

Abstract
Progestins play a deleterious role in the onset of breast cancer, yet their influence on existing breast cancer and tumor progression is not well understood. In luminal estrogen receptor (ER) and progesterone receptor (PR) positive breast cancer, progestins induce a fraction of cells to express cytokeratin 5 (CK5), a marker of basal epithelial and progenitor cells in the normal breast. CK5+ cells lose expression of ER and PR and are relatively quiescent, increasing their resistance to endocrine and chemotherapy compared to intratumoral CK5−ER+PR+ cells. Characterization of live CK5+ cells has been hampered by a lack of means for their direct isolation. Here we describe optical (GFP) and bioluminescent (luciferase) reporter models to quantitate and isolate CK5+ cells in luminal breast cancer cell lines utilizing the human KRT5 gene promoter and a viral vector approach. Using this system, we confirmed that the induction of GFP+/CK5+ cells is specific to progestins, is dependent on PR, can be blocked by antiprogestins, and does not occur with other steroid hormones. Progestin-induced, FACS isolated CK5+ cells had lower ER and PR mRNA, were slower cycling, and were relatively more invasive and sphere-forming than their CK5− counterparts in vitro. Repeated progestin treatment and selection of GFP+ cells enriched for a persistent population of CK5+ cells, suggesting that this transition can be semi-permanent. These data support that in PR+ breast cancers, progestins induce a subpopulation of CK5+ER−PR− cells with enhanced progenitor properties and has implications for treatment resistance and recurrence in luminal breast cancer.
Keywords: progesterone, progesterone receptor, cytokeratin 5, breast cancer, progenitor cells
Introduction
Progesterone is a multifunctional female hormone critical for development and maintenance of the breast. However, progesterone action manifests into breast cancer risk under some circumstances. Most notably, hormone therapies (HT) that include progestins increase breast cancer incidence in post-menopausal women compared to non-progestin containing HT (1, 2). These occurrences may be mediated in part by progestin-mediated reactivation of dormant cancer stem cells that, in turn, accelerate the appearance of latent breast tumors in progestin HT users (3, 4). Progesterone and progesterone receptors (PR) are also necessary for, and accelerate the development of, carcinogen-induced rodent mammary tumors (5–7). How progestins affect existing breast cancer is less clear - studies have focused on altered tumor cell growth, cell signaling, and gene regulation (reviewed in 8, 9, 10). In the last several years, it was discovered that progesterone regulates the number of mouse mammary stem cells (MaSCs) (11, 12), increases the number of cells with progenitor cell properties in normal breast tissue models (13), and increases the number of cells with cancer stem cell properties in estrogen receptor (ER) and PR positive breast cancer cell lines and xenografts (14).
The mechanism of progesterone-mediated expansion of stem cells appears to be different between the normal and malignant breast. In normal breast, progesterone-regulated paracrine factors mediate the signal from luminal PR+ cells to basal-located PR− stem cells to divide, although the precise paracrine factors may differ between human and mouse (13, 15, 16). The isolated progestin-expanded MaSC fraction in the mouse has significantly greater mammary repopulating capability than the nascent MaSC fraction (11, 12), suggesting that progesterone not only expands, but augments the regenerative capacity of MaSCs. As mammary epithelial cells undergo malignant transformation, it is hypothesized that they transition from paracrine to autocrine-mediated signaling (4, 10). In line with this, progestins induce the appearance of otherwise undetectable cytokeratin 5 (CK5)+ ER−PR− cells in luminal ER+PR+ breast cancer cell lines within 24 h (17, 18). The short time frame, coupled with the near uniformity of ER and PR expression in several breast cancer cell lines tested, strongly suggests a direct conversion of CK5−ER+PR+ to CK5+ER−PR− cells. In 3D culture models, this is evidenced by a fraction of cells detected in a temporary intermediate CK5+ER+PR+ state following progestin treatment (14). The precise biology and significance of progesterone-induced CK5+ cells in luminal breast cancer is unknown.
CK5+ cells are found in both the luminal and basal epithelium of the normal human breast (19, 20), in contrast to other tissues where CK5 is considered a strict basal epithelial cell marker. CK5 is expressed in progenitor cells that can give rise to luminal lineages (luminal progenitor) (21) or both luminal and myoepithelial lineages (bi-potent progenitor) (22). Thus, there are potentially two pools of CK5+ cells in the breast: luminal-committed and bi-potent progenitors. Aberrant CK5+ luminal progenitors are the potential origin of basal-like breast cancers in BRCA1 deficient women (21). In breast malignancies, CK5 is a signature marker of poor prognosis basal-like breast tumors (23) and an independent indicator of relapse-free survival (24). Latent CK5+ER−PR− cells exist in up to half of all luminal ER+ tumors (17, 25), and we have previously demonstrated that these cells are more resistant to conventional endocrine and chemotherapies than CK5−ER+PR+ cells (17). Luminal tumor CK5+ cells express higher levels of the tumor initiating cell marker CD44 and the basal breast cancer marker epidermal growth factor receptor (EGFR) compared to CK5− cells (14), although overlap between these markers is not complete. The precise tumor initiating cell signatures CD44+CD24−/low (26) and ALDH1+ (27) are low or absent in luminal tumors and cell lines, and more prevalent in triple negative (TN, ER−PR−HER2−) tumors and cell lines (28, 29). Progestin-inducible CK5+ cells may therefore represent a pool of hormone-regulated stem-like cells specific to luminal breast cancer.
Direct isolation and characterization of CK5+ luminal breast cancer cells is difficult, due to its cytoplasmic location, without using surrogate cell surface markers such as CD44 and EGFR. These markers, however, do not share uniform overlap with CK5. Furthermore, CK5+ breast cancer cells are more consistently ER−PR− than CD44+ cells (14). Herein we describe a novel engineered reporter system to quantitate, track, and isolate CK5+ cells from luminal breast cancer cell lines and prospectively from tumors. We define here for the first time that progestin-induced CK5+ cells have enhanced progenitor properties compared to CK5− cells. Further study of this dynamic population will advance our understanding of progesterone regulation of breast cancer stem/progenitor cells and help identify means to prevent their acquisition.
Materials and Methods
Cell lines and reagents
Breast cancer cell lines (T47D and MCF7) were obtained from the University of Colorado Cancer Center Tissue Culture Core Laboratory. SCC-15 human squamous carcinoma cells were obtained from the ATCC. T47D-PRnull cells were a gift from Dean Edwards (30, 31). Breast cancer cell lines were maintained in DMEM + 10% fetal bovine serum (FBS, Hyclone), 10 nM insulin, non-essential amino acids (Sigma), penicillin G (100 U/ml), and streptomycin (100 µg/ml). For maintenance of virally transduced cell lines, puromycin (200 ng/ml) was added to the medium. All experiments, unless otherwise noted, were carried out in DMEM + 10% FBS without added supplements or antibiotics. Monoclonal antibodies were used to recognize β-actin (AC-15, Sigma Aldrich, 1:10,000 Western blot), CK5 (NCL-L-CK5, Leica Microsystems, 1:500 Western blot, 1:250 immunofluorescence), ER (SP1, Thermo-Fisher, 1:5,000 Western blot, 1:100 immunofluorescence), GFP (MAB3580, Millipore, 1:2,500 Western blot), PR (1294, DAKO, 1:10,000 Western blot, 1:500 immunofluorescence), and BrdU (B44, BD Biosciences, 1:50 immunofluorescence). Infrared secondary antibody IRDye 800CW (Li-Cor, 1:10,000) was used for Western blot detection following incubation with primary antibodies. Alexa Fluor 488 goat anti-rabbit and 594 goat anti-mouse secondary antibodies (Invitrogen, 1:250) were used for immunofluorescence. R5020 was purchased from Perkin Elmer; all other hormones were purchased from Sigma.
CK5 promoter-reporter cloning and stable cell line engineering
Promoter-reporter cloning
A 6034 bp fragment (−6002 to +32) of the proximal promoter for the human KRT5 gene (32) (K5p) was amplified by PCR and cloned into either the pA3 luciferase vector (33) or the pCDH1 HIV vector in place of the CMV promoter as described (34). Reportable markers enhanced green fluorescent protein (GFP) and luciferase ((luc)2p fragment, Promega) were inserted downstream of the promoter using Swa1/Not1 sites (luc2P) and EcoRI/NotI sites (GFP) within the multiple cloning site of the pCDH1 HIV vector to produce pCDH1-K5pGFP and pCDH1-K5pLuc vectors. All PCR products and plasmids were confirmed by sequencing.
Stable transduction of cell lines
To produce virus, HEK 293T cells were transfected with plasmids pCMV-VSV-G, pHR-8.2 ΔR, and pCDH1-K5pGFP or -K5pLuc using liposomal LT 1 transfection reagent (Mirus Bio). The virus-containing medium was collected after 48 h and filtered through a 0.45 µm filter. Cell lines (T47D, T47D-PRnull and MCF7) were incubated with viral particle-containing supernatant supplemented with polybrene (Santa Cruz Biotechnology, 8 µg/mL) for 24 h. After 2 days, transduced cells (designated as - K5pGFP or -K5pLuc) were selected with 1 µg/mL puromycin for seven days, then maintained as stably transduced pools in 200 ng/mL puromycin.
Luciferase Assays
Nontransduced T47D and MCF7 cells were plated in triplicate at 1 × 105 cells per well in 12-well plates and treated with vehicle (EtOH), 100 nM progesterone (P4) (T47D), or 10 nM 17β-estradiol (E2) + 100 nM P4 (MCF7) for 24 h. 1 × 105 T47DK5pLuc cells were plated in triplicate in 12-well plates. 24 hours later, cells were treated with various progestins and steroid hormones for 24 h: vehicle (ethanol, EtOH), 1 µM RU486 (RU), 100 nM P4, 100 nM P4 + 1 µM RU486, 100 nM MPA, 100 nM MPA + 1 µm RU486, 100 nM DHT, 100 nM R1881, 100 nM dexamethasone (Dex), 10 nM E2. 18 hours later, cells were rinsed with cold PBS and harvested in 300 µl cold harvest buffer (20 mM K2PO4 (pH 7.8), 5 mM MgCl2, 0.5% Triton X-100). 50 µl lysate was added to 350 µl assay buffer (100mM dibasic K2PO4 (pH 7.8), 1mM DTT, 5mM ATP, 15mM MgSO4), in duplicate, and 100 µl 10 mM D-luciferin (AnaSpec) added to each sample. Luciferase activity was measured using a Monolight 3010 Luminometer (Analytical Luminescence Laboratory), normalized to total protein per lysate as determined by standard Bradford assay, and plotted as fold induction over vehicle control.
Western blot analysis
T47DK5pGFP cells were seeded into 6-well dishes at 3 × 105 cells per well in 6-well plates. 18 hours later, cells were treated with vehicle control (EtOH) or 100 nM P4 for 24 h. Cells were harvested on ice in cold PBS containing 0.2 mM EDTA and protease inhibitors (Halt Protease Inhibitor Cocktail, Thermo Scientific) and centrifuged at 1,500 × g for 5 min at 4°C. Total cellular extracts were prepared by resuspending cell pellets in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 7.5) containing protease inhibitors. Lysates were incubated on ice for 10 min, then centrifuged at 10,000 × g for 15 min. Protein concentrations of collected supernatants were determined by standard Bradford assay and proteins (0.1 mg) resolved on 4%–12% Bis-Tris NuPAGE gels (Invitrogen) and transferred to PVDF membrane. Membranes were probed with antibodies to CK5, GFP, PR, and β-actin as described above. Signals were detected using the Li-Cor Odyssey infrared imaging system and bands quantified using Image J software (NIH) and normalized to β-actin loading controls.
Immunofluorescence
Cells (T47DK5pGFP, MCF7K5pGFP or T47DK5PGFP 3xPhigh) were seeded onto sterile glass coverslips in 6-well dishes at 3 × 105 cells/well. The following day, cells were treated as indicated with vehicle (EtOH), 100 nM P4 (T47DK5pGFP), 10 nM E2 + 100 nM P4 (MCF7 K5pGFP) or 100 nM P4 + 1 µM RU486 for 24 h (T47DK5pGFP). Cells were fixed with 1 mL 4% paraformaldehyde in PBS for 20 min at −20°C prior to permeabilization in 0.25% Triton X-100 in PBS for 15 min at RT with gentle agitation. Cells were blocked with 10% normal goat serum (NGS, Jackson ImmunoResearch) for 30 min followed by incubation with primary (1 h) and secondary (1 h) antibodies (diluted in PBS + 1% NGS) at RT. For dual immunofluorescence, rabbit and mouse generated primary antibodies were used simultaneously and detected with appropriate secondary antibodies listed above as indicated. Nuclei were counterstained with DAPI (1 mg/mL in PBS), and coverslips mounted onto slides with Fluorogel (Electron Microscopy Sciences). Images were taken using a Nikon Eclipse TiE fluorescent microscope equipped with NIS elements software.
Quantitative PCR analysis
Total RNA was isolated using miRNeasy Plus (Qiagen) according to the manufacturer’s instructions. One µg total RNA was reverse transcribed in a total volume of 20 µL using MMLV reverse transcriptase (Promega), then brought to a total volume of 50 µL in sterile water. Real-time reverse transcriptase-PCR for CK5, GFP, ERα, PR and GAPDH was performed on 1/50 of the synthesized cDNA using SYBR Universal Master Mix (Applied Biosystems) and the following primer sets: CK5 forward 5’-GGAGAAGGAGTTGGACCAGTCAAC-3’; CK5 reverse 5’-CTACCTCCGGCAAGACCTCCAC-3’; GFP forward 5’-CACATGAAGCAGCACGACTTCT-3’; GFP reverse 5’-AACTCCAGCAGGACCATGTGAT-3’; ERα forward 5’-TGGGCTTACTGACCAACCTG-3’; ERα reverse 5’- CCTGATCATGGAGGGTCAAA-3’; PR forward: 5’-TCGAGCTCACAGCGTTTCTA-3’; PR reverse 5’-CCCGGGACTGGATAAATGT-3’; GAPDH forward 5’-GGTATCGTGGAAGGACTC-3’; GAPDH reverse 5’-GGATGATGTTCTGGAGAGC-3’. Amplification signals were detected with an ABIprism 7500 Fast Sequence Detection system (Applied Biosystems). Fold change in expression was calculated using the comparative CT method (35) following confirmation that the amplification efficiencies of the targets (CK5, GFP, ERα, PR and GFP) and endogenous reference control (GAPDH) were approximately equal. Values were calculated according to the following equation: Fold change = 2−ΔΔCT where ΔΔCT = ΔC T 1 − ΔC T 2. ΔC T 1 = (C T, CK5, GFP, ERα, PR, treated - C T, GAPDH, treated) and ΔC T 2 = (ΔC T, CK5, GFP, ERα, PR, control - ΔC T, GAPDH, control).
Cell cycle analysis and proliferation
Cell cycle analysis
To measure the relative cell cycle distribution of T47DK5pGFP cells, cells were treated with P4 or vehicle (EtOH) for 24 h, trypsinized, resuspended in DMEM + 10% FBS, and stained with live cell permeable VyBrant DyeCycle Violet (Invitrogen) for 30 minutes at 37°C according to the manufacturer’s recommendations. The DNA content of P4-treated GFP+/− (CK5+/−) cells was measured by flow cytometry and compared to vehicle-treated control cells.
Invasion and sphere forming assays
Invasion assays
CK5+ and CK5− (GFP+ and GFP−) cells were isolated by fluorescence-activated cell sorting (FACS) from T47DK5pGFP cells treated with 100 nM P4 for 24 h and compared with vehicle-treated, unsorted cells. Collected cells (5 × 104) were suspended in 0.5 mL DMEM + 0.1% FBS and added to each upper invasion or migration chamber (BD Biosciences) in triplicate, then placed into a 24-well plate, each well containing 750 µL DMEM + 10% FBS and incubated for 24 h at 37°C. Following incubation, chambers were removed from wells, rinsed in PBS, and cells in the upper chamber removed using a sterile cotton swab. Membranes were fixed with 95% EtOH and stained with haematoxylin and eosin, then dried and mounted onto cover slips and imaged using an Aperio digital slide scanner and quantitated using Image J software (NIH).
Sphere formation assays
T47DK5pGFP cells (P-treated FACS isolated CK5+ and CK5− cells, and vehicle-treated unsorted cells) were suspended in mammosphere medium (MammoCult base medium, proliferation supplement, 0.2% heparin, 200 µg/mL hydrocortisone, Stem Cell Technologies) containing 5% Matrigel (BD Biosciences). Cells were seeded into ultra low attachment 96-well plates (Costar) at a density of 3 × 102 cells per well and incubated for seven days. Cells were stained with 10 µM Hoechst 33342 for 30 min and mammospheres (≥50 µm in diameter) in 16 fields per well were counted using the Operetta imaging system (Perkin Elmer) with the following settings: 10X long working distance, non-confocal mode, 50% excitation, 0% transmission and 100 ms exposure for Hoechst 33342.
Statistics
Graphpad Prism 5.04 was used to plot data and to determine statistical significance using either a student’s unpaired t-test or One-Way ANOVA followed by a Tukey post-test, where noted.
Results
A promoter-reporter model to isolate progesterone-inducible CK5+ breast cancer cells
The isolation of live cells based on intracellular markers is technically challenging and requires partial permeabilization of the cell membrane, which affects cell viability and function. In the past we have utilized co-expression of cell surface markers (CD44, EGFR) to isolate breast cancer cell subpopulations enriched for CK5+ cells, although these cell pools are not pure as there is not uniform overlap with CK5. We reasoned that the human CK5 promoter was used successfully for targeted transgene expression in CK5+ murine epithelial cells (32), and thus could be utilized for tracking and isolating CK5+ human breast cancer cells. To test this, a 6 kb fragment of the proximal human KRT5 gene promoter was cloned upstream of the luciferase coding sequence in the pA3-Luc vector (pA3-K5pLuc) and transiently transfected into SCC-15 squamous cell carcinoma cells, or human breast cancer cell lines T47D and MCF7, treated with vehicle (EtOH), 100 nM P4, or 10 nM E2 + 100 nM P4 (Figure 1A). In MCF7 cells, a combination of E2 + P4 treatment is necessary for CK5 induction, as PR expression is E2-dependent. SCC-15 cells (a CK5 positive cell line) displayed constitutive expression of pA3-K5pLuc in the absence of hormone treatment. T47D and MCF7 cells showed 17 and 1.7 fold induction, respectively, of pA3-K5pLuc upon P4 treatment, compared to negligible activity with vehicle alone. This is consistent with previously observed P4-dependent CK5 expression in these cell lines, which is relatively higher in T47D compared to MCF7 (18). The 6 kb KRT5 promoter fragment was subsequently inserted into the pCDH1 HIV vector, upstream of either the GFP or Luc coding sequence and stably transduced cell lines developed as described. To verify P4 induction of GFP and appropriate overlap between endogenous CK5- and GFP-expressing cells, immunofluorescence was performed on vehicle (EtOH), 100 nM P4, and 100 nM P4 + 1 µM RU486 treated T47DK5pGFP cells (Figure 1B). Both CK5 and GFP were undetectable in vehicle-treated cells, and expressed in ~20% of cells in P4-treated samples. Overlap between CK5 and GFP was ~85%, determined by quantification of multiple fields. The antiprogestin RU486 abolished P4-induced CK5 and GFP expression, supporting that this is a P4-dependent phenomenon. A Western blot using extracts from T47DK5pGFP cells treated as described above demonstrated P4-dependent induction of both CK5 and GFP, which were both abolished by RU486 treatment. These data confirm that P4 induction of GFP correlates with CK5 expression in pCDH1-K5pGFP-transduced luminal breast cancer cells.
Figure 1. KRT5 promoter reporter constructs effectively mark P4-inducible CK5+ breast cancer cells.

A. SCC-15 squamous carcinomas cells, and T47D and MCF7 breast cancer cells, were transiently transfected with a fragment of the human KRT5 promoter (K5p) fused to luciferase (K5pLuc) and treated with vehicle control (EtOH), 100 nM P4 (T47D), or 10 nM E2 + 100 nM P4 (MCF7) for 24 h (SCC-15 were treated with vehicle only). Data shown as fold induction normalized to empty vector. *p<0.05, **p<0.01, one-way ANOVA/Tukey post test. B. Immunofluorescence for CK5 (red) and GFP (green) in T47D cells stably transduced with K5pGFP virus (T47DK5pGFP) and treated with vehicle (EtOH), 100 nM P4, or 100 nM P4 + 1 µM RU486 for 24 h. Representative fields of cells are shown for CK5, GFP, and merged images plus DAPI counterstaining. Scale bar, 50 µm. C. T47DK5pGFP cells were treated as in panel B and extracts prepared and analyzed by Western blot with antibodies against GFP, CK5, PR, or β-actin. GFP and CK5 bands were undetectable by quantitation in EtOH, RU486, or P4 + RU486 lanes, with a relative density of 1.47 and 1.19 (normalized to β-actin), respectively, in the P4-treated sample.
P4 shifts breast cancer cells towards CK5+ and CD44+ phenotypes
To quantitatively measure P4-induced CK5+ cells in breast cancer cell lines and compare their overlap with CD44+ cells, T47DK5pGFP and MCF7K5pGFP cells were treated with vehicle (EtOH) or 100 nM P4 (T47DK5pGFP) or 100 nM P4 + 10 nM E2 (MCF7K5pGFP) for 24 h, and analyzed by flow cytometry. A fraction of the cells was incubated with an Alexa Fluor 647-conjugated antibody to CD44 prior to analysis (Figure 2A and 2B, lower panels). P4 treatment induced CK5 expression in T47DK5pGFP (10.6 fold P4 compared to control) and in MCF7K5pGFP cells (2.5 fold E2 + P4 compared to control, Figures 2A and 2B, upper panels). P4 treatment also increased the fraction of CD44+ cells in both cell lines (>30 fold T47DK5pGFP, 6.8 fold MCF7K5pGFP, Figures 2A and 2B, lower panels), as previously reported (18). Over half the CK5+ cells in T47DK5pGFP were CD44+ whereas in MCF7K5pGFP 12% were CD44+ (Figure 2A and 2B, lower panels). We previously demonstrated that CD44 expression is enriched in microdissected CK5+ compared to CK5− T47D cells (14). Conversely, CK5 expression was enriched in CD44+ cells isolated from primary human breast tumors (36). Thus, while there is considerable overlap, we demonstrate here that the CK5+ population contains both CD44+ and CD44−/low fractions. It should be noted that the plasticity of these populations is unknown and therefore CK5 and CD44 expression may fluctuate. T47D cells lacking expression of PR (T47D-PRnull) (30, 31) were also transduced with the K5pGFP virus, treated with vehicle or P4, and analyzed by flow cytometry (Figure 2C). GFP+/CK5+ cells were undetectable, signifying that P4-induced expression of CK5 is PR dependent. These data support a P4 and PR-dependent increase in the expression of CK5 and CD44 in luminal breast cancer cell lines, but demonstrate that the two populations do not overlap completely.
Figure 2. P4 shifts breast cancer cells towards a more CK5+ and CD44+ phenotype.

K5pGFP cells were treated with vehicle (EtOH) or 100 nM P4 (T47DK5pGFP), or 10 nM E2 + 100 nM P4 (MCF7K5pGFP) for 24 h. Cells were either unlabeled (A, B upper panel) or labeled with an Alexa Fluor 647 conjugated antibody to CD44 (A, B lower panel) and analyzed by flow cytometry. Dead cells were omitted via addition of 10 nM DAPI and gates were drawn based on vehicle-treated samples. The percent of cells in each quadrant is indicated. C. T47D-PRnull cells were treated with vehicle (EtOH) or 100 nM P4 and analyzed by flow cytometry as in panel A.
P4-induced CK5+ cells have reduced ER and PR mRNA expression levels
CK5+ breast cancer cells are near ubiquitously absent for ER (α) and PR; this holds true for CK5+ cells in basal-like TN breast cancers and for rare CK5+ cells found in luminal tumors (17). Since P4 treatment induces a rapid but temporary downregulation of PR protein through a ubiquitin/26S proteosome-mediated mechanism (37), we determined whether P4-induced CK5+ cells would also repress ER and PR expression at the message level. To accomplish this, qPCR was performed on cDNA prepared from sorted CK5+ and CK5− populations and unsorted T47DK5pGFP and MCF7K5pGFP cells that were treated with vehicle (unsorted controls), P4 (T47DK5pGFP), or E2 + P4 (MCF7K5pGFP) for 24 h (Figure 3A and 3B). As an assay control, qPCR was performed for CK5 and GFP in each fraction. CK5 mRNA was near absent in both vehicle-treated GFP− and GFP+ fractions, consistent with its strict P4-dependency in luminal breast cancer cell lines (Figure 3A and 3B). The P4 (or E2 + P4)-treated GFP− populations expressed low levels of CK5 mRNA, whereas the P4 (or E2 + P4)-treated GFP+ fractions expressed significantly higher CK5 mRNA levels (5 fold T47DK5pGFP and 10 fold MCF7K5pGFP). GFP mRNA levels significantly increased in the P4 or E2 + P4-treated GFP+ compared to the GFP− populations and generally correlated with CK5 mRNA levels. The small GFP+ fraction in vehicle-treated cells (<5%) did express correlative CK5 mRNA, suggesting low background GFP expression using this model system. Therefore, vehicle-treated GFP− and GFP+ fractions are both CK5−, thus unsorted vehicle-treated cells were used as a negative control in subsequent experiments for consistency. These results validate that hormone-treated, FACS isolated GFP− and GFP+ cell fractions adequately correspond to CK5− and CK5+ populations, respectively.
Figure 3. P4-treated CK5+ cells have reduced ER and PR mRNA levels.

A and B. T47DK5pGFP cells (A) were treated with vehicle (EtOH, green) or 100 nM P4 (red) and MCF7 cells (B) were treated with vehicle (EtOH, green) or 10 nM E2 + 100 nM P4 (red) for 24 h and GFP+/− cells sorted by FACS. Diagram indicates where the 10% GFP− and GFP+ fractions were collected. Relative levels of CK5 (center panel) and GFP (right panel) mRNA were measured by qPCR and normalized to GAPDH in GFP− and GFP+ fractions from each treatment. C. Relative levels of ER and PR mRNA in T47DK5pGFP (upper panel) or MCF7K5pGFP (lower panel) measured by qPCR and normalized to GAPDH in unsorted vehicle-treated and GFP− and GFP+ fractions of P4-treated cells. qPCR values represent mean δδCT normalized to vehicle-treated unsorted and GAPDH, ± SD (n=3). ** p<0.01, ***p<0.001 compared to vehicle-treated GFP−, one-way ANOVA/Tukey post test. D. Dual immunofluorescence for CK5 (green)/ER (red) or CK5 (green)/PR (red) in P4-treated T47D cells treated with EtOH or 100 nM P4 for 24 h. Scale bar, 50 µm.
We next assessed ER and PR mRNA levels by qPCR in CK5− and CK5+ samples from vehicle and P4-treated T47DK5pGFP, and vehicle and E2 + P4-treated MCF7K5pGFP cells (Figure 3C). As noted above, both the GFP− and GFP+ vehicle-treated fractions lack CK5 mRNA expression (Figure 3A and 3B), therefore ER and PR mRNA levels in P4-treated, GFP+/− sorted fractions were compared to those of unsorted, vehicle-treated control cells. In P4-treated T47DK5pGFP cells, the CK5+ population had significantly lower PR and ER mRNA levels relative to the CK5− population. PR mRNA increased significantly in the E2 + P4-treated MCF7 CK5− cell fraction, corresponding to the E2-dependent PR expression in this cell line. However, the E2 + P4-treated CK5+ population did not have increased PR mRNA, suggesting that E2-dependent PR expression is blocked specifically in CK5+ cells when P4 is given simultaneously. ER mRNA was decreased in the E2 + P4-treated CK5+ population relative to the E2 + P4-treated CK5− fraction, although this did not reach statistical significance (Figure 3C). These results support that ER and PR are partially repressed at the transcriptional level in the P4-induced CK5+ population, compared to transient receptor downregulation that occurs in the bulk CK5− cells.
To verify that P4-induced CK5+ cells are ER/PR−/low at the protein level, nontransduced T47D cells were treated with 100 nM P4 for 24 h and immuofluorescence for ER/CK5 or PR/CK5 performed (Figure 3D). P4-induced CK5+ cells were mainly ER−PR−, compared to the many surrounding CK5−ER+PR+ cells (Figure 3D). Occasional dual CK5+ER+ were observed, whereas CK5+PR+ cells were not detected.
Induction of CK5+ cells is specific to P4 and does not occur with other steroids
To determine steroid hormone-specific regulation of the CK5 promoter, we utilized stably transduced T47DK5pLuc cells treated with progestins (100 nM each of P4, MPA, or R5020), antiprogestins (1 µM RU486 alone or with progestins), androgen (100 nM dihydrotestosterone, DHT), glucocorticoid (100 nM Dex), and estrogen (10 nM E2) for 24 h and measured luciferase activity (Figure 4A). Both the natural hormone, P4, and synthetic progestins MPA and R5020, significantly activated the CK5 promoter (>100 fold induction over vehicle). The pure androgen DHT did not activate the CK5 promoter, but the synthetic androgen, R1881, which has partial binding affinity for PR (38), showed approximately 50% activation compared to the other progestins. Glucocorticoids (Dex) and estrogens (E2) were unable to activate the CK5 promoter. It is possible that AR and GR levels are nonfunctional or too low in T47D cells to measure activity (39), although other reports suggest AR and GR are biologically active in these cells (40, 41). These data suggest that induction of the CK5 promoter is specific to progestins (or partial progestins) and PR (Figure 2) in breast cancer cells.
Figure 4. Induction of the KRT5 promoter is specific to progestins, and not other steroids.

T47DK5pLuc cells were treated with various progestins and steroid hormones for 24 h: vehicle (EtOH), 1 µM RU486 (RU), 100 nM P4, 100 nM P4 + 1 µM RU486, 100 nM MPA, 100 nM MPA + 1 µm RU486, 100 nM DHT, 100 nM R1881, 100 nM Dex, or 10 nM E2. Luciferase activity was measured, normalized to total protein per lysate, and plotted as fold induction over vehicle control, ± SEM (n=3). ***p<0.001, one-way ANOVA/Tukey post test.
P4 treatment produces a population of slower cycling CK5+ cells
Cancer stem cells are thought to be slower cycling and thus able to avoid anti-proliferative therapeutics and later regenerate new tumors (42). We have previously demonstrated that nascent CK5+ cells in 3D cultures of T47D and MCF7 cells are less proliferative and subsequently less responsive to chemotherapeutics targeting rapidly dividing cells (17). To measure the relative cell cycle distribution of CK5+ vs. CK5− populations, T47DK5pGFP cells were treated with vehicle or 100 nM P4 for 24 h, and the cell cycle of GFP+/− fractions was measured by flow cytometry using a live cell permeable DNA intercolating dye. P4-treated T47DK5pGFP cells had increased accumulation in G1, as is expected given that P4 causes cell cycle arrest (43). Notably, P4-treated GFP+/CK5+ cells had less cells in S phase (2.2%) compared to P4-treated GFP−/CK5− cells (19.5%, Figure 5A). To verify the proliferation status of P4-induced CK5+ and CK5− cells, T47D and MCF7 cells were treated with 100 nM P4 (T47D) or 10 nM E2 + 100 nM P4 (MCF7) for 24 h and stained by immunofluorescence for CK5 and BrdU (Figure 5B). In both cell lines, CK5+ cells had less S-phase BrdU incorporation (4% of T47D, 0% of MCF7) than CK5− cells (22% of T47D, 20% of MCF7) (Figure 5B).
Figure 5. P4-induced CK5+ cells have decreased cell cycle compared to CK5− cells.

A. T47DK5pGFP cells were treated with vehicle (EtOH) or 100 nM P4 for 24 h, incubated with VyBrant DyeCycle Violet (Invitrogen), and cell cycle distribution of total vehicle-treated and P4-treated GFP−/+ (CK5−/+) cells measured by flow cytometry. Bars indicate percent of cells in each phase plus/minus SEM. **p<0.01, CK5+ (GFP+) compared to CK5− (GFP−), two-way ANOVA/Tukey post test. B. T47D cells were treated with 100 nM P4 for 24 h (+ E2 for MCF7 cells), incubated with BrdU, and immunofluorescence for CK5 (green) and BrdU (red) performed. Cells were counterstained with DAPI. Scale bar, 50 µm.
P4-induced CK5+ cells are more invasive and have higher self-renewal capacity than CK5− cells
CK5+ cells in T47D tumors express genes consistent with basal-like breast cancer cells (CK17, EGFR) and cancer stem cells (CD44, ATP-binding cassette sub-family G member 2 (ABCG2)) (14, 44). We therefore examined whether P4-induced CK5+ cells have enhanced features of basal or cancer stem cells, compared to CK5− cells from the same culture. To determine the invasive capacity of P4-converted CK5+ cells in luminal breast cancer cell lines, we performed Boyden invasion chamber assays on FACS-isolated CK5+ and CK5− populations from P4-treated T47DK5pGFP cells, compared with unsorted vehicle-treated cells. Cells (5 × 104 each fraction) were placed in invasion chambers for 24 h, then fixed and counterstained. Counterstains of representative invasion chamber wells are shown in Supplemental Figure 1A., The P4-treated GFP+/CK5+ population was significantly more invasive than its GFP−/CK5− counterpart (Figure 6A). Therefore, P4-induced CK5+ cells within luminal breast cancer cultures have increased invasive potential relative to intra-culture CK5−ER+PR+ luminal cells.
Figure 6. P4-induced CK5+ cells are more invasive and sphere-initiating than CK5− cells.

T47DK5pGFP cells were treated with vehicle (EtOH) or 100 nM P4 for 24 h and the GFP−/+ (CK5−/+) fractions isolated by FACS as described. A. Sorted cell fractions (50,000 each) were placed in invasion chambers for 24 h and the number of invading cells per field measured by Image J analysis (NIH). Graph of mean number of invasive cells per field for each condition. Boxes represent range and lines represent means. All sets were compared by one-way ANOVA/Tukey; *p<0.05, P4-treated CK5+ vs. CK5−. B. Sorted cells (300 each) were placed in mammosphere/3D-Matrigel culture, and the number of spheres ≥50 µm measured by automated counter after seven days. Graph of mean number of spheres per field for each condition. Boxes represent range and lines represent means. All data sets were compared by one way ANOVA/Tukey; **p<0.01, ***p<0.001, P4-treated CK5+ compared to all other groups.
We utilized sphere formation assays (often referred to as mammosphere assays) (45) to measure the relative ability of CK5+ cells to self-renew and generate spherical colonies in 3D mammosphere culture relative to CK5− cells. FACS isolated P4-treated CK5+ or CK5− cells and unsorted, vehicle-treated T47DK5pGFP cells were placed into 3D mammosphere culture and allowed to form spheres, then fixed, counterstained and imaged via a high content imaging apparatus. Representative wells containing spheres are depicted in Supplementary Figure 1B. All cell fractions were capable of forming spheres with some capacity (Figure 6B). However, the P4-treated CK5+ cells were more efficient at sphere formation (greater number of spheres ≥50 µm in diameter) compared to unsorted vehicle-treated cells, or P4-treated CK5− cells (Figure 6B). These data indicate that P4 imparts enhanced self-renewal function on a subpopulation of luminal breast cancer cells.
Repetitive P4 treatment produces an intrinsically more stem-like phenotype
To test the transiency of the P4-induced CK5−ER+PR+ cells, P4-treated FACS isolated T47DK5pGFP GFP+/high cells were reseeded in culture, and the P4 treatment/sorting protocol repeated 3 times (referred to as “3xPhigh”). In the parental cells (Figure 7A, green), P4-treatment produced the expected shift to GFP+/CK5+ (Figure 7A, yellow). After three rounds of P4-treatment and GFP sorting, the cells were inherently more GFP+/CK5+, even in the absence of P4 (3xPhigh + EtOH, Figure 7A, orange), although P4 treatment led to a further shift in both the intensity and percentage of cells that were GFP+/CK5+ (3xPhigh + P4, Figure 7A, red). To confirm that serial GFP selection appropriately selects for CK5+ cells, immunofluorescence for CK5 was performed comparing 3xPhigh T47DK5pGFP to parental T47DK5pGFP cells. 3xPhigh selected T47DK5pGFP cells contained a significantly higher proportion of CK5+ cells (48%, lower panel, Figure 7B), even in the absence of hormone, than parental T47D cells treated with P4 for 24 h (9%, upper panel, Figure 7B). To determine PR protein expression in serially sorted CK5+ cells, immunofluorescence was performed on untreated, serially selected cells to measure overlap between CK5 and PR. CK5+ cells were near uniformly absent for PR (Figure 7C). To test their relative self-renewal capacity, 3xPhigh selected and parental T47DK5pGFP cells were placed in spheroid culture as described above in the absence of hormone and their ability to generate spheres tested. T47DK5pGFP 3xPhigh cells (high endogenous CK5+ cells) formed significantly more spheres than parental (non-selected) T47DK5pGFP cells (Figure 7D). These results suggest that repeated P4-treatment can imprint the CK5+ phenotype on a subpopulation of cells, even after the hormone has been removed, and has implications for chronic progestin use in women with breast cancer risk.
Figure 7. Serial selection of GFP+/CK5+ stem-like cells from T47DK5pGFP.

A. Diagram depicting a shift in the number and intensity of GFP+ cells upon triplicate P4 treatment and FACS isolation of the GFP+ fractions (3xPhigh). B. Immunofluorescence for CK5 (red) and DAPI (blue) in serial sorted cell line in absence of P4 (lower panel, “3xPhigh”), compared with parental control + 100 nM P4 for 24 h (parental (“par”), upper panel). Scale bar, 50 µm. C. Immunofluorescence for CK5 (red), PR (green) and DAPI (blue) in serial sorted cell line in absence of P4. Scale bar, 50 µm. D. Parental and serial sorted cells as in A were placed in mammosphere/3D-Matrigel culture (300 per well), and the number of spheres ≥50 µm measured by automated counter after seven days. Graph of mean number of spheres per field for each condition. Boxes represent range and lines represent means. *p<0.05, unpaired t-test.
Collectively, these data impart a reprogramming role for P4 in luminal ER+PR+ breast cancers in converting a subfraction of cells to a more de-differentiated phenotype. The P4-induced CK5+ER−PR− cells have enhanced properties akin to basal (invasiveness) and stem/progenitor cells (quiescence, self-renewal). It can be speculated that P4 exposure during various stages of tumor formation and propagation may alter the behavior of some receptive tumors by converting a subpopulation of cells to a more survival-fit CK5+ tumor stem/progenitor cell phenotype.
Discussion
Hormonal regulation of hierarchical cell populations occurs in normal breast tissue, and is necessary to foster and maintain development of the gland. This was elegantly demonstrated by recent studies showing fluctuation of MaSC numbers in murine mammary glands during normal estrous and pregnancy, and that this could be mimicked by manipulating E2 and P4 levels (11, 12). Hormone regulation of a tumor cell hierarchy per se has not, however, been fully explored in breast cancer. Although P4 initiates a subpopulation of CK5+ cells in breast cancers, the differential behavior of isolated CK5+ vs. CK5− cells has thus far not been demonstrated. Here, using a novel optical reporter system, we show that P4 induces an invasive, self-renewing CK5+ER−PR− subpopulation in luminal ER+PR+ breast cancer cell lines. Coupled with our findings that CK5+ cells are more resistant to endocrine and chemotherapies (17), these studies provide important insight into a role for P4 in the progression and recurrence of luminal breast cancers.
The main purpose of this study was to define the characteristics of P4-induced CK5+ cells in luminal breast cancer cell models. CK5 is a characteristic marker of basal-like breast cancers (23). The latter tumors constitute approximately 50% of all TN breast cancers (46), and were so named based on expression of basal-like cytokeratins (CK5, CK14, CK17) in addition to EGFR. Expression profiling studies and hierarchical clustering suggest that basal-like breast cancers (referred to as Basal A) are more closely related to luminal than TN mesenchymal-like breast cancers (Basal B) (44). The basal-like breast cancer nomenclature, however, has been challenged due to the fact that CK5+ cells are frequently found in the luminal epithelial layer of the human breast (19). Many of these luminal CK5+ cells also express CK18 (CK5+CK18+) and were identified as potential luminal progenitors (47). Of note is that in normal prostate, intermediate CK5+CK18+ cells are progenitors to CK18+AR+ luminal cells (48, 49). CK5 is expressed in both the normal human breast stem (lineage(Lin)− epithelial cell adhesion molecule (EpCAM)−CD49f+) and progenitor (Lin−EpCAM+CD49f+) cell fractions (21, 50). Notably, P4 increases the colony forming potential of isolated luminal progenitor cells (21), and increases progenitor cell function of human breast organoids (13). Here we demonstrate that in luminal breast cancer cell lines, P4 induces CK5+ cells with enhanced progenitor cell function, including increased mammosphere formation and relative quiescence, compared to CK5− cells. Since breast cancer stem cells can arise spontaneously or by exogenous stimuli (51, 52), P4 is therefore one factor that seemingly facilitates this switch within hormone dependent cancers.
A major question is whether P4-induced CK5 cells are transitory, or if they persist in culture long-term once converted. We have observed that two weeks after initial P4 exposure, some CK5+ cells persist, while the majority have returned to a CK5− state. However, further selection of cells that were sensitive to the initial P4-induced transformation and subjected to repeated P4 treatment become stably enriched for a population of cells that retain the CK5+ phenotype, up to several weeks post-treatment. In previous reports, we have also demonstrated that in 3D cultures prepared from xenograft tumors, CK5+ cells persist for weeks (17). This has implications for post-menopausal women where exposure to progestins could occur through hormone therapy, or through other natural supplements. P4-induced CK5+ are slower cycling than the majority CK5− cells, at least initially. The fact that some cultures contain a propagatable population of CK5+ cells suggests they can re-enter the cell cycle. Luminal ER+ tumors have increased tendencies towards dormancy (53) and are more prone to long term (> 10 year) recurrence (53, 54). Although the factors regulating dormant tumor cells are relatively unknown, it is conceivable that hormones could play a role in influencing a quiescent tumor cell state. Long term in vivo studies are necessary to follow up on the observed quiescence of CK5+ cells.
Induction of CK5+ cells in breast cancer is restricted to progestins/PR as opposed to other steroids and their receptors. E2 alone does not alter CK5 expression, but is necessary for P4-induced CK5 expression in cell lines where PR is E2-dependent (18). In mouse models it was implied that E2 may be increasing PR expression while P4 was the critical hormone signaling MaSC expansion (12). We have previously shown that CK5 expression increases in breast cancer cells following prolonged tamoxifen treatment or estrogen withdrawal, and increases in breast tumors following neoadjuvant endocrine therapy (17), suggesting that E2 signaling may actually suppress the CK5 phenotype. E2 increases the CD44+CD24− population in MCF7 breast cancer cells via an autocrine mechanism involving growth factors (55), suggesting that the CD44+CD24− and CK5+ populations are differentially regulated by hormones. In other studies we have shown that P4-regulated microRNAs facilitate the inception of CK5+ cells, partially by relieving repression of reprogramming transcription factors such as Krüppel-like factor 4 (KLF4) (18). Additionally, using a small molecule screen, retinoids were identified as potent inhibitors of P4-mediated induction of CK5+ cells (34). Crosstalk between P4 and retinoid signaling has been observed in earlier studies (56, 57). Whether retinoids specifically block P4-mediated de-differentiation of cancer cells, or impart a general blockade on all P4 action requires further exploration. Retinoids are involved in maintenance of stem cell differentiation in many tissues (58–60) and have been explored as agents for prevention and treatment of breast cancer (61–63).
It is important to note that P4-dependent induction of CK5+ cells does not occur in all PR+ breast cancer cell lines and xenograft tumors. Two ER+PR+ cell lines, one isolated from a pleural effusion and one from a primary tumor, have no detectable CK5+ cells basally or upon P4 or E2 + P4 treatment.1 We have previously observed that CK5+ cells exist latently in approximately 36% of all luminal tumors (17). We have also generated patient-derived breast cancer xenografts where CK5+ expression varies from absent (0%) in some tumors to near 30% of cells in others (64). Since only some luminal tumors express CK5+ cells, we therefore speculate that there are “pure” luminal breast tumors that are not prone to spontaneous, hormone, or perhaps treatment-induced CK5+ cells. Indeed this was observed in a large study measuring cytokeratin expression patterns, where tumors containing mixed luminal and basal cytokeratins (CK5, CK14, CK17) had poorer outcome than tumors expressing only luminal cytokeratins (CK8, CK18, CK19) (24). Therefore, only a subset of luminal tumors may be susceptible to P4/PR reprogramming and the molecular reasons for these differences need to be explored. This may be the case with P4-containing hormone therapy users, where occult tumors were accelerated in only a small subset of women (65).
In summary, in many luminal breast cancer cell lines, P4 targets a susceptible population of cells to undergo a transition to a more progenitor cell phenotype, void of ER and PR. These reprogrammed cells have enhanced properties of progenitors relative to the bulk non-reprogrammed cells. Although this state is transient in many cells, some converted cells remain transitioned, which has implications for tumor cell survival and disease recurrence. Further analysis of P4-sensitive cells in heterogeneous patient tumor models may pinpoint a subset of luminal tumors that are prone to P4 reprogramming and may benefit from additional agents that block or revert this phenotype. In particular, clinical trials on antiprogestins were ceased mainly due to toxicity. Newer compounds in development with prospectively less toxicity may be effective on this particular profile of luminal tumors.
Supplementary Material
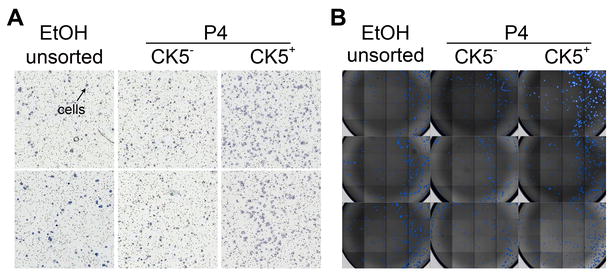
Supplemental Figure 1. P4-induced CK5+ cells are more invasive and sphere-initiating than CK5− cells. T47DK5pGFP cells were treated with vehicle (EtOH) or 100 nM P4 for 24 h and the GFP−/+ (CK5−/+) fractions isolated by FACS as described. A. Sorted cell fractions (50,000 each) were placed in invasion chambers for 24 h and the number of invading cells per field measured by Image J analysis (NIH). Picture denotes representative filters. B. Sorted cells (300 each) were placed in mammosphere/3D-Matrigel culture, and the number of spheres ≥50 µm measured by automated counter after seven days. Merged bright field/DAPI stained images of wells are shown.
{kind=link}
Acknowledgements
The authors wish to thank Elaine Fuchs for the gift of the human CK5 promoter fragment, Dean Edwards for the T47D-PRnull cells, Bill Wood for the pA3Luc plasmid, Storey Wilson for assistance with quantitative software analysis, the Molecular Pathology Core and the University of Colorado Flow Cytometry Core for their expert technical assistance and services. This work was supported by grants from the Cancer League of Colorado ((fellowship to S.D.A., pilot award to D.V.L. and C.A.S.), the Wendy Will Case Foundation (C.A.S.), and the National Institutes of Health R01 CA140985 (C.A.S.).
Footnotes
B.M. Jacobsen, personal communication; C.A. Sartorius unpublished data
References
- 1.Beral V, Reeves G, Bull D, Green J. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J Natl Cancer Inst. 2011 Feb 16;103(4):296–305. doi: 10.1093/jnci/djq527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chlebowski RT, Anderson GL, Gass M, Lane DS, Aragaki AK, Kuller LH, et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA. 2010 Oct 20;304(15):1684–1692. doi: 10.1001/jama.2010.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horwitz KB, Sartorius CA. Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: a hypothesis. J Clin Endocrinol Metab. 2008 Sep;93(9):3295–3298. doi: 10.1210/jc.2008-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axlund SD, Sartorius CA. Progesterone regulation of stem and progenitor cells in normal and malignant breast. Mol Cell Endocrinol. 2012 Jun 24;357(1–2):71–79. doi: 10.1016/j.mce.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huggins C, Moon RC, Morii S. Extinction of experimental mammary cancer. I. Estradiol-17beta and progesterone. Proc Natl Acad Sci U S A. 1962;48:379–386. doi: 10.1073/pnas.48.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldaz CM, Liao QY, LaBate M, Johnston DA. Medroxyprogesterone acetate accelerates the development and increases the incidence of mouse mammary tumors induced by dimethylbenzanthracene. Carcinogenesis. 1996 Sep;17(9):2069–2072. doi: 10.1093/carcin/17.9.2069. [DOI] [PubMed] [Google Scholar]
- 7.Lydon JP, Ge G, Kittrell FS, Medina D, O'Malley BW. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Res. 1999 Sep 1;59(17):4276–4284. [PubMed] [Google Scholar]
- 8.Hagan CR, Daniel AR, Dressing GE, Lange CA. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol. 2012 Jun 24;357(1–2):43–49. doi: 10.1016/j.mce.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. 2012 Jun 24;357(1-2):18–29. doi: 10.1016/j.mce.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obr AE, Edwards DP. The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol Cell Endocrinol. 2012 Jun 24;357(1-2):4–17. doi: 10.1016/j.mce.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010 Jun 10;465(7299):798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 12.Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. 2010 Jun 10;465(7299):803–807. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- 13.Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI, et al. DNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology. 2009a Jul;150(7):3318–3326. doi: 10.1210/en.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA. Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci U S A. 2008 Apr 15;105(15):5774–5779. doi: 10.1073/pnas.0706216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010 Nov 4;468(7320):103–107. doi: 10.1038/nature09495. [DOI] [PubMed] [Google Scholar]
- 16.Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010 Nov 4;468(7320):98–102. doi: 10.1038/nature09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A, et al. Cytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat. 2011 Jul;128(1):45–55. doi: 10.1007/s10549-010-1078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. 2012 Jul 2; doi: 10.1038/onc.2012.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gusterson B. Do 'basal-like' breast cancers really exist? Nat Rev Cancer. 2009 Feb;9(2):128–134. doi: 10.1038/nrc2571. [DOI] [PubMed] [Google Scholar]
- 20.Gusterson BA, Ross DT, Heath VJ, Stein T. Basal cytokeratins and their relationship to the cellular origin and functional classification of breast cancer. Breast Cancer Res. 2005;7(4):143–148. doi: 10.1186/bcr1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009 Aug;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 22.Boecker W, Moll R, Poremba C, Holland R, Van Diest PJ, Dervan P, et al. Common adult stem cells in the human breast give rise to glandular and myoepithelial cell lineages: a new cell biological concept. Lab Invest. 2002;82(6):737–746. doi: 10.1097/01.lab.0000017371.72714.c5. [DOI] [PubMed] [Google Scholar]
- 23.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000 Aug 17;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 24.Abd El-Rehim DM, Pinder SE, Paish CE, Bell J, Blamey RW, Robertson JF, et al. Expression of luminal and basal cytokeratins in human breast carcinoma. J Pathol. 2004;203(2):661–671. doi: 10.1002/path.1559. [DOI] [PubMed] [Google Scholar]
- 25.Haughian JM, Pinto MP, Harrell JC, Bliesner BS, Joensuu KM, Dye WW, et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc Natl Acad Sci U S A. 2012 Feb 21;109(8):2742–2747. doi: 10.1073/pnas.1106509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003 Apr 1;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell. 2007 Nov 15;1(5):555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009 Feb 15;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, et al. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10(3):R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol. 2007 Feb;21(2):359–375. doi: 10.1210/me.2006-0337. [DOI] [PubMed] [Google Scholar]
- 31.Pink JJ, Bilimoria MM, Assikis J, Jordan VC. Irreversible loss of the oestrogen receptor in T47D breast cancer cells following prolonged oestrogen deprivation. Br J Cancer. 1996 Oct;74(8):1227–1236. doi: 10.1038/bjc.1996.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrne C, Fuchs E. Probing keratinocyte and differentiation specificity of the human K5 promoter in vitro and in transgenic mice. Mol Cell Biol. 1993 Jun;13(6):3176–3190. doi: 10.1128/mcb.13.6.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gordon DF, Woodmansee WW, Lewis SR, James RA, Wood WM, Ridgway EC. Cloning of the mouse somatostatin receptor subtype 5 gene: promoter structure and function. Endocrinology. 1999 Dec;140(12):5598–5608. doi: 10.1210/endo.140.12.7165. [DOI] [PubMed] [Google Scholar]
- 34.Yoo BH, Axlund SD, Kabos P, Reid BG, Schaack J, Sartorius CA, et al. A High-Content Assay to Identify Small-Molecule Modulators of a Cancer Stem Cell Population in Luminal Breast Cancer. J Biomol Screen. 2012 Jun 29; doi: 10.1177/1087057112452138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 36.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007 Mar;11(3):259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Shen T, Horwitz KB, Lange CA. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol. 2001 Sep;21(18):6122–6131. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cowan RA, Cowan SK, Grant JK. Binding of methyltrienolone (R1881) to a progesterone receptor-like component of human prostatic cytosol. J Endocrinol. 1977 Aug;74(2):281–289. doi: 10.1677/joe.0.0740281. [DOI] [PubMed] [Google Scholar]
- 39.Nordeen SK, Kuhnel B, Lawler-Heavner J, Barber DA, Edwards DP. A quantitative comparison of dual control of a hormone response element by progestins and glucocorticoids in the same cell line. Mol Endocrinol. 1989 Aug;3(8):1270–1278. doi: 10.1210/mend-3-8-1270. [DOI] [PubMed] [Google Scholar]
- 40.Ariazi EA, Leitao A, Oprea TI, Chen B, Louis T, Bertucci AM, et al. Exemestane's 17-hydroxylated metabolite exerts biological effects as an androgen. Mol Cancer Ther. 2007 Nov;6(11):2817–2827. doi: 10.1158/1535-7163.MCT-07-0312. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Wong J, Tsai SY, Tsai MJ, O'Malley BW. Progesterone and glucocorticoid receptors recruit distinct coactivator complexes and promote distinct patterns of local chromatin modification. Mol Cell Biol. 2003 Jun;23(11):3763–3773. doi: 10.1128/MCB.23.11.3763-3773.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011 Mar;17(3):313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 43.Musgrove EA, Lee CS, Sutherland RL. Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor alpha, epidermal growth factor receptor, c-fos, and c-myc genes. Mol Cell Biol. 1991 Oct;11(10):5032–5043. doi: 10.1128/mcb.11.10.5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 2010 Jun;15(2):235–252. doi: 10.1007/s10911-010-9175-z. [DOI] [PubMed] [Google Scholar]
- 45.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003 May 15;17(10):1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008 Mar 1;14(5):1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- 47.Boecker W, Buerger H. Evidence of progenitor cells of glandular and myoepithelial cell lineages in the human adult female breast epithelium: a new progenitor (adult stem) cell concept. Cell Prolif. 2003 Oct;36(Suppl 1):73–84. doi: 10.1046/j.1365-2184.36.s.1.7.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran CP, Lin C, Yamashiro J, Reiter RE. Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Mol Cancer Res. 2002;1(2):113–121. [PubMed] [Google Scholar]
- 49.Uzgare AR, Xu Y, Isaacs JT. In vitro culturing and characteristics of transit amplifying epithelial cells from human prostate tissue. J Cell Biochem. 2004 Jan 1;91(1):196–205. doi: 10.1002/jcb.10764. [DOI] [PubMed] [Google Scholar]
- 50.Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L, Gudjonsson T, Rank F, LaBarge MA, et al. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol. 2007 Apr 9;177(1):87–101. doi: 10.1083/jcb.200611114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011 May 10;108(19):7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011 Jan 25;108(4):1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim RS, Avivar-Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre-Ghiso JA, et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS One. 2012;7(4):e35569. doi: 10.1371/journal.pone.0035569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guedj M, Marisa L, de Reynies A, Orsetti B, Schiappa R, Bibeau F, et al. A refined molecular taxonomy of breast cancer. Oncogene. 2011 Jul 25; doi: 10.1038/onc.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010 Dec 14;107(50):21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke CL, Graham J, Roman SD, Sutherland RL. Direct transcriptional regulation of the progesterone receptor by retinoic acid diminishes progestin responsiveness in the breast cancer cell line T-47D. J Biol Chem. 1991 Oct 5;266(28):18969–18975. [PubMed] [Google Scholar]
- 57.Roman SD, Clarke CL, Hall RE, Alexander IE, Sutherland RL. Expression and regulation of retinoic acid receptors in human breast cancer cells. Cancer Res. 1992 Apr 15;52(8):2236–2242. [PubMed] [Google Scholar]
- 58.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol. 2011 Feb;226(2):322–330. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tulachan SS, Doi R, Kawaguchi Y, Tsuji S, Nakajima S, Masui T, et al. All-trans retinoic acid induces differentiation of ducts and endocrine cells by mesenchymal/epithelial interactions in embryonic pancreas. Diabetes. 2003 Jan;52(1):76–84. doi: 10.2337/diabetes.52.1.76. [DOI] [PubMed] [Google Scholar]
- 60.Baldassarre G, Boccia A, Bruni P, Sandomenico C, Barone MV, Pepe S, et al. Retinoic acid induces neuronal differentiation of embryonal carcinoma cells by reducing proteasome-dependent proteolysis of the cyclin-dependent inhibitor p27. Cell Growth Differ. 2000 Oct;11(10):517–526. [PubMed] [Google Scholar]
- 61.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 62.Koay DC, Zerillo C, Narayan M, Harris LN, DiGiovanna MP. Anti-tumor effects of retinoids combined with trastuzumab or tamoxifen in breast cancer cells: induction of apoptosis by retinoid/trastuzumab combinations. Breast Cancer Res. 2010;12(4):R62. doi: 10.1186/bcr2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zanardi S, Serrano D, Argusti A, Barile M, Puntoni M, Decensi A. Clinical trials with retinoids for breast cancer chemoprevention. Endocr Relat Cancer. 2006 Mar;13(1):51–68. doi: 10.1677/erc.1.00938. [DOI] [PubMed] [Google Scholar]
- 64.Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J, et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen dependent gene signatures. Breast Cancer Res Treat. 2012 doi: 10.1007/s10549-012-2164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santen RJ, Yue W, Heitjan DF. Modeling of the Growth Kinetics of Occult Breast Tumors: Role in Interpretation of Studies of Prevention and Menopausal Hormone Therapy. Cancer Epidemiol Biomarkers Prev. 2012 Jun 13; doi: 10.1158/1055-9965.EPI-12-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. P4-induced CK5+ cells are more invasive and sphere-initiating than CK5− cells. T47DK5pGFP cells were treated with vehicle (EtOH) or 100 nM P4 for 24 h and the GFP−/+ (CK5−/+) fractions isolated by FACS as described. A. Sorted cell fractions (50,000 each) were placed in invasion chambers for 24 h and the number of invading cells per field measured by Image J analysis (NIH). Picture denotes representative filters. B. Sorted cells (300 each) were placed in mammosphere/3D-Matrigel culture, and the number of spheres ≥50 µm measured by automated counter after seven days. Merged bright field/DAPI stained images of wells are shown.