

Abstract
Amidst controversy, the cellular form of the prion protein PrPc has been proposed to mediate oligomeric Aβ-induced deficits. In contrast, there is consistent evidence that the Src kinase Fyn is activated by Aβ oligomers and leads to synaptic and cognitive impairment in transgenic animals. However, the molecular mechanism by which soluble Aβ activates Fyn remains unknown. Combining the use of human and transgenic mouse brain tissue as well as primary cortical neurons, we demonstrate that soluble Aβ binds to PrPc at neuronal dendritic spines in vivo and in vitro where it forms a complex with Fyn, resulting in the activation of the kinase. Using the antibody 6D11 to prevent oligomeric Aβ from binding to PrPc, we abolished Fyn activation and Fyn-dependent tau hyperphosphorylation induced by endogenous oligomeric Aβ in vitro. Finally we showed that gene dosage of Prnp regulates Aβ-induced Fyn/tau alterations. Altogether, our findings identify a complete signaling cascade linking one specific endogenous Aβ oligomer, Fyn alteration and tau hyperphosphorylation in cellular and animal models modeling aspects of the molecular pathogenesis of Alzheimer’s disease.
Keywords: amyloid-beta, oligomer, prion protein, Fyn, Alzheimer’s disease, transgenic
INTRODUCTION
According to our current knowledge, Alzheimer’s disease (AD) may result from abnormal changes in soluble amyloid-β (Aβ) and tau proteins prior to the formation of amyloid plaques and neurofibrillary tangles in the cerebral parenchyma. With evidence indicating that soluble forms of Aβ, also called Aβ oligomers, may be the real culprits for AD (Walsh et al., 2002; Lesne et al., 2006; Shankar et al., 2008), there has been a paradigm shift in the approach to studying disease-associated proteins. Following these reports, synthetic oligomeric forms of Aβ have been proposed to inhibit long-term potentiation (LTP) upon binding to the cellular form of the prion protein, PrPc (Lauren et al., 2009). Several groups, however, failed to reach the conclusions that Aβ-induced LTP inhibition/cognitive impairment is dependent on PrPc (Balducci et al., 2010; Calella et al., 2010; Kessels et al., 2010) fueling an intense debate regarding the role of PrPc in AD (Benilova and De Strooper, 2010). More recently, antibodies targeting the 94-104 domain of PrPc blocked the inhibition of LTP triggered by soluble extracts of AD brain (Barry et al., 2011; Freir et al., 2011). In the latter study, both synthetic Aβ oligomers, or ADDLs, and soluble extracts of AD brain were unsuccessful at blocking LTP in PrP-null hippocampal slices (Freir et al., 2011). In addition, insoluble monomeric Aβ was also proposed to bind to PrPc (Zou et al., 2011). Thus, PrPc appears to be required to mediate plasticity impairments triggered by certain Aβ species, which remain to be identified.
In contrast, there is evidence documenting the implication of the Src tyrosine kinase Fyn in Aβ-induced neuronal dysfunction: (i) genetic ablation of Fyn protects oligomeric Aβ-mediated toxicity (Lambert et al., 1998), (ii) alterations of Fyn expression and activation induced by Aβ plays a role in synaptic and cognitive impairment in AD mouse models (Chin et al., 2004; Chin et al., 2005; Roberson et al., 2011), (iii) Fyn can mediate Aβ/tau-induced toxicity (Williamson et al., 2002; Williamson et al., 2008; Haass and Mandelkow, 2010; Ittner et al., 2010). Despite these studies linking Aβ and Fyn, the molecular mechanism by which endogenous soluble extracellular Aβ species activate the intracellular Fyn kinase remains unknown.
A recent study demonstrated that synthetic Aβ oligomers bind to postsynaptic PrPc resulting in Fyn activation and neuronal demise (Um et al., 2012). However, while the authors described that their interaction relate to a “subset of Aβ peptide” and that different Aβ oligomeric species might elicit different mechanisms, the type of endogenous Aβ molecule interacting with PrPc:Fyn is still an enigma. Resolving this mystery is particularly important if one hopes to use this biological event as a possible diagnostic and therapeutic tool.
In this study, we report that affinity purified human Aβ oligomers form a complex with PrPc and Fyn ex vivo, in situ and in vitro, leading to the abnormal phosphorylation of Fyn and tau in a PrPc-dependent manner. Finally, Prnp gene dosage differentially regulates Aβ-induced Fyn and tau phosphorylation in vivo.
MATERIALS AND METHODS
Human brain tissue
Brain tissue from the inferior temporal gyrus (Brodman Area 20) from 85 subjects enrolled in the Religious Order Study underwent biochemical analyses. One brain specimen showed signs of protein degradation and was discarded from all analyses resulting in a total number of 84 brain tissue samples used in this study. The demographic and clinical characteristics of the cases selected for these analyses were representative of all ROS cases available at the time.
Each participant had undergone uniform structured baseline clinical evaluation and annual follow-up evaluation until death (Bennett et al., 2012). Briefly, dementia and AD diagnosis required evidence of meaningful decline in cognitive function and impairment on at least 2 cognitive domains (for AD, 1 domain had to have been episodic memory), based on the results of 21 cognitive performance tests and their review by a clinical neuropsychologist and expert clinician. “MCI” refers to participants with cognitive impairment as assessed by the neuropsychologist but without a diagnosis of dementia, as determined by the clinician. “NCI” refers to those individuals without dementia or MCI. At death, a neurologist, blinded to all postmortem data, reviewed all available clinical data and rendered a summary diagnostic opinion regarding the clinical diagnosis at the time of death (Bennett et al., 2002; Bennett et al., 2006).
Following death, each case was assigned a Braak score based on neuronal neurofibrillary tangle pathology (Braak and Braak, 1991), a neuritic plaque score based on the modified Consortium to Establish a Registry for Alzheimer Disease (CERAD) criteria (excluding age and clinical diagnosis), and an AD pathologic diagnosis based on the National Institute on Aging - Reagan criteria by examiners blinded to all clinical data (Bennett et al., 2005; Schneider et al., 2009). Neuritic plaques, diffuse plaques, and neurofibrillary tangles in the inferior temporal cortex were counted after Bielschowsky silver staining, as previously described (Bennett et al., 2003).
The characteristics of the three clinical diagnostic groups are summarized in Table 1.
Table 1.
Characteristics of the Religious Order Study Participants.
| Group | NCI (n = 26) | MCI (n = 34) | AD (n = 24) | P values* |
|---|---|---|---|---|
| Age of death (years), Mean ± SD | 82.97 ± 7.53 | 86.33 ± 5.69 | 90.27 ± 7.20 | 0.108 |
| No. of M/F (%) | 12/14 (46.1%) | 14/20 (41.2%) | 9/15 (37.5%) | >0.99 |
| Last MMSE Score, Mean ± SD | 28.35 ± 1.38 | 26.41 ± 2.96 | 12.33 ± 8.79 | <0.0001 |
| PMI (hours), Mean ± SD (Range) | 5.57 ± 2.25 (2-10) | 4.90 ± 2.56 (1-9) | 4.48 ± 1.66 (2-9) | 0.191 |
| Amyloid Burden (% of area), Mean ± SD | 1.63 ± 0.40 | 1.57 ± 0.35 | 3.12 ± 0.42 | 0.0099 |
| Tangle density (#/mm2), Mean ± SD | 4.92 ± 3.93 | 5.81 ± 5.43 | 11.18 ± 10.08 | 0.002 |
Abbreviations: NCI, non cognitive impairment, MCI, mild-cognitive impairment, AD, Alzheimer’s disease, MMSE, mini-mental status examination, M/F, male/female ratio, PMI, post-mortem interval.
Kruskal-Wallis test followed by Bonferroni-adjusted test for multiple comparisons.
Transgenic animals
Mice from the APP line Tg2576, which express the human amyloid precursor protein with the Swedish mutation (APPKM670/671NL), directed by the hamster PrP promoter (Hsiao et al., 1996), were purchased from Taconic Farms, Inc. and bred to obtain wild-type and hemizygous transgenic littermates.
APPPS1 mice (Radde et al., 2006) (kindly provided by Dr. Mathias Jucker), coexpressing the same mutant APP and mutant presenilin-1 (PS1L166P) were crossed with mice lacking (Prnp-/-) or overexpressing PrPc under the mouse PrP promoter (tga20) as described previously (Calella et al., 2010).
Both male and female mice were used in all experiments.
Primary cell cultures
Mouse cortical cultures of neurons were prepared from 14- to 15-d-old embryos as described previously (Lesne et al., 2005) using 5×105 cells/dish. After 3 d in vitro (DIV), neurons were treated with 10 μM AraC to inhibit proliferation of non-neuronal cells. All experiments were performed on near pure neuronal cultures (> 98% of microtubule associated protein-2 immunoreactive cells) after 12-14 DIV. Three to six 35mm dishes per culture per condition were used across three independent experiments.
Following treatment(s), cells were harvested in an ice-cold lysis solution containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Triton X-100 (Sigma) with 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM 1,10-phenanthroline monohydrate (1,10-PTH), 1% (v/v) mammalian protease inhibitor cocktail (Sigma), 0.1% (v/v) phosphatase inhibitor cocktails A (Santa Cruz Biotechnology, Inc.) and 2 (Sigma-Aldrich). Cell lysates were centrifuged for 10 minutes at 13,000 × g, supernatants were isolated, and corresponding pellets were resuspended with the protease/phosphatase inhibitor-containing lysis buffer to extract membrane-bound proteins. Plasma membranes were solubilized in RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 1 mM EGTA, 3% SDS, 1% deoxycholate, 1 mM PMSF, 2 mM 1,10-PTH, 1% (v/v) mammalian protease inhibitor cocktail (Sigma), 0.1% (v/v) phosphatase inhibitor cocktails A (Santa Cruz Biotechnology, Inc.) and 2 (Sigma-Aldrich). Membrane lysates were then subjected to centrifugation for 10 minutes at 16,000 × g, and the soluble fraction was removed and stored for analysis.
Protein extractions
For analyzing Aβ species, we used two extractions protocols described elsewhere (Lesne et al., 2006; Shankar et al., 2008; Sherman and Lesne, 2011). In particular, membrane-enriched protein extracts (or MB extracts) refer to protein lysates obtained following the third step of a serial extraction with a lysis RIPA buffer comprised of 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 3% SDS and 1% deoxycholate. As detailed in methodology chapter recently published (Sherman & Lesne, 2011), samples are then centrifugated at 16,100 × g for 90 minutes. Supernatants are collected and pellets further extracted with formic acid to analyze fibrillar/deposited proteins. It is possible that the use of the RIPA lysis buffer might strip loosely bound Aβ from plaques. Protein amounts were determined by the Bradford protein assay (BCA Protein Assay, Pierce). All supernatants were ultra-centrifuged for 60 min at 100,000 × g. Finally, before analysis, fractions were endogenous immunoglobulins were depleted by sequentially incubating extracts for one hour at room temperature with 50 μL of Protein A-Sepharose, Fast Flow® followed by 50 μL of Protein G-Sepharose, Fast Flow® (GE Healthcare Life Sciences).
Affinity purification of human Aβ oligomers
One milligram of total proteins from AD brain TBS extracts (Shankar et al., 2008) were incubated for 3 hours at 4°C with Protein G-coupled magnetic beads (MagG beads, GE Life Sciences) previously crosslinked with 200 μg of purified 6E10 antibody (Covance) or with 200 μg of Mab13.1.1 and Mab2.1.3 (100 μg of each). Immunocaptured proteins were eluted from the immune complexes using 1% n-octyl beta-D-thioglucopyranoside (OTG; Sigma Aldrich) in 100 mM Glycine, pH 2.8 for 1 minute (3 rounds).
Relative amounts of purified oligomeric Aβ were calculated based on synthetic Aβ1-42 standards (0.001, 0.025, 0.05, 0.1, 0.25, 0.5, 1 and 2.5 ng) ran alongside the samples used for experiments.
Size-exclusion chromatography (SEC)
Immunoaffinity purified protein extracts were loaded on Tricorn Superdex® 75 columns (Amersham Life Sciences, Piscataway, NJ, USA) and run at a flow rate of ~0.3 mL/min. Fractions of 250 μL of eluate in 50 mM Tris-HCl, 150 mM NaCl, 0.01% Triton X-100, pH 7.4, were collected using a BioLogic DuoFlow QuadTec 40 system (Bio-Rad) coupled to a microplate-format fraction collector. A280 was determined live during the experiments and confirmed following each run on a DTX800 Multimode microplate reader (Beckman Coulter).
Western blotting and quantification
SDS-PAGE
Electrophoreses were done on pre-cast 10-20% SDS-polyacrylamide Tris-Tricine gels, 10.5-14% and 4-10.5% Tris-HCl gels (Bio-Rad). Protein levels were normalized to 2-100 μg of protein per sample (depending on targeted protein) and resuspended with 4X Tricine loading buffer.
Transfer
Thereafter, proteins were transferred to 0.2 μm nitrocellulose membrane (Bio-Rad).
Blotting
Nitrocellulose membranes were boiled in 50 mL PBS by microwaving for 25 and 15 sec with 3 min intervals. Membranes were blocked in TTBS (Tris-Buffered Saline-0.1%Tween®20) containing 5% bovine serum albumin (BSA) (Sigma) for 1-2 hours at room temperature, and probed with appropriate antisera/antibodies diluted in 5%BSA-TTBS. Primary antibodies were probed either with anti-IgG immunoglobulins conjugated with biotin or InfraRed dyes (Li-Cor Biosciences, USA). When biotin-conjugated secondary antibodies were used, IR-conjugated Neutravidin® (Pierce) was added to amplify the signal. Blots were revealed on an Odyssey platform (Li-Cor Biosciences).
Stripping
When required, membranes were stripped using Restore™ Plus Stripping buffer (Pierce) for 30-180 min at room temperature depending on antibody affinity.
Quantification
Densitometry analyses were performed using OptiQuant software (Packard Bioscience, Meriden, CT) or using the Odyssey software (Li-Cor). Each protein of interest was probed in three individual experiments under the same conditions, and quantified by software analysis, following determination of experimental conditions ascertaining linearity in the detection of the signal, and expressed as Density Light Units (DLU). The method used allows for a dynamic range of ~100-fold above background (0.01×106 DLU). Respective averages were then determined across the triplicate western blots. Normalization was performed against the neuron-specific nuclear protein NeuN, also performed in triplicate. None of the protein brain levels measured correlated with postmortem interval, arguing against potential protein degradation within human tissues (data not shown).
Immunoprecipitations
Aliquots (100 μg) of protein extracts were diluted to 1 mL with dilution buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl) and incubated with appropriate antibodies (10 μg of either C20 or 8B4; 5 μg of 6E10 or Mab2.1.3/13.1.1 antibodies) overnight at 4°C, and added 50 μL of Protein G-Sepharose, Fast Flow® (GE Life Sciences) 1:1 (v:v) slurry solution with dilution buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, pH 7.4) for two hours. The beads were washed twice in 1mL of dilution buffer and proteins were eluted in 25 μL of loading SDS-PAGE buffer by boiling.
Antibodies
The following primary antibodies were used in this study: 6E10 [1:2,500], 4G8 [1:2,500], biotinylated-6E10 [1:2,500], Tau5 [1:2,000], 6D11 [10 μg] (Covance, USA), DW6 antiserum [1:1,000] (kind gift of Dr. Dominic Walsh), anti-α-tubulin [1:100,000] (Sigma, USA), anti-PrP(C-20) [1:200], anti-PrP(8B4) [1:1,000], anti-Fyn(Fyn3) [1:200], anti-PSD95 [1:200] (Santa Cruz Biotechnology, USA), anti-Fyn [1:1,000] (BD Biosciences, USA), anti-pSrc (Y416) [1,1,000] (Cell Signaling, USA), anti-pY18 [1:2,500] (kind gift from Gloria Lee), 40-/42-end specific Mab2.1.3 and Mab13.1.1 [1:1,000] (kind gift from Pritam Das), anti-NeuN [1:5,000] (Chemicon, USA), anti-MAP2 [1:500] (Novus Biologicals, USA).
Confocal imaging
Triple or Quadruple-label immunofluorescence was performed as previously described (Lesne et al., 2005) using Alexa Fluor 488, 555, 635-conjugated secondary antibodies (Molecular Probes, Invitrogen). Human brain sections were treated for autofluorescence with 1% Sudan Black solution (Schnell et al., 1999) and coverslipped with ProLong-DAPI mounting medium (Molecular Probes). Digital images were obtained using an Olympus IX81 FluoView1000 microscope with laser intensities ranging from 7-11%. Raw image z-stacks (0.2 μm intervals) were analyzed using Bitplane’s Imaris7.x software suite. Frame size was maintained at 1024 × 800 and optical zoom of 1.00 was utilized to allow for maximum distribution of pixel size to tissue dimensions without over sampling. Six regions of interest (ROIs) per brain section (6 sections/case) per case (3 cases per group) were used for voxel quantification.Z stacks were reconstructed using the Surpass module of the Imaris software package (version 7.x, Bitplane Inc., USA). Dendritic shafts and spines were manually traced in the xy plane using the AutoDepth function of the Filament module. After tracing, accurate reconstruction of the diameter of the dendritic shaft and spines was made possible using the diameter function with a constant contrast threshold.
Statistical Analyses
Since many variables were non-normally distributed, nonparametric statistics were used (Spearman rho correlation coefficients, Kruskal-Wallis nonparametric analysis of variance followed by Bonferroni-corrected two-group posthoc Mann-Whitney U tests). Analyses were performed using StatView software, version 5.0.1 and JMP 8.0.1 (SAS Institute, USA).
RESULTS
Abnormal expression of PrPc associated with Fyn phosphorylation in AD brains
Using an extremely well characterized cohort from the Religious Order Study (Table 1), we first sought to measure the protein expression of PrPc, Fyn and phosphorylated Fyn (pY416-Fyn) in human brain tissues by western blot/immunoprecipitation (Fig. 1A). To take into consideration neuronal cell loss across clinical groups, we also determined tissue levels of the neuron-specific nuclear protein NeuN (data not shown) and protein expressions of PrPc, Fyn and pFyn were normalized to NeuN values (Fig. 1B-D). Our results indicate an elevation of membrane-bound PrPc in AD brain tissue compared to MCI and NCI (Fig. 1B). Total Fyn levels (Fig. 1C) were unchanged across clinical groups, whereas pY416-Fyn brain levels rose in AD compared to NCI (2.15-fold) and MCI (1.38-fold) (Fig. 1D). When testing whether these modulations of protein expression were associated with each other, we found that PrPc and Fyn were highly correlated (Fig. 1E) in human brains regardless of clinical status (data not shown). The observed relationship was not caused by the normalization to NeuN as regression analyses using absolute data still indicated a positive correlation between PrPc and Fyn levels (Spearman Rho = 0.383, P = 0.0004). Interestingly, pY416-Fyn and PrPc protein expressions correlated positively in the AD group (Fig. 1F) but not in NCI or MCI groups, even though a weak trend for a possible positive correlation between PrPc levels and pFyn levels in MCI subjects was noticed (Spearman Rho = 0.144, P = 0.0769) (data not shown). Of note, the accumulation of PrPc in AD was not linked to astroglial activation as indicated by the absence of correlation between GFAP and PrPc levels (R = 0.056, P not significant, data not shown).
Figure 1. Increased membrane-bound PrPc levels are associated with Fyn activation in AD.
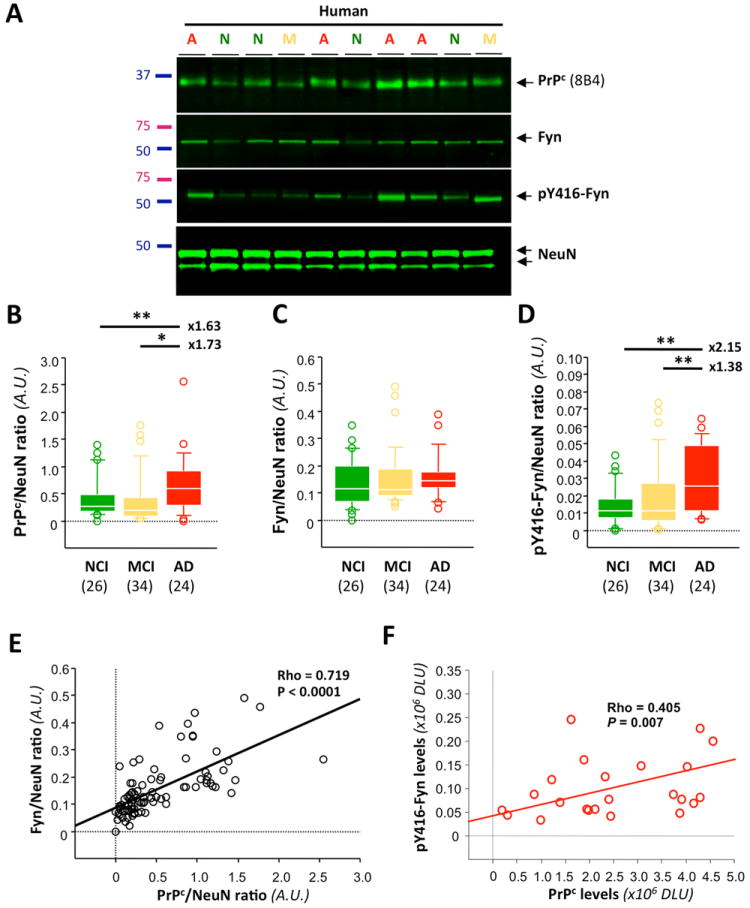
(A) Representative western blot (WB) for PrPc, Fyn, pY416-Fyn, and NeuN in 10 out of 84 specimens of NCI (N), MCI (M) and AD (A) composing our cohort. PrPc, Fyn, and α-tubulin were measured by direct WB, while pY416-Fyn levels were estimated following total Fyn immunoprecipitation. (B-D) Box plots for PrPc (B), Fyn (C) and pY416-Fyn (D) protein levels in the MB fraction of NCI, MCI and AD groups. Total Fyn levels were unchanged whereas the levels of its active phosphorylated form, pY416-Fyn, were increased in the AD group compared to NCI and MCI (Kruskal-Wallis followed by Mann-Whitney’s U test and Bonferroni correction (P < 0.05)). (E,F) Linear regression analyses between PrPc/Fyn (E) and PrPc/pY416-Fyn (F) protein levels (Spearman rank correlation). PrPc and total Fyn showed a strong association regardless of clinical status (E). In contrast, a significant positive correlation was only found in the AD group linking activated Fyn and PrPc levels.
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
Soluble Aβ binds to a PrPc/Fyn complex in vivo
To examine whether PrPc interacts with Fyn in AD brain tissue, we performed co-immunoprecipitations (co-IP) using membrane-enriched protein extracts (Sherman and Lesne, 2011; Larson et al., 2012). Following pulldown of PrPc with either 8B4 (N-terminus PrP antibody) or C20 (C-terminus PrP antibody), we observed that using C20 yielded better capture efficiencies. Despite this difference, Fyn and caveolin-1 co-immunoprecipitated with PrPc indicating the presence of a PrPc/Fyn/Caveolin-1 complex in AD brain protein extracts (Fig. 2A). Our results suggest that the observation reported in cell lines by Mouillet-Richard and coworkers (2000) may also be true using human brain tissue. We then tested whether PrPc could form a putative complex with Fyn and endogenous soluble Aβ species in brains of subjects with AD or without cognitive impairment (Fig. 2B). Throughout this report, the term “soluble Aβ” refers to Aβ molecules that remain soluble in aqueous buffers after tissue homogenization and ultracentrifugation (see Methods). Following PrPc immunoprecipitation, Fyn was detected readily in AD brain extracts but not in age-matched controls (top insert, left panel). To identify potential Aβ species, nitrocellulose membranes were stripped and probed with 6E10. Under these conditions, Aβ dimers were the only oligomeric Aβ assembly detected. Neither Aβ*56 nor large Aβ protofibrils (~150 kDa) were found co-immunoprecipitating with PrPc using brain lysates from subjects with AD (data not shown). These results were reproduced across a selection of 6 cases per group (5 additional cases are shown) and importantly, without stripping the membranes (Fig. 2B, right panel). To confirm the specificity of our observations, we incubated human brain extracts containing PrPc but with no detectable Aβ as assessed by western blot (Fig. 2C, lane 1) with affinity purified Aβ oligomers (2.85 ng) from Tris-buffered saline (TBS) fractions of human AD brains (Fig. 2C, lane 2 and Fig. 2D, lane 1 for left and right panels). PrPc was then immunoprecipitated and potential Aβ binding was determined with either 40-/42-end specific Aβ antibodies or 6E10. In both cases, dimeric Aβ was the only assembly found to interact with PrPc despite the clear presence of Aβ monomers, trimers and larger assemblies (including Aβ*56 and ~150kDa protofibrils) in the exogenous mixture (Fig. 2D). Our results suggested that endogenously produced Aβ dimers are specifically binding to PrPc/Fyn in lysed AD brain extracts. While forms composed of SDS-stable Aβ dimers appear to interact with PrPc/Fyn, it is conceivable that higher molecular weight assemblies of Aβ existing in the absence of denaturing agents may also interact with this pathway.
Figure 2. PrPc immunoprecipitates specifically with human soluble Aβ dimers.
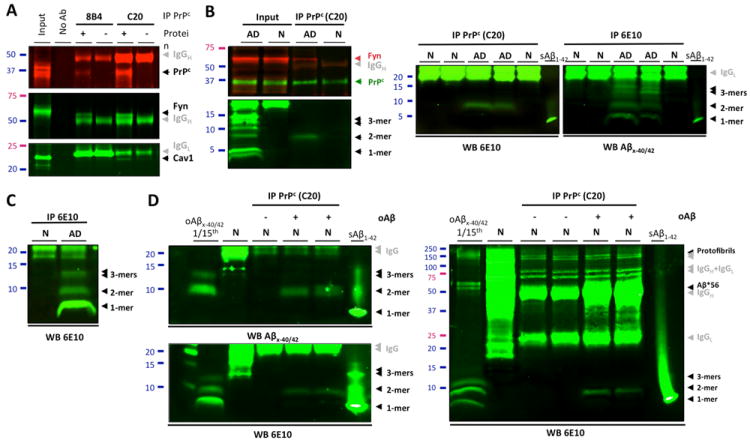
(A) Potential interaction of PrPc with Fyn in AD brain tissues was assessed by SDS-PAGE analysis of the PrPc/caveolin-1/Fyn complex following immunoprecipitation of PrPc (with 8B4 or C20 antibodies) or Fyn. Grey arrowheads indicate exogenous antibodies used for IP. (B) PrPc forms a putative complex with Fyn and soluble Aβ dimers using AD brain tissue. Western blots for Aβ were performed with 6E10. Following membrane stripping, PrPc and Fyn were revealed with PrP C20 and Fyn3 antibodies. Additional IPs with different cases are shown on the right panels to illustrate the reproducibility of the findings. (C) Western blot analysis of TBS-soluble extracts from selected brains of subjects with AD or no cognitive impairment (N). Monomers, dimers and trimers are readily detected using 6E10. (D) Immunoprecipitation of PrPc with exogenous human Aβ dimers in a cell-free assay. Affinity-purified human soluble Aβx-40/42 species (lane 1, left and right panels, an estimated total of 2.85 ng relative to Aβ1-42 standards) were added to brain protein extracts with no detectable Aβ (N; same as panel A); PrPc was immunoprecipitated using the C20 antibody and bound Aβ species were detected with 40/42-end specific antibodies (top left panel) or 6E10 (lower left and right panels). Full western blots images indicated that isolated large soluble Aβ assemblies (e.g. Aβ*56 and protofibrils) were not pulled down by C20 in our assay (right panel).
Abbreviations: IP: immunoprecipiration; No Ab: no antibody; N: non-impaired age-matched control brain; AD: Alzheimer’s disease brain; oAβ: endogenous oligomeric Aβ species purified from human AD brain tissue; sAβ1-42: 0.5 ng of human synthetic Aβ1-42 (Sigma).
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
To test whether this potential association occurs in situ, we analyzed PrPc and Aβ cellular distribution in AD brains by confocal immunofluorescence imaging (Fig. 3). While PrPc was largely predominant in the ER in non-neuronal cells, PrPc was also present at apparent specific synaptic sites and co-localized with Aβ (Fig. 3A,B), consistent with the notion that Aβ is a ligand for PrPc. At those areas (Fig. 3A, panels b-e and Fig. 3B), the respective fluorescence for PrPc and for Aβ was overlapping possibly indicating that Aβ binding to PrPc at the neuronal surface occurs in vivo. In contrast, we did not observe any apparent dendritic puncta with PrPc and Aβ colocalizing in age-matched control brain sections (Fig. 3A, panels g-h). We went on similarly to determine whether Fyn was also co-localized with PrPc and Aβ by performing a quadruple-channel image analysis (Fig. 3B). Fyn (in magenta) was preferentially observed at spines along the dendritic shaft (in blue), some of which were also positive for PrPc and Aβ. Software-based voxel analyses revealed that while ~35% of Aβ colocalized with the postsynaptic protein Fyn at dendritic spines, less than 10% of Aβ (8.86 ± 1.27) colocalized with PrPc (Fig. 3C). These numbers are compatible with the data from Lauren and colleagues (2009) indicating that only a fraction of synthetic Aβ oligomers binds to PrPc and with the knowledge that soluble Aβ molecules bind to synapses. Altogether, the colocalization of Aβ-PrPc-Fyn further supports our hypothesis that synaptic PrPc might act as a potential receptor for endogenous soluble Aβ species in AD.
Figure 3. PrPc and soluble Aβ colocalize at dendritic spines in human AD brain tissues.
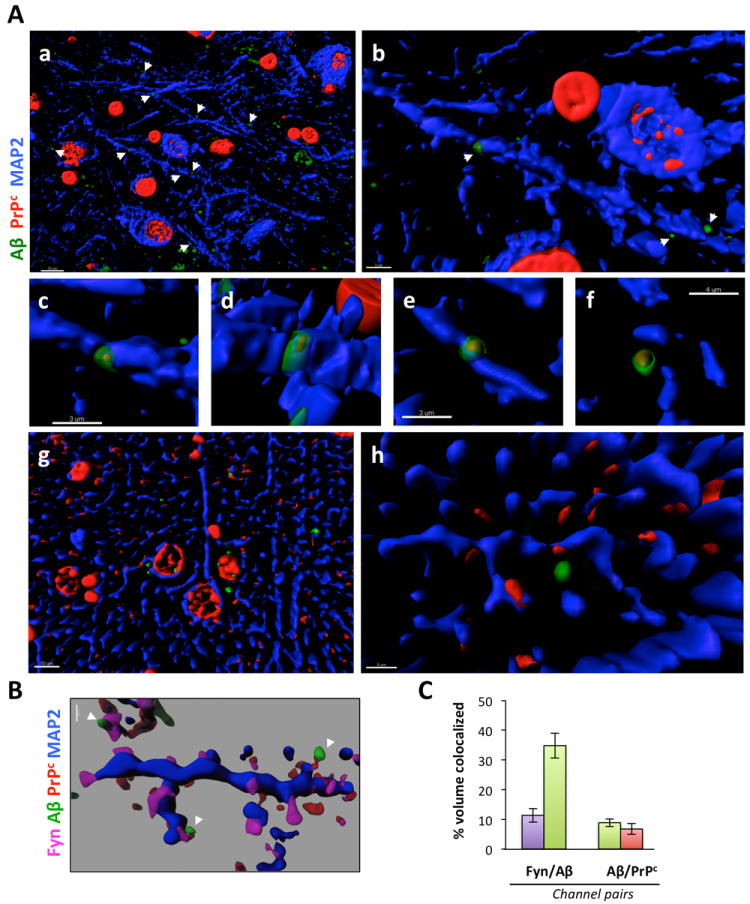
Surface rendering of triple-channel confocal immunofluorescence reveals that PrPc (in red, labeled using the C20 antibody) and Aβ (green, labeled using the DW6 antiserum) colocalize with the postsynaptic protein Fyn (magenta) along neuronal dendritic shafts (in blue, labeled using a MAP-2 antibody) in human brain tissues (AD: a-f; Age-matched NCI: g-h). Images were acquired using oil immersion 60x or 100x objectives and processed with Imaris7.0 software. Transparency of the 405, 488, 536 nm channels (magenta, green, red) was increased to 40% in order to visualize the rendered volumes of all other fluorescent probes conjointly. (A) Low- and high-power images of PrPc/Aβ puncta along dendritic shafts of inferior temporal gyrus neurons. Examples of colocalization between PrPc and Aβ at higher magnification are shown in b-f. Please note the lack of colocalization of PrPc and Aβ on dendrites of NCI brain sections (g,h). Scale bars = 10 (a, g), 3 μm (b,c,d, e), 4 μm (f) and 5 μm (h). (B) PrPc:Aβ complexes colocalize with the dendritic spine protein Fyn in vivo. Scale bar = 1 μm. (C) Software-assisted quantification of colocalized voxels (N = 3 subjects/ 6 sections/ 18 R.O.I.s). Bars represent the mean ± standard deviation.
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
Soluble Aβ oligomers bind to a PrPc/Fyn complex in vitro
To confirm these results, we opted to test whether Aβ could activate Fyn in primary cortical neurons from the widely used AD mouse model Tg2576 (Hsiao et al., 1996). The soluble Aβ species produced and secreted by Tg2576 neurons have been previously described (Lesne et al., 2006). To illustrate the oligomeric Aβ content in conditioned medium of primary cortical Tg2576 neurons, Aβ molecules were analyzed by western blot/immunoprecipitation (Fig.4A). Apparent SDS-resistant Aβ tetramers (18 kDa), trimers (13-14 kDa), dimers (8-9 kDa) and monomers (4 kDa) were readily detected, with trimeric Aβ seemingly the most abundant oligomeric Aβ species as detected with 6E10. This observed profile is similar to the one previously reported (Lesne et al., 2006) at the exception of dimers, which were then not detected likely due to the increased sensitivity in our current system (lower detection limit of ~2 pg). We first confirmed the presence of soluble Aβ and PrPc at postsynaptic sites identified with the labeling of the postsynaptic density protein 95, PSD95 (Fig. 5A). Quantification of dendritric sections (6 regions of interest, or R.O.I., per culture dish per experiment) indicated that 46.83% and 36.69% of Aβ colocalized with PSD95 and PrPc respectively in Tg2576 neurons (Fig.5B). If PrPc activates Fyn upon binding of soluble Aβ, we postulated that Aβ, PrPc and pFyn should be localized at the same postsynaptic spines. Fig.5C shows that using antibodies specific to Aβx-42, pY416-Src and PrPc, soluble Aβ42/pSrc/PrPc are actually sequestered at the same subcellular space in 14-day-old cortical Tg2576 neurons. Interestingly, those sites also included enlarged varicosities (Fig.5C, arrow heads), seen when the tubulin/tau axis is abnormally disrupted by low molecular weight Aβ oligomers (Jin et al., 2011).
Figure 4. Biochemical characterization of soluble Aβ species present in Tg2576 conditioned medium and in human brain tissues.
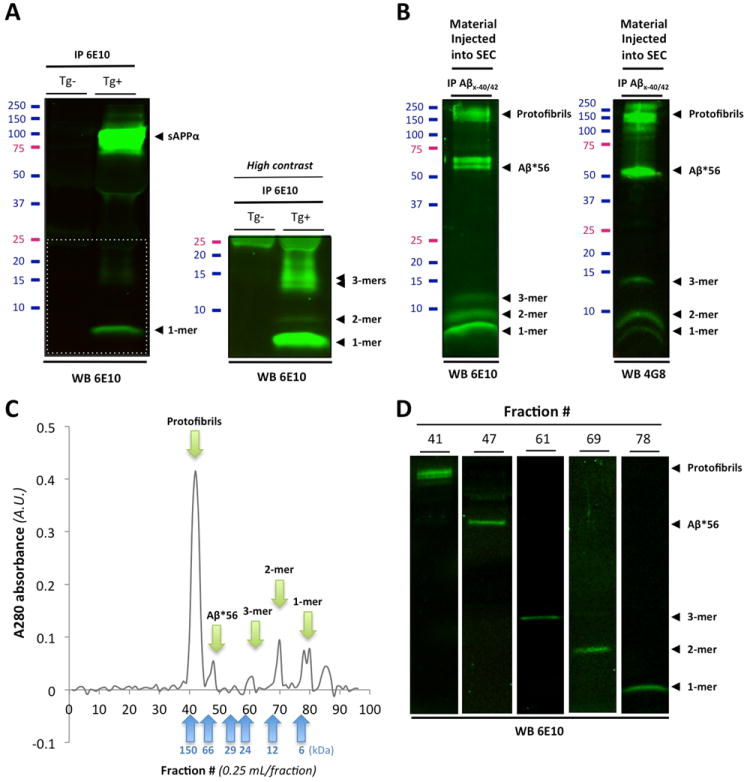
Western blot analyses of affinity-purified soluble Aβ species present in conditioned media (CM) of Tg2576 primary cortical neurons and in soluble TBS extracts human AD brain. (A) Immunoprecipitation of soluble APP/Aβ molecules present in CM of Tg2576 primary neurons (DIV12-14) using 6E10. A high-intensity laser scan is also shown to better visualize Aβ monomers, dimers and trimers (lower insert). (B) Representative profile of soluble Aβ oligomers affinity-purified with 40- and 42-end specific antibodies (Mab 2.1.3 and Mab13.1.1, kindly provided by Dr. Pritam Das) to prevent capturing APP molecules and detected with 6E10 (left panel) or 4G8 (right panel). Several soluble Aβ assemblies are readily observed: monomers, dimers, trimers, Aβ*56, a larger 60kDa oligomer and protofibrils > 150kDa. (C) Typical size-exclusion chromatogram of the endogenous human Aβ oligomers shown in (B) using a Tricorn Superdex™ 75 column. Five peaks (green arrows) corresponding to the 5 Aβ species seen by western blot are clearly observed with elutions at predicted molecular weights (blue arrows correspond to the 6 molecular weight standards used). (D) Representative western blot images of the respective SEC fractions isolated in (C) using 6E10.
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
Figure 5. Specific PrPc antibodies prevent soluble Aβ-induced Fyn activation in primary neurons.
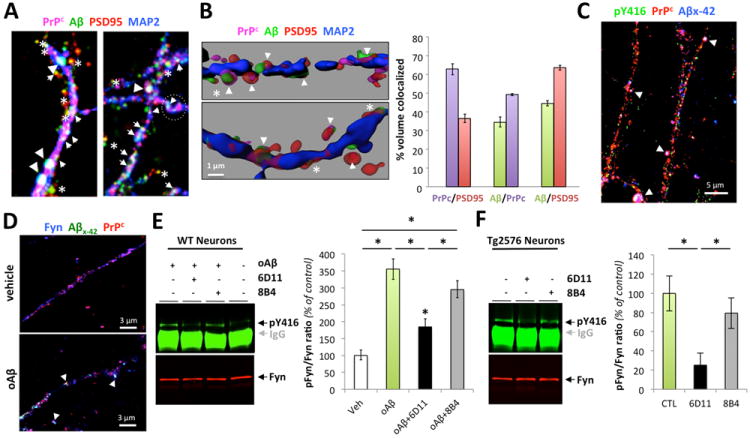
(A) Quadruple channel confocal immunofluorescence reveals that PrPc (labeled using the C20 antibody) and Aβ (labeled using the DW6 antiserum, a kind gift from Dr. Walsh) colocalize on neuronal dendritic spines (labeled using a PSD95 antibody) in 14 day-old primary cortical Tg2576 neurons. (B) Quantification of colocalized voxels for each channel pair of interest. Representative 3D-rendered images used for voxel counts are shown in the left panel. Results for channel pairs are shown in the histogram. (C) Triple channel confocal imaging shows that Aβ42 species partly co-localize with pY416-Src and PrPc in Tg2576 primary neurons. (D) Exogenous Aβ species purified from AD brain tissue colocalized with PrPc and Fyn at dendritic spines after application onto non-transgenic primary cortical neurons. (E-F) Fyn activation induced by human oligomeric Aβ is partially blocked by the antibody 6D11 targeting the 95-105 domain of PrPc. Preincubation with the N-terminus PrPc antibody 8B4 failed to inhibit Fyn phosphorylation. 6D11 (10 μg) and 8B4 (10 μg) were pre-incubated for 2 hours onto cells prior to acute oAβ exposure (1 hour at 37°C). 6D11 reduced Fyn activation in wild-type neurons acutely exposed to human-brain purified Aβ oligomers (E) and in Tg2576 neurons chronically exposed to low-n Aβ oligomers (F). Images were acquired using an oil immersion 60x or 100x objective and processed with Imaris7.1 software.
Arrowheads indicate examples of colocalization between Aβ/PrPc while white asterisks correspond to colocalization between Aβ and PSD95 in absence of PrPc. Bars represent the mean ± standard deviation (n > 3-6 dishes / experiment; independent experiments were performed in triplicate). *, P < 0.05, ANOVA followed by student t test.
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
Since the oligomeric Aβ molecules secreted by Tg2576 neurons may differ from human brain-derived Aβ oligomers, we cultured primary cortical neurons from non-transgenic animals under the same conditions used for Tg2576 neurons and exposed them briefly (1 hour) to nanomolar concentrations (5-6 nM) of affinity-purified oligomeric Aβ species from TBS AD brains extracts (Fig.4B). These purified assemblies were characterized using multiple Aβ antibodies (6E10, 4G8 and 40-/42-end specific Mab2.1.3 and Mab13.1.1, kind gifts from Dr. Pritam Das) as well as by size-exclusion chromatography (Fig.4C). Recovered fractions were then reanalyzed by western blotting to ensure that the absorbance peaks observed indeed contained the respective human Aβ oligomers (Fig. 4D). Both methods confirmed the presence of five predominant species including monomers, dimers, trimers, Aβ*56 and large (~150 kDa) soluble protofibrils (PF). Cells were then fixed and analyzed with confocal microscopy and biochemistry techniques. Using a 42-end specific antibody (Mab2.1.3) to avoid cross-reactivity with APP, we found that soluble Aβ species colocalized with PrPc/Fyn at synaptic spines (Fig.5D), similarly to our previous observations in Tg2576 neurons. To demonstrate that Fyn activation induced by affinity-purified human oligomeric Aβ molecules (oAβ) depends on PrPc/Aβ interaction, we used the differential ability of two anti-PrPc antibodies to prevent Aβ binding to PrPc (Lauren et al., 2009). We therefore pre-incubated non-transgenic cortical neurons with either 6D11 (detecting the region 93-109 of PrPc) or 8B4 (N-terminus PrPc antibody) for 2 hours at 37°C, then applied oAβ for 1 hour at 37°C. Following this acute exposure with human brain-derived Aβ oligomers, Fyn was immunoprecipitated and pFyn levels were determined (Fig.5E). Upon quantification, pY416-Fyn was elevated in neurons treated with human oAβ (a ~300% increase compared to controls). Pretreatment with 6D11, an antibody preventing Aβ from binding to the 95-105 domain of PrPc, blocked Aβ-induced Fyn activation, whereas pretreatment with 8B4 had no effect. Importantly either pretreatment did not modulate Fyn phosphorylation on its own (data not shown). Finally, we examined whether 6D11 could lower pY416-Fyn levels in a more chronic model of Aβ exposure using Tg2576 neurons (Fig.5F). Similarly to the acute exposure paradigm, a 2-hour application of 6D11 but not 8B4 was sufficient to reduce Aβ-induced phosphorylation of Fyn to ~30% (28.58 ± 7.14) of control cells.
PrPc-dependent activation of Fyn leads to tau hyperphosphorylation in vitro
Several reports suggested that Aβ, tau, and Fyn cooperate in AD-related pathogenesis (Lambert et al., 1998; Lee et al., 1998; Chin et al., 2004; Chin et al., 2005; Lee, 2005; Roberson et al., 2007). In follow-up studies, the genetic ablation of tau prevented cognitive decline induced by synergistic effects of Aβ and Fyn, indicating that Aβ, Fyn and tau jointly impair synaptic function (Roberson et al., 2011). Since affinity-purified human oAβ species containing Aβ dimers induced Fyn activation in a PrPc-dependent manner, we examined possible changes in tau phosphorylation at Y18, a Fyn-specific site (Lee, 2005), in primary cortical neurons (Fig. 6). Because the dendritic role of tau regulates the postsynaptic targeting of Fyn conferring Aβ toxicity (Ittner et al., 2010) and because hyperphosphorylated tau abnormally accumulates in dendritic spines (Hoover et al., 2010), we also determined tau missorting to the postsynaptic density by determining its biochemical segregation in vitro. Two postsynaptic proteins, the glutamatergic N-Methyl D-Aspartate receptor subunit 2B, GluN2B, and the scaffolding protein PSD95 were used as internal controls to ensure proper biochemical separation (Fig. 6A). In presence of oAβ (5 nM for 1 hour), phosphorylation at Y18 sharply increased. In addition, tau compartmentalization was altered compared to controls (Fig. 6A,B). In addition to changes its phosphorylation status, neuronal tau levels also showed a remarkable alteration of its cellular distribution (Fig.6C), consistent with earlier observations (Ittner et al., 2010; Zempel et al., 2010). Both changes were dependent on oAβ-PrPc interaction as 6D11 pre-treatment partially reduced pY18-tau and total tau levels in both cytosolic and PSD-containing protein fractions. As shown for Fyn activation, pre-treatment with 8B4 did not reverse tau alterations induced by oligomeric Aβ.
Figure 6. PrPc-dependent phosphorylation of tau induced by endogenous Aβ oligomers in primary neurons.

(A) Protein levels of hyperphosphorylated tau at Y18 (pY18-Tau) and total tau as assessed by western blot using PY18 and Tau5 antibodies in wild-type primary neurons exposed to human-brain purified oligomeric Aβ (5 nM for 1 hour). The glutamatergic NMDA receptor subunit 2B (GluN2B) and the scaffolding protein PSD95 were used as internal control. (B, C) Quantitation of pY18-Tau (B) and total tau (C) across conditions and protein fractions (IC and MB) revealed an significant increase in tau hyperphosphorylation and mislocalization following oAβ exposure. This effect was partially inhibited by 6D11 but not 8B4 (Bars represent the mean ± standard deviation; *, P < 0.05, ANOVA followed by student t test).
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
To determine which oligomeric Aβ species isolated from AD brain tissue was responsible for altering the Fyn/tau axis, isolated Aβ species (monomers, dimers, trimers, Aβ*56 and PFs; Fig. 4C,D) were then applied to primary neurons for 1 hour at equivalent concentration (5nM) calculated based on synthetic Aβ1-42 standards (not shown). While monomeric Aβ, Aβ*56 and PF preparations did not appear to induce Fyn activation under these conditions, phosphorylated Fyn levels were markedly increased in neurons treated with either dimers or trimers (Fig.7A,B). When we examined the phosphorylation status of two downstream targets of Fyn, pY18-Tau and pY1472-GluN2B, we observed increases in tau phosphorylation in dimer- and trimer-treated neurons but no change in total GluN2B levels in membrane extracts and in Y1472 status (Fig. 7B). Consistent with our co-immunoprecipitation results identifying Aβ dimers as an endogenous ligand to a PrPc:Fyn complex, these findings indicate that purified Aβ oligomers from AD brain tissue (identified as Aβ dimers under denaturing conditions) alterthe Fyn/tau axis and suggest that the signaling pathway induced by Aβ trimers converges onto Fyn likely through a PrPc-independent mechanism.
Figure 7. Human AD brain-purified Aβ dimers and trimers activate Fyn in primary cortical neurons.
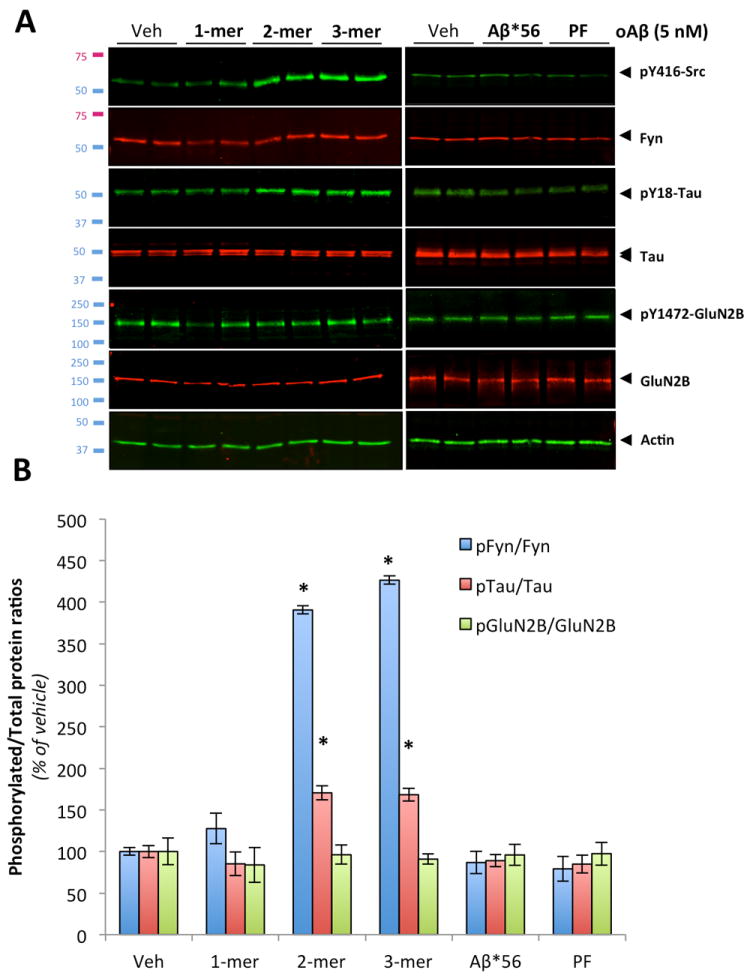
(A) Application of purified soluble Aβ dimers and trimers selectively phosphorylate Fyn and tau as assessed by quantitative western blotting (duplicates are shown; n = 4 per experiment). Sixty-minute long treatments were performed at the calculated concentration of 5 nM for all species (estimated based on monomeric synthetic Aβ levels). Total protein levels of GluN2B and phosphorylation of GluN2B at Y1472 were also examined. Actin levels are shown as control. (B) Quantification of the ratios for pY416-Src/Fyn (labeled as pFyn/Fyn), pY18-Tau/total tau (pTau/Tau) and pY1472-GluN2B/GluN2B (pGluN2B/GluN2B) after one hour-long exposure with the human oligomeric Aβ species listed. Both brain-purified Aβ dimers and trimers activate Fyn in vitro. Isolated monomeric Aβ, Aβ*56 and soluble Aβ protofibrils (PF) did not induce Fyn phosphorylation under similar conditions. After 60 minutes, neither membrane-bound levels of GluN2B nor Y1472-GluN2B phosphorylation were altered by AD brain-derived Aβ species. (Bars represent the mean ± standard deviation; *, P < 0.05, ANOVA followed by student’s t test with Bonferroni correction).
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
Prnp gene deletion uncouples oAβ and the Fyn/tau axis in vivo
To demonstrate that PrPc is mediating the deleterious effects induced by Aβ dimers, we used a genetic approach to examine the potential effects of Prnp gene dosage on Fyn/tau phosphorylation in vivo. Based on our previous findings, we hypothesized that Prnp gene ablation would attenuate oAβ-induced Fyn activation and tau hyperphosphorylation. In contrast we anticipated that Prnp overexpression would potentiate Fyn and tau phosphorylation in APPPS1+ mice.
We crossed APPPS1 transgenic mice (Radde et al., 2006) with PrPc deficient mice (Prnp-/-) or with PrPc overexpressing mice (tga20tg/-Prnp-/- thereafter called tga20+). All lines used here were previously described (Bueler et al., 1992; Fischer et al., 1996; Calella et al., 2010). However, the respective levels of low molecular weight Aβ oligomers in these lines were not documented. Since these lines were previously used at 2-4 month of age (Calella et al., 2010), we first measured oligomeric Aβ levels in young and old mice (respectively 2 and 14 months of age) when amyloid deposition is either quite modest or very abundant (Radde et al., 2006). Fig.8A illustrates the respective levels of low-n Aβ oligomers across multiple animals per genotype. Importantly, we found no overall differences in Aβ levels resulting from the reduction of the Prnp copy number at each age studied ((Calella et al., 2010) and data not shown). Aβ monomers and trimers were faintly present in 2-month-old APPPS1+xPrnp+/+, APPPS1+xPrnp+/-, APPPS1+xPrnp-/- mice (Fig.8A, lanes 1-6). In contrast, Aβ dimers were readily visible in aged APPPS1+xPrnp+/- or APPPS1+xPrnp-/- as well as abundant monomers and trimers (Fig.8A, lanes 7-12). Following the determination of the relative amounts of soluble Aβ oligomers in each animal, we examined the protein expression of Fyn and its activated phosphorylated form. Total Fyn levels were unchanged across genotypes at 2 and 14 months of age. As previously shown for other APP transgenic mouse models, pFyn levels were markedly increased in old APPPS1+xPrnp+/- mice. Phosphorylated Fyn levels were ~10-fold lower in young APPPS1+ mice compared to aged APPPS1+xPrnp+/- animals (Figure 7, lanes 1-6 vs. 7-9). When both copies of Prnp were ablated in 14 month-old APPPS1+xPrnp-/-, pY416-Fyn levels dropped to 19.25 ± 8.01 % compared to APPPS1 mice heterozygous for Prnp (Fig.8B). Since the inhibition of Fyn phosphorylation was not restored to baseline levels (8-9% compared to 14-month-old APPPS1+xPrnp+/-), this result indicated that PrPc was required, in part, to mediate Fyn activation induced by the various oligomeric forms of Aβ present in aged APPPS1 mice.
Figure 8. Prnp gene ablation attenuates Aβ-induced activation of Fyn and tau hyperphosphorylation in vivo.
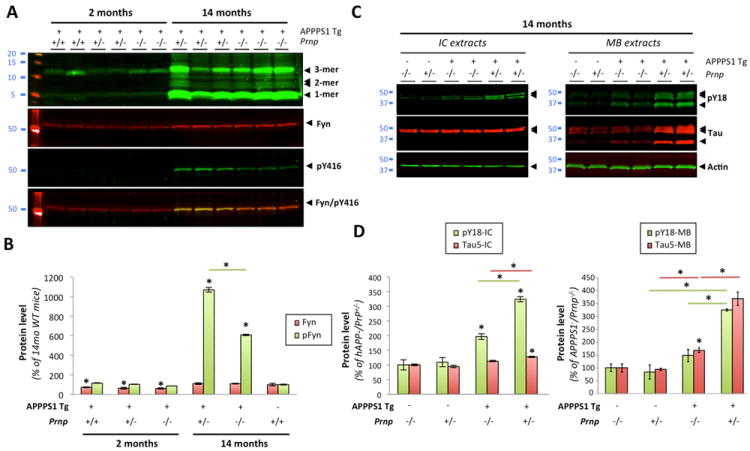
Gene ablation of Prnp modulates Fyn activation and tau phosphorylation at pY18 in brains of aged transgenic APPPS1+xPrnp animals. (A) Protein levels of low-molecular weight Aβ oligomers, total Fyn and pY416-Src were assessed by quantitative western blot (WB). 6E10 was used for Aβ. A marked reduced of pY416 was revealed in APPPS1+xPrnp-/- compared to APPPS1+xPrnp+/- mice. (B) Relative protein levels for total Fyn and pY416-Fyn determined by densitometry analyses. (C, D) Prnp gene deletion limits tau hyperphorphorylation and missorting in vivo. PY18-tau and total tau levels were measured by WB using either intracellular-enriched or membrane-enriched protein extracts (C) and quantified by software analysis (D). APPPS1+xPrnp-/- mice showed a >2-fold reduction in tau phosphorylation at Y18 compared to APPPS1+xPrnp+/- littermates. (Bars represent the mean ± standard deviation; *, P < 0.05, ANOVA followed by student’s t test, n ≥ 3 animals/genotype/age/experiment).
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
To determine whether the reduction of Fyn activation observed in aged APPPS1+xPrnp-/- mice had any effect on tau phosphorylation and tau missorting, we assessed total tau and pY18-Tau levels in intracellular- (IC) and membrane- (MB) enriched fractions of aged APPPS1+xPrnp mice (Fig. 8C,D). Under physiological conditions (i.e. when the APPPS1 transgene is not expressed), soluble tau is nearly exclusively found in the IC fraction and tau is not phosphorylated at Y18 (Fig. 8C, lanes 1-2 of right and left panels). In the presence of soluble Aβ/hAPP (Fig. 8A), hyperphosphorylated tau abnormally accumulated within MB extracts (Fig.8C, lanes 5-6) similarly to our in vitro findings (Fig. 7). When both copies of Prnp were ablated (Fig. 8C, lanes 3-4), both hyperphosphorylation at Y18 and tau mislocalization were reduced by 2.18- and 2.19-fold compared to APPPS1+xPrnp+/- littermates (Fig.8C,D).
These data thus indicate that lowering Prnp gene expression reduces the activation of Fyn and hyperphosphorylation of tau at Y18 induced by Aβ/APP molecules in aged APPPS1+ mice.
PrPc-overexpression potentiates oAβ-induced Fyn/tau toxicity in vivo
To provide further evidence that PrPc is involved in mediating the effects triggered by dimeric Aβ in vivo, we crossed PrPc overexpressing mice (thereafter called tga20+) with APPPS1+ mice. Calella and coworkers (2010) previously described these mice. Analogously to the absence of alterations in Aβ levels across APPPS1+xPrnp genotypes, we did not observe overt changes in low-molecular Aβ levels by quantitative western blotting in APPPS1+xtga20+ mice compared to age-matched APPPS1+xtga20-animals (Fig.9A and data not shown).
Figure 9. Aβ-induced activation of Fyn and tau hyperphosphorylation is enhanced in aged APPS1 mice overexpressing PrP.
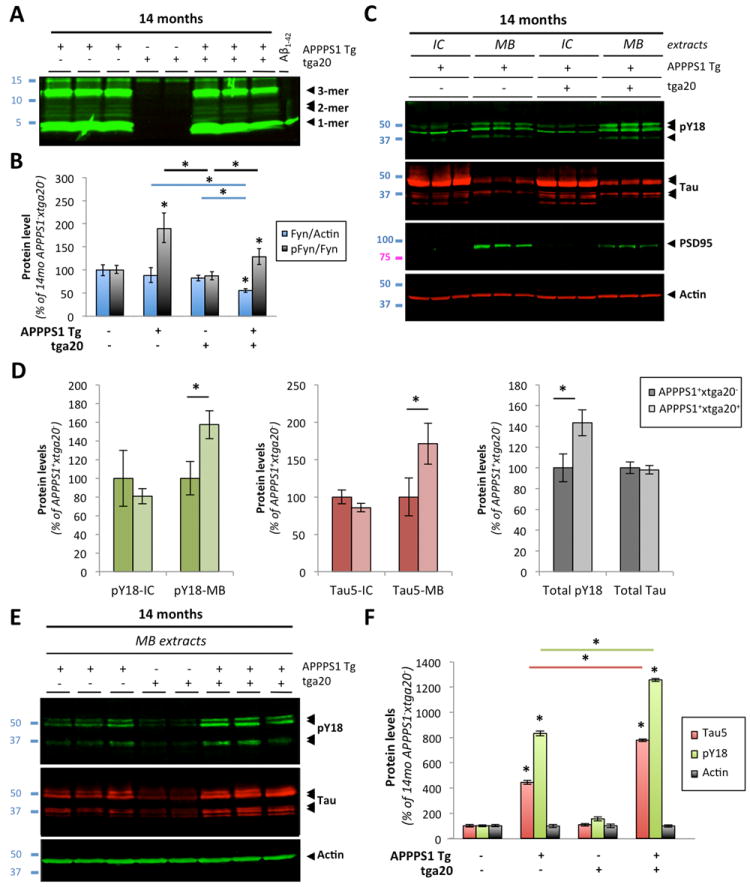
(A) Soluble brain levels of low-molecular weight Aβ oligomers in 14-month-old APPPS1xtga20 mice was assessed by quantitative western blot (WB) using 6E10. (B) Quantification of total Fyn, pY416-Src and actin protein levels in 14-month-old mice (APPPS1-xtga20-, APPPS1+xtga20-, APPPS1-xtga20+ and APPPS1+xtga20+) following WB. A marked elevation of pY416-Fyn was observed in APPPS1+xtga20+ compared to APPPS1-xtga20+ mice. (C) Tau missorting was illustrated by biochemical segregation of pY18-Tau, total tau, PSD95 in IC vs. MB extracts using western blotting. (D) Quantification of PY18-tau and total tau immunoreactivity across protein fractions and genotypes. Aged APPPS1+xtga20+ mice showed a ~1.8-fold accumulation of tau and a ~1.6-fold increase in phosphorylation at Y18 compared to age-matched APPPS1+xtga20- mice. (E) Enhanced accumulation of full-length tau and cleavage products phosphorylated at Y18 in PSD95-containing extracts of old APPPS1+xtga20+ mice. Actin was used as internal standard. (F) Quantification of tau species immunoreactive for Tau5 and PY18 in 14-month-old APPPS1-xtga20-, APPPS1+xtga20-, APPPS1-xtga20+ and APPPS1+xtga20+ mice. (Bars represent the mean ± standard deviation; *, P < 0.05, ANOVA followed by student t test, n ≥ 3 animals/genotype/age/experiment).
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
We then measured total Fyn, pY416-Fyn and actin brain levels by quantitative western blotting (data not shown). Following image analyses, the pFyn/Fyn ratio was elevated in old APPPS1+xtga20+ mice compared to APPPS1-xtga20+ (Fig.9B). There was no statistical difference between APPPS1+xtga20- and APPPS1+xtga20+ animals at 14 months of age. Importantly, an abnormal elevation of the pFyn/Fyn ratio in aged APPPS1+xtga20+ mice was observed despite a ~33% reduction of total Fyn in these mice (Fig.9B).
Since exogenous application of AD brain-purified Aβ oligomers induced tau missorting and hyperphosphorylation at Y18 in primary cortical neurons, we determined whether similar tau changes were found in vivo and whether PrPc overexpression led to a potentiation of these pathological alterationsal ready present in APP transgenic mice (Ittner et al., 2010). As described previously (Lesne et al., 2006; Larson et al., 2012) and earlier in this manuscript (Fig. 7), MB extracts selectively contain postsynaptic density proteins as illustrated in Fig. 9C by the presence of PSD95 in MB but not in IC fractions. Comparing the biochemical distribution of pY18-tau and total tau (reflected by Tau5 immunoreactivity) in IC/MB brain lysates from APPPS1+xtga20-or APPPS1+xtga20+ mice (Fig. 9C), it was readily visible that pY18-tau levels were elevated in MB extracts of APPPS1+xtga20+ mice while intracellular pY18 levels remained equivalent (Fig. 9D). Soluble total tau levels across IC and MB fractions also displayed a similar profile (Fig. 9C,D). Of interest, despite possible changes in the respective amounts of full-length and cleaved tau products, the overall levels of total tau were comparable between APPPS1 transgenic mice (Fig. 9D, right panel).
When focusing on tau molecules present in the PSD95 containing protein fraction, we confirmed a potentiation of phosphorylation at Y18 for tau species of ~50-52 kDa and ~35 kDa in APPPS1+xtga20+ compared to APPPS1+xtga20- mice (Fig. 9E,F). In contrast, very little tau immunoreactivity was detected in APPPS1-xtga20+ littermates (Fig. 9E, lanes 4-5). Total actin levels remained unchanged across all genotypes tested (Fig.9E,F).
Intrigued by the apparent reduced expression of total Fyn in aged APPPS1+xtga20+ mice, we postulated that the reduction of this postsynaptic protein might simply reflect a potentiation of the synaptotoxic effects of Aβ dimers previously reported (Shankar et al., 2008; Jin et al., 2011). To test this hypothesis, we measured two additional well-known pre- and post-synaptic proteins, synaptophysin and PSD95 alongside Fyn in our 14-month-old groups (Fig.10A). Quantification of the data revealed a selective decrease in postsynaptic proteins in APPPS1+xtga20+ mice compared to APPPS1+xtga20- and APPPS1-xtga20+animals (Fig.10B), suggesting that PrPc overexpression in aged APPPS1 mice may potentiate Aβ dimer-induced synaptotoxicity.
Figure 10. PrPc overexpression in aged APPPS1 mice triggers a selective decrease in postsynaptic proteins.
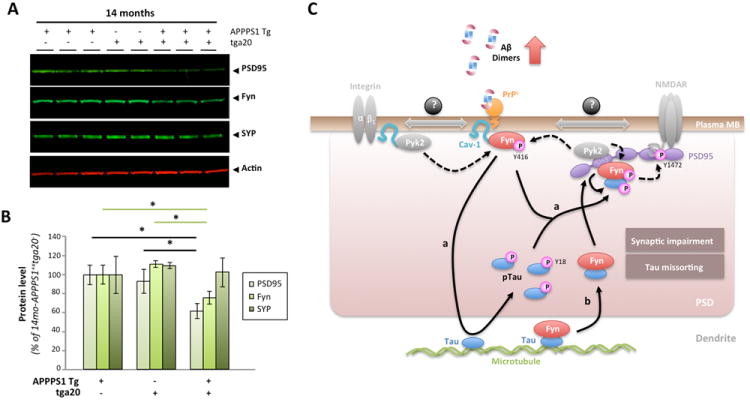
(A) Apparent selective decrease in postsynaptic proteins PSD95 and Fyn in 14-month-old APPPS1+xtga20+ mice. Representative WBs for the postsynaptic proteins PSD95 and Fyn as well as for the presynaptic vesicle protein synaptophysin (SYP) are shown. Actin was used as internal standard. (B) Quantification of PSD95, Fyn and Synaptophysin in 14-month-old APPPS1+xtga20-, APPPS1-xtga20+ and APPPS1+xtga20+ mice revealed significant reductions in postsynaptic proteins in APPPS1 mice overexpressing PrPc. (Bars represent the mean ± standard deviation; *, P < 0.05, ANOVA followed by student t test, n ≥ 3 animals/genotype/age/experiment). (C) Proposed model of tau regulation by the triad oAβ-PrPc-Fyn. In the presence of accumulating Aβ dimers, PrPc, Fyn and Cav-1 form a complex at the plasma membrane (likely with receptors such GluN or integrins known to interact with Cav-1 and Fyn). Upon phosphorylation of Fyn at Y416 (possibly by the kinase Pyk2), this complex becomes biologically active. Two scenarios are possible depending on the status of Fyn with respect to tau: (a) Fyn is not bound to tau and (b) Fyn is bound to tau. In (a), activated Fyn causes the hyperphosphorylation of tau at Y18 and its aberrant accumulation at the postsynaptic density (PSD). It is possible that pFyn might be bound to pTau at this stage and binds the SH3 domain of PSD95, where it is ideally located to modulate GluN/NMDA receptor subunits (i.e. GluN2B at Y1472). In the model (b), Fyn is already bound to tau in the dendrite, translocate to the PSD to interact with PSD95. There, Pyk2 could phosphorylate Fyn at Y416 resulting in Fyn activation and tau phosphorylation at Y18.
Italicized numbers between parentheses indicate group sizes. NCI is shown in green, MCI in yellow/orange and AD in red. In box plots, the bar inside the box indicates the median; the upper and lower limits of boxes represent the 75th and 25th percentiles, respectively, and bars flanking the box represent 95th and 5th percentiles. Asterisks (*) indicate P< 0.05 while (**) indicates P< 0.01.
Abbreviations: NCI: no cognitive impairment, MCI: mild cognitive impairment, AD: Alzheimer’s disease, EC: extracellular-enriched fraction, IC: intracellular-enriched fraction, MB: membrane-associated fraction, Tg: transgenic, AD, Alzheimer’s disease, DLU: Densitometry Light Units, A.U.: arbitrary units.
Altogether our genetic data provide further evidence for a cellular signaling pathway linking soluble Aβ, PrPc, Fyn and tau in AD pathogenesis.
DISCUSSION
The Src kinase Fyn has been proposed to mediate oligomeric Aβ neurotoxicity since Klein and coworkers first reported that synthetic globular Aβ species (or ADDLs) required the expression of Fyn to be deleterious to cells (Lambert et al., 1998). Following this initial report, several in vivo lines of evidence demonstrated that Aβ, Fyn and tau might constitute a deleterious triad in AD physiopathology (Chin et al., 2004; Chin et al., 2005; Ittner et al., 2010; Zempel et al., 2010; Roberson et al., 2011). With the recent discoveries that the cellular form of PrP can act as a surface binding partner for large synthetic Aβ oligomers (Lauren et al., 2009), that PrPc is required for memory impairment in APP transgenic mice (Gimbel et al., 2010) and that PrPc can form a complex with Fyn (Mouillet-Richard et al., 2000; Um et al., 2012), these accumulating observations point out Fyn as a crucial mediator of synaptotoxicity triggered by soluble Aβ molecules.
Controversy between PrPc and Aβ oligomers
The controversy surrounding the potential interaction between PrPc and Aβ molecules (2011) may result from the source, state of aggregation and manipulation of Aβ oligomers. Dr. Strittmatter’s group previously established the potential functional relevance of PrPc in Alzheimer’s disease (Gimbel et al., 2010) using APPPS1xPrPnull mice (APPswe/PS1ΔE9 line). Gimbel and coworkers showed that synaptotoxicity and behavior was rescued by Prnp deletion, thereby demonstrating the functional relevance of PrPc in mediating Aβ/APP-induced deficits. This particular point was recently further validated by using electroencephalogram recordings (Um et al., 2012). The crucial questions that needed to be addressed by the field were to determine: (i) what endogenous soluble form of Aβ is binding to PrPc?, (ii) what signaling pathway is activated upon complex formation (if complex there is)? (iii) is PrPc coupling oAβ and tau pathology? and (iv) why did some groups observed an mediation of oAβ toxicity by PrPc and why others did not?.
In this study, we deliberately chose to use Aβ oligomers endogenously produced or secreted in human brain tissue or in primary neurons. Since we do not possess oligomer-specific tools to visualize each putative soluble Aβ form in situ at the present time, we define soluble endogenous Aβ oligomers as Aβ molecules that (i) remain soluble in aqueous buffers following ultracentrifugation, (ii) are SDS-resistant following tissue lysis, (iii) are affinity-purified, (iv) are separated in liquid phase chromatography and (v) are immunoreactive to at least 2 different Aβ antibodies. For these reasons, the terms “Aβ dimer” and “Aβ trimer” are used here to describe species that migrate as apparent dimer/trimer on SDS-PAGE even though it is conceivable that these species may not necessarily exist or function as dimers/trimers in vivo (O’Nuallain et al., 2010). All preparations used in our experimental paradigms are biochemically characterized and documented (Fig.4). Under these conditions, we report that Fyn expression and Fyn activation are correlated with PrPc levels in AD brain. The elevation of PrPc observed in our AD cohort is further supported by recent studies indicating that PrPc accumulates in dystrophic neurites in AD (Takahashi et al., 2011) and that PrPc expression at the cell surface is increased by synthetic Aβ oligomers (Caetano et al., 2011). We also show that endogenous soluble Aβ species, specifically dimers, interact with PrPc and Fyn in vivo and ex vivo combining biochemical approaches and confocal imaging. These findings were supported by colocalization studies performed in Tg2576 primary cortical neurons used as a model of chronic exposure with endogenously produced Aβ oligomers. In parallel, we applied affinity purified Aβ oligomers from Tris buffered saline (TBS) extracts of AD brain tissue to non-transgenic primary cortical neurons as a model of acute exposure. Again, under these parameters, exogenous human Aβ oligomers colocalized with PrPc and Fyn in certain dendritic spines. Moreover, we demonstrated that pretreatment or treatment with 6D11 prevented oAβ-induced activation of Fyn. Finally, we showed that Prnp gene dosage regulates Aβ-induced phosphorylation of Fyn and tau in vivo at ages when dimers are present. Altogether, these results support the earlier report that PrPc acts as a receptor for oligomeric Aβ and links oAβ to disease-relevant tau changes. In contrast to previous studies though (Lauren et al., 2009; Balducci et al., 2010; Barry et al., 2011; Freir et al., 2011; Um et al., 2012), we directly show that AD brain-purified Aβ dimers are specifically binding to PrPc, activate Fyn which in turn triggers tau aberrant missorting and hyperphosphorylation. Our results are in contrast with recent findings indicating that insoluble monomeric Aβ is interacting with insoluble PrPc conformers, called iPrP, in the detergent-insoluble protein fraction of AD brain tissue (Zou et al., 2011). Using five antibodies raised against PrPc (8B4, C20, 6D11, M20 and 7D11) and four antibodies against Aβ (6E10, 4G8 and 40-/42-end specific Mab2.1.3 and Mab13.1.1), we did not detect Aβ monomers coimmunoprecipitating with PrPc (data not shown and Fig.2). Given the aggregation propensities of Aβ and PrP, it is conceivable that detergent-insoluble fibrillar forms of both proteins might interact with each other. This notion is supported by previous reports indicating that PrPc accumulates in amyloid deposits (Ferrer et al., 2001) and Aβ co-depositing with PrPSc aggregates in Creutzfeldt-Jakob disease (Debatin et al., 2008).
In human studies, apparent endogenous Aβ dimers were only found in brains of subjects with AD (Shankar et al., 2008; Mc Donald et al., 2010) when comparing individuals with extensive amyloid and tau pathology and subjects with no detectable amyloid accumulation. In animal models of AD such as Tg2576 (Kawarabayashi et al., 2004) and J20 (Meilandt et al., 2009; Shankar et al., 2009), Aβ dimers can be detected at 10-12 and 10-14 months of age, respectively. Interestingly, these ages correspond to amyloid plaques depositing in cortical areas (Mucke et al., 2000; Kawarabayashi et al., 2001). Altogether, these findings suggest that Aβ dimers are closely associated with fibrillar Aβ. Moreover, Aβ dimers can be extracted from insoluble Aβ deposits (Shankar et al., 2008; Mc Donald et al., 2010), further suggesting that amyloid burden and brain levels of Aβ dimers might be related. Our findings and the point mentioned above could explain the lack of PrP-dependence in Aβ-induced LTP impairment seen in young,2 to 4-month-old APPPS1 mice (Calella et al., 2010) and 4 to 7-month-old J20 mice (Cisse et al., 2011a), which only display fair amyloid deposition at the age tested (Mucke et al., 2000; Radde et al., 2006). Our own studies in 2-month-old APPPS1 mice seem to confirm this notion as we demonstrated that Aβ dimers were beyond the sensitivity of our detection assay currently limited to less than 5 pg of human Aβ (Fig. 8A), that Fyn was not activated at that age therefore suggesting that the PrPc:Fyn complex is not engaged. Based onour findings, we also speculate that ≥14-month-old APPPS1+xPrnp-/- mice would display a significant LTP rescue compared to age-matched APPPS1+ mice. In agreement with our results, both synaptotoxicity and cognitive deficits were rescued in aged 12-month-old APP transgenic mice with abundant deposited amyloid (APPswe/PS1ΔE9 mice) lacking PrPc compared to APPswe/PS1ΔE9 littermates (Gimbel et al., 2010). We therefore predict that the spatial reference memory deficit characterizing J20 mice would also be rescued (in part) by the ablation of the Prnp gene in old J20 animals, when Aβ dimers are readily detected (10-14 months of age). By extension, the mediation of endogenous oligomeric Aβ-induced LTP/cognitive dysfunction by PrPc would greatly depend on age and plaque-associated dimeric Aβ levels. Finally, while SDS-stable Aβ dimers or forms composed of Aβ dimers appear to interact with PrPc/Fyn, it is also possible that higher molecular weight assemblies of Aβ existing in the absence of denaturing agents may also interact with this pathway.
PrPc:Fyn links Aβ oligomers and tau
Several other receptors for Aβ oligomers have been proposed including mGluR5 (Renner et al., 2010), NMDA receptors (Decker et al., 2010) and EphB2 (Cisse et al., 2011b). However, none of these studies showed that specific, soluble, endogenously produced Aβ assemblies interacted with these receptors by combining co-immunoprecipications and imaging techniques using AD or APP transgenic brain tissue. Importantly, we used medically relevant specimens to purify soluble Aβ oligomers in order to apply nanomolar concentrations of these proteins for in vitro and ex vivo paradigms. Of notable interest, picomolar concentrations of the same Aβ assemblies did not trigger Fyn activation (data not shown).
In addition, the identification of PrPc as a molecular link between Aβ oligomers and Fyn may reveal new potential strategies to slow down the progression of AD. Contrary to Fyn, PrPc is a membrane-anchored protein, thus relatively accessible. Several PrP antibodies binding to the 95-105 domain of the protein have been shown to block Aβ binding (Lauren et al., 2009; Barry et al., 2011; Freir et al., 2011). Among them, the antibody 6D11 was recently used as a novel treatment for cognitive deficits in APP transgenic mice (Chung et al., 2010). We further extended these findings by reporting here that 6D11 also inhibits Fyn activation induced by acute and chronic exposure to human Aβ oligomers in primary neurons. Even though the activation status of Fyn in APPPS1 mice used by Chung and coworkers is unknown, we would expect pY416-Fyn to be downregulated in 6D11-treated animals based on our findings.
Lastly, we demonstrated that PrPc-dependent activation of Fyn induced by Aβ dimers leads to tau missorting and tau hyperphosphorylation at tyrosine 18 both in vitro and in vivo. PY18-tau is found in neurofibrillary tangles in AD brain (Lee, 2005) and tangle-forming tau transgenic brains (Bhaskar et al., 2005; Bhaskar et al., 2010). Until now however, a direct link between this hyperphosphorylated tau species and oligomeric Aβ was only speculative or fragmented. Despite the demonstration that tau and Fyn are mediating Aβ-induced toxicity in experimental mouse models (Roberson et al., 2007; Roberson et al., 2011), the status of Y18 phosphorylation had not been investigated in these studies. Our work characterized a signaling pathway coupling endogenous oAβ, Fyn and tau thereby providing a mechanistic link for the studies by Dr. Mucke’s group. In addition, we showed that tau localization to dendritic spines is exacerbated in the presence of oAβ in a PrPc:Fyn-dependent fashion in vitro and in vivo, thereby further documenting the importance of the dendritic positioning of tau (Hoover et al., 2010; Ittner et al., 2010; Zempel et al., 2010).
To summarize, we believe that the findings presented here are the first to describe a signaling cascade induced by a specific, endogenous Aβ oligomer. Importantly, this study is directly relevant to the disease process as we used endogenous Aβ oligomers from AD brain. Finally, we think this is the first description of a complete signaling pathway, including ligand/receptor/messenger/target, linking apparent Aβ dimers to tau hyperphosphorylation. Overall, our data suggest that PrPc-mediated Fyn activation might play a determinant role in late stages of AD, when Aβ dimers levels are at their highest in the brain.
Acknowledgments
This work was supported in part by NIH grants P30AG10161 and R01AG15819 to D.A.B. and startup funds from the University of Minnesota Medical Foundation to S.E.L. M.N. was partially supported by an investigator fellowship from Collegio Ghislieri, Pavia, Italy. We thank Gloria Lee for the PY18 antibody, Michael K. Lee for critical discussions, Harry Orr for comments on the manuscript and Kenji Kanamura, and Hoa Nguyen for technical help. We are indebted to the participants in the Religious Orders Study.
Footnotes
The authors have no conflicts of interests in relation to this manuscript.
References
- State of aggregation. Nat Neurosci. 2011;14:399. doi: 10.1038/nn0411-399. [DOI] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer’s Disease Brain-Derived Amyloid-{beta}-Mediated Inhibition of LTP In Vivo Is Prevented by Immunotargeting Cellular Prion Protein. J Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, De Strooper B. Prion protein in Alzheimer’s pathogenesis: a hot and controversial issue. EMBO Mol Med. 2010;2:289–290. doi: 10.1002/emmm.201000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the religious orders study. Curr Alzheimer Res. 2012;9:628–645. doi: 10.2174/156720512801322573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–841. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Mendes de Leon CF, Arnold SE, Barnes LL, Bienias JL. Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology. 2003;60:1909–1915. doi: 10.1212/01.wnl.0000069923.64550.9f. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Aggarwal NT, Arvanitakis Z, Shah RC, Kelly JF, Fox JH, Cochran EJ, Arends D, Treinkman AD, Wilson RS. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology. 2006;27:169–176. doi: 10.1159/000096129. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Hobbs GA, Yen SH, Lee G. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol Appl Neurobiol. 2010;36:462–477. doi: 10.1111/j.1365-2990.2010.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Caetano FA, Beraldo FH, Hajj GN, Guimaraes AL, Jurgensen S, Wasilewska-Sampaio AP, Hirata PH, Souza I, Machado CF, Wong DY, De Felice FG, Ferreira ST, Prado VF, Rylett RJ, Martins VR, Prado MA. Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem. 2011;117:538–553. doi: 10.1111/j.1471-4159.2011.07225.x. [DOI] [PubMed] [Google Scholar]
- Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2:306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPc monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci. 2011a;31:10427–10431. doi: 10.1523/JNEUROSCI.1459-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu GQ, Mucke L. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011b;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debatin L, Streffer J, Geissen M, Matschke J, Aguzzi A, Glatzel M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener Dis. 2008;5:347–354. doi: 10.1159/000121389. [DOI] [PubMed] [Google Scholar]
- Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Puig B, Ribera R, Rey MJ, Ribalta T. Prion protein expression in senile plaques in Alzheimer’s disease. Acta Neuropathol. 2001;101:49–56. doi: 10.1007/s004010000271. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. Embo J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Mandelkow E. Fyn-tau-amyloid: a toxic triad. Cell. 2010;142:356–358. doi: 10.1016/j.cell.2010.07.032. [DOI] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–4. doi: 10.1038/nature09217. discussion E4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, Lesne SE. Soluble alpha-Synuclein Is a Novel Modulator of Alzheimer’s Disease Pathophysiology. J Neurosci. 2012;32:10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G. Tau and src family tyrosine kinases. Biochim Biophys Acta. 2005;1739:323–330. doi: 10.1016/j.bbadis.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111(Pt 21):3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lesne S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, Plotkine M, Marchand-Verrecchia C, Vivien D, Buisson A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci. 2005;25:9367–9377. doi: 10.1523/JNEUROSCI.0849-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain. 2010;133:1328–1341. doi: 10.1093/brain/awq065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K, Cheng IH, Yu GQ, Mucke L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–1986. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, Collinge J, Walsh DM. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, Selkoe DJ, Lemere CA, Walsh DM. Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MA, Lesne SE. Detecting abeta*56 oligomers in brain tissues. Methods Mol Biol. 2011;670:45–56. doi: 10.1007/978-1-60761-744-0_4. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Tobiume M, Sato Y, Sata T, Gouras GK, Takahashi H. Accumulation of cellular prion protein within dystrophic neurites of amyloid plaques in the Alzheimer’s disease brain. Neuropathology. 2011;31:208–214. doi: 10.1111/j.1440-1789.2010.01158.x. [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012 doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Williamson R, Usardi A, Hanger DP, Anderton BH. Membrane-bound beta-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. Faseb J. 2008;22:1552–1559. doi: 10.1096/fj.07-9766com. [DOI] [PubMed] [Google Scholar]
- Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci. 2002;22:10–20. doi: 10.1523/JNEUROSCI.22-01-00010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, Zhu X, McGeer PL, McGeehan J, Kneale G, Rincon-Limas DE, Fernandez-Funez P, Lee HG, Smith MA, Petersen RB, Guo JP. Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem. 2011;286:15095–15105. doi: 10.1074/jbc.M110.199356. [DOI] [PMC free article] [PubMed] [Google Scholar]