVAD-fmk was replenished every 10−12 hours as suggested in the literature (Bendfeldt et al., 2003). Cells were then further incubated for an intended duration of time in the presence or absence of the inhibitor.
3) Cell viability
MDBK cells were plated at a density 1−2×103 cells/well in 96 well plates. On the following day, after initial infection for one hour, cells were washed and medium containing required inhibitors were added to cells and incubated for needed period of time. Then, cell viability was assessed using the Resaruzin (Almar blue) indicator dye (Mazzio & Soliman, 2004). Quantitative analysis of dye conversion was measured using a fluorescent plate reader with ex/em = 550/580. Cell viability was expressed as percentage of untreated cells.
4) Light and Fluorescent Microscopy
Detection of cytoplasmic vacuolization
Mock-infected, cpBVDV-infected, and NY-1 infected MDBK cells were grown on 96- or 24-well plates in defined medium for 6, 16, or 24 h post infection. At these time points cells were subjected to the light microscopy using a Zeiss fluorescent microscope and photographed. All images were processed and analyzed by counting total number of cells and number of vacuolized cells. Very often those cells were also examined for signs of apoptosis including chromatin condensation (Hoechst 33342) and caspase activation (FLICA (for details, see below). This allowed us to superimpose light- and fluorescent microscopic images to determine if vacuolization and characteristics of apoptosis co-exist in the same cell. All experiments were done in triplicates and each “n” represents 3−6 fields from each of three different wells for each time point (6, 16, and 24 hours post infection) in the presence or absence of cpBVDV.
Detection and cellular localization of viral NS3 in MDBK cells
To determine cellular localization of the nonstructural BVDV protein NS3 while preserving cellular architecture infected or control cells were fixed in 0.1% gluteraldehyde for 10 minutes. Cells were then incubated in 1% Triton X100 for 30 minutes, washed in PBS, and stained for NS3 using BVDV Mab 20.1.6 according to published methodology(Deregt et al., 2005).
Detection of apoptosis by Hoechst Staining
MDBK cells were grown on 96-or 24-well plates. Cells were then stained with 1 μg/ml Hoechst 33342 for 20 min, washed with cell culture medium, and imaged using a Zeiss fluorescent microscope equipped with a camera. Nuclear morphology was evaluated using an excitation wavelength of 350±10 nm and a longpass filter of 400 nm for emission. All images were processed and analyzed by counting total number of cells, vacuolized cells and cells with apoptotic nuclei. Uniformly stained nuclei were scored as healthy, viable cells, while condensed or fragmented nuclei were scored as apoptotic. All experiments were done in triplicate and each “n” represents 3−6 fields from 3 different wells for each time point (6, 16, and 24 hours post infection) in the presence or absence of cpBVDV.
Detection of apoptosis by FLICA Staining
Mock-infected and cpBVDV-infected MDBK cells were grown on 96-or 24-well plates for 24 hours post infection were assayed for the caspase activity using a commercial kit based on fluorochrome-labeled caspase inhibitors (FLICA, Immunochemistry Technologies LLC, Bloomington, MN), according to manufacturer's instructions. Cells were imaged by fluorescence microscopy (ex/em = 488/520 nm) and the images were processed and analyzed by counting total number of cells, vacuolized cells and FLICA-stained cells. All experiments were done in triplicate and each “n” represents 3−6 fields from 3 different wells.
Detection of necrosis by Propidium-iodide Staining
Propidium iodide (2.5 μg/ml) (Immunochemistry Technologies LLC, Bloomington, MN) was incubated with infected MDBK cells and nuclear localization was determined using a Zeiss fluorescent microscope as described above. Cellular morphology was evaluated using ex/em wavelengths of 552/570 nm. All images were processed and light and fluorescent microscopy images were superimposed to determine propidium iodide staining in highly vacuolized cells.
Detection of lysosomes by LysoTracker Red Staining
MDBK cells were grown on 24-well plates. All treatments were carried out in quadruplicates. Cells were then stained with 100 nM LysoTracker Red for 20 min, washed in cell culture medium, and imaged using a Zeiss fluorescent microscope as described above. Cellular morphology was evaluated using ex/em wavelengths of 552/570 nm. All images were processed and light and fluorescent microscopy images were superimposed to determine lysosomal staining in highly vacuolized cells.
5) Electron microscopy
Cells were grown in 6-well plates for 24 hours prior to infection with cpBVDV. 16 hours post-infection cells were washed with serum-free medium and fixed with fixation buffer (2.5% glutaraldehyde, 4% paraformaldehyde, 0.02% picric acid in 0.1M buffer) for 60 minutes and then washed with the same buffer. Post-fixation was done in 1% OsO4− 1.5%K-ferricyanide (aqueous) for 60 minutes and then washed. After staining with 1.5% uranyl acetate for 30 min, dehydration was done through graded ethanol series. Samples were then infiltrated and embedded in LX-112 resin (Ladd Research Industries, Burlington, VT) by inserting tubes made from BEEM capsules into the resin prior to polymerization. Sections were cut at 55−60 nm (silver-gold) using a Diatome diamond knife-(Diatome, USA, Hatfield, PA) on an RMC MT-7000 Ultramicrotome. (Boeckeler-RMC Instruments, Tucson, AZ). Sections were then contrasted with lead citrate and viewed on a JSM 100 CX-II electron microscope (JEOL, USA, Inc., Peabody, MA) operated at 80 kV. Images were recorded on Kodak 4489 Electron Image film then digitized on an Epson Expression1600 Pro scanner at 900dpi.
RESULTS
The morphology of MDBK cells infected with cpBVDV (NADL) was practically indistinguishable from mock-infected control cells up to 6 h post-infection. However, by 16 and 24 h after infection, about 15 and 25% of total number of cells, respectively, were found to have extensive vacuolization of cytoplasm (Fig 1A and B). In addition, by 24 h post infection, vacuolized cells started to round up and detach, suggesting that virus-induced dynamic cellular pathology in whole population of cells have to be investigated within first 24 hours. By 24−36 h post-infection, about 50% of monolayers in all wells inspected for this study were partially destroyed. Further incubation of infected cells beyond 36 h post infection resulted in a significant number of detached cells, making it difficult to determine the mode of the cell death. Infection of MDBK cells with the noncytopathic strain of BVDV (NY-1) did not result in cytoplasmic vacuolization or disruption of the cellular monolayer (Fig 1A). In addition, ncpBVDV-infected MDBK cells could be easily cultured without any morphological signs of infection. Infection of FBK cells with NADL also resulted in appearance of vacuolized cells, which were not observed in uninfected cells (data not shown).
Figure 1.
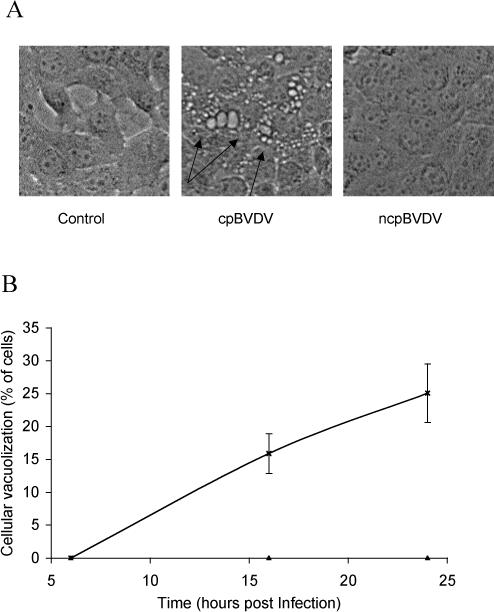
cpBVDV infection induces cytoplasmic vacuolization in MDBK cells in a time-dependent manner. A. Microscopic analysis of a representative control, cpBVDV, and ncpBVDV-infected MDBK cells at 24 hours post infection. Arrows indicate representative vacuolized cells in slides 24h post infection. B. Vacuolized cells in mock-infected (▲) and cpBVDV-infected (*) samples were analyzed and counted as described in Experimental Procedures. Error bars indicate standard deviation for each time point and specified condition (n=3).
Infection of MDBK cells with cpBVDV was confirmed by immunohistochemistry using a monoclonal antibody to viral NS3 protein 24 h post infection (Fig 2, right panel). Cells which stained for the viral protein also showed extensive cytoplasmic vacuolization. NS3 staining was observed throughout the cytoplasm, but the nucleus and cytoplasmic vacuoles were unstained. Uninfected cells were always NS3 negative (Fig 2, left panel). These data clearly show that the vacuolated cells were not simply responding to being next to an infected cell.
Figure 2.
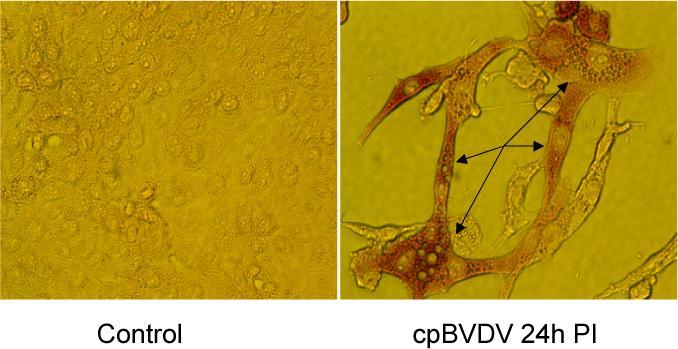
Cytoplasmic vacuolization in MDBK cells expressing NS3 viral protein. Microscopic analysis of a representative control and cpBVDV-infected MDBK cells at 24 hours post infection, stained for NS3 viral protein. Arrows indicate representative vacuolized cells in slides 24h post infection.
To demonstrate that virus-induced cytoplasmic vacuoles were not due to degeneration of plasma membrane and necrosis, we stained MDBK cells with propidium iodide (PI) at 24 hours post infection. No PI staining was observed in any cells, including those with a large number of cytoplasmic vacuoles (indicated by arrows) (Fig 3A). In control experiments, treatment of the same cells with 0.1 mM of diethyl pyrocarbonate (DEPC) (0.1 mM) for 10 minutes to permeabilize the cell plasma membrane resulted in a rapid uptake of PI into cells as evidenced by the stained nuclei (Fig 3B). In addition, on the ultrastructural level, abnormal and swollen mitochondria, which are commonly found during necrosis and necroptosis (Degterev et al., 2005), were not detected in infected cells with extensive cytoplasmic vacuolization (Figures 6B and 7).
Figure 3.
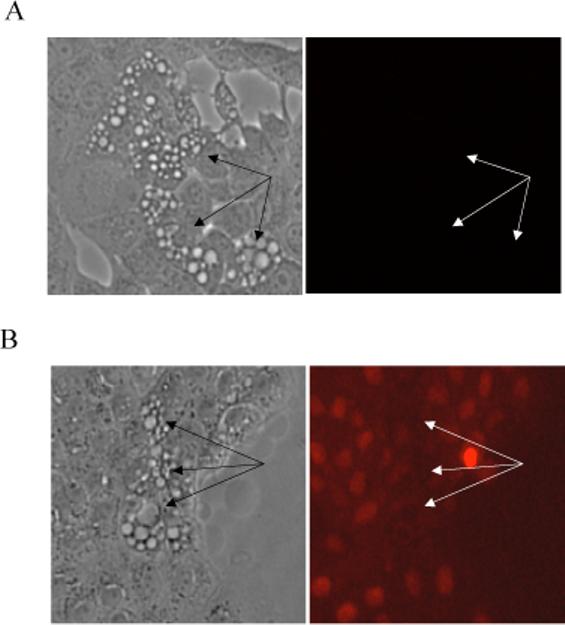
cpBVDV-induced cytoplasmic vacuolization is not associated with loss of plasma membrane integrity. A. Infected cells (24 hours post infection) were stained with propidium iodide(PI) and analyzed using fluorescent microscopy., Vacuolated areas (arrows) show no sign of PI localization in nuclei. B. Infected cells (24 hours post infection) were treated with 0.1 mM DEPC for 10 minutes and stained with propidium iodide and analyzed using fluorescent microscopy, as described in Experimental Procedures. Arrows indicated vacuolated cells.
Figure 6.
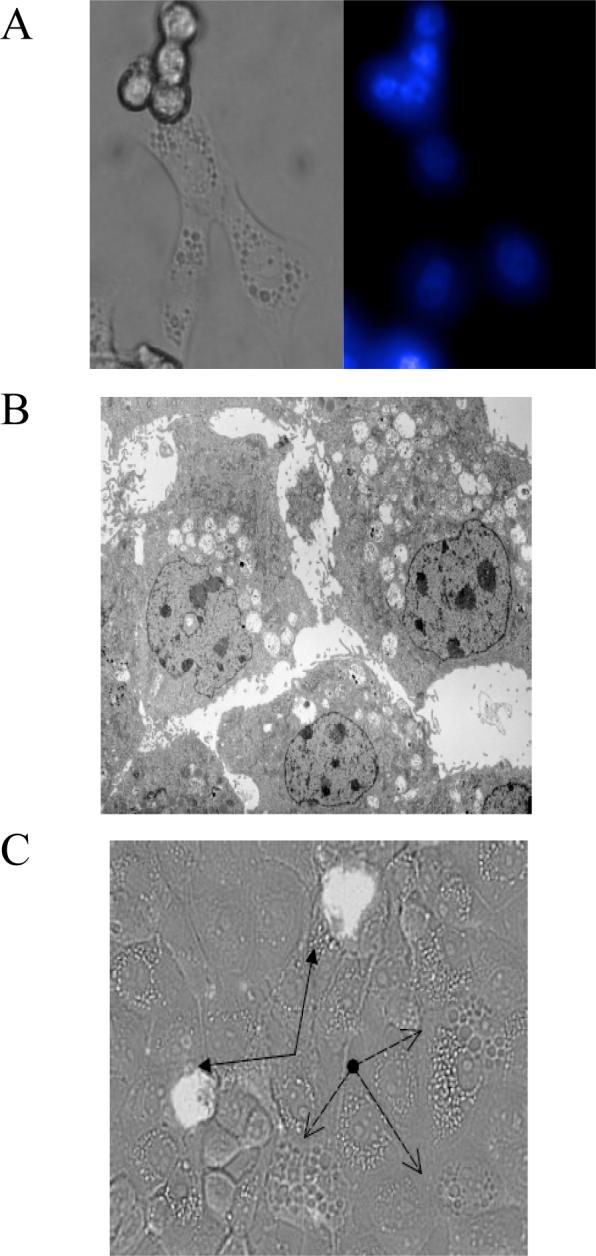
Cytoplasmic vacuolization in cpBVDV-infected cells is not associated with apoptotic markers. (A) Microscopic analysis of Hoechst dye staining in cpBVDV-infected MDBK cells 48 hours post infection demonstrates uniformly low intensity stained nuclei in vacuolized cells and brightly stained nuclei in detached cells. Left panel correspond to light microscopy of representative cells and right represents fluorescent microscopy of the same cells. (B) Electron microscopy of infected MDBK cell (16 hours post infection) demonstrates the absence of chromatin condensation in nuclei of highly vacuolized cells. (C) Microscopic analysis of FLICA dye staining in cpBVDV-infected MDBK cells 24 hours post infection. Light microscopic and fluorescent microscopic images for FLICA staining were superimposed demonstrating no staining of vacuolized cells (dashed arrows) and brightly stained apoptotic cells (solid arrows, filled arrow heads).
Figure 7.

A) Electron micrographic analysis of representative control, ncpBVDV and cpBVDV-infected MDBK cell 24 hours post infection. Large vacuoles (V), encapsulated by single membranes and containing cellular debris, are found only in the cytoplasm of infected cells. Mitochondria and nuclei appear to be normal in infected cells. B) Ultrastructural analysis of representative cytoplasmic vacuoles found in infected cells. Large encapsulated vacuoles were found to fuse with each other, forming larger vacuoles (left panel). Some of cytoplasmic autophagic vacuoles were found to be in a process of engulfing cytoplasmic material (as indicated by arrows) (right panel).
Because previous studies have reported cpBVDV-induced apoptotic cell death (Grummer et al., 2002a, Grummer et al., 1998, Jordan et al., 2002, Yamane et al., 2005), we compared the proportion of cells expressing characteristics of apoptosis, such as chromatin condensation and caspase activation, with those undergoing extensive cytoplasmic vacuolization. A hallmark of apoptosis, chromatin condensation, can be observed using the Hoechst 33824 reagent, which brightly stains condensed chromatin. Chromatin condensation was determined to be in only 4±0.9 % of cells at 24 hours post infection (Fig 4) whereas vacuolization was >25%. Caspase activation, quantified using Flica staining, was detected in only 15.3±1.7 % of all cells at 24 hours post infection (Fig 4).
Figure 4.
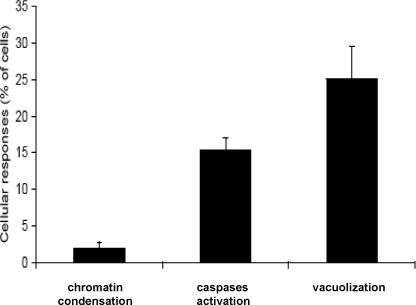
Cytoplasmic vacuolization and apoptotic responses in cpBVDV-infected MDBK cells. Control and cpBVDV-infected cells were stained with Hoechst and FLICA dyes, for chromatin condensation and caspases activation, respectively, at 24 hour post infection. Vacuolized and dye-stained cells, were then analyzed and counted. Error bars indicate standard deviation for each specified analysis (n=3).
To determine the involvement of caspase activation in cpBVDV-induced cytoplasmic vacuolization, we used the general caspase inhibitor, zVAD-fmk (100μM), which was shown to inhibit apoptosis in BVDV-infected cells (Bendfeldt et al., 2003). We found that, although addition of 100 μM zVAD-fmk to MDBK cells prior to and during viral infection improved cell viability from 6.7±2.2% to 18.8±2.2%, (Fig 5B), caspase inhibition had no effect on the appearance of vacuoles (Fig 5A) in infected cells 24 hours post infection, suggesting that virus-induced cytoplasmic vacuolization is caspase-independent.
Figure 5.

Inhibition of caspase activation in infected cells partially prevents cpBVDV-induced cell death but does not block cytoplasmic vacuolization. A) Light microscopy of infected MDBK cell (16 hours post infection) demonstrates that cpBVDV-induced cytoplasmic vacuolization is not affected by treatment with 100 μM zVAD-fmk. (B) Effect of zVAD-fmk (100 μM) on cell viability in cpBVDV infected cells.
We further investigated whether apoptotic markers were localized to the infected cells with extensive cytoplasmic vacuolization. We demonstrated that at 48 h post infection, the nuclei of cells, which were still attached with extensive vacuolization showed no evidence of bright staining with Hoechst 33324 and chromatin condensation (Fig 6A). On the other hand, detached cells were labeled by the Hoechst dye, producing bright fluorescence, and nuclei in those cells were very poorly defined (Fig 6A). To further characterize the apoptosis-vacuolization association on the ultrastructural level, we examined the nuclei of highly vacuolated cells at 16 hours post infection using electron microscopy. We found that none of the highly vacuolized cells had apoptotic bodies or condensed chromatin in their nuclei (Fig 6B). Moreover, infected cells with extensive cytoplasmic vacuolization did not have FLICA staining, a marker for caspase activation (Fig 6C).
Further examination of the cytoplasmic vacuoles observed after cpBVDV infection on ultrastructural level, demonstrated that the vacuoles are surrounded by a clearly-defined single membrane and contain remains of organelles and cell debris (Fig 7). The virus-induced vacuoles were larger than 500 nm in diameter, with an average of 10±1 vacuoles per cell per thin section examined. Control cells had some cells with cytoplasmic vacuoles smaller than 100nm in diameter, averaging 1−2 vacuoles per cell (Fig 7A). Cells infected with noncytopathic BVDV had cytoplasmic vacuolization similar to control cells (Fig 7A). In addition, we were able to identify vacuoles that can fuse with each other(Fig 7B, left panel) and can engulf surrounding cytoplasm and organelles (Fig 7B, right panel), which could be a pathway for the formation of enlarged vacuoles.
Interestingly, in all infected cells examined by electron microscopy, there were no signs of autophagosomes, i.e. dense vacuoles surrounded by a double membrane, suggesting that macroautophagy is unlikely to be responsible for cpBVDV-induced cytoplasmic vacuolization. To further rule out the involvement of macroautophagy in virus-induced cytoplasmic vacuolization, we demonstrated that wortmannin, an PI3-kinase and macroautophagy inhibitor, at concentrations between 0.1 to 10 μM had no effect on cytoplasmic vacuolization induced by cpBVDV at 24h post-infection (Fig 8 A). Treatment of cells with wortmannin also had no effect on cell viability either in control cell or infected cells (Fig 8B).
Figure 8.
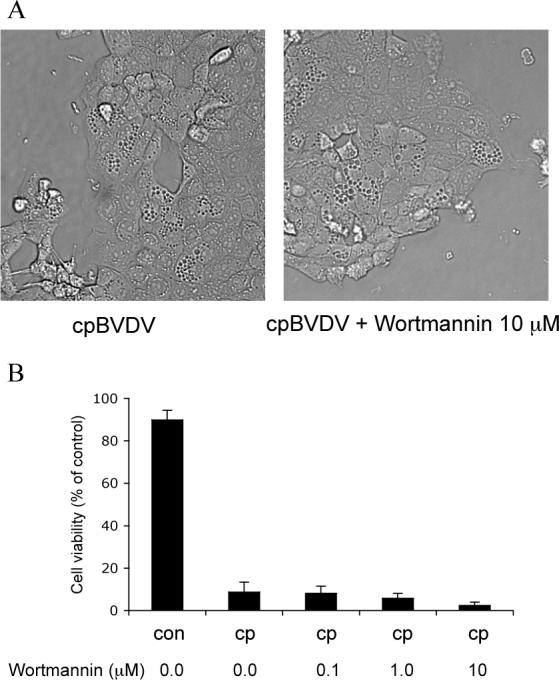
Cytoplasmic vacuolization induced by cpBVDV is insensitive to treatment with wortmannin. (A) Light microscopy of infected MDBK cell (16 hours post infection) demonstrates that cpBVDV-induced cytoplasmic vacuolization is not affected by treatment with 10 μM wortmannin. (B) Wortmannin (0.1−10 μM) had no effect on decrease in cell viability mediated by cpBVDV infection
The presence of cytoplasmic and organelle debris in cpBVDV-induced vacuoles might be suggestive of lysosomal involvement. Thus, we investigated lysosomal involvement in cpBVDV-induced vacuolization using the lysosome-specific dye, LysoTracker Red. We found that cells with extensive vacuolization of cytoplasm demonstrated a partial loss of typical punctuated LysoTracker Red fluorescence staining compared to cells without vacuolization. (Fig 9), and it was found to be distributed throughout cytoplasm of infected cells. Interestingly, LysoTracker Red, which labels acidic cellular compartments including macroautophagy-associated autophagolysosomes, did not label cytoplasmic vacuoles in cpBVDV (Fig 9).
Figure 9.
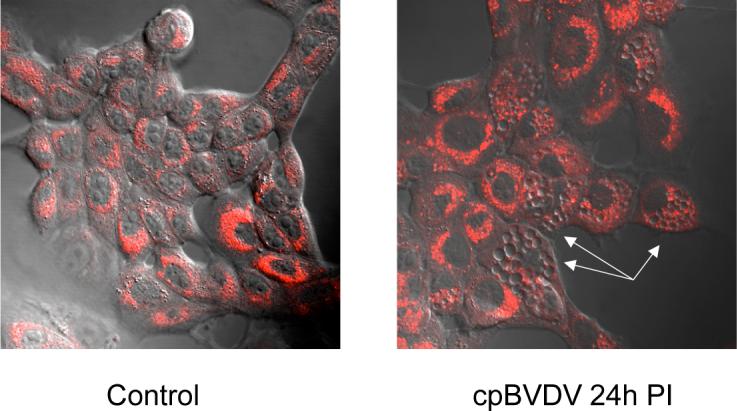
Microscopic analysis of LysoTracker Red fluorescence in cpBVDV-infected MDBK cells, demonstrating bright fluorescence in morphologically normal cells and depletion of fluorescence in vacuolized cells (arrow).
DISCUSSION
Cytoplasmic vacuolization is a commonly noted response in cells infected with the cytopathic biotype of BVDV. However, there are no detailed reports directly linking the vacuoles to the presence of virus in the infected cell. In this work, using a monoclonal antibody to viral protein, NS3, to identify BVDV-infected cells (Deregt et al., 2005),we demonstrated that NS3 can be easily detected in infected cells with cytoplasmic vacuolization, suggesting the virus induces vacuolization of infected MDBK cells. Interestingly, ncpBVDV does not produce any morphological changes in infected cells, and can actually block CPE when co-infected with cpBVDV (Bendfeldt et al., 2003, Yamane et al., 2005). Thus, vacuolization is unlikely to be important for viral replication. In support of this hypothesis, we demonstrated that NS3, which is required for viral replication (Gu et al., 2000), was identified throughout the cytoplasm of infected cells, but the lumen of vacuoles were free of this protein.
Extensive cytoplasmic vacuolization is thought to be associated with necrosis and necroapoptosis (Degterev et al., 2005, Kitanaka & Kuchino, 1999, Kroemer et al., 2005). Here we demonstrated that infected cells with extensive cellular vacuolization are impermeable to propidium iodide, suggesting virus-induced vacuolization does not alter plasma membrane integrity, a hallmark of necrosis (Kroemer et al., 2005). In addition, mitochondria in BVDV-infected cells appear to be normal, unlike mitochondrial abnormalities, such as swelling, that are observed during necrosis and necroptosis (Degterev et al., 2005). This also suggests that necrotic events are unlikely to be primary reasons for cytoplasmic vacuolization in infected cells.
To address the question of apoptosis versus cellular vacuolization during cpBVDV infection, we found that cellular vacuolization is a major early response to viral infection as compared to the late apoptotic response of chromatin condensation. In addition, cells with extensive cytoplasmic vacuolization have morphologically normal nuclei, which are not brightly stained with the Hoechst dye. These data are in agreement with our electron microscopy study demonstrating that highly vacuolized cells do not show evidence of apoptotic bodies or chromatin condensation. Although, in agreement with other publications (Grummer et al., 2002b, Jordan et al., 2002, Schweizer & Peterhans, 1999), caspase activation was also observed at 24h post infection, but cytochemical examination showed an absence of caspase activation in cells with extensive cytoplasmic vacuolization (Fig 6C). These data suggest that caspase activation is not involved in virus-induced cytoplasmic vacuolization responses.
A recent study of cells infected with the coronavirus mouse hepatitis virus (MHV), another cytoplasmic positive sense RNA virus, showed extensive vacuolization of the cytoplasm thought to be associated with autophagy (Prentice et al., 2004). This does not appear to be the case for BVDV. Three classes of autophagy have been described: macroautophagy, chaperone-mediated autophagy, and microautophagy. Macroautophagy and chaperone-mediated autophagy are normal cellular stress responses designed to remove and recycle misfolded proteins and damaged organelles (Cuervo, 2004, Shintani & Klionsky, 2004). Electron microscopy of macroautophagic responses reveals two subtypes of electron-dense macroautophagic vacuoles: autophagosomes and autophagolysosomes (Eskelinen, 2005, Lawrence & Brown, 1992) both of which are double membrane-bound structures. These macroautophagic structures are also labeled with LysoTracker Red which is easily detected by wide-field and confocal fluorescent microscopy (Moriyasu et al., 2003, Rideout et al., 2004). Pharmacologically, macroautophagy can be distinguished from the other types of autophagy by its sensitivity to wortmannin and other phosphoinositide 3-kinases (PI3-kinase) inhibitors (Finn et al., 2005, Petiot et al., 2000). The BVDV-induced vacuoles are single membrane structures not labeled with LysoTracker Red.
Recently, microtuble-associated protein lightchain 3 (LC3), a mammalian homolog of yeast Atg8, has been put forth as a specific marker to monitor macroautophagy. LC3 was found to be co-localized with LysoTracker-stained macroautophagic structures (Bampton et al., 2005). However, further work on LC3 and macroautophagy indicates that LC3 could be incorporated into protein aggregates independent of autophagy (Kuma et al., 2007). In addition, detection of macroautophagy–associated LC3 conversion (LC3 I to LC3 II) could be problematic because LC3 II tends to be much more sensitive to detection by immunoblotting than LC3 I (Mizushima & Yoshimori, 2007). We chose not to pursue LC3 staining for the above reason and because the EM morphological studies, LysoTracker staining, and insensitivity to wortmannin clearly demonstrated that virus-induced vacuolization is inconsistent with macroautophagy. Chaperone-mediated autophagy does not exhibit cytoplasmic vacuoles and can only be detected with specific probes (Kaushik & Cuervo, 2006, Kaushik et al., 2006). Microautophagy (Kunz et al., 2004, Muller et al., 2000, Sattler & Mayer, 2000) is defined as the sequestration of cytoplasmic components by direct invagination of lysosomal or vesicular membrane, followed by budding of engulfed small vesicular structures into the lumen of the autophagolysosomes containing cellular debris. This process is topologically equivalent to membrane invagination during multivesicular body formation and to the budding of enveloped viruses (Kunz et al., 2004). Functionally, microautophagy is associated with macroautophagy, thus, counterbalancing macroautophagy-mediated membrane influx, which is essential for resumption of cell growth(Dubouloz et al., 2005).
We did not find electron-dense double-membraned autophagosomes and autophagolysosomes, typical hallmarks of macroautophagy (Cuervo, 2004), suggesting that macroautophagy is unlikely to be involved in cellular response to BVDV. Furthermore, we determine that the PI-3 kinase inhibitor, wortmannin (0.1−10 μM), known to block macroautophagy even at 0.1 μM (Finn et al., 2005) , had no effect on either cytoplasmic vacuolization or cell survival. These data would also suggest that vacuolization induced by BVDV is PI-3 kinase-independent. Furthermore, we demonstrated that the cytoplasmic vacuoles were not stained with LysoTracker Red. Thus, the absence of three out of four parameters for determination of macroautophagy suggests that BVDV-induced vacuolization is different from macroautophagy.
The ultrastructural morphological nature of BVDV-induced cytoplasmic vacuoles was examined by electron microscopy. We determined that the vacuoles in infected cells contain organelles' and cytoplasmic debris. Further examination of cytoplasmic vacuoles revealed that they are capable of fusing with each other and engulfing surrounding cytoplasmic components, suggesting that virus-induced vacuoles possess phagocytic properties. This morphological observation correlates well with extensive growth of vacuoles and disappearance of cytoplasm in infected cells, demonstrated at the level of light microscopy. This morphology is consistent with previously described microautophagy, where the cytoplasmic vacuoles seem to be the primary mechanism for sequestering cytoplasm and organelles in cells (Tuttle & Dunn, 1995, Tuttle et al., 1993, Veenhuis et al., 1983, Yokota et al., 1993). However, the phagocytic vacuolization response induced by the virus seems to be cytotoxic, which is different from postulated functional role of microautophagy for cell survival (Dubouloz et al., 2005). In addition, because microautophagy is thought to be in a close association with macroautophagy, the absence of BVDV-induced macroautophagy could also suggest the absence of virus-induced microautophagy. Therefore, cpBVDV-induced cytoplasmic phagocytic vacuolization appears to be morphologically similar to microautophagy, but functionally it is different from previously described macroautophagy, chaperon-mediated autophagy, and microautophagy.
Although not extensively covered by this study, it would be worthwhile to point out that LysoTracker Red staining throughout the cytoplasm of infected cells is indicative of lysosomal abnormality (Caruso et al., 2005). If lysomal dysfunction occurs, an appearance of lysosomal enzymes such as cathepsins and serine proteases in cytoplasm is likely to result in activation of number of caspase-dependent and caspase-independent pathways in addition to general cell lysis, as was initially described in serum-starved PC12 cells (Uchiyama, 2001). Interestingly, a significant proportion of viral NS3, produced during infection with cytopathic BVDV strains, was found to be associated with smooth endoplasmic reticulum (Zhang et al., 2003), known to be involved in the formation of lysosomes (Novikoff, 1976). Thus, it is possible that the stress induced by NS3 on smooth endoplasmic reticulum in cells infected with cytopathic strains of BVDV could be the cause of lysosomal abnormalities and induction of formation of cytoplasmic vacuoles with phagocytic properties.
In recent years, there has been much interest in the role of cellular structures in the replication of numerous viruses. Evidence has been presented to suggest that autophagy is induced by viruses to provide intracellular compartments and scaffolds to assist the replication process (Novoa et al., 2005, Wileman, 2006). BVDV provides some unique insight into this area in that two biotypes of the virus exist, one that induces extensive vacuole formation (cpBVDV) and the other which does not (ncpBVDV). Both biotypes replicate equally well to produce infectious progeny, which indicates that the formation of vacuoles is irrelevant to virus replication. The autophagy-like response in this instance may be more related to a cellular defense mechanism of an innate or adaptive immune response (Deretic, 2005).
In conclusion, we demonstrate here that the autophagy-like cellular response is an early response to cpBVDV infection. This cellular response to the virus is different from apoptosis, necrosis, and recently described necroapoptosis. Virus-induced vacuoles possess phagocytic properties, responsible for digestion of cytoplasm and cytoplasmic components, suggesting that this form of vacuole-mediated autolysis could be named phagoptosis.
ACKNOWLEDGMENTS
We thank Dr. Joel D. Pardee for the excellent and very useful discussions. This work was supported by NIH/NIDA grant,K01 DAO18262, and a seed grant from Cornell University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Bendfeldt S, Grummer B, Greiser-Wilke I. No caspase activation but overexpression of Bcl-2 in bovine cells infected with noncytopathic bovine virus diarrhoea virus. Vet Microbiol. 2003;96:313–26. doi: 10.1016/j.vetmic.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Bielefeldt Ohmann H, Bloch B. Electron microscopic studies of bovine viral diarrhea virus in tissues of diseased calves and in cell cultures. Arch Virol. 1982;71:57–74. doi: 10.1007/BF01315175. [DOI] [PubMed] [Google Scholar]
- Bolin SR, McClurkin AW, Cutlip RC, Coria MF. Severe clinical disease induced in cattle persistently infected with noncytopathic bovine viral diarrhea virus by superinfection with cytopathic bovine viral diarrhea virus. Am J Vet Res. 1985;46:573–6. [PubMed] [Google Scholar]
- Brownlie J. The pathways for bovine virus diarrhoea virus biotypes in the pathogenesis of disease. Arch Virol Suppl. 1991;3:79–96. doi: 10.1007/978-3-7091-9153-8_10. [DOI] [PubMed] [Google Scholar]
- Brownlie J, Clarke MC, Howard CJ. Experimental production of fatal mucosal disease in cattle. Vet Rec. 1984;114:535–6. doi: 10.1136/vr.114.22.535. [DOI] [PubMed] [Google Scholar]
- Caruso JA, Mathieu PA, Reiners JJ,, Jr. Sphingomyelins suppress the targeted disruption of lysosomes/endosomes by the photosensitizer NPe6 during photodynamic therapy. Biochem J. 2005;392:325–34. doi: 10.1042/BJ20050313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett MS, Anderson DK, Retzel E. Comparisons of the pestivirus bovine viral diarrhoea virus with members of the flaviviridae. J Gen Virol. 1988;69(Pt 10):2637–43. doi: 10.1099/0022-1317-69-10-2637. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Deregt D, Dubovi EJ, Jolley ME, Nguyen P, Burton KM, Gilbert SA. Mapping of two antigenic domains on the NS3 protein of the pestivirus bovine viral diarrhea virus. Vet Microbiol. 2005;108:13–22. doi: 10.1016/j.vetmic.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Deretic V. Autophagy in innate and adaptive immunity. Trends Immunol. 2005;26:523–8. doi: 10.1016/j.it.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Finn PF, Mesires NT, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy. 2005;1:141–5. doi: 10.4161/auto.1.3.2000. [DOI] [PubMed] [Google Scholar]
- Grummer B, Bendfeldt S, Greiser-Wilke I. Apoptosis inhibitors delay the cytopathic effect of bovine viral diarrhoea virus (BVDV). J Vet Med B Infect Dis Vet Public Health. 2002a;49:298–303. doi: 10.1046/j.1439-0450.2002.00573.x. [DOI] [PubMed] [Google Scholar]
- Grummer B, Bendfeldt S, Wagner B, Greiser-Wilke I. Induction of the intrinsic apoptotic pathway in cells infected with cytopathic bovine virus diarrhoea virus. Virus Res. 2002b;90:143–53. doi: 10.1016/s0168-1702(02)00150-8. [DOI] [PubMed] [Google Scholar]
- Grummer B, Moennig V, Greiser-Wilke I. [Cytopathogenic bovine viral diarrhea viruses induce apoptosis in bovine cell cultures]. Dtsch Tierarztl Wochenschr. 1998;105:29–31. [PubMed] [Google Scholar]
- Gu B, Liu C, Lin-Goerke J, Maley DR, Gutshall LL, Feltenberger CA, Del Vecchio AM. The RNA helicase and nucleotide triphosphatase activities of the bovine viral diarrhea virus NS3 protein are essential for viral replication. J Virol. 2000;74:1794–800. doi: 10.1128/jvi.74.4.1794-1800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R, Wang L, Graczyk TM, Block TM, Romano PR. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J Virol. 2002;76:9588–99. doi: 10.1128/JVI.76.19.9588-9599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27:444–54. doi: 10.1016/j.mam.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Massey AC, Cuervo AM. Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. Embo J. 2006;25:3921–33. doi: 10.1038/sj.emboj.7601283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanaka C, Kuchino Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999;6:508–15. doi: 10.1038/sj.cdd.4400526. [DOI] [PubMed] [Google Scholar]
- Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463–7. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–8. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- Kunz JB, Schwarz H, Mayer A. Determination of four sequential stages during microautophagy in vitro. J Biol Chem. 2004;279:9987–96. doi: 10.1074/jbc.M307905200. [DOI] [PubMed] [Google Scholar]
- Lawrence BP, Brown WJ. Autophagic vacuoles rapidly fuse with pre-existing lysosomes in cultured hepatocytes. J Cell Sci. 1992;102(Pt 3):515–26. doi: 10.1242/jcs.102.3.515. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, Soliman KF. Glioma cell antioxidant capacity relative to reactive oxygen species produced by dopamine. J Appl Toxicol. 2004;24:99–106. doi: 10.1002/jat.954. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy. 2007:3. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Moriyasu Y, Hattori M, Jauh GY, Rogers JC. Alpha tonoplast intrinsic protein is specifically associated with vacuole membrane involved in an autophagic process. Plant Cell Physiol. 2003;44:795–802. doi: 10.1093/pcp/pcg100. [DOI] [PubMed] [Google Scholar]
- Muller O, Sattler T, Flotenmeyer M, Schwarz H, Plattner H, Mayer A. Autophagic tubes: vacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J Cell Biol. 2000;151:519–28. doi: 10.1083/jcb.151.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikoff AB. The endoplasmic reticulum: a cytochemist's view (a review). Proc Natl Acad Sci U S A. 1976;73:2781–7. doi: 10.1073/pnas.73.8.2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol Cell. 2005;97:147–72. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–8. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279:10136–41. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–62. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Sattler T, Mayer A. Cell-free reconstitution of microautophagic vacuole invagination and vesicle formation. J Cell Biol. 2000;151:529–38. doi: 10.1083/jcb.151.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer M, Peterhans E. Oxidative stress in cells infected with bovine viral diarrhoea virus: a crucial step in the induction of apoptosis. J Gen Virol. 1999;80(Pt 5):1147–55. doi: 10.1099/0022-1317-80-5-1147. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle DL, Dunn WA,, Jr. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J Cell Sci. 1995;108(Pt 1):25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- Tuttle DL, Lewin AS, Dunn WA,, Jr. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur J Cell Biol. 1993;60:283–90. [PubMed] [Google Scholar]
- Uchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol. 2001;64:233–46. doi: 10.1679/aohc.64.233. [DOI] [PubMed] [Google Scholar]
- Veenhuis M, Douma A, Harder W, Osumi M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch Microbiol. 1983;134:193–203. doi: 10.1007/BF00407757. [DOI] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and autophagy generate sites for virus replication. Science. 2006;312:875–8. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- Yamane D, Nagai M, Ogawa Y, Tohya Y, Akashi H. Enhancement of apoptosis via an extrinsic factor, TNF-alpha, in cells infected with cytopathic bovine viral diarrhea virus. Microbes Infect. 2005;7:1482–91. doi: 10.1016/j.micinf.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Yokota S, Himeno M, Roth J, Brada D, Kato K. Formation of autophagosomes during degradation of excess peroxisomes induced by di-(2-ethylhexyl)phthalate treatment. II. Immunocytochemical analysis of early and late autophagosomes. Eur J Cell Biol. 1993;62:372–83. [PubMed] [Google Scholar]
- Zhang G, Aldridge S, Clarke MC, McCauley JW. Cell death induced by cytopathic bovine viral diarrhoea virus is mediated by apoptosis. J Gen Virol. 1996;77(Pt 8):1677–81. doi: 10.1099/0022-1317-77-8-1677. [DOI] [PubMed] [Google Scholar]
- Zhang G, Flick-Smith H, McCauley JW. Differences in membrane association and sub-cellular distribution between NS2−3 and NS3 of bovine viral diarrhoea virus. Virus Res. 2003;97:89–102. doi: 10.1016/s0168-1702(03)00223-5. [DOI] [PubMed] [Google Scholar]