

Abstract
Growing evidence suggests that a physiological activity of the cellular prion protein (PrPC) plays a crucial role in several neurodegenerative disorders, including prion and Alzheimer’s diseases. However, how the functional activity of PrPC is subverted to deliver neurotoxic signals remains uncertain. Transgenic mice expressing PrP with a deletion of residues 105–125 in the central region (referred to as ΔCR PrP) provide important insights into this problem. Tg(ΔCR) mice exhibit neonatal lethality and massive degeneration of cerebellar granule neurons, a phenotype that is dose-dependently suppressed by the presence of wild-type PrP. When expressed in cultured cells, ΔCR PrP induces large, ionic currents that can be detected by patch-clamping techniques. Here, we have tested the hypothesis that abnormal ion channel activity underlies the neuronal death seen in Tg(ΔCR) mice. We find that ΔCR PrP induces abnormal ionic currents in neurons in culture and in cerebellar slices, and that this activity sensitizes the neurons to glutamate-induced, calcium-mediated death. In combination with ultrastructural and biochemical analyses, these results demonstrate a role for glutamate-induced excitotoxicity in PrP-mediated neurodegeneration. A similar mechanism may operate in other neurodegenerative disorders due to toxic, β-rich oligomers that bind to PrPC.
INTRODUCTION
Although the notion that prion propagation involves conformational conversion of PrPC into PrPSc is now widely accepted, the mechanism by which prions cause neurodegeneration is still uncertain (Caughey and Baron, 2006; Harris and True, 2006; Aguzzi et al., 2007). Multiple lines of evidence indicate that the presence of functionally active PrPC on the cell membrane is required for PrPSc molecules to exert their neurotoxic effects (Brandner et al., 1996; Mallucci et al., 2003; Chesebro et al., 2005; Mallucci et al., 2007). Recently, PrPC has also been shown to mediate the toxicity of other pathological protein aggregates, including oligomers of the amyloid beta (Aβ) peptide, which are associated with Alzheimer’s disease (Laurén et al., 2009; Balducci et al., 2010; Chen et al., 2010; Resenberger et al., 2011). Collectively, these studies suggest that the normal function of PrPC could be corrupted by several kinds of β-rich protein oligomers to generate neurotoxic effects (Gunther and Strittmatter, 2010; Biasini et al., 2012a).
As a way of elucidating the neurotoxic potential of PrPC, we have been studying an artificial mutant PrP (designated Δ CR) that carries a deletion of 21 amino acids in the conserved central region of the protein (residues 105–125). Expression of ΔCR PrP in transgenic (Tg) mice causes a neonatal lethal phenotype characterized by spontaneous degeneration of cerebellar granule neurons, as well as white matter pathology in the brain and spinal cord (Li et al., 2007b). Importantly, this ΔCR-induced phenotype is suppressed by co-expression of wild-type (WT) PrP, suggesting that mutant and WT forms can somehow interact at a functional level.
An important insight into the toxicity of Δ CR PrP is provided by our observation that PrP molecules carrying deletions or disease-associated point mutations in the central region induce large, spontaneous ionic currents when expressed in transfected cell lines (Solomon et al., 2010b). We also discovered that cells expressing these mutants are hypersensitive to the toxic effects of several cationic drugs, a phenomenon that is likely related to enhanced current activity (Massignan et al., 2010). WT PrP suppresses ΔCR-induced currents and drug hypersensitivity, paralleling the ability of the WT protein to reverse the neurodegenerative phenotype of Tg(ΔCR) mice. Our interpretation of these results is that ΔCR PrP molecules form ion channels or pores in the plasma membrane, or else activate endogenous ion channels, and that these channels are silenced by co-expression of WT PrP (Solomon et al., 2012).
These results raise the possibility that abnormal ion channel activity is a major cause of neurodegeneration in Tg(ΔCR mice). Excessive excitation of CNS neurons, due to activating mutations in ion channel proteins, or to increased stimulation by the neurotransmitter glutamate, are well-known causes of neuronal death in animal models and human diseases (Michaelis, 1998). In this paper, we report that ΔCR PrP induces spontaneous current activity in several kinds of neuronal preparations, and also sensitizes neurons to glutamate-mediated excitotoxic death. These effects are suppressed by co-expression of WT PrP, or by co-treatment with an NMDA antagonist. Our results provide a likely explanation for the toxicity of ΔCR PrP in vivo, and raise the possibility that a similar excitotoxic mechanism may occur in infectious prion diseases, and possibly other neurodegenerative conditions resulting from binding of β-rich protein oligomers to PrPC.
MATERIALS AND METHODS
Mice
All experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. For experiments involving CGNs and cerebellar slice cultures, ΔCR-A+/−/Prn-p+/− mice (Li et al., 2007b) were used. ΔCR-A+/−/Prn-p+/−/Tga20+/− (Fischer et al., 1996) and Prn-p+/− (Büeler et al., 1992) littermates served as controls. C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, Maine). Mice were genotyped by PCR analysis of tail DNA, prepared using the Puregene DNA Isolation Kit (Gentra Systems, Minneapolis, MN). In order to obtain neural stem cells (NSCs) of different genotypes, ΔCR-A+/−/ Tga20+/−/Prn-p+/− mice were mated to Prn-p−/− mice on the C57BL/6 background, as described previously (Massignan et al., 2010; Biasini et al., 2012b). E13.5 mouse embryos were genotyped by PCR analysis of limb DNA.
Cerebellar granule neurons (CGNs)
CGNs were cultured from P5 mice as previously described (Li et al., 2007b). Cerebella were removed from Prn-p−/− mice or Tg(ΔCR)-A mice, and neurons were isolated by mechanical disruption and trypsinization. Neurons (350,000 cells/cm2) were plated on poly-L-lysine coated coverslips in Basal Media Eagle (BME) with Earle’s salts supplemented with 10% dialyzed fetal calf serum, 2 mM glutamine, 25 mM KCl, and 0.02 mg/mL gentamycin. Aphidicholin (3.3 μg/mL) was added to cultures one day after plating. Two days after plating, PrP−/− CGNs were transduced with recombinant lentiviruses.
Lentiviruses were constructed according to published procedures (White et al., 2008). cDNAs encoding either enhanced GFP alone or WT or ΔCR PrP followed by an internal ribosomal entry site and enhanced GFP were cloned into the transfer vector pRRLsinCMV. 293T packaging cells were co-transfected with the resulting transfer plasmid, along with the plasmids pMD-Lg, pCMV-G, and RSV-REV. Virus was collected from the medium and concentrated.
Viral transductions were performed by incubating cells with purified virus at a multiplicity of infection of 0.1–2.0. Two days after lentiviral transduction, CGNs were collected for Western blotting or stained by immunofluorescence. Western blots were probed with anti-PrP antibody 6D11 followed by goat anti-mouse IgG (Pierce) and developed with ECL (GE Healthcare). Surface immunofluorescence staining was performed by incubating cells on ice with 6D11, after which they were fixed in 4% paraformaldehyde in PBS and labeled with Alexa-488 goat anti-mouse IgG (Molecular Probes). Cells were viewed with a 40X objective on a Nikon TE-2000E inverted fluorescence microscope, and images were captured with METAMORPH software (Molecular Devices, Sunnyvale, CA).
Neural Stem Cells
NSCs were obtained and cultured following a procedure described previously, with minor modifications (Massignan et al., 2010; Biasini et al., 2012b). Brains dissected from E13.5 mouse embryos were triturated in 5 ml of NeuroCult NSC basal medium containing NeuroCult NSC proliferation supplement (StemCell Technologies, Vancouver, BC) along with 20 ng/ml EGF. Once formed, neurospheres were differentiated by pipetting a 0.1–1 ml suspension (containing approximately 30–40 mature neurospheres) into each well of an 8-well chamber slide (Ibidi GmbH, München, Germany) containing NeuroCult NSC basal medium with NeuroCult NSC differentiation supplement (StemCell Technologies) along with 10 μg/ml retinoic acid. NSCs differentiated for 10 days were treated with 500 μM glutamate for 24 hrs, stained with propidium iodide (PI; Sigma-Aldrich, St. Louis, MO, USA) to estimate the number of dead cells, and with DAPI to detect cell nuclei.
Ca2+ measurement in differentiated NSCs
Cells were washed with Krebs-Ringer-Hepes (KRH) buffer (128 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, 25 mM Hepes, pH 7.4) and incubated with 10 μM Fura-2-acetoxymethyl ester (Fura-2AM) in KRH buffer plus 1% BSA, for 30 min at 37 °C. After washing once with KRH buffer, chamber slides were transferred to the recording chamber of an Olympus IX81 inverted microscope equipped with a Ca2+ imaging unit (Cell R, Olympus). Fluorescence was measured at 37°C in 5% CO2/95% air by recording 669-ms frames for 100 cycles, each cycle alternating excitation at 340 nm and 380 nm and monitoring emission at 510 nm. Ca2+ response was measured after exposure to 0.5 mM glutamate beginning at cycle 20. The fluorescence ratio F340:380 was measured for each cycle and kinetic analysis of F340/380 was done by CellR software (Olympus). Data are reported as ΔF340/380, the difference between F340/380 before and after the stimulus, which is proportional to the glutamate-induced Ca2+ influx.
Neuronal and non-neuronal cells were distinguished based on expression of microtubule associated protein 2 (MAP2). After acquiring data for Ca2+ influx, the position of the microscope stage was recorded in order to allow exact re-alignment of each chamber slide. Cells were immediately fixed with 4% paraformaldehyde in 200 mM Hepes/NaOH pH 7.4 for 30 minutes at RT, washed in PSB and incubated in blocking solution (1% bovine serum albumin, 50 mM NH4Cl, 0.1% saponin in PBS pH 7.4) containing 10% normal goat serum (NGS) for 30 minutes at RT. Cells were then incubated with primary antibody anti-MAP2 (Sigma) diluted 1:1000 in blocking solution overnight at 4°C. After washing in PBS, cells were incubated for 1 h at RT with AlexaFluor 594-conjugated secondary antibody (1:500), then reacted with 300 nM DAPI (4′,6 -diamidino-2-phenylindole, Invitrogen) in PBS for 10 min at RT to stain nuclei. Finally, the chamber slide was re-aligned under the microscope and Ca2+ measurements were assigned to MAP2-positive or -negative cells.
Organotypic slice cultures of mouse cerebellum
After decapitation, brains were removed quickly and the cerebellum was transferred to a container with liquid agarose [low-melting point, prepared freshly each day at 2% in Hanks’ Balanced Salt Solution (HBSS); both reagents obtained from Invitrogen/Life Technologies, Carlsbad, CA, USA]. After the agarose was cooled on ice, agarose blocks were glued onto a vibratome disc, which was then placed in ice-cold HBSS in the buffer reservoir of a Vibratome 1000Plus Sectioning system (Vibratome Company, St. Louis, MO, USA). Several 300 μm sagittal slices were cut at medium speed and amplitude. Slices were collected in a Petri dish containing ice-cold HBSS with 0.5% glucose and released from the agarose under a stereomicroscope using fine forceps. All subsequent steps were carried out under sterile conditions in a cell culture hood.
Each slice was placed in a Millicell Cell Culture insert (Millipore, Billerica, MA, USA) in a 24-well cell culture plate. Culture wells contained 200 μl of slice culture medium consisting of 50% minimum essential medium (MEM; with Earle’s salts, 25 mM Hepes, without L-glutamine; Invitrogen), 22.5% HBSS, 25% horse serum (heat-inactivated; Gibco/Life Technologies), 50 U/ml penicillin/50 μg/ml streptomycin (Invitrogen), 0.5% glucose, and 2 mM L-glutamine (Invitrogen). Slice cultures were kept under standard cell culture conditions (37°C, 5% CO2) in a humidified atmosphere with the medium being changed every other day.
Toxicity assays were performed by adding to the culture media for 24 hours: L-glutamic acid (500 μM), kainic acid, N-Methyl-D-aspartic acid (NMDA), and 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid (AMPA) (all at 10 μM; purchased from Sigma-Aldrich); or Zeocin™ (500 μg/ml; Invitrogen). To assess cell death, cerebellar slices were kept in culture for varying time spans, after which they were incubated with 2 μM PI in culture medium for 30 minutes in a cell culture incubator. Red fluorescence was imaged on a Nikon TE2000E2 microscope with a 20x long-working range objective. In some case, nuclear labeling was performed by staining with Hoechst 34580 (2 μg/ml; Invitrogen). Several images of the cerebellar granule layer of each slice were obtained using the auto-expose function of the camera. Positive nuclei were counted with the same settings for brightness and diameter for each image using MetaMorph software (Molecular Devices, Sunnyvale, CA).
For characterization and documentation of the quality of each slice culture, some of the slices were fixed in 4% buffered formalin for 1 hour at 4°C, then washed with phosphate buffered saline (PBS) several times, subjected to paraffin embedding and hematoxylin and eosin (H&E) staining, or immunofluorescence staining. For paraffin embedding, slices were carefully removed from the membranes, placed between two biopsy foam pads in an embedding cassette, and processed according to standard histology procedures. Four μm sections were stained with H&E and assessed using a conventional light microscope.
All incubation steps for immunofluorescence staining were performed with the slices still sitting on the membranes. First, slices were immersed in a blocking solution containing 0.3% Triton X-100 and 10% fetal calf serum (FCS; heat-inactivated; Gibco/Life Technologies) for 24 hours at 4°C. On the following day, an antibody against neuronal nuclei (mouse monoclonal anti-NeuN; clone A60, Chemicon/Millipore) was added at a 1:1000 dilution in blocking solution and left on for 48 hours at 4°C. After 3 washes in PBS/0.3% Triton X-100, an Alexa Fluor 488 rabbit anti-mouse antibody (Molecular Probes/Life Technologies) was added at a 1:200 dilution in PBS/0.3% Triton X-100 for 4 hours at room temperature in the dark. Finally, slices were washed again three times in PBS and imaged using a Nikon TE2000E2 inverted fluorescence microscope equipped with a CCD camera.
All reagents for electron microscopic analysis of organotypic cellebellar slices were purchased from Ted Pella, Inc. (Redding, CA, USA). Slices were washed twice with cacodylic acid buffer and fixed for 24 hours at 4°C in 4% glutaraldehyde in cacodylic acid buffer. After another two washes in cacodylic acid buffer, the specimens were dehydrated through an ascending alcohol series. An epoxy resin consisting of Dodecenyl Succinic Anhydride (DDSA) and Araldite 502 in a 1:1 relation was added to the membrane inserts, and the inserts left on a shaker at room temperature for 2 days. Membrane patches with slices were punched out and transferred to glass vials containing epoxy resin with 2% N,N-Dimethylbenzylamin Benzyldimethylamin (BDMA), which were left on a rotating shaker for 1 hour. Membranes were then placed in electron microscopy molds with resin and left to solidify in a vacuum cabinet. Ultrathin sections were prepared from each block and stained with lead citrate according to standard electron microscopy protocols.
Electrophysiology
Whole-cell patch clamp recordings were made from CGNs after 4 days in culture, or 4 days after lentiviral transduction. Pipettes were pulled from borosilicate glass, coated with Sylgard, and polished to an open resistance of 1–10 megaohms. Experiments were conducted at room temperature with the following solutions in the patch pipette and extracellular medium: internal, 140 mM Cs-glucuronate, 5 mM CsCl, 4 mM MgATP, 1 mM Na2GTP, 10 mM EGTA, and 10 mM HEPES (pH 7.4 with CsOH); external, 150mM NaCl, 4mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10mM glucose, and 10 mM HEPES (pH 7.4 with NaOH). Current signals were collected from an Axopatch 200B amplifier and digitized with a Digidata 1330 interface (Axon Instruments) or with an EPC-10 amplifier controlled by PatchMaster acquisition software (HEKA Elektronik) and were saved to disc for analysis with PClamp 9 software. Current activity was plotted as the proportion of total recording time that a cell exhibited inward current ≥ 450 pA.
To prepare cerebellar slices for acute recordings, Prn-p+/− ΔCR-A−/− and Prn-p+/− ΔCR-A+/− mice at P10 were sacrificed by decapitation, and their brains were immediately submerged in oxygenated (95% O2; 5% CO2) ice-cold Ringer’s solution [25 mM NaHCO3, 124 mM NaCl, 1 mM KCl, 2 mM KH2PO4, 10 mM Glucose, 2.5 mM CaCl2, and 1.3 mM MgCl2 (pH 7.4)]. Cerebella were affixed to an agar slab with cyanoacrylate glue and placed in a tissue holder for cutting. Four to 6 slices (300 μm thick) were cut into ice-cold Ringer’s solution with a vibrating microtome, and then equilibrated for at least 1 h at room temperature in oxygenated Ringer’s solution. Individual slices were positioned in a submersion-type recording chamber (Harvard Apparatus, Holliston, MA) on the stage of a Nikon E600 infrared-differential interference contrast microscope (IR-DIC; Micro Video Instruments, Avon, MA), and were continuously perfused with room temperature with oxygenated Ringer’s solution (2– 2.5 ml/min). Whole-cell patch clamp recordings were obtained from cerebellar granule neurons at a potential of −80 mV. Patch electrode pipettes were pulled from capillary tubes on a Flaming and Brown horizontal pipette puller (Model P87, Sutter Instrument, Novato, CA) to a resistance of 3–6 MΩ, and were filled with potassium glucuronate (KGlu) internal solution [122 mM KGlu, 2 mM MgCl2, 5 mM EGTA, 10 mM NaHEPES, 2 mM MgATP, 0.3 mM NaGTP, and 1% biocytin, (pH 7.4)]. PatchMaster software was used for data acquisition with EPC-9 and EPC-10 amplifiers, and data were saved to disc for analysis with PClamp 9 software.
RESULTS
ΔCR PrP induces spontaneous ionic currents in cerebellar granule neurons
We recently reported that ΔCR PrP induces spontaneous ionic currents in a variety of transformed cell lines (Solomon et al., 2010b; Solomon et al., 2011). Here, we sought to determine whether ΔCR PrP also exerts such an activity in neurons. First, CGNs were cultured from postnatal day 5 (P5) PrP-null mice and transduced with lentiviruses encoding WT or ΔCR PrP. Each construct included an internal ribosome entry site (IRES) and a GFP gene, allowing us to monitor protein expression in individual cells by visualizing the intrinsic green fluorescence of the GFP. Cells transduced with “GFP-only” lentiviruses were used as controls. Surface immunofluorescence staining showed the lack of PrP signal in the GFP-only control cells, and both PrP and GFP signals in CGNs transduced with WT or ΔCR PrP-encoding lentiviruses (Figure 1A). These analyses also confirmed that PrP molecules, either WT or ΔCR, were correctly expressed at the neuronal surface. Whole-cell patch clamp recordings were then collected 48 h after lentiviral transduction (four days after culturing) from GFP-positive cells, at a holding potential of −80 mV (Figure 1B, C). ΔCR PrP-expressing CGNs, but not WT or GFP-only controls, exhibited large, spontaneous inward currents similar to those previously observed in transformed cell lines (Solomon et al., 2010b).
FIGURE 1. ΔCR PrP induces spontaneous ionic currents in cerebellar granule neurons.

(A–C) CGNs isolated from P5 PrP−/− mice were transduced with recombinant lentiviruses encoding GFP alone (panel i), or GFP plus either WT (panel ii) or ΔCR PrP (panel iii). (A) Surface immunofluorescence staining shows PrP expression (red) in GFP (green) containing neurons (panels ii and iii). Coincidence of PrP fluorescence (on the cell membrane) and EGFP fluorescence (in the cytoplasm) is most evident in neuronal processes, where PrP is preferentially expressed. Scale bar = 50 μm. (B) Whole-cell patch clamp recordings were made at a holding potential of −80 mV from GFP positive cells 48 h after transduction. (C) Quantitation of the currents recorded in panel B, plotted as the percentage of total time the cells exhibited inward current at 450 pA (mean ± S.E.M., n = 5 cells). Asterisks (*) indicate statistically significant differences in spontaneous channel activity (p < 0.05, one-tailed Student’s t-test). (D) CGNs were cultured from transgenic mice with the following genotypes: PrP+/−; ΔCR+/−/PrP+/− (ΔCR/PrP); or ΔCR+/−/Tga20+/−/PrP+/− (ΔCR/Tga). Whole-cell patch clamp recordings were made at a potential of −80 mV. (E) Quantitation of the currents recorded in panel D, plotted as the percentage of total time the cells exhibited inward current of 450 pA (mean ± S.E.M., n = 5 cells). Asterisks (*) indicate statistically significant differences in spontaneous channel activity (p < 0.05, one-tailed Student’s t-test). (F) Expression of ΔCR PrP induces spontaneous currents and increases fragility of CGNs in acute cerebellar slices. Cerebellar slices dissected from P10 mice of the indicated genotypes were allowed to equilibrate in external recording buffer for 1 h prior to whole-cell patch clamp recordings at −80 mV. Unlike CGNs from ΔCR/Tga mice, which remain stable for >10 min of recording without current activity, the majority of CGNs in ΔCR/PrP slices exhibited spontaneous inward currents which returned to baseline, or were unable to be observed for 10 min due to cell death or instability of the patch. The pie charts indicate the number and proportion cells in each category.
To determine whether neurons derived from Tg(ΔCR) mice also exhibited spontaneous currents, CGNs were cultured from P5 mice expressing ΔCR PrP on the PrP+/− background (ΔCR+/−/ Prn-p+/−, referred to as ΔCR/PrP). The presence of endogenous PrP (0.5X) was required for recovery of sufficient numbers of postnatal animals, since expression of ΔCR PrP on a PrP-null background induces neonatal lethality, with mice often dying within the first week after birth, while Tg(ΔCR+/−/Prn-p +/−) mice survive approximately 3 weeks (Li et al., 2007b). CGNs from Tg(ΔCR) mice overexpressing WTPrP from a second transgene (ΔCR+/−/Tga20+/−/ Prn-p+/−, referred to as ΔCR/Tga) or non-transgenic C57Bl6 mice expressing one copy of endogenous PrP (PrP+/−) were used as controls. Four days after culturing, whole-cell patch clamp recordings were collected at a potential of −80 mV (Fig. 1D, E). ΔCR cells exhibited spontaneous inward currents, which were absent in ΔCR/Tga and PrP+/− control cells. These currents were of similar magnitude to those observed in lentivirus-transduced CGNs. However, we noted that the frequency and duration of the currents were less, most likely due to the partial channel silencing effect of endogenous PrP expression.
Taken together, these results demonstrate that ΔCR PrP-dependent channel activity is not restricted to transformed cell lines, but can also be detected in primary neurons, either virally transduced or taken from Tg(ΔCR) mice.
Expression of ΔCR PrP causes spontaneous ionic currents and increased neuronal fragility in acute cerebellar slices
In order to extend the observations made in cultured CGNs, we sought to determine whether ΔCR PrP-dependent ionic currents can be detected in neurons in cerebellar slices. Acute slice cultures were made from P10 ΔCR/PrPand ΔCR/Tga mice. Whole-patch clamp recordings were collected at −80 mV from neurons in the granule cell layer (Figure 1F). As expected, no currents were detected in ΔCR/Tga cells for the entire duration of the recordings (>10 min). In contrast, several ΔCR/PrP neurons exhibited large, spontaneous inward currents similar to those observed in cultured CGNs. In a few cases, however, no currents were detected. As discussed for CGNs, this assortment of phenotypes could possibly be explained by the presence of endogenous PrP in ΔCR/PrP slices. We found that the majority of ΔCR cells were lost shortly after initial patching, frequently after detection of an initial inward current that did not return to baseline. This problem, which was never encountered with ΔCR/Tga cells, occurred in slices from several ΔCR/PrPmice derived from multiple litters. The phenomenon, which may reflect an intrinsic fragility of the neuronal membrane, suggests that ΔCR-induced channel activity is deleterious for primary neurons.
ΔCR PrP sensitizes neurons in differentiated cultures of neural stem cells to glutamate-evoked Ca2+ influx and cell death
In vivo, CGNs receive glutamate-mediated excitatory input from mossy fibers originating in the deep cerebellar nuclei (Martin, 1996). Therefore, we decided to investigate whether the channel activity generated by ΔCR PrP can alter neuronal sensitivity to glutamate. For these experiments, we utilized differentiated neural stem cells (NSCs). We have previously shown that these cells are susceptible to the toxic effect of ΔCR PrP, which makes them hypersensitive to the cationic antibiotics Zeocin and G418 (Biasini et al., 2012b). NSCs provide several advantages over CGNs. Since NSCs are derived from mice at E13.5, they can be obtained from Tg(ΔCR) mice on a Prn-p−/− background, eliminating the confounding effects due to expression of endogenous PrP. Moreover, upon differentiation, NSCs give rise to mixed cultures of mature astrocytes, oligodendrocytes and neurons, allowing us to determine the effect of glutamate on both neuronal and non-neuronal cells within the same culture.
To test the hypothesis that ΔCR PrP alters glutamate-mediated excitability, we preloaded NSCs with the ratiometric Ca2+ indicator Fura-2AM in order to monitor influx of Ca2+ through glutamate receptor channels, predominantly of the NMDA sub-type. We derived NSCs from several mouse lines, including non-transgenic C57Bl6 (C57), Prn-p −/− (KO), Tga20+/+/Prn-p−/− (Tga), Tg(ΔCR/Prn-p−/−)(ΔCR) and Tg(ΔCR/Tga20/Prn-p−/−) (ΔCR/Tga) mice. Ca2+ influx before and after stimulation with 0.5 mM glutamate was measured by ratiometric quantification of fluorescence corresponding to free or Ca2+-bound Fura-2 (F340/380), on both neurons (MAP2-positive) and glia (MAP2-negative) cells (see Material and Methods for details). These analyses revealed that a significantly higher concentration of Ca2+ accumulates in the cytoplasm of ΔCR neurons in response to glutamate, as compared to C57, KO or Tga neurons (Figure 2A, B). We also found that that intracellular Ca2+ concentrations returned to baseline with slower kinetics in ΔCR neurons compared to control cells (Figure 2A). Importantly, the kinetics of glutamate-evoked Ca2+ influx and recovery were restored to normal when WT PrP was co-expressed with the ΔCR mutant (ΔCR/Tga NSCs). Moreover, no difference was detected between ΔCR and controls when glutamate-dependent Ca2+ influx was recorded in glial cells (Figure 2C and D). These results indicate that expression of ΔCR PrP causes an enhanced Ca2+ response to glutamate stimulation selectively in neuronal cells, an effect that is rescued by WT PrP.
FIGURE 2. ΔCR PrP causes abnormal, glutamate-induced Ca2+ influx in NSC-derived neurons.

NSCs dissected from mouse embryos at 13.5 days were cultured and propagated as neurospheres, differentiated for 7 days, and then incubated with the Ca2+-sensitive dye Fura 2-AM prior to imaging analysis. Ca2+ influx in response to stimulation with 0.5 mM glutamate was detected by alternating excitation at 340 nm and 380 nm and monitoring emission at 510 nm. Raw data were collected as the ratio F340/380, which is directly proportional to the amount of Ca2+ in the cytosol. Neuronal cells (A–B) were discriminated from non-neuronal cells (C–D) on the basis of the expression of the microtubule associated protein 2 (MAP2), which was detected by immunostaining after acquiring Ca2+ influx recordings. Examples of recordings from individual neuronal (A) and non-neuronal (C) cells from the indicated genotypes are shown. (B and D) Bar graphs show quantitation of the Ca2+ burst in the different cells, calculated as the difference between F340/380 before and after glutamate stimulation. Data are reported as percentage of the C57 control. ** p < 0.01, one-tailed Student’s t-test.
Excessive influx of Ca2+ in response to glutamate stimulation can cause excitotoxic neuronal death. In order to test whether ΔCR cells are hypersensitive to glutamate-induced excitotoxicity, we chronically exposed C57, KO, Tga, ΔCR and ΔCR/Tga differentiated NSCs to glutamate, and scored cell death by propidium iodide (PI) staining (Figure 3A). In all the experimental groups, we found that the vast majority of PI-positive cells (>90%) were also positive for MAP2 staining, suggesting that neuronal cells are more susceptible to excitotoxicity than glial cells (data not shown). However, we detected a significantly higher (>two-fold increase) number of PI-positive cells in cultures of ΔCR NSCs, as compared to cultures of control cells (Figure 3B). Glutamate sensitivity in ΔCR cells was restored to normal levels by co-expression of WT PrP (Figure 3B, green bar), or by co-treatment with MK-801, a non-competitive NMDA receptor antagonist (Figure 3C, red stripes bar). These results, which are consistent with the observed increase in Ca2+ influx after glutamate stimulation, provide direct evidence that ΔCR PrP hypersensitizes neurons to glutamate-induced excitotoxicity, and that this effect requires activation of NMDA receptors.
FIGURE 3. Differentiated NSCs expressing ΔCR PrP are hypersensitive to glutamate-induced excitotoxicity.

(A) Differentiated NSCs of the indicated genotypes were treated for 24 hrs with 0.5 mM glutamate, and then stained by PI (red) to reveal cell death. Scale bar: 10 μm. (B) The bar graph shows the number of PI-positive cells after treatment with 0.5 mM glutamate, expressed as a percentage of the untreated cells, which was determined in 5 fields for each sample group. Bars show means ± SEM (n = 5 independent experiments). The number of PI-positive cells was significantly higher in ΔCR cells than in controls (**p< 0.01, one-tailed Student’s t-test). (C) Differentiated NSCs of the indicated genotypes were treated for 24 hrs with 0.1 mM of MK-801, 0.5 mM glutamate, or a mixture of the two, and then stained with propidium iodide to reveal cell death. The graph shows the number of PI-positive cells, expressed as a percentage of the untreated cells, determined in 3 fields for each sample group. Bars show means ± SEM (n = 3 independent experiments). The number of PI-positive cells was significantly higher in ΔCR cultures treated with glutamate (red bar), compared to those co-treatment with glutamate and MK-801 (red stripes bar) (*p< 0.01, one-tailed Student’s t-test).
We previously reported that granule cell death in Tg(ΔCR) mice occurs by a non-apoptotic mechanism that is independent of caspase or Bax activation (Christensen et al., 2010). To test the involvement of caspase-3 in glutamate-induced cell death of differentiated ΔCR NSCs, we chronically exposed C57, KO, Tga and ΔCR differentiated NSCs to glutamate, and scored cell death by PI staining or by staining for activated caspase-3 (Figure 4). As expected, a much greater number of ΔCR cells were positive for PI staining, as compared to cells from control genotypes (Figure 4, red bar). In contrast, we observed no difference in the number of cells positive for activated caspase-3 (Figure 4, stripe bars). These results are consistent with previous studies indicating that NMDA-induced excitotoxicity is not associated with activation of classical apoptotic markers (Forloni et al., 1997).
FIGURE 4. Cell death in glutamate-treated, ΔCR-expressing NSCs is not caspase-3-dependent.
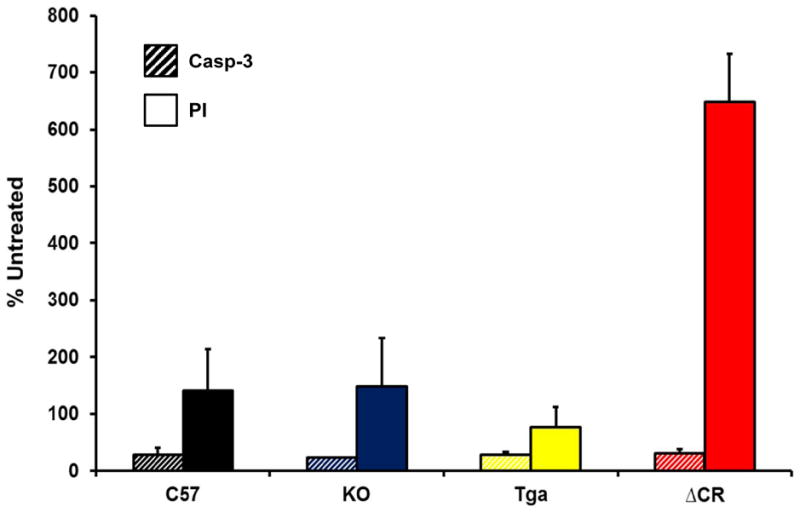
Differentiated NSCs of the indicated genotypes were treated for 24 hrs with 0.5 mM glutamate, and then stained for activated caspase-3 (Casp-3) (stripes bars) or propidium iodide (PI) (red bars). The bar graph shows the number of positively stained cells, expressed as a percentage of the untreated cells, determined in 3 fields for each sample group. Bars show means ± SEM (n = 3 independent experiments). The number of PI-positive cells, but not the number of activated caspase 3-positive cells, was significantly higher in ΔCR cultures than in control cultures (**p< 0.01, one-tailed Student’s t-test).
Characterization of organotypic cerebellar slice cultures derived from Tg(ΔCR) mice
To confirm the data obtained with NSCs, we employed organotypic cerebellar slice (OCS) cultures. For these experiments, we cut sagittal OCSs from PrP+/−, ΔCR/PrP+/−, and ΔCR/Tga mice between postnatal days 10 and 12. Slices were then kept in culture for approximately 15 days. Slice integrity and morphology were confirmed by H&E staining after fixation and embedding in paraffin (Figure 5A), and by immunofluorescence staining for the neuronal marker NeuN (Figure 5B–C). Detection of cell nuclei by Hoechst staining revealed that the total number of granule neurons in each slice at the time it was placed in culture was not detectably different in OCSs from different genotypes (data not shown).
FIGURE 5. Morphology of organotypic slice cultures of mouse cerebellum.
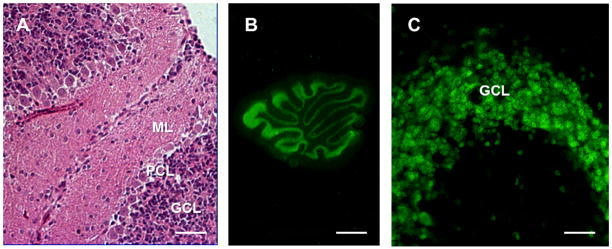
WT cerebellar slices were kept in culture for 15 days prior to assessment of slice morphology and integrity (slices were dissected and cultured on post-natal day 10). (A) Hematoxylin & eosin staining of a formalin-fixed, paraffin-embedded slice cut at 4 μm. (B, C) Immunofluorescence staining for NeuN (neurons) in formalin-fixed slices. Scale bars: 20 μm (A and C) and 800 μm (B). ML = molecular layer, PCL = Purkinje cell layer, GCL = granule cell layer.
As described previously, despite the potent neurotoxicity of ΔCR PrP in vivo, only a small increase in spontaneous cell death can be detected in transformed cell lines transfected to express ΔCR PrP (Christensen and Harris, 2009; Massignan et al., 2010). However, the number of dying ΔCR cells dramatically increases after treatment with the cationic antibiotics Zeocin, G418 or hygromycin. In order to confirm these observations in OCSs, we quantitated the proportion of PI-positive cells in slices from PrP+/−, ΔCR/PrP+/−, or ΔCR/Tga mice, before and after treatment with Zeocin. In absence of Zeocin, we detected a small but significantly higher percentage of PI-positive cells in OCSs derived from ΔCR/PrP+/− mice, as compared to PrP+/−controls (2.6% vs. 5.2%, p<0.01) (Figure 6A and B). As expected, ΔCR/PrP+/− OCSs showed hypersensitivity to Zeocin, as the proportion of PI-positive cells was increased by more than 10-fold after treatment (Figure 6C and D), compared to 2-fold for PrP+/−control cells. Importantly, both the spontaneous cell death and the hypersensitivity to Zeocin were fully suppressed by over-expression of WT PrP from the Tga20 transgene (Figure 6A–D). These results demonstrate that, similar to cell lines expressing ΔCR PrP, OCSs from ΔCR/PrP+/− mice display hypersensitivity to Zeocin, an effect which can be rescued by the presence of WT PrP.
FIGURE 6. Granule neurons expressing ΔCR PrP in cerebellar slices are hypersensitive to Zeocin.

(A) Propidium iodide labeling of slices obtained from ΔCR/PrP+/−, ΔCR/Tga20+/−/PrP+/−, and PrP+/− animals was evaluated 15 days after slices were placed in culture. (B) Quantitation of labeled cells (as a percentage of DAPI-positive nuclei) revealed a significant difference between untreated slices from ΔCR/PrP+/− and control animals (** indicates p<0.01). (C) Propidium iodide labeling of slices treated with 500 μg/ml Zeocin for 24 hours beginning on day 15 in culture. D) Quantitation of the proportion of labeled cells in Zeocin-treated slices (as a percentage of those in untreated slices) revealed a significant difference between ΔCR/PrP+/− and control animals in Zeocin-induced toxicity (** indicates p<0.01). Scale bars: 10 μm.
We have previously reported that cerebellar granule neurons in Tg(ΔCR) mice undergo a distinctive, non-apoptotic form of cell death that is morphologically and biochemically similar to that seen in certain types of glutamate excitotoxicity (Christensen et al., 2010). The most striking ultrastructural feature of dying neurons in Tg(ΔCR) mice is a heterogeneous condensation of the nuclear matrix, which is distinct from discrete chromatin aggregates typical of classical apoptosis. To further validate OCS cultures as an experimental system to study the ΔCR toxicity, we determined whether similar morphological features were seen in granule neurons in OCS preparations from Tg(ΔCR) mice. Electron micrographs of ΔCR OCSs prepared on postnatal day P11, and kept in culture for 14 more days, revealed granule neurons displaying marked condensation of the nuclear matrix (Figure 7B), analogous to that observed in Tg(ΔCR) mice. These neurons presumably correspond to the PI-positive granule neurons observed in untreated ΔCR slices (Figure 6A). Conversely, no signs of nuclear condensation were detected in control slices (Fig. 7A, C). These results suggest that ΔCR neurons dye in a similar fashion in Tg mice and slices, and reinforce the idea that OCS cultures represent a reliable experimental system for studying ΔCR toxicity.
FIGURE 7. ΔCR neurons in cerebellar slices show ultrastructural features reminiscent of excitotoxic cell death.
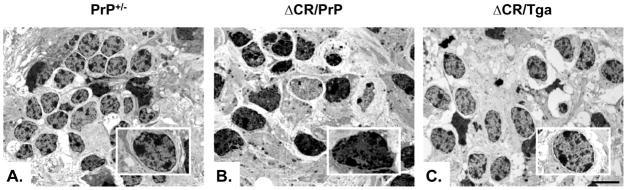
Electron micrographs of the cerebellar granule layer in slices cultures kept in culture for 15 days prior to fixing and embedding. Note the heterogeneously condensed chromatin in granule cell nuclei of ΔCR/PrP+/− mice (B) that is not present in granule cell nuclei of PrP+/− (A) or ΔCR/Tga+/−/PrP+/− (C) animals (see boxes). Scale bars: 2 μm
ΔCR-expressing OCSs are hypersensitive to glutamate-. NMDA- and kainate-induced excitotoxicity
In order to test whether OCSs from ΔCR/PrP+/− mice are also hypersensitive to excitotoxic stimuli, we chronically exposed slices to 0.5 mM glutamate for 24 h, and scored cell death by PI staining (Figure 8A). We detected a markedly higher (>two fold) percentage of PI-positive cells in ΔCR/PrP+/− OCSs, as compared to PrP+/− slices (Figure 8B). PI-positive cells were almost exclusively localized in the cerebellar granule layer, consistent with the conclusion that glutamate induces excitotoxicity preferentially in ΔCR PrP-expressing granule neurons and not in glia (Figure 8A) [Purkinje cells do not express ΔCR PrP, since the transgenic vector lacks a Purkinje cell-specific enhancer (Li et al., 2007b)]. As expected, the hypersensitizing effect of the PrP mutant was fully abrogated in ΔCR/Tga slices. These data confirm that, as seen in NSCs, expression of ΔCR PrP enhances the susceptibility of neuronal cells to glutamate-induced excitotoxic stress.
FIGURE 8. ΔCR granule neurons in cerebellar slices are hypersensitive to glutamate, NMDA, and kainate.

(A) Propidium iodide labeling of slices after treatment with 500 μM glutamate for 24 hours shows that cells within the cerebellar granule cell layer of ΔCR/PrP+/−animals are significantly more susceptible to glutamate-induced toxicity than controls. Overlay of brightfield images and propidium iodide fluorescence images confirms that the dying cells localize to the granule cell layer (Scale bar: 20 μm). (B) Quantitation of labeled cells revealed significant differences in glutamate-induced toxicity between ΔCR/PrP+/− animals and controls (** indicates p<0.01). (C) Quantitation of propidium iodide positive cells after treatment with receptor sub-type agonists reveals that ΔCR/PrP+/− slices contain significantly more labeled cells compared to controls after treatment with NMDA and kainate, but not AMPA (*, ** indicate p<0.05, and p<0.01, respectively).
To dissect the receptor specificity of glutamate-dependent cell death in ΔCR neurons, we used specific agonists of different subtypes of ionotropic glutamate receptors, including N-methyl-D-aspartic acid (NMDA), kainic acid, and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA). We exposed PrP+/−, ΔCR/PrP+/−, or ΔCR/Tga OCSs to NMDA, kainate and AMPA, and estimated the number of PI-positive granule neurons before and after treatment (Figure 8C). We found that granule neurons in ΔCR/PrP+/− OCSs were significantly more susceptible to both kainate and NMDA than those in PrP+/− or ΔCR/Tga control slices, with NMDA having a more marked effect. There was no significant difference between ΔCR/PrP+/−and control slices in their response to AMPA.
DISCUSSION
One of the most puzzling questions in the field of prion diseases is how prions and other abnormal forms of PrP cause neurodegeneration (Mallucci, 2009). To address this question, we have been studying ΔCR PrP, a deleted form of PrP that causes a lethal neurodegenerative phenotype when expressed in Tg mice. This phenotype is dose-dependently suppressed by wild-type PrP, suggesting that the ΔCR mutation subverts a normal, functional activity of PrPC. We are interested in understanding the cellular pathways underlying the potent toxicity of ΔCR PrP, with the expectation that these mechanisms will be relevant to naturally occurring prion disorders, such as those caused by dominantly inherited PrP mutations or infectious PrPSc.
Previous studies of ΔCR PrP expressed in transfected cell lines revealed that this mutant induces large, spontaneous inward currents that can be recorded by whole cell patch clamping, and also hypersensitizes cells to the toxic effects of several cationic antibiotics, probably by facilitating drug entry (Massignan et al., 2010; Solomon et al., 2010b). Biophysical and cell biological analysis suggested that ΔCR PrP currents are caused by formation of non-selective cationic channels in the plasma membrane, and that the N-terminal domain of PrP and attachment to the membrane are crucial for this activity (Solomon et al., 2010a; Solomon et al., 2011). However, whether ΔCR PrP could exert these effects in neurons and cause neurotoxicity was unknown.
In this manuscript, we demonstrate that ΔCR PrP induces spontaneous currents in neuronal cells from primary cultures and cerebellar slices. This activity, although insufficient by itself to induce extensive cell death, makes neurons unusually susceptible to glutamate-dependent, Ca2+-mediated excitotoxicity. Importantly, WT PrP suppresses all these phenotypes, demonstrating a tight connection between the neurotoxicity of ΔCR PrP and the physiological activity of PrPC. Our results suggest a model in which ΔCR PrP-induced current activity combines with endogenous, glutamate-mediated synaptic activity to unleash a Ca2+-dependent excitotoxic cascade, leading to neuronal death as observed in Tg(ΔCR) mice. Because of the emerging role of PrPC as a receptor for oligomeric forms of the Alzheimer’s Aβ peptide, and possibly other β-rich protein aggregates, our conclusions suggest that a similar PrP-dependent, excitotoxic mechanism may be operative in other neurodegenerative disorders.
ΔCR PrP induces ionic currents in neurons
To gain insights into the electrophysiological consequences of ΔCR PrP expression, we analyzed ΔCR-expressing neurons by whole-cell patch clamping. Our results show that ΔCR PrP generates spontaneous inward currents in cerebellar granule neurons in several different preparations, including dissociated neurons that have been lentivirally transduced, as well as isolated neurons and cerebellar slices derived from Tg(ΔCR) mice. These currents are similar in their properties to those we have described in a variety of non-neuronal cell types, including those of mammalian as well as insect origin (Solomon et al., 2010b). Considering the wide variety of cell types in which ΔCR currents have been observed, these data indicate either that mutant PrP molecules activate a ubiquitously expressed, endogenous ion channel, or else that the mutant protein itself forms pores in the cell membrane.
Downstream pathways activated by ΔCR PrP
How might the spontaneous current activity associated with ΔCR PrP lead to the striking degeneration of cerebellar granule neurons observed in Tg(ΔCR) mice? Several pieces of data have helped pinpoint downstream pathways that may be involved. In vivo, cell death occurring in ΔCR-expressing CGNs does not involve activation of caspase-3 or caspase-8 (Li et al., 2007b; Christensen et al., 2010). Moreover, genetic deletion of the pro-apototic factor Bax had no effect on the disease progression or neuropathological signs in Tg(ΔCR) mice (Li et al., 2007a). Consistent with these results, we failed to detect activated caspase-3 in ΔCR NSCs treated with glutamate. Collectively, our data indicate that neither the intrinsic nor extrinsic apoptotic pathways are involved in ΔCR PrP-dependent neuronal loss. The possibility that ΔCR CGNs die by an autophagic cell death pathway is also ruled out by the lack of increased LC3-II reactivity, a marker for autophagy, in the cerebellum of Tg(ΔCR) mice (Christensen et al., 2010). Ultrastructural analyses further support these conclusions. In particular, degenerating granule neurons in Tg(ΔCR) mice displayed a heterogeneous compaction of chromatin with preservation of the nuclear envelope, features distinct from neurons undergoing classical apoptosis (Christensen et al., 2010).
The distinctive ultrastructure of degenerating CGNs in Tg(ΔCR) mice, which was also seen in the OSCs studied here (Figure 7), is strikingly reminiscent of neurons undergoing excitotoxic death induced by the neurotransmitter glutamate (Christensen et al., 2010). This form of neuronal death is non-apoptotic and depends on activation of a pathway involving PARP-1 and AIF (Yu et al., 2003). Thus, our data suggest that ΔCR neurons degenerate by a non-apoptotic process that resembles glutamate-mediated excitotoxic death both biochemically and morphologically.
A role for glutamate in ΔCR PrP-dependent neuronal death
We present here several different experiments that directly implicate glutamate-mediated excitotoxicity in the neuronal death caused by ΔCR PrP. First, we observed that expression of ΔCR PrP induced neuron-specific, Ca2+-mediated hypersensitivity to glutamate stimulation in differentiated NSCs. This phenomenon, which was characterized by a higher amount of Ca2+-influx and a slower rate of intracellular Ca2+ recovery in ΔCR PrP-expressing neurons, as compared to control neurons, was accompanied by increased neuronal death in cultures that were chronically treated with glutamate. ΔCR neurons in OCS cultures were also highly susceptibile to glutamate-induced cell death. When treated with different glutamate agonists, these cells showed a pattern of sensitivity (NMDA>kainate>AMPA) that paralleled the ability of each agonist to induce Ca2+-influx. Consistently, hyper-susceptibility to glutamate treatment of ΔCR NSCs was abrogated by interfering with the activation of NMDA receptors. Taken together, these results provide strong support for the notion that expression of CR PrP sensitizes neuronal cells to glutamate-induced, Ca2+-mediated excitotoxicity.
How does ΔCR PrP enhance glutamate sensitivity?
One possible explanation for our data is that ΔCR PrP directly activates endogenous, ionotropic glutamate receptors in neurons. PrPC was previously reported to suppress the activity of NMDA receptors through a direct interaction with NR2D subunits, thereby protecting neurons from glutamate excitotoxicity (Khosravani et al., 2008; Stys et al., 2012; You et al., 2012). In addition, increased kainate-dependent neuronal death was observed in mice lacking PrPC (Rangel et al., 2007). We report here that WT PrP fully rescues ΔCR PrP-induced hypersensitivity to glutamate in differentiated NSCs and OCS cultures. These results suggest that ΔCR PrP, instead of silencing NMDA receptors like WT PrP, activates them, possibly via a physical interaction with NR2D or other channel subunits. Alternatively, ΔCR PrP could also directly or indirectly affect the activity of other ion channels, such as nAChRs (Petrakis et al., 2008; Beraldo et al., 2010), to increase neuronal excitability. A number of electrophysiological abnormalities have been reported in neurons from PrP knock-out mice (Linden et al., 2008), suggesting that PrPC might normally modulate several kinds of ion channels, a function that could be corrupted by ΔCR or other central region mutations.
In our initial characterization of CR-associated currents, we reported a lack of inhibitory effect of different ion channel blockers, including TEA, tetrodotoxin, and MK-801 (blockers of voltage-gated potassium channels, voltage-gated sodium channels, and NMDA receptors, respectively) (Solomon et al., 2010a). In the same study, we also showed that ΔCR currents can be detected in a variety of cell lines, including HEK293 and Sf9 cells. These latter cell lines are known to have limited expression of endogenous ion channels, including undetectable levels of NMDARs (Larsen et al., 1996; Thomas and Smart, 2005). Moreover, the presence of ΔCR currents in cell types from evolutionary distant species (HEK293 are a human cell line, while Sf9 are insect cells), suggeststhat any channel involved in mediating the currents of mutant PrP must be highly conserved across cell types and species. There are examples of such channels, for example connexin (gap junction) or pannexin hemichannels, but it remains to be determined if such channels play a role in ΔCR PrP-induced currents.
An alternative model is based on the idea that the ΔCR PrP itself forms an ion channel or pore in the cell membrane. In this scenario, the spontaneous current activity induced by ΔCR PrP is insufficient on its own to cause significant neuronal death, but it primes the neuron to degenerate in response to endogenous synaptic inputs such as those mediated by glutamate. This model is consistent with the striking observation that, although ΔCR PrP induces massive death of cerebellar granule neurons in Tg(ΔCR) mice, expression of the mutant protein has minimal effect on the viability of neurons in differentiated stem cell cultures (Massignan et al., 2010) and in cerebellar slices (Figure 6A–B), situations in which these neurons are deprived of their normal synaptic input. During normal cerebellar development, CGNs receive excitatory glutamatergic input from mossy fibers at the time they complete their migration from the external to the internal granule layer (Kandel, 2012). Consistent with this timeline, we have observed that granule cell degeneration in Tg(ΔCR) mice is absent from the external layer, and occurs in a highly synchronous fashion in the internal layer at P13–15 in Tg(ΔCR)/Prn-p+/− mice, coincident with mossy fiber innervation (Christensen et al., 2010). Moreover, our patch clamping analyses of acute cerebellar slices revealed an unexpected fragility of ΔCR granule neurons. This phenomenon, although difficult to dissect experimentally, indicates that ΔCR PrP is deleterious to the integrity of the neuronal plasma membrane, and may prime neurons to degenerate in response to increased levels of endogenous synaptic activity, or perhaps other physiological stresses.
As mentioned above, it was previously reported that WT neurons were less susceptible to glutamate-induced excitotoxicity than neurons from PrP-null mice (Khosravani et al., 2008). These data apparently conflict with our observation that NSCs lacking PrPC don’t show higher sensitivity to glutamate treatment than control NSCs (Figures 2 and 3). However, the discrepancy may easily be related to the use of different experimental models (differentiated NSCs vs. primary neuronal cultures), or to the strain background of the mice used for the experiments.
Relevance for prion diseases and other neurodegenerative disorders
Recent evidence that PrPC binds to Aβ oligomers and other misfolded protein isoforms suggests that the underlying mechanism for several neurodegenerative disorders may converge on PrPC (Biasini et al., 2012a). We have hypothesized that binding of several oligomeric protein assemblies, including PrPSc and Aβ oligomers, causes a conformational change in the PrPC molecule, with consequent insertion of its N-terminal polybasic domain across the membrane and generation of “ΔCR-like” channels or pores (Solomon et al., 2012). Deletions within the central region of PrP would constitutively induce such a topological change, and produce the strongest channel activity. In support of this model, the same two regions on PrP that determine ΔCR PrP channel activity (residues 23–31 and 105–125), overlap with or are adjacent to regions involved in binding of PrPC to PrPSc, Aβ oligomers and other β-rich aggregates. Based on the results presented here, we suggest that other oligomeric protein assemblies might sensitize neurons to glutamate-mediated excitotoxicity via a PrPC-dependent mechanism. If so, then glutamate channel blockers, as well as drugs that prevent binding of β-sheet rich oligomers to PrPC, or that block the topological transition of PrPC, may show therapeutic benefits for several neurodegenerative diseases (Biasini and Harris, 2012). Indeed, memantine, a glutamate channel blocker, is currently used for treatment of Alzheimer’s disease (Maggiore et al., 2007).
Acknowledgments
We thank Joe Amatrudo in the Leubke laboratory for advice and technical assistance with patch clamping. We also thank Jorge De Castro for mouse maintenance and genotyping, and Nada Husic for lentiviral preparation. Lentiviral constructs were created by the Hope Center Viral Vectors Core at Washington University, which is supported by a Neuroscience Blueprint Core grant (Grant P30 NS057105) from the N.I.H.
This work was supported by grants from the N.I.H. to D.A.H. (NS040975 and NS052526) and J.L. (AG00001 and AG025062), from the Fondazione Telethon to R.C. (TCR08005) and V.B. (TCR08002), and by a fellowship from the Fondazione Monzino to E.B. U.U. was supported by an Erwin Schroedinger fellowship (J2840), from the Austrian Science Fund (FWF). I.H.S was supported by an N.I.H. Pre-doctoral Fellowship NS063547 and by the Medical Scientist Training Program at Washington University (N.I.H. Grant T32GM007200). V.B and R.C. are Associate Telethon Scientists (Dulbecco Telethon Institute, Fondazione Telethon). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors declare no competing financial interests
References
- Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–561. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA, Martins VR. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem. 2010;285:36542–36550. doi: 10.1074/jbc.M110.157263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini E, Harris DA. Targeting the cellular prion protein to treat neurodegeneration. Future Med Chem. 2012;4:1655–1658. doi: 10.4155/fmc.12.114. [DOI] [PubMed] [Google Scholar]
- Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012a;35:92–103. doi: 10.1016/j.tins.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini E, Turnbaugh JA, Massignan T, Veglianese P, Forloni G, Bonetto V, Chiesa R, Harris DA. The toxicity of a mutant prion protein is cell-autonomous, and can be suppressed by wild-type prion protein on adjacent cells. PLoS One. 2012b;7:e33472. doi: 10.1371/journal.pone.0033472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443:803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Christensen HM, Harris DA. A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J Neurochem. 2009;108:44–56. doi: 10.1111/j.1471-4159.2008.05719.x. [DOI] [PubMed] [Google Scholar]
- Christensen HM, Dikranian K, Li A, Baysac KC, Walls KC, Olney JW, Roth KA, Harris DA. A highly toxic cellular prion protein induces a novel, nonapoptotic form of neuronal death. Am J Pathol. 2010;176:2695–2706. doi: 10.2353/ajpath.2010.091007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Forloni G, Lucca E, Angeretti N, Chiesa R, Vezzani A. Neuroprotective effect of somatostatin on nonapoptotic NMDA-induced neuronal death: role of cyclic GMP. J Neurochem. 1997;68:319–327. doi: 10.1046/j.1471-4159.1997.68010319.x. [DOI] [PubMed] [Google Scholar]
- Gunther EC, Strittmatter SM. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J Mol Med (Berl) 2010;88:331–338. doi: 10.1007/s00109-009-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Kandel ER. Principles of neural science. 5. New York: McGraw-Hill; 2012. [Google Scholar]
- Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen EH, Gabriei SE, Stutts MJ, Fullton J, Price EM, Boucher RC. Endogenous chloride channels of insect sf9 cells. Evidence for coordinated activity of small elementary channel units. The Journal of general physiology. 1996;107:695–714. doi: 10.1085/jgp.107.6.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Barmada SJ, Roth KA, Harris DA. N-terminally deleted forms of the prion protein activate both Bax-dependent and Bax-independent neurotoxic pathways. J Neurosci. 2007a;27:852–859. doi: 10.1523/JNEUROSCI.4244-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007b;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- Maggiore C, Locatelli L, Grandi FC, Pizzolato G. Memantine in the treatment of dementia. Neuroepidemiology. 2007;28:118–119. doi: 10.1159/000101511. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mallucci GR. Prion neurodegeneration: starts and stops at the synapse. Prion. 2009;3:195–201. doi: 10.4161/pri.3.4.9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron. 2007;53:325–335. doi: 10.1016/j.neuron.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Martin JH. Neuroanatomy: text and atlas. 2. Stamford, Conn: Appleton & Lange; 1996. [Google Scholar]
- Massignan T, Stewart RS, Biasini E, Solomon IH, Bonetto V, Chiesa R, Harris DA. A novel, drug-based, cellular assay for the activity of neurotoxic mutants of the prion protein. J Biol Chem. 2010;285:7752–7765. doi: 10.1074/jbc.M109.064949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54:369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- Petrakis S, Irinopoulou T, Panagiotidis CH, Engelstein R, Lindstrom J, Orr-Urtreger A, Gabizon R, Grigoriadis N, Sklaviadis T. Cellular prion protein co-localizes with nAChR beta4 subunit in brain and gastrointestinal tract. Eur J Neurosci. 2008;27:612–620. doi: 10.1111/j.1460-9568.2008.06037.x. [DOI] [PubMed] [Google Scholar]
- Rangel A, Burgaya F, Gavin R, Soriano E, Aguzzi A, Del Rio JA. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J Neurosci Res. 2007;85:2741–2755. doi: 10.1002/jnr.21215. [DOI] [PubMed] [Google Scholar]
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. Embo J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon IH, Schepker JA, Harris DA. Prion neurotoxicity: insights from prion protein mutants. Curr Issues Mol Biol. 2010a;12:51–61. [PMC free article] [PubMed] [Google Scholar]
- Solomon IH, Huettner JE, Harris DA. Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem. 2010b;285:26719–26726. doi: 10.1074/jbc.M110.134619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon IH, Biasini E, Harris DA. Ion channels induced by the prion protein: mediators of neurotoxicity. Prion. 2012;6:40–45. doi: 10.4161/pri.6.1.18627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem. 2011;286:14724–14736. doi: 10.1074/jbc.M110.214973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, You H, Zamponi GW. Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. The Journal of physiology. 2012;590:1357–1368. doi: 10.1113/jphysiol.2011.225276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Smart TG. HEK293 cell line: a vehicle for the expression of recombinant proteins. Journal of pharmacological and toxicological methods. 2005;51:187–200. doi: 10.1016/j.vascn.2004.08.014. [DOI] [PubMed] [Google Scholar]
- White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci U S A. 2008;105:10238–10243. doi: 10.1073/pnas.0802759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]