

Abstract
Neuronal network hyperexcitability underlies the pathogenesis of seizures and is a component of some degenerative neurological disorders such as Alzheimer’s disease (AD). Recently, the microtubule binding protein tau has been implicated in the regulation of network synchronization. Genetic removal of Mapt, the gene encoding tau, in AD models overexpressing amyloid-beta (Aβ) decreases hyperexcitability and normalizes the excitation/inhibition imbalance. Whether this effect of tau removal is specific to Aβ mouse models remains to be determined. Here we examined tau as an excitability modifier in the non-AD nervous system using genetic deletion of tau in mouse and Drosophila models of hyperexcitability. Kcna1−/− mice lack Kv1.1 delayed rectifier currents and exhibit severe spontaneous seizures, early lethality, and megencephaly. Young Kcna1−/− mice retained wild-type levels of Aβ, tau, and tau phospho-Thr231. Decreasing tau in Kcna1−/− mice reduced hyperexcitability and alleviated seizure-related comorbidities. Tau reduction decreased Kcna1−/− video-EEG recorded seizure frequency and duration as well as normalized Kcna1−/− hippocampal network hyperexcitability in vitro. Additionally, tau reduction increased Kcna1−/− survival and prevented megencephaly and hippocampal hypertrophy, as determined by MRI. Bang-sensitive Drosophila mutants display paralysis and seizures in response to mechanical stimulation, providing a complementary excitability assay for epistatic interactions. We found that tau reduction significantly decreased seizure sensitivity in two independent bang-sensitive mutant models, kcc and eas. Our results indicate that tau plays a general role in regulating intrinsic neuronal network hyperexcitability independently of Aβ overexpression and suggest that reducing tau function could be a viable target for therapeutic intervention in seizure disorders and antiepileptogenesis.
Introduction
The microtubule binding protein tau is implicated in cytoskeletal and intracellular trafficking functions including microtubule stability, transport, and signal transduction (Dixit et al., 2008, Wang and Liu, 2008, Ittner et al., 2010). This multifaceted protein figures prominently in the pathogenesis of neurodegenerative disorders such as Alzheimer’s disease (AD) and frontotemporal dementia, where abnormally phosphorylated tau proteins aggregate to form neurofibrillary tangles (Goedert and Spillantini, 2006, Morris et al., 2011).
Although the underlying molecular mechanisms are unclear, recent studies have implicated tau in the regulation of excitability and synchronization of neuronal networks in AD mouse models. Genetic removal of tau decreases interictal spiking and spontaneous seizures in the J20 human amyloid precursor protein (hAPP) AD mouse, which overexpresses amyloid-beta (Aβ) (Roberson et al., 2011). In this model, tau knockout normalizes the inhibitory/excitatory imbalance and rescues both the early lethality and cognitive dysfunction (Roberson et al., 2007, Roberson et al., 2011). Tau protein is also linked to Aβ induced axonal transport deficits and synaptic long term potentiation alterations in hAPP AD mice, both of which are rescued by tau knockout (Vossel et al., 2010, Shipton et al., 2011). Expression of ApoE4, an AD risk factor allele, in mice also leads to network hyperexcitability (Hunter et al., 2012). ApoE4 mice exhibit GABAergic interneuron loss accompanied by learning and memory deficits due to high levels of toxic hyperphosphorylated tau that are rescued by tau reduction (Li et al., 2009, Andrews-Zwilling et al., 2010).
While the interaction between tau and network excitability is robust in AD models with Aβ or tau pathology, the ability of tau to modulate neuronal excitability in models without aberrant amyloid expression or tau hyperphosphorylation remains unknown. Tau knockout mice themselves show decreased seizure severity following activation by convulsants (Roberson et al., 2007, Ittner et al., 2010). If tau removal plays a more general role in limiting excessive network firing, the interaction could be a viable target for treatment of other central excitability disorders. To address this possibility, we examined the effect of genetically decreased tau expression in mouse and Drosophila ion channelopathy mutants. The Kcna1−/− mouse is a model of temporal lobe epilepsy bearing a null allele for the alpha subunit of the Kv1.1 voltage-gated potassium channel (Smart et al., 1998). Humans with loss-of-function mutations in this gene have hyperexcitability phenotypes including epilepsy, myokymia, and episodic ataxia (Adelman et al., 1995, Zuberi et al., 1999, Liguori et al., 2001). Kcna1−/− mice exhibit severe spontaneous seizures beginning in the third week of life that are accompanied by early lethality and megencephaly, making this model a robust test for the effects of tau on hyperexcitability (Smart et al., 1998, Glasscock et al., 2007, Persson et al., 2007). Bang-sensitive (BS) Drosophila mutants display behavioral seizure susceptibility following mechanical stimulation that is characterized by a period of paralysis followed by seizure-like limbshaking movements. BS mutants represent a genetically tractable model system for evaluating modifiers of human seizure disorders (Pavlidis et al., 1994, Kuebler and Tanouye, 2000, Kuebler et al., 2001, Parker et al., 2011).
Materials and Methods
Animals
Double-mutant mice were produced carrying various combinations of Kcna1- and Tau-alleles. Kcna1- mice carry a null mutation in the Kcna1 gene on chromosome 6 (Smart et al., 1998). Tau- mice contain a targeted knockout mutation in the Mapt gene on chromosome 11 (Dawson et al., 2001). Heterozygous F1 mice (Kcna1+/−Tau+/−), derived by crossing heterozygous Kcna1−/+ mice (Tac:N:NIHS-BC) with homozygous Tau−/−(C57BL/6) mice, were intercrossed to produce F2 double mutants of mixed Black Swiss (BlSw, Tac:N:NIHS-BC) and C57BL/6 background. Mice of either sex were used for all experiments unless otherwise stated. Mice were housed at 22°C with a 12 h light/dark cycle and fed ad libitum. All procedures were carried out in accordance with the guidelines of the National Institute of Health, as approved by the Animal Care and Use Committee of Baylor College of Medicine.
Genotyping
Genomic DNA was isolated from tail clips using DirectPCR Lysis Reagent (Viagen Biotech). Genotype was determined using PCR amplification for specific alleles as described previously for Kcna1- (Smart et al., 1998) and Tau- (Dawson et al., 2001). PCR products were separated on a 1.5% agarose gel by electrophoresis.
In vivo video-electroencephalography (EEG) recordings
Kcna1−/− mice bred with varying tau alleles (Tau+/+, Tau+/−, and Tau−/−) were anesthetized by Avertin and surgically implanted with bilateral silver wire electrodes (0.005 inch diameter) attached to a microminiature connector. EEG wires were inserted into the subdural space over the temporal cortex through cranial burr holes. Mice were allowed to recover for 24 or more hours before analysis. EEG and behavioral activity in freely moving mice were analyzed using simultaneous video-EEG monitoring (Haramonie software version 6.1c, Stellate Systems). All EEG signals were filtered using a 0.3 Hz high-pass filter, 70 Hz low-pass filter, and 60 Hz notch filter. Eight mice of each genotype were monitored at 4–6 weeks of age and were each evaluated for a total of 9 or more hours. Mice were recorded in one or more 3–9 hour monitoring sessions over several days to mitigate the effect of seizure clustering. Seizure activity defined by EEG waveform and corresponding video-recorded behavior was quantified by visual inspection.
Hippocampal slice preparation and electrophysiology
Transverse hippocampal slices (300 μm thickness) from double-mutant mice of genotypes Kcna1+/+Tau+/+, Kcna1+/+Tau−/−, Kcna1−/−Tau+/+, and Kcna1−/−Tau−/− (6–11 weeks old) were prepared using a vibrotome. Slices were sectioned in cutting solution containing (in mM) 100 sucrose, 30 NaCl, 3 KCl, 0.5 CaCl2, 28 NaHCO3, 7 MgCl2, 1.4 NaH2PO4, and 11 D-glucose and were constantly gassed with 95%O2/5%CO2 to maintain a constant pH of 7.4. After incubation in artificial CSF (ACSF) at 32°C for 1 h, slices were transferred into a submerged recording chamber for electrophysiological recordings. Brain slices were constantly perfused with ACSF containing (in mM) 125 NaCl, 2.5 KCl, 2 CaCl2, 25 NaHCO3, 1 MgCl, 1.25 NaH2PO4, and 11 D-glucose. Recording chamber temperature was controlled at 32–33°C. Recording pipettes (4–6 Mohm) were pulled from borosilicate glass and filled with 2M NaCl. CA3 pyramidal neurons have been previously shown to exhibit altered in vitro network excitability in Kcna1−/− mice (Smart et al., 1998, Lopantsev et al., 2003, Glasscock et al., 2007). Therefore, field recordings were made from the CA3 pyramidal somata identified visually in the stratum pyramidale using a Getting Instruments Model 5A amplifier, digitized by a Digidata 1322A and collected using Clampex (Molecular Devices). A low pass filter was set at 5 kHz. Slices were perfused with ACSF containing 7.5 mM KCl (Glasscock et al., 2007) and began synchronously discharging within a period of 15–25 minutes. To calculate the burst frequency, slices were allowed to equilibrate to 7.5 mM KCl for an additional 5–15 minutes and the burst frequency was then calculated over a 5 minute period. The burst duration was defined as the interval between baseline crossings and analyzed as the average of 10 bursts per slice. Data analysis was performed using Clampfit (Molecular Devices) and Origin 7.5 (OriginLab).
3-Dimensional Magnetic Resonance Imaging (3D-MRI) and brain volumetry
MRI of the brain was performed on 12-week-old male mice of genotypes Kcna1−/−Tau+/+, Kcna1−/−Tau−/−, Kcna1+/+Tau+/+, and Kcna1+/+Tau−/− (n=3). Mice were overdosed with isoflourane, placed in a 50mL conical tube, and all imaged identically within 1 h. To mitigate post mortem delay, mice were cooled to 20°C during imaging. MRI images were obtained using a 9.4T, 21 cm bore horizontal scanner with a 35 mm volume resonator (Bruker, BioSpin). The imaging parameters used to obtain three dimensional (3D) rapid acquisition with relaxation enhancement (RARE) images of the mouse brain were as follows: TR = 2000 ms; TE = 11.713 ms; Effective TE = 46.85 ms; FOV = 25 mm3; matrix = 256 × 256 × 164; RARE Factor = 8; Number of Averages = 5; Total Scan Time = 14 h 34 m 40 s. Images were obtained using Paravision software version 4 (Bruker, BioSpin).
MRI images were analyzed while blinded to genotype using Amira 3.1 software (Visage Imaging). The hippocampal border was segmented manually in both coronal and sagittal planes and the hippocampus volume measurement for each mouse was computed as the average of the volumes in the two planes. The forebrain volume was segmented in the sagittal plane.
Fly stocks and behavioral testing
Drosophila melanogaster were maintained on standard media at 25–26°C. The bang-sensitive (BS) strains used were easily shocked (eas), which encodes an ethanolamine kinase, and kazachoc (kcc), which encodes a K+/Cl− cotransporter, and were obtained from M. Tanouye at University of California, Berkley (Pavlidis et al., 1994, Hekmat-Scafe et al., 2006). The BS alleles used in this study were easPC80 and kccDHS1. Tau P-element (tauEP3203) and deficiency (Df(3R) MR22) alleles were obtained from Bloomington Stock Center and D. St. Johnston at University of Cambridge, respectively (Doerflinger et al., 2003). Male and female kcc flies and male eas flies were analyzed for bang sensitivity 1–2 days post eclosion and not exposed to CO2 within 2 hours preceding testing. To test, no more than 10 flies were placed in a clean vial and allowed to rest for 30 minutes. Flies were vortexed (VWR) at maximum strength for 10 seconds and visually monitored for the presence of paralysis and seizure in each fly.
Western blotting
Kcna1−/− and Kcna1+/+ forebrain hemisphere samples (4.5 weeks old) were extracted and flash frozen in liquid nitrogen. Samples were homogenized on ice using a Tissue Tearor (VWR) in lysis buffer with phosphatase inhibitors (20 mM Tris, pH 7.5, 138 mM NaCl, 3 mM KCl, 1% Tx-100, 1 mM EGTA, 2 mM EDTA, 1 mM Benzamidine, 5 μg/ml Aprotinin, 5 μg/ml Leupeptin, 5 μg/ml Pepstatin A, 1 mM PMSF, 1 mM DTT, 50 mM NaF, 1 mM Na3VO4) and protein concentration determined by Pierce BCA Protein Assay Kit (Thermo Scientific). 20 μg of protein was separated on 12% Tris-Glycine-SDS polyacrylamide gels (Thermo Scientific) and transferred to nitrocellulose membrane. Membranes were blocked overnight at 4°C in 5% milk in TBST, and incubated with primary antibodies including mouse anti-Tau (Tau-5, 1:1000, Millipore), rabbit anti-Tau phospho-Threonine 231 (Tau-pT231, 1:1000, Millipore), and mouse anti-GAPDH (6C5, 1:5000, Advanced ImmunoChemical) for 2 h at room temperature. Membranes were then rinsed in TBST and incubated with appropriate HRP conjugated secondary antibody, either donkey anti-mouse or goat anti-rabbit (1:10,000, Santa Cruz Biotechnology), for 1 h at room temperature. Protein was detected using SuperSignal chemiluminescent substrate (Pierce Thermo Scientific) and quantified by ImageJ (NIH). Membranes were stained sequentially for Tau-5 and GAPDH, stripped in stripping buffer (2% SDS, 100 mM β-mercaptoethanol, 50 mM Tris, pH 6.8) at 50°C for 30 minutes, and stained for Tau-pT231. Results were replicated 3 times to ensure accuracy.
Aβ ELISA
Forebrain hemispheres were sonicated in 0.2% diethylamine (DEA) in 50 mM NaCl with protease inhibitor (Sigma) at a volume of 1 ml per 100 mg of tissue. Samples were spun at 100,000 x g (53,000 rpm) for 30 minutes at 4°C and supernant collected, neutralized in 0.5 M Tris-HCl (pH 6.8), and analyzed by ELISA as previously described (Kawarabayashi et al., 2001). The pellet was then sonicated in 70% formic acid, centrifuged, and supernant analyzed by ELISA. Mouse Aβ levels were analyzed in Kcna1−/− and Kcna1+/+ mice using capture antibody BNT77, which specifically recognized rodent Aβ11–28, and BA27 and BC05 antibodies to detect Aβ40 and Aβ42, respectively.
Statistical analysis
Data was analyzed for statistical significance using SPSS 16.0 (IBM). Survival analysis was completed by Kaplan-Meier log rank (Mantel-Cox). One-way ANOVA, with Tukey post hoc when necessary, was used to compare tau protein expression, abnormal EEG discharge duration, hippocampal volume, and forebrain volume between genotypes. Comparisons of seizure frequency between genotypes were made using the non-parametric Kruskal-Wallis test. Electrophysiology data was analyzed by One-way ANOVA with Bonferroni post hoc and Aβ level comparisons made by t-test (two-tailed) using GraphPad Prism 5.0 (Graphpad Software). All analysis of Drosophila results was completed by Chi-Squared analysis. Error bars represent standard deviation (SD) unless otherwise stated with the exception of in vitro electrophysiology results, which are reported as standard error of the mean (SEM).
Results
Young Kv1.1-deficient mice maintain wild-type Aβ, tau and tau phospho-Thr231 protein levels in the forebrain
Since electrically induced seizures are known to acutely stimulate Aβ secretion, and kainic acid injections induce tau phosphorylation in mice, we first evaluated the Kcna1−/− brain for evidence of AD-like molecular pathology (Cirrito et al., 2005, Crespo-Biel et al., 2007, Liang et al., 2009). To determine whether chronic spontaneous seizures in young Kcna1−/− mice alter Aβ levels or tau levels and phosphorylation, 4.5-week-old Kcna1−/− and Kcna1+/+ (n=8) littermate mouse forebrain samples were analyzed by ELISA for Aβ40 and Aβ42 as well as western blot for total tau (Tau-5) and tau phosphorylated at threonine 231 (Tau-pT231). ELISA analysis showed that soluble, DEA-extracted Aβ40, Aβ42, and total Aβ levels in young Kcna1−/− mice were not significantly different from Kcna1+/+ wild-type controls (Aβ40: Kcna1−/−: 108.44 ± 8.26 pmol, Kcna1+/+: 96.91 ± 14.84 pmol, p=0.08, Aβ42: Kcna1−/−: 27.07 ± 3.05 pmol, Kcna1+/+: 24.28 ± 3.60 pmol, p=0.12, total Aβ: Kcna1−/−: 135.51 ± 11.22 pmol, Kcna1+/+: 121.19 ± 18.37 pmol, p=0.08, n=8). There was also no difference in the ratio of Aβ40/Aβ42 between Kcna1−/− and wild-type mice (Kcna1−/−: 4.02 ± 0.18, Kcna1+/+: 3.99 ± 0.13, p=0.69, n=8). No aggregated Aβ was detected in formic acid isolated samples from mice of either genotype.
Next, we analyzed Tau levels and phosphorylation at threonine 231. We selected this phosphorylation site since Thr231 phosphorylation is increased within 3 days after kainic acid-induced status epilepticus in mice, but the effect of spontaneous seizures on this site is not currently known (Crespo-Biel et al., 2007). Additionally, tau phosphorylation at Thr231 occurs early in the pre-NFT stage of human AD pathogenesis, and a high level of tau phospho-Thr231 in patients with mild cognitive impairment correlates with subsequent development of AD (Augustinack et al., 2002, Buerger et al., 2002), making it an informative biomarker to test for early changes in tau phosphorylation status in young Kv1.1-null mice. Western blot analysis (Fig. 1A) showed that Kcna1−/− total tau and tau phospho-Thr231 levels, normalized to GAPDH, were not significantly different from those in wild-type Kcna1+/+ mice (Fig. 1B, Tau-5: Kcna1+/+: 1.84 ± 0.74, Kcna1−/−: 1.58 ± 0.70, p=0.49, Tau-pT231: Kcna1+/+: 1.76 ± 0.52, Kcna1−/−: 1.34 ± 0.42, p=0.10, n=8). The ratio of tau phospho-Thr231 to total tau also did not significantly differ between Kcna1−/− and Kcna1+/+ mice (Kcna1+/+: 1.06 ± 0.32, Kcna1−/−: 0.92 ± 0.23, p=0.33, n=8). These results demonstrate that hyperexcitability and spontaneous epileptic seizures in the forebrain of young Kv1.1-null mice do not appreciably affect Aβ and tau levels, or induce sustained abnormal tau phosphorylation at Thr231. Thus, young Kcna1−/− mice do not display signs of AD-like molecular pathology despite the occurrence of spontaneous seizures.
Figure 1.

Tau protein levels and phosphorylation at Thr231 in 4.5-week-old Kcna1−/− mouse forebrain do not significantly differ from Kcna1+/+ mice. (A) Representative western blots of total tau (Tau-5), tau phospo-Thr231 (Tau-pT231), and GAPDH loading control for Kcna1−/− and Kcna1+/+ mice, as well as a tau knockout, which showed no tau or tau phospho-Thr231 staining. (B) Quantification of Tau-5 and Tau-pT231 normalized to GAPDH showed no significant difference in total tau or tau phospho-Thr231 levels between Kcna1−/− and Kcna1+/+ mice. p>0.05; One-way ANOVA; n=8; error bars represent SD.
Decreasing tau reduces hyperexcitability in Kv1.1-deficient mice
To determine if tau loss affects hyperexcitability and spontaneous seizures we recorded Kcna1−/− Tau double-mutant male and female mice (4–6 weeks old) with in vivo video-EEG for 9 or more hours. Electrocorticograms of Kcna1−/− mice showed frequent and severe spontaneous seizures characterized by abnormal, generalized, high-frequency hypersynchronous EEG discharges (200–500 mV amplitude) with abrupt cessation, followed by a period of EEG amplitude depression (Fig. 2A). Seizures in Kcna1−/− mice occurred up to 1.27 times an hour with an average of 0.51 ± 0.19 seizures/hour (Fig. 2B, ±SEM). Decreasing tau by heterozygous and homozygous tau knockout significantly decreased seizure frequency in Kcna1−/− mice to 0 and 0.03 ± 0.03 seizures/hour, respectively (Fig. 2B, ±SEM, Kcna1−/−Tau+/+ vs. Kcna1−/−Tau+/−: p=0.011, Kcna1−/−Tau+/+ vs. Kcna1−/−Tau−/−: p=0.035, n=8). None of the 8 Kcna1−/−Tau+/− mice and only 1 of 8 Kcna1−/−Tau−/− mice displayed seizures during recording, compared to 5 of 8 Kcna1−/−Tau+/+ mice (Fig. 2B). Individual Kcna1−/−Tau+/+ mice showed a variable range of seizure frequency that was not due to sex or litter. The range was most likely due to phenotypic variation of the Kcna1−/− model within a mixed background (C57Bl/6/BlSw). Importantly, this variability was not observed in Kcna1−/−Tau+/− or Kcna1−/−Tau−/− mice, which both had significantly decreased seizure frequency compared to Kcna1−/−Tau+/+.
Figure 2.
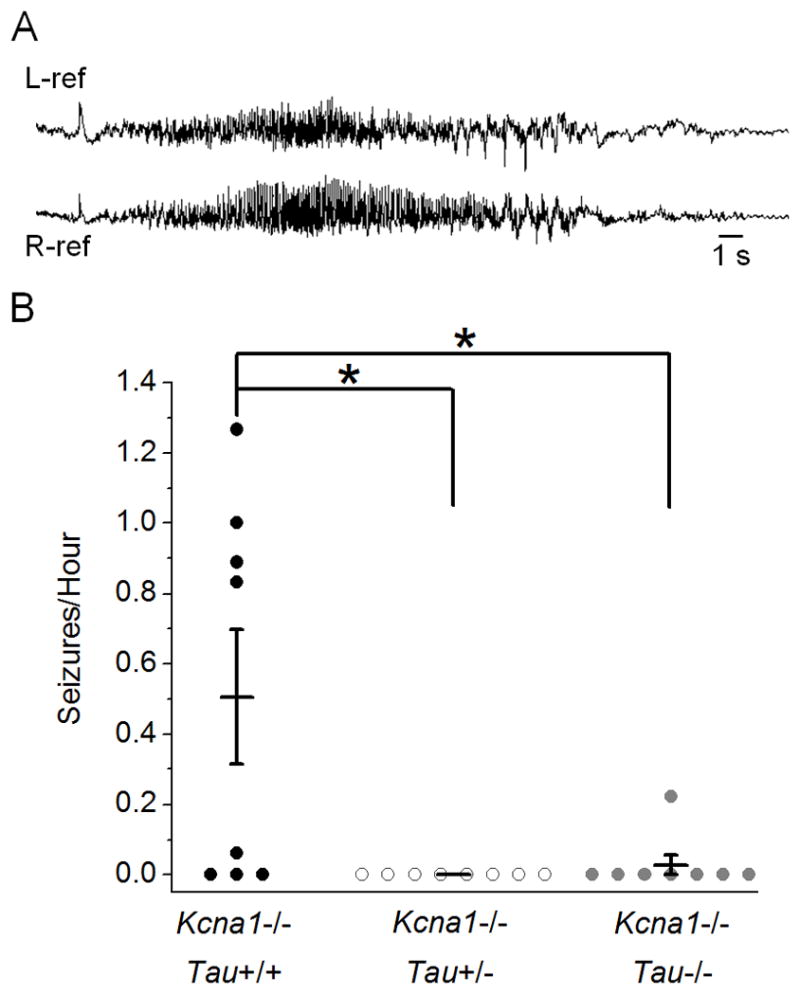
Tau reduction significantly decreases seizure frequency in Kcna1−/− mice. (A) Representative spontaneous cortical seizure recorded bilaterally in a Kcna1−/− mouse during chronic in vivo EEG monitoring. (B) Analysis of seizures/hour in double-mutant mice recorded for 9 or more hours. Both tau reduction and loss significantly decreased seizure frequency in Kcna1−/−Tau+/− (n=8) and Kcna1−/−Tau−/− (n=8) double mutants compared to Kcna1−/−Tau+/+ (n=8). Seizures were observed in 5/8 Kcna1−/−Tau+/+ mice, 0/8 Kcna1−/−Tau+/− mice, and only 1/8 Kcna1−/−Tau−/− mice. Total deletion of tau reduced the average seizure frequency by over 94%. * p<0.05; Kruskal-Wallis; error bars represent SEM.
In addition to decreasing the number of behavioral seizures in Kcna1−/− mice, tau loss also significantly decreased the duration of abnormal electrographic cortical discharge activity by approximately 60%. This measure includes Kcna1−/− seizures (Fig. 2A) as well as abnormal spike bursts that consist of repetitive, distinct spikes and/or spike-wave complexes occurring at a maximum frequency of 3–9 Hz. These aberrant electrographic discharges in Kcna1−/−Tau+/+ mice averaged 43 ± 32 seconds in duration (n=54) compared to Kcna1−/−Tau−/− mice which averaged only 18 ± 8 seconds (n=11) (p=0.013). Taken together, these results demonstrate that decreasing tau dosage, even to heterozygous levels, can decrease cortical hyperexcitability in Kcna1−/− seizure mutants.
Kcna1−/− mice have altered in vitro network excitability in the CA3 pyramidal region of the hippocampus when exposed to elevated potassium levels (Smart et al., 1998, Lopantsev et al., 2003, Glasscock et al., 2007). To further examine the effect of tau loss on hyperexcitability, we looked at in vitro hippocampal network excitability in the CA3 pyramidal region of Kcna1Tau double mutants. When extracellular K+ was raised from 2.5 mM to 7.5 mM, we observed spontaneous discharges that stabilized at frequencies after 5–15 minutes in all slices, but frequency and duration of bursts varied by genotype. Kcna1−/−Tau+/+ burst, on average, almost 3 times faster than Kcna1+/+Tau+/+ wild-type controls (Fig. 3A, B, Kcna1−/−Tau+/+: 0.52 ± 0.04 Hz, n=22, Kcna1+/+Tau+/+: 0.17 ± 0.02 Hz, n=17, p<0.0001). Conversely, burst duration was significantly decreased in Kcna1−/−Tau+/+ mice compared to Kcna1+/+Tau+/+ controls (Fig. 3C, D, Kcna1−/−Tau+/+: 94.4 ± 5.0 ms, n=22, Kcna1+/+Tau+/+: 118.6 ± 5.4 ms, n=17, p<0.05).
Figure 3.

Tau loss decreases network hyperexcitability in Kcna1−/− hippocampal slices exposed to increased extracellular K+ levels. (A) Spontaneous discharges in CA3 pyramidal cells were observed in 6–11 week-old mouse brain slices when K+ was raised from 2.5 to 7.5 mM. Representative 30 second traces illustrate differences in burst frequency between genotypes. (B) Analysis of burst frequency during 5 minute periods of spontaneous bursting per slice. Kcna1−/−Tau+/+ hippocampus (n=22) had significantly increased burst frequency compared to Kcna1+/+Tau+/+ wild-type controls (n=17), while Kcna1−/−Tau−/− slices (n=19) were not significantly different from Kcna1+/+Tau+/+. Kcna1+/+Tau−/− burst frequency (n=15) was also indistinguishable from wild type or Kcna1−/−Tau−/− double mutants. (C) Representative 200 ms traces illustrate burst duration differences between genotypes, as defined by baseline crossings (arrows). (D) Quantification of burst duration in 10 spontaneous bursts per brain slice. Kcna1−/−Tau+/+ hippocampus (n=22) generated significantly shorter bursts than Kcna1+/+Tau+/+ (n=17), Kcna1+/+Tau−/− (n=15), and Kcna1−/−Tau−/− slices (n=19), which were not significantly different. Loss of tau normalized Kcna1−/− spontaneous burst frequency and duration to wild-type levels. * p<0.05, ** p<0.01, *** p<0.0001; One-way ANOVA; error bars represent SEM.
Tau loss significantly decreased burst frequency in Kcna1−/− mice and Kcna1−/−Tau−/− mice were not significantly different from Kcna1+/+Tau+/+ controls. Tau loss in Kcna1−/− mice reduced burst frequency to wild-type levels (Fig. 3A, B, Kcna1−/−Tau−/−: 0.26 ± 0.03 Hz, n=19, vs. Kcna1−/−Tau+/+: p<0.0001, vs. Kcna1+/+Tau+/+: p>0.05). Interestingly, tau loss did not alter burst frequency in wild-type slices (Fig. 3A, B, Kcna1+/+Tau−/−: 0.18 ± 0.02 Hz, n=15, vs. Kcna1+/+Tau+/+: p>0.05, vs. Kcna1−/−Tau−/−: p>0.05). Similarly, burst duration in Kcna1−/−Tau−/− mice is significantly increased compared to Kcna1−/−Tau+/+ and not significantly different from Kcna1+/+Tau+/+ controls. Loss of tau rescued burst duration in Kcna1−/− mice to wild-type levels (Fig. 3C, D, Kcna1−/−Tau−/−: 134.8 ± 8.1 ms, n=19, vs. Kcna1−/−Tau+/+: p<0.0001, vs. Kcna1+/+Tau+/+: p>0.05). Kcna1+/+Tau−/− burst duration was also not significantly different from Kcna1+/+Tau+/+ controls or Kcna1−/−Tau−/− mice (Fig. 3C, D, Kcna1+/+Tau−/−: 123.8 ± 5.8 ms, n=15, vs. Kcna1+/+Tau+/+: p>0.05, vs. Kcna1−/−Tau−/−: p>0.05). These results demonstrate that tau loss can prevent in vitro hippocampal network hyperexcitability in the epileptic Kcna1−/− brain, but does not alter wild-type hippocampal excitability.
Decreasing tau dosage increases survival in Kv1.1-deficient mice
Kv1.1-null mice die prematurely and are an established model of sudden unexpected death in epilepsy (SUDEP) (Glasscock et al., 2010, Glasscock et al., 2012). Since tau loss in Kcna1−/− mice decreased cortical hyperexcitability and seizures, we examined whether tau loss could prolong lifespan in Kcna1−/− mice by daily monitoring of Kcna1−/−Tau double-mutant survival. Kcna1−/−Tau+/+ mice (n=27) died prematurely beginning in the third week of life with only 30% surviving to 10 weeks (Fig. 4). However, when tau dosage is decreased by heterozygous tau deletion, Kcna1−/−Tau+/− mice (n=37) had a significant two-fold increase in survival, with 59% alive at 10 weeks of age (Fig. 4, p=0.013). Kcna1−/−Tau−/− mice (n=23) exhibited an even more striking decrease in mortality rate, with 74% surviving until 10 weeks (Fig. 4, p=0.003). This increase in survival was also demonstrated in video-EEG monitoring studies, where episodes of status epilepticus followed by death were observed in Kcna1−/−Tau+/+ and Kcna1−/−Tau+/− mice but not Kcna1−/−Tau−/− mice while undergoing video-EEG recording. These results support a robust role for tau loss in suppressing central hyperexcitability, and demonstrate the positive effect of decreasing tau dosage, even by 50%, on seizures and premature lethality.
Figure 4.

Decreasing tau dosage significantly increases survival of Kcna1−/− mice. Kaplan-Meier survival curves show that Kcna1−/−Tau+/+ mice (n=27) exhibited severe early lethality beginning in the third week of life, with only 30 percent surviving to ten weeks of age. Decreasing tau dosage in Kcna1−/− mice by heterozygous (n=37) and homozygous (n=23) tau knockout significantly increased survival, with 59% and 74% survival, respectively, to ten weeks of age. * p=0.013, ** p=0.003, all significant with respect to Kcna1−/−Tau+/+; Kaplan-Meier log rank test.
Tau loss prevents megencephaly in Kv1.1-deficient mice
In addition to seizures and early lethality, Kcna1−/− mice exhibit abnormal enlargement of the hippocampus and ventral cortex (Persson et al., 2007). To determine if tau loss can influence megencephaly in Kcna1−/− mice, double-mutant mouse brains were imaged by MRI and analyzed for total hippocampal and forebrain volume (Fig. 5). At 12 weeks of age, Kcna1−/− mice had noticeably larger hippocampal and forebrain volumes that were significantly increased relative to Kcna1+/+Tau+/+ controls by 43.3% and 33.1%, respectively (Fig. 5Q, R, hippocampus: Kcna1−/−Tau+/+: 31.65 ± 1.42 mm3, Kcna1+/+Tau+/+: 22.08 ± 2.48 mm3, p=0.001, forebrain: Kcna1−/−Tau+/+: 379.86 ± 13.24 mm3, Kcna1+/+Tau+/+: 285.38 ± 27.64 mm3, p=0.001, n=3). Removal of tau resulted in a significant decrease in both hippocampal and forebrain volume by 25.7% and 22.4%, respectively, for Kcna1−/−Tau−/− mice compared to Kcna1−/−Tau+/+ (Fig. 5Q, R, Kcna1−/−Tau−/−: hippocampus: 23.53 ± 2.01 mm3, p=0.003, forebrain: 294.59 ± 20.88 mm3, p=0.003, n=3). Kcna1−/−Tau−/− hippocampal and forebrain volumes did not differ from Kcna1+/+Tau−/− or Kcna1+/+Tau+/+ controls (Fig. 5Q, R, hippocampus: Kcna1−/−Tau−/− vs. Kcna1+/+Tau−/−: p=0.43, Kcna1−/−Tau−/− vs. Kcna1+/+Tau+/+: p=0.78, forebrain: Kcna1−/−Tau−/− vs. Kcna1+/+Tau−/−: p=0.49, Kcna1−/−Tau−/− vs. Kcna1+/+Tau+/+: p=0.93, n=3). Loss of tau rescued Kcna1−/− megencephaly and decreased brain volume to wild-type levels. This observed decrease in volume was not due to tau loss alone, since hippocampal and forebrain volumes in Kcna1+/+Tau−/− mice did not differ from those of Kcna1+/+Tau+/+ wild-type controls (Fig. 5Q, R, Kcna1+/+Tau−/−: hippocampus: 21.07 ± 1.30 mm3, p=0.91, forebrain: 271.35 ± 10.87 mm3, p=0.81, n=3). Light microscopic examination of cresyl violet-stained sections revealed that tau loss had no discernible neurocytological effects on regional brain structure or organization, including cortical or hippocampal lamination (not shown). These findings suggest that tau loss not only decreases network hyperexcitability, but also prevents megencephaly associated with epilepsy in the Kv1.1 model.
Figure 5.
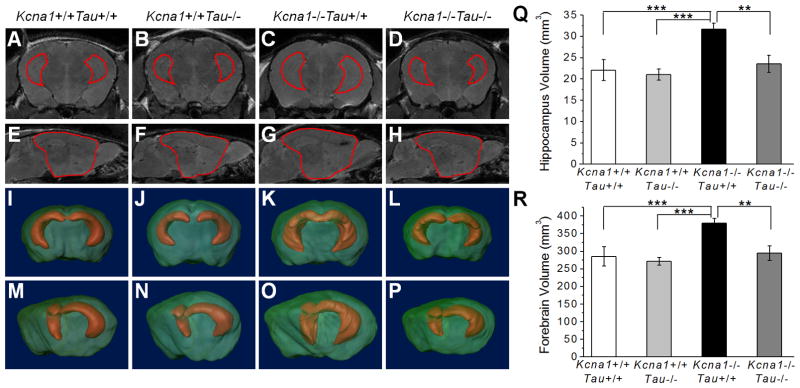
Tau loss prevents megencephaly in Kcna1−/− mouse brain. (Left) MRI images of Kcna1+/+Tau+/+, Kcna1+/+Tau−/−, Kcna1−/−Tau+/+ and Kcna1−/−Tau−/− mouse brain were manually segmented and hippocampus (A–D) and forebrain (E–H) borders drawn to allow 3D reconstruction. 3D reconstructions shown in both coronal (I–L) and partial sagittal (M–P) views demonstrate the enlargement of the hippocampus and forebrain in Kcna1−/−Tau+/+ (K, O) mice relative to other genotypes. (Right) Quantification of hippocampus (Q) and forebrain (R) volume in double-mutant mice. Kcna1−/−Tau−/− mice had significantly decreased hippocampus and forebrain volume compared to the megencephalic Kcna1−/−Tau+/+. Loss of tau in Kcna1−/− mice decreased hippocampus and forebrain volume to wild-type levels. ** p=0.003, *** p=0.001; One-way ANOVA; n=3; error bars represent SD.
Reducing tau decreases hyperexcitability in bang-sensitive Drosophila mutants
Bang-sensitive (BS) Drosophila mutants define a class of functional excitability phenotypes that display behavioral seizure susceptibility following mechanical or electrical shock. Upon stimulation, these mutants exhibit intense activation (limbshaking) followed by a period of paralysis and a second period of hyperexcitability characterized by uncoordinated seizure-like movements (Pavlidis et al., 1994), and are an established model system for human seizure disorders (Kuebler and Tanouye, 2000, Kuebler et al., 2001, Song and Tanouye, 2008). To determine if tau reduction is sufficient to decrease hyperexcitability in Drosophila BS mutants, double-mutant flies generated by decreasing tau gene expression in several different BS models were tested for paralysis and seizure phenotypes following 10 seconds of a vortex stimulus. Drosophila with homozygous tau deficiency are non-viable. Therefore, to reduce tau levels in Drosophila two previously reported alleles were used. The first, a P-element disruption of the tau gene, tauEP3203, has a P-element insertion in the first intron of the gene (Doerflinger et al., 2003). The second is a tau deficiency allele, Df(3R)MR22 (MR22), which has a 62 kb deletion caused by recombination of EP3203, removing most of the tau locus and resulting in homozygous lethality (Doerflinger et al., 2003).
Kcc BS Drosophila (kcc DHS1) carry a partial loss-of-function mutation in the K+/Cl−cotransporter gene kazachoc (Hekmat-Scafe et al., 2006). When tau was reduced in kcc flies by tauEP3203, the percentage of flies that exhibited bang-sensitive behavior and paralysis was significantly decreased by over 40% with homozygous tauEP3203 and 34% with tauEP3203/+ (Fig. 6, kcc;+/+: 47% BS, kcc; tauEP3203: 28% BS, kcc; tauEP3203/+: 31% BS, kcc;+/+ vs. kcc;tauEP3203: p<0.01, kcc;+/+ vs. kcc;tauEP3203/+: p<0.05, n≥ 87). Additionally, we decreased tau levels in the more seizure-sensitive eas mutant, which displays hyperexcitability due to reduced ethanolamine kinase activity, using the deficiency allele MR22 (Pavlidis et al., 1994, Doerflinger et al., 2003). Tau reduction by MR22 significantly decreased bang sensitivity in eas Drosophila by over 13% in eas;MR22/+ and 15% in eas;MR22/tauEP3203 flies (Fig. 6, eas;+/+: 97% BS, eas;MR22/+: 84% BS, eas;MR22/tauEP3203: 82% BS, p<0.001, n≥98). Tau reduction therefore decreased bang sensitivity in multiple Drosophila models, suggesting a broader role of tau in regulation of hyperexcitability due to a variety of molecular pathologies.
Figure 6.
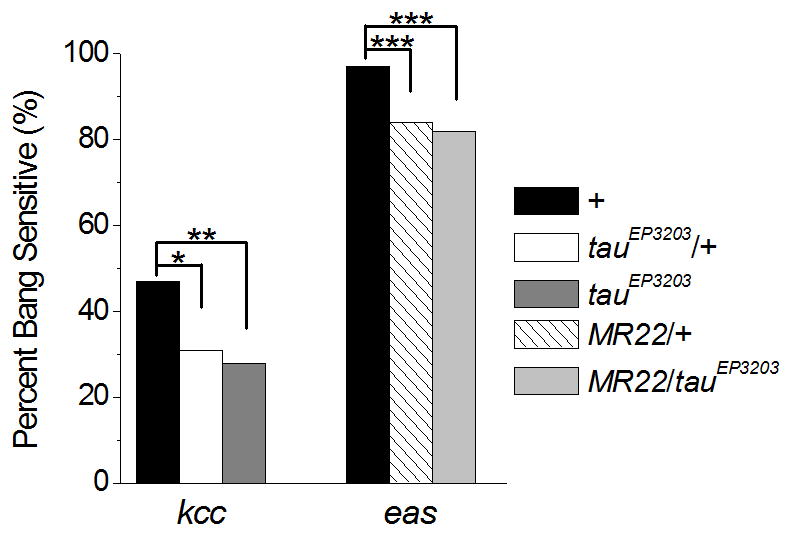
Tau reduction significantly decreases hyperexcitability in bang-sensitive Drosophila mutants kcc and eas. Quantification of the percentage of flies that were bang sensitive in response to a vortex stimulus. Decreasing tau dosage by the tauEP3203 allele significantly decreased bang sensitivity in kcc mutants by 34% in kcc;tauEP3203/+ and 40% in kcc;tauEP3203 compared to kcc flies with wild-type tau (n ≥87). Decreasing tau by the deficiency allele MR22 significantly decreased bang sensitivity in eas mutants by 13% (eas;MR22/+) and 15% (eas;MR22/tauEP3203) compared to eas flies with wild-type tau (n ≥98). * p<0.05, ** p<0.01, *** p<0.001; Chi-squared; Genotypes: kccDHS1, kccDHS1;tauEP3203/+, kccDHS1;tauEP3203, easPC80/Y, easPC80/Y;Df(3R)MR22/+, easPC80/Y;Df(3R)MR22/tauEP3203.
Discussion
The removal of tau and consequent suppression of epilepsy in AD mouse models established a critical epistatic role for tau in regulating cortical network excitability (Ittner et al., 2010, Roberson et al., 2011). Here we demonstrate that the beneficial effects of tau loss on aberrant synchronization extend beyond the prevention of Aβ induced network excitability to encompass hyperexcitability phenotypes due to other, non-AD related causes. Genetic reduction of tau dosage, even to heterozygous levels, significantly decreased spontaneous seizure frequency and duration in the Kcna1−/− mouse model of temporal lobe epilepsy. Tau knockout also decreased Kcna1−/− hippocampal network hyperexcitability in response to elevated K+ levels in vitro. In addition to attenuating hyperexcitability, tau loss decreased the early brain and vagal nerve-driven lethality in this model (Glasscock et al., 2010, Glasscock et al., 2012) and prevented Kv1.1-related megencephaly, with hippocampal and forebrain volumes decreased to wild-type levels. These findings demonstrate a robust and beneficial effect of tau removal on hyperexcitability-related brain phenotypes in the absence of AD-related molecular pathology. As further confirmation, we also found a decrease in experimentally evoked hyperexcitability behaviors in Drosophila kcc and eas BS mutants when tau levels were reduced, consistent with findings that tau loss decreases chemically evoked seizure severity in mice (Roberson et al., 2007, Ittner et al., 2010).
Aβ, tau, and network excitability in young Kcna1−/− mice
Hyperphosphorylation of tau at Thr231 is an early event in the progression of tau phosphorylation and NFT production in the human AD brain (Augustinack et al., 2002). Although only one of many tau phosphorylation sites, Thr231 phosphorylation is increased in response to kainic acid-induced seizures, and is thus a suitable indicator of tau phosphorylation in our spontaneous seizure mouse model (Crespo-Biel et al., 2007). Additionally, electrically induced synaptic activity and seizures stimulates Aβ secretion (Cirrito et al., 2005). Despite chronic spontaneous seizures, young Kcna1−/− mice had no changes in forebrain Aβ production or tau levels and phosphorylation at Thr231. Therefore, rescue of the Kcna1−/− hyperexcitability phenotype by tau loss in 4–6 week-old mice is unlikely due to prevention of a neurotoxic hyperphosphorylated tau species, as seen in AD ApoE4 knockin mice, but rather to a loss or reduction of an intrinsic molecular function of tau (Andrews-Zwilling et al., 2010). Transgenic hAPP mice, including the J20 model, also lack hyperphosphorylated tau yet are protected from hyperexcitability by tau loss, supporting the possibility that tau regulates excess excitability in developing or adult brain (Roberson et al., 2007, Ittner et al., 2010, Roberson et al., 2011).
Tau loss alleviates downstream seizure-induced comorbidities
Longevity
Tau removal increases survival in two hAPP transgenic mouse strains as well as kainic acid injection seizure models (Roberson et al., 2007, Ittner et al., 2010, Roberson et al., 2011). Our results show that tau loss is similarly effective in a genetic model of epilepsy. Even heterozygous reduction of tau was sufficient to significantly increase survival in the Kcna1−/− mutant, as it did in hAPP AD models. In contrast, tau deletion has no effect on survival of SOD1 mutants, a model of ALS with excitotoxic neurodegeneration, indicating that tau loss does not confer resistance to premature mortality in all neurological disorders (Roberson et al., 2011).
Megencephaly
A dramatic enlargement of the hippocampus and ventral cortex in Kcna1−/− BALB. C3HeB mice is detectable by 12 weeks of age (Persson et al., 2007). We confirmed this pathology in Kcna1−/− mutants on a different genetic background (mixed C57Bl/6/BlSw), where we found a 43% and 33% increase in hippocampal and forebrain volume, respectively, compared to wild-type mice. Since the original megenchephaly (Mceph) mouse model containing a spontaneous Kcna1 truncation mutation shows a comparable increase in brain volume, the megencephaly phenotype is likely caused by loss of Kcna1 function (Persson et al., 2007). The Mceph mutation increases neuronal and astrocytic proliferation, leading to slow postnatal brain enlargement (Almgren et al., 2007, Yang et al., 2012). This hyperplasia can be suppressed by early treatment with the antiepileptic drug carbamazepine (Lavebratt et al., 2006). The mechanism linking impaired Kv1.1 current, seizures, and the multifactorial megencephaly phenotype has not been fully explored, however recent findings indicate that Kv1.1 currents can play a cell autonomous role in precursor cell neurogenesis (Yang et al., 2012).
We found that tau loss prevented Kv1.1 megencephaly, with both hippocampal and forebrain volume decreased in 12-week-old Kcna1−/−Tau−/− mice to wild-type levels. This was not due to the simple absence of tau, since hippocampal and forebrain volume in pure tau knockout mice were indistinguishable from wild type at 12 weeks of age. A previous report also shows that Tau−/− mice exhibit wild-type brain size at 6 months of age, although beyond one year brain atrophy is observed (Lei et al., 2012). Based on these lines of evidence, early rescue of megencephaly in our model is likely due to the decreased seizures and hyperexcitability in developing Kcna1−/−Tau−/− mice.
Tau reduction decreases hyperexcitability in Drosophila seizure models
We have further shown that partial tau reduction is sufficient to significantly ameliorate the BS phenotype in two different Drosophila hyperexcitability mutants. Kcc encodes the fly K+/Cl− cotransporter, and a partial loss-of-function allele increases seizure susceptibility due to reduction of the Cl− gradient and impaired GABAergic transmission (Hekmat-Scafe et al., 2010). Loss of KCC2, the mammalian homologue of kcc, in mice also results in seizures and hyperexcitability (Woo et al., 2002). Truncation of the eas gene reduces ethanolamine kinase activity, interfering with metabolism of phosphatidylethanolamine, the predominant membrane lipid, which may alter neuroblast development in the Drosophila mushroom body (Pavlidis et al., 1994, Pascual et al., 2005). The ability of tau reduction to decrease seizure susceptibility in all three mutant models demonstrates that tau is a genetic modifier of hyperexcitability that is not specific to a single pathway or molecular mechanism of excitability.
Is tau a possible therapeutic target in epilepsy?
While our results reflect the absence of tau during nervous system development rather than reversal of a preexisting seizure disorder, the robust effect of even partial genetic tau reduction on hyperexcitability phenotypes supports future examination of pharmacological tau reduction as a possible strategy for early disease modification in epilepsy. Although the potentially adverse effects of tau loss have not been fully evaluated, they so far appear to be limited. Despite delays in neuronal migration and process length in culture, synaptic connectivity in Tau−/− mouse brain is not apparently altered, as evident by normal synaptophysin and GAP-43 distribution as well as normal GFAP staining patterns (Dawson et al., 2001). In addition, we saw no visible differences in cellular lamination patterns in cresyl violet stained Tau−/− sections. Tau−/− mice also reportedly show no learning or memory deficits, as well as relatively normal hippocampal electrophysiological properties through 6 months of age (Ikegami et al., 2000, Roberson et al., 2007, Dawson et al., 2010, Ittner et al., 2010, Roberson et al., 2011, Lei et al., 2012). Tau−/− mice do show increased sIPSCs in dentate granule cells and less network bursting in disinhibited hippocampal slices, providing one candidate mechanism for decreasing network hyperexcitability in the brain (Morris et al., 2011, Roberson et al., 2011). Tau loss may also be neuroprotective, since knockout leads to defective postsynaptic localization of fyn kinase and decreased excitotoxic damage in response to neuronal activity (Ittner et al., 2010). Some deleterious consequences of early and complete genetic tau loss were found in aged, 12-month-old Tau−/− mice including brain atrophy, iron accumulation, substantia nigra cell loss, and axonal spheroids. (Dawson et al., 2010, Lei et al., 2012).
We have shown that tau loss decreases excitability in the non-AD, epileptic brain. Interestingly, even heterozygous reduction of tau was sufficient to significantly lower seizure frequency, suggesting the potential of tau regulation as a therapeutic target. This is supported by evidence in AD mouse models where heterozygous tau loss decreases hyperexcitability and also improves cognition (Roberson et al., 2007, Ittner et al., 2010, Roberson et al., 2011). Importantly, to our knowledge, no neurological deficits have been shown in Tau+/− mice at any age studied (Ikegami et al., 2000, Roberson et al., 2007, Roberson et al., 2011). Additionally, pharmacological tau reduction by methylene blue, which reduces soluble tau and improves cognition in transgenic tau mice, does not impair Morris Water Maze memory recall in wild-type mice (O’Leary et al., 2010). These results suggest that while complete loss of tau throughout life is not inconsequential, there are potential therapeutic benefits of decreasing tau at an early stage of epileptogenesis.
Overall, these data reflect the genomic absence of tau throughout brain development rather than the reversal of an established seizure disorder. Further studies utilizing developmentally delayed reduction of tau levels will be necessary to distinguish whether the observed changes in hyperexcitability are due to early, synaptic reorganization of the brain or to an ongoing intrinsic regulatory role of tau in adult neuronal firing properties and synaptic transmission.
Acknowledgments
This work is supported by the Epilepsy Foundation Predoctoral Fellowship (JKH), NIH grants R01NS29709 (JLN), P01 AG022074 (JLN, PI: L Mucke), NS042179 (JB), R01AG029977 (RGP), P30DK079638 (BCM DERC, MRI Core), NIA06656 (SY) and the Blue Bird Circle Foundation. We would like to thank Mark Tanouye and Daniel St. Johnston for supplying the drosophila strains used in this study and Joanna Jankowski for her assistance with amyloid analysis.
Footnotes
The authors declare no competing financial interests.
References
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Almgren M, Persson AS, Fenghua C, Witgen BM, Schalling M, Nyengaard JR, Lavebratt C. Lack of potassium channel induces proliferation and survival causing increased neurogenesis and two-fold hippocampus enlargement. Hippocampus. 2007;17:292–304. doi: 10.1002/hipo.20268. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinack J, Schneider A, Mandelkow E, Hyman B. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Buerger K, Teipel SJ, Zinkowski R, Blennow K, Arai H, Engel R, Hofmann-Kiefer K, McCulloch C, Ptok U, Heun R, Andreasen N, DeBernardis J, Kerkman D, Moeller H, Davies P, Hampel H. CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology. 2002;59:627–629. doi: 10.1212/wnl.59.4.627. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Crespo-Biel N, Canudas A, Camins A, Pallàs M. Kainate induces AKT, ERK and cdk5/GSK3beta pathway deregulation, phosphorylates tau protein in mouse hippocampus. Neurochem Int. 2007;50:435–442. doi: 10.1016/j.neuint.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Dawson H, Ferreira A, Eyster M, Ghoshal N, Binder L, Vitek M. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, Laskowitz DT. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger H, Benton R, Shulman J, St Johnston D. The role of PAR-1 in regulating the polarised microtubule cytoskeleton in the Drosophila follicular epithelium. Development. 2003;130:3965–3975. doi: 10.1242/dev.00616. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Kole MJ, Noebels JL. Transcompartmental reversal of single fiber hyperexcitability in juxtaparanodal Kv1.1-deficient vagus nerve axons by activation of nodal KCNQ channels. J Physiol. 2012 doi: 10.1113/jphysiol.2012.235606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Yoo J, Noebels J. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Hekmat-Scafe D, Lundy M, Ranga R, Tanouye M. Mutations in the K+/Cl-cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Mercado A, Fajilan AA, Lee AW, Hsu R, Mount DB, Tanouye MA. Seizure sensitivity is ameliorated by targeted expression of K+-Cl-cotransporter function in the mushroom body of the Drosophila brain. Genetics. 2010;184:171–183. doi: 10.1534/genetics.109.109074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JM, Cirrito JR, Restivo JL, Kinley RD, Sullivan PM, Holtzman DM, Koger D, Delong C, Lin S, Zhao L, Liu F, Bales K, Paul SM. Emergence of a seizure phenotype in aged apolipoprotein epsilon 4 targeted replacement mice. Brain Res. 2012;1467:120–132. doi: 10.1016/j.brainres.2012.05.048. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett. 2000;279:129–132. doi: 10.1016/s0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. Modifications of seizure susceptibility in Drosophila. J Neurophysiol. 2000;83:998–1009. doi: 10.1152/jn.2000.83.2.998. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Zhang H, Ren X, Tanouye MA. Genetic suppression of seizure susceptibility in Drosophila. J Neurophysiol. 2001;86:1211–1225. doi: 10.1152/jn.2001.86.3.1211. [DOI] [PubMed] [Google Scholar]
- Lavebratt C, Trifunovski A, Persson AS, Wang FH, Klason T, Ohman I, Josephsson A, Olson L, Spenger C, Schalling M. Carbamazepine protects against megencephaly and abnormal expression of BDNF and Nogo signaling components in the mceph/mceph mouse. Neurobiol Dis. 2006;24:374–383. doi: 10.1016/j.nbd.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, Halabisky B, Deng C, Mahley RW, Huang Y. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–645. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J Alzheimers Dis. 2009;17:531–539. doi: 10.3233/JAD-2009-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori R, Avoni P, Baruzzi A, Di Stasi V, Montagna P. Familial continuous motor unit activity and epilepsy. Muscle Nerve. 2001;24:630–633. doi: 10.1002/mus.1048. [DOI] [PubMed] [Google Scholar]
- Lopantsev V, Tempel BL, Schwartzkroin PA. Hyperexcitability of CA3 pyramidal cells in mice lacking the potassium channel subunit Kv1.1. Epilepsia. 2003;44:1506–1512. doi: 10.1111/j.0013-9580.2003.44602.x. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary JC, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, Jones JR, Kraft C, Peters M, Abisambra JF, Duff KE, Weeber EJ, Gestwicki JE, Dickey CA. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker L, Howlett IC, Rusan ZM, Tanouye MA. Seizure and epilepsy: studies of seizure disorders in Drosophila. Int Rev Neurobiol. 2011;99:1–21. doi: 10.1016/B978-0-12-387003-2.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A, Chaminade M, Préat T. Ethanolamine kinase controls neuroblast divisions in Drosophila mushroom bodies. Dev Biol. 2005;280:177–186. doi: 10.1016/j.ydbio.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Persson AS, Westman E, Wang FH, Khan FH, Spenger C, Lavebratt C. Kv1.1 null mice have enlarged hippocampus and ventral cortex. BMC Neurosci. 2007;8:10. doi: 10.1186/1471-2202-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson E, Scearce-Levie K, Palop J, Yan F, Cheng I, Wu T, Gerstein H, Yu G, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart S, Lopantsev V, Zhang C, Robbins C, Wang H, Chiu S, Schwartzkroin P, Messing A, Tempel B. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- Song J, Tanouye MA. From bench to drug: human seizure modeling using Drosophila. Prog Neurobiol. 2008;84:182–191. doi: 10.1016/j.pneurobio.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Yang SB, McLemore KD, Tasic B, Luo L, Jan YN, Jan LY. Kv1.1-dependent Control of Hippocampal Neuron Number as Revealed by Mosaic Analysis with Double Markers (MADM) J Physiol. 2012 doi: 10.1113/jphysiol.2012.228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JB, Stephenson JP, Kullmann DM, Hanna MG. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122 (Pt 5):817–825. doi: 10.1093/brain/122.5.817. [DOI] [PubMed] [Google Scholar]