

Abstract
In a large cohort of osteogenesis imperfecta type V (OI type V) patients (17 individuals from 12 families), we identified the same mutation in the 5' UTR of the IFITM5 gene by whole exome and Sanger sequencing (IFITM5 c.-14C>T) and provide a detailed description of their phenotype. This mutation leads to the creation of a novel start codon adding 5 residues to IFITM5 and was recently reported in several other OI type V families. The variability of the phenotype was quite large even within families. Whereas some patients presented with the typical calcification of the forearm interosseous membrane, radial head dislocation and hyperplastic callus (HPC) formation following fractures, others had only some of the typical OI type V findings. Thirteen had calcification of interosseous membranes, fourteen had radial head dislocations, ten had HPC, nine had long bone bowing, eleven could ambulate without assistance, and one had mild unilateral mixed hearing loss. The bone mineral density varied greatly, even within families. Our study thus highlights the phenotypic variability of OI type V caused by the IFITM5 mutation.
Introduction
Osteogenesis imperfecta (OI) is a genetically and clinically heterogeneous disorder of connective tissues (1). Although originally classified by phenotype and inheritance as four distinct types by Sillence et al. in 1979, recent advances in mutation analysis and the recognition of new phenotypes has resulted in an extended classification (2,3). First reported by Glorieux et al. in 2000, OI type V is a non-lethal autosomal dominant form of OI (4). There are an estimated 25,000 cases of OI in the US, 4–5% of which may be type V (3,5). As with other non-lethal forms of the disorder, such as Sillence types I, III and IV, OI type V has been characterized by moderate bone fragility, scoliosis, long-bone deformities, and diminished stature. However, type V demonstrates highly variable expressivity. It is distinguished from other OI types by the frequent occurrence of HPC following fracture or surgery in approximately 65% of the affected individuals, the presence of bilateral radial head dislocation, and ossification of the interosseous membrane in the forearm and lower extremity. However, type V patients typically do not have discoloration of sclerae or dentinogenesis imperfecta (DI) (4).
At the molecular level, OI type V patients lack detectable mutations in the COL1A1 and COL1A2 genes that are found in more than 90% of cases of OI types I through IV or in recessive OI genes (6). In 2012, it was published that a mutation in the 5'UTR of the IFITM5 gene (IFITM5 c.-14C>T) can cause OI type V (7,8). Interferon-inducible transmembrane protein 5 (IFITM5), otherwise known as bone restricted ifitm-like protein (BRIL), is a two-transmembrane domain membrane protein that presents its termini extracellularly. Its expression is limited to osteoblasts and shows a pattern similar to the transcription factor osterix (SP7) (9–11). It has been shown that IFITM5 plays a role in mineralization in vitro and is involved in bone growth during prenatal development in a mouse model (10,11).
Our report confirms the presence of the same mutation (IFITM5 c.-14C>T) in our cohort and describes the clinical and radiological manifestations of the disease in seventeen patients, approximately half of whom have inherited the disorder from an affected parent.
Materials and Methods
Patients
Permission to report this retrospective chart analysis was granted by the Institutional Review Board at the Kennedy Krieger Institute/Johns Hopkins University. Molecular testing was performed according to the specifications of the Baylor College of Medicine Institutional Review Board.
Thirteen OI type V patients were identified in a review of approximately 350 OI patients at the Kennedy Krieger Institute, thus, OI type V patients constitute 3.7% of the center's OI population. Four other patients were enrolled at the Skeletal Dysplasia Clinic of the Texas Children's Hospital. Ten patients had Sanger sequencing at CLIA-certified laboratories by Sanger sequencing for COL1A1 and COL1A2 and were not found to harbor mutations in these genes.in clinical laboratories. Three patients did not have any molecular studies prior to enrollment (patients 6, 7, and 13); they were diagnosed by their archetypical clinical and radiographic presentation.
Radiographic images and reports were reviewed for the presence of radial head dislocation and mineralized interosseous membranes between the radius and ulna, and/or the tibia and fibula. Evidence was sought for excessive callus formation post-injury which is defined as a relatively large, radio-opaque lesion originating from the surface of the bone, showing unusual features such as sun-ray spicules or a “butterfly-like” appearance (12). Also, spine x-rays were reviewed for presence of scoliosis. Comparative z-scores for height and weight were obtained from the Center for Disease Control; 2004 Advanced Data Vital and Health Statistics (13). Dual Energy X-ray Absorptiometry (DXA) bone density measurements of lumbar vertebrae were obtained using a Hologic DelphiA machine (software version 11.2:3, Hologic, Bedford, MA).
Exome sequencing
Before the publication of the gene for OI type V, exome sequencing and analysis was conducted as previously described to try to identify the causative gene(14). Briefly, exomes were captured on Nimblegen's SeqCap EZ V2.0 library and sequencing was conducted on Illumina HiSeq2000. Reads were aligned with BWA (Burrows-Wheeler Aligner) to human reference genome GRCh37/hg19 and locally realigned with GATK (Genome Analysis Toolkit) around potential INDELs. SNPs were called using Samtools Pileup (version 0.1.17) (15) and short indels were called using Samtools (15), Atlas-INDEL (16), and GATK (17). Variants were annotated with ANNOVAR (18); protein-impacting variants that were rare (minor allele frequency <5%) or novel were preferentially explored.
Sanger sequencing
Amplicons were generated using 1 ng/microliter of genomic DNA using TaqMan polymerase (ABI, Life Technologies, Carlsbad, CA) using the manufacturer's protocol with an annealing temperature of 60 °C and an amplification of 1 minute. Primers used were forward: 5'-AGGGCGACAGGGCTATAAGTGAG-3' and reverse: 5'-GAAGCCGAGGCAACACAGATTCAGGTAG-3'. Products were verified by agarose gel then sequenced by Sanger sequencing using the same primers at Beckman Coulter Genomics (Danvers, MA).
Results
Demographic data and treatment modalities are summarized in Table 1. Four families with multiple individuals with OI type V are grouped in this table; the remaining eight non-familial cases were the result of spontaneous mutations. The average age at diagnosis in our cohort of 8 males and 9 females was one year of age. Height z-scores averaged −2.9 (±3.4)for males and −4.5 (±3.6)for females (mean values are presented with standard deviations throughout this work). Weight z-scores averaged −1.5 (±1.4) for males and −1.3 (±1.0)for females. Body mass index (BMI) values averaged 29 (±8) kg/m2 for men and 29 (±2) kg/m2 for women over the age of 18 years.
Table 1.
Demographic Overview of Cohort
| PT | Gender | Age at OI Dx (years) | Age (years) at Examination | Ambulatory Status | Height (cm) | Height z-score | Weight (kg) | Weight z-score | BMI (kg/m2) | Racei | Familial Association | Bisphosphonate Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Birth | 7 | Ambulatory | 118 | −0.4 | 19 | −1.1 | 13 | A/C | Son of patient 3 | Pamidronate (IV) |
| 2 | M | 1.5 | 12 | Ambulatory | 149 | 0.2 | 36 | −0.4 | 16 | A/C | Son of patient 3 | Pamidronate (IV) |
| 3 | F | 0.5 | 48 | Ambulatory w/ Assistance | 147 | −2.9 | 61 | −0.4 | 28 | C | Mother of patients 1 and 2 | Alendronate (PO) |
| 4 | F | 2.0 | 43 | Wheelchair | 97 | −11.9 | 24 | −1.6 | 26 | C | - | - |
| 5 | F | 2.0 | 33 | Ambulatory | 115 | −6 | 41 | −0.9 | 31 | C | - | - |
| 6 | F | In Utero (27wks) | 2 months | n/a | 53 | −2.4 | 5 | −0.8 | 17 | C | Daughter of patient 7 | - |
| 7 | M | Birth | 32 | Ambulatory | 147 | −3.6 | 43 | −1.2 | 20 | C | Father of patient 6 | - |
| 8 | M | 0.5 | 60 | Wheelchair | 98 | −10.4 | 33 | −3.6 | 34 | C | Father of patient 9 | Pamidronate (IV) & Alendronate (PO) |
| 9 | M | 0.7 | 24 | Ambulatory | 173 | −0.5 | 68 | −0.4 | 23 | A/C | Son of patient 8 | Pamidronate (IV) |
| 10 | F | 2.0 | 23 | Ambulatory | 151 | −1.5 | 74 | −0.4 | 32 | C | - | - |
| 11 | M | 3.0 | 23 | Wheelchair | 152 | −3.4 | 91 | 0.5 | 39 | C | - | - |
| 12 | F | 1.3 | 5 | Ambulatory | 86 | −4.3 | 10 | −1.8 | 13 | C | - | Pamidronate (IV) |
| 13 | M | 0.7 | 47 | Wheelchair | 150 | −2.5 | 54 | −2.2 | 24 | C | - | - |
| 14 | M | N.A | 6 | Ambulatory | 101 | −3.0 | 14 | −3.2 | 14 | C | - | Alendronate |
| 15 | F | Birth | 36 | Ambulatory | 140 | 56 | 28 | C | - | Alendronate, Ibandronate, zoledronic acid Calcitonin, Teriparatide |
||
| 16 | F | 1 | 4 | Ambulatory | 91 | −2.7 | 12 | −3.1 | 14 | C | Daughter of patient 17 | Pamidronate IV |
| 17 | F | 0.5 | 21 | Ambulatory | 147 | 64 | 29 | C | Mother of patient 16 | None |
C=Caucasian; A=Asian; H=Hispanic.
Clinical features are summarized in Table 2. Two patients exhibited blue sclerae. Three patients had pes planus. Eleven patients were described as having bilateral hyperextensible joints in at least one extremity, typically in digits and elbows. Fifteen patients exhibited limited/absent ability to supinate and pronate the forearm due to dislocation of the radial head. With regard to other common OI findings: triangular facies were observed in ten patients. DI was not observed in any patient. Hearing loss, either conductive or mixed, was generally absent from the cohort; only one patient (Patient 2) was noted to have mild (8 kHz) unilateral mixed hearing loss.
Table 2.
Clinical Features of Cohort
| PT | Blue Sclera | Triangular Facies | Pes Planus | Hyperextensible Joints | Limited Range Supination or Pronation | COL1A1/COL1A2 Analysis for Mutation | IFITM5 c.-14C>T mutation |
|---|---|---|---|---|---|---|---|
| 1 | - | + | + | - | Limited | Negative | + |
| 2 | - | + | + | + (Knees, Elbows) | Limited | Negative | + |
| 3 | - | + | - | + (Digits) | Absent | Negative | + |
| 4 | - | + | - | + (Digits) | Absent | Negative | + |
| 5 | - | + | - | + | Limited | Negative | + |
| 6 | - | - | - | - | Normal | Not Performed | + |
| 7 | - | + | - | - | Limited | Not Performed | + |
| 8 | - | + | - | - | Absent | Negative | + |
| 9 | - | - | - | + (Digits) | Limited | Negative | + |
| 10 | - | + | + | - | Limited | Negative | + |
| 11 | - | - | - | + (Elbows) | Limited | Negative | + |
| 12 | - | + | - | + (Ankles, Hips) | Limited | Negative | + |
| 13 | - | - | - | + (Elbows) | Absent | Not Performed | + |
| 14 | - | - | - | + (elbows) | Normal | Negative | + |
| 15 | - | + | - | + | Absent | Negative | + |
| 16 | + | - | - | + | Limited | Negative | + |
| 17 | + | - | - | - | Limited | Not performed | + |
Radiographic findings characteristic of OI Type V patients are summarized in Table 3. Thirteen patients exhibited interosseous membrane calcification (Figure 2). One patient (patient 5) was incidentally noted to have calcification of her left lower extremity interosseous membrane. Also, fourteen patients were observed to have dislocated radial heads bilaterally. Thirteen patients had scoliosis; all patients over the age of five exhibited mild to severe scoliosis. Additionally, nine patients exhibited bowing of long bones: six had anterior bowing of the tibias and/or fibulas, three had bowing of the femurs, and six had bowing of the forearms. Ten patients experienced one-or-more episodes of HPC formation (Figure 2) after trauma-related fractures.
Table 3.
Radiographic Findings of Cohort
| Patient | Calcification of Interosseous Membranes | Radial Head Dislocation | HPC | Scoliosis |
|---|---|---|---|---|
| 1 | + | + | + (Humerus) | + |
| 2 | N.A | + | - | + |
| 3 | + (Fusion) | + | - | + |
| 4 | + | + | + (Femur) | + |
| 5 | + | + | - | + |
| 6 | - | - | + (Ribs) | - |
| 7 | + (Fusion) | + | + (Femur) | + |
| 8 | + | + | - | + |
| 9 | + | + | - | + |
| 10 | + | + | + (Femur) | + |
| 11 | N.A | + | - | + |
| 12 | + | + | + | - |
| 13 | + | + | + (Humerus) | + |
| 14 | N.A. | - | + | - |
| 15 | + | No X-ray but supination/pronation absent | - | + |
| 16 | + | + | + (Femur) | + |
| 17 | + | + | + (Femur) | - |
N.A.= not available
Figure 2.

Radiographic features in OI type V. A and B) Radial head dislocation (arrows on the right) and forearm interosseous membrane calcification (middle arrows) in patients 1 and 3 respectively. C) HPC in patient 14 at 1.5 years.
An unusual opportunity permitted us to follow the course of pregnancy in the first child of a 35 year old man with OI type V. The ultrasound scans at 22 weeks of gestation permitted the detection of skeletal abnormalities (bowing of the femur illustrated in Figure 3), and at 33 weeks a thin calvarium and angulated ribs suggesting intrauterine fracture. At birth, the infant had two healing rib fractures, bowing of the femurs and a thin calvarium but no other abnormalities.
Figure 3.

Prenatal ultrasound at 22 weeks showing bowing of the femur (the crosshairs show the extremities of the femur).
Extra-skeletal findings of note include two patients with mitral valve prolapse (Patients 8 and 10), two patients (Patients 7 and 8) with histories of symptomatic nephrolithiasis and one patient with hypertension and depression (Patient 15). Orthopedic surgical histories include seven patients who had undergone at least one long-bone intramedullary rodding (typically tibiae and femurs). Additionally, three patients underwent posterior spinal fusion for scoliosis. Surgical resection of dislocated radial heads were performed in two patients from the same family (Patients 2 and 3) with only marginal improvements noted in elbow range of motion and no increase in range of motion at the wrist. There was no HPC formation following rodding or other orthopedic surgeries in our cohort.
Of the 17 patients with OI reported here, 10 received treatment with bisphosphonates: five received only pamidronate intravenously (Patients 1, 2, 9, 12 and16), one adult (Patient 3) and one pediatric patient (Patient 14) received only oral alendronate, and another 2 adults (Patient 8 and 15) received both oral and intravenous treatments in succession (alendronate and pamidronate or zoledronic acid).
The results of clinical laboratory studies of serum calcium, phosphorus, 25 (OH) vitamin D, and insulin-like growth factor were within normal limits for all patients. Alkaline phosphatase activity, although elevated during fracture or HPC formation, were typically within normal limits with one notable exception: serum total and bone-specific alkaline phosphatase activities were persistently elevated in three patients.
Exome sequencing was performed for 17 individuals before the reports on the gene identification were published. Over 90% of the target region obtained over 20× coverage, with an average coverage between 80–130× for coding regions. Exome sequencing identified on average 574 rare variants per individual predicted to impact proteins (missense, nonsense, indels, and splice site mutations). No mutations were identified in the OI genes COL1A1, COL1A2, CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1, SERPINF1, PLOD2 or SP7. When considering only coding regions and variants within 5 positions of splice sites, exome sequencing identifies on average 300–500 rare or novel coding variants in any individual (19). In our cohort, no gene showed rare or novel variants in more than 3 families, suggesting either great genetic heterogeneity or that the causative mutation was not captured by the sequencing or was missed by our analysis, which did not include untranslated regions. After the publication of a de novo mutation introducing an early start site in IFITM5 (7,8), this region was analyzed on the exome data. The average coverage was 19× within 5'UTRs and 12× within 3'UTRs. Exome sequencing data showed the same mutation in our cohort of OI type V individuals. We confirmed the mutation by Sanger sequencing and the same mutation was present in 12 families (17 individuals). This experience highlights the importance of considering UTRs in analyzing exome data.
Discussion
In 2000, Glorieux et al. described OI type V as a phenotype different from Sillence types I–IV (4). Definition for this subtype was based on the recognition of the frequent occurrence of HPC in a group of patients that were initially categorized as Sillence type IV disease. Subsequent literature includes a cohort study on twelve Korean patients (20), a summary of the response to treatment with bisphosphonates (21), a description of the natural history of HPC in type V patients (22), and just recently, the identification of the gene by two groups (7,8).
Clinical and radiological findings in our cohort of seventeen patients strengthen the case for considering this as a separate phenotype within the OI family. Although hearing loss—a symptom reportedly experienced by approximately 50% of type I OI adults—appears to occur infrequently in OI type V, its real frequency cannot be estimated in the absence of audiological testing or without a larger number of patients (23,24). Only one patient in our cohort experienced minimal unilateral sensorineural hearing loss.
Variable expressivity, both within and between affected families, is characteristic of OI type V. Illustrative of the diverse phenotype, over one-half of these patients were able to ambulate unassisted while others required walking-aids or wheelchairs. A photograph depicting the remarkably varied phenotypic expression of the same mutation in two affected members of the same family is seen in Figure 1.
Figure 1.
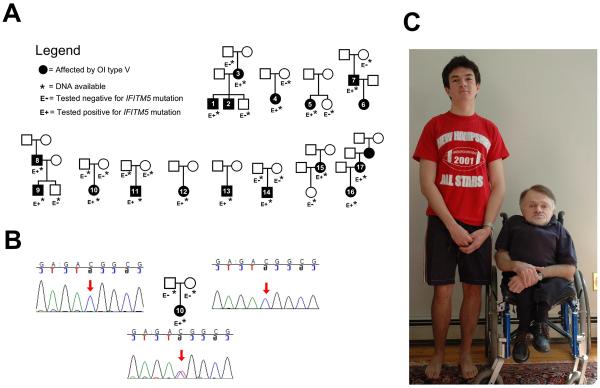
A) Pedigrees of the individuals described. B) Example of Sanger sequencing results for the IFITM5 mutation, here in patient 10 and her parents. C) Two affected members of the same family: patients 8 and 9. The father is aged 52, and the son 18 at the time of the photograph. Although the son was treated at an earlier age with bisphophonates, this does not explain the important difference in phenotype.
HPC is described as a post-operative or post-fracture soft-tissue mass that may be confused with osteosarcoma because of an accompanying inflammatory component. CT and MRI findings characterize HPC as exuberant bone formation, disproportionate to the size of the affected bone with extensive expansion beyond the region of fracture (12). The differential diagnosis includes intraosseous osteosarcoma with aggressive periosteal reaction, periosteal osteosarcoma, juxtacortical myositis ossificans and large osteochondromata (12). However, in the 10 type V patients who experienced one-or-more occurrence of HPC the hyperplastic response did not appear as a tumor mass. Rather, the hyperplastic response was usually confined to localized overgrowth of bone at the fracture site seen on x-ray rather than presenting as a soft tissue mass (Figure 2). Our patients experienced callus formation in their femurs, humeri, and ribs. Similar to our findings, Cheung et al. reported a 65% incidence of HPC in OI type V patients, with half of the reactions occurring on, or around, the femurs (25). HPC has also been reported to involve the pelvis and forearms (12). In our cohort, HPC appeared to start from the periosteal region. In one patient (Patient 5), post-fracture calcification involved the plane between leg muscle bundles.
Another common radiographic finding in OI type V patients is the idiopathic calcification of the forearm interosseous membranes. The majority of our patients exhibited the mineralization phenomenon. Ossification was radiographically apparent in one patient by the age of four. Images of the child's arm can be seen in Figure 2. Although it may be assumed that calcification at these sites follows trauma, the relationship, if any, of the mechanism underlying interosseous membranes mineralization and hyperplasic callus is unknown. In this series, the two processes were not linked, in that ossification of the interosseous membrane between the radius and ulna and tibia and fibula were not coincident with the existence of HPC in the same patients. A question arises as to why HPC occurs in only about 50% of type V patients and, further, why is this absent in other OI syndromes?
Congenital radial head dislocation (CRHD) can occur as an isolated abnormality or in at least fourteen syndromes (26). Previously, it has been reported that the incidence of one or more radial head dislocations is 58% in type V patients, compared to an incidence of 1.5%, 8.5% and 6.5% for types I, III, and IV respectively (27). Dislocation of the radial head was observed in 0–29 % of OI patients other than type V and in 86 % of type V patients by Fassier et al. (27). Bilateral dislocation of the radial heads was observed in 88% of our patients. These patients have restricted pronation/supination of the forearm, a decreased flexion arc, and variable degrees of functional impairment. It is of interest that radial head dislocation was not present in the infant (Patient 6) at age 4 months. Dislocation of the radial head is associated with significant functional impairment: Resection of the radial head has been proposed to improve function. However, only minimal improvement was observed in the flexion arc at the elbow, and no improvement occurred in the degrees of pronation or supination in the two patients who underwent radial head resection. Lack of improvement in forearm function following radial head excision was also reported by Lee et al. (20). Thus, whether radial head excision should be recommended, remains to be decided.
Glorieux et al. described the presence of appendicular metaphyseal bands as a “constant feature in growing type V patients” (4). These radio-dense bands are visualized on plain films located immediately adjacent to the growth plate, typically involving the proximal tibia, distal femur or radius. No metaphyseal banding pattern was observed in this group. Last, it is worth noting that metaphyseal banding is described in OI type V patients should not be confused with the bisphosphonate-induced epiphyseal “zebra lines,” as are typically seen with cyclical IV pamidronate therapy.
Three OI type V patients were observed to have chronically elevated alkaline phosphatase values. In the case of two patients who were brothers, elevated alkaline phosphatase levels were seen even during bisphosphonate therapy, while their affected mother and normal father had normal alkaline phosphatase values. Serum alkaline phosphatase results were not reported by Lee et al. in 12 OI type V patients from three Korean families (20). Glorieux et al. observed that serum alkaline phosphatase increased markedly during periods of active HPC formation (4). However, Zeitlin et al. in reporting the effect of intravenous pamidronate treatment in type V patients observed initial alkaline phosphatase values to be above the upper level of normal (300 U/l) at 436 ± 160 U/l (21). Following 2 years of pamidronate treatment alkaline phosphatase values declined approximately 50% from baseline, but less than the post-treatment decline in urinary N- telopeptide. Thus, the clinical significance of alkaline phosphatase elevation remains to be elucidated.
Numerous reports have suggested that pediatric-age OI patients increase vertebral bone mineral density and decrease fracture rate in response to bisphosphonate treatment (28,29). In a recent report of 11 type V pediatric patients undergoing two years of cyclic pamidronate treatment, Zeitlin et al. reported a statistically-significant lower incidence of fracture, increases in transiliac cortical thickness, and increases in lumbar vertebral body size and volumetric bone mass density (21).
The last decade has seen remarkable progress in both our clinical and molecular understanding of osteogenesis imperfecta (30). Case finding of OI type V has been hampered by the marked phenotypic variability and until recently by the absence of a defined molecular cause. In turn, these have limited our ability to understand the mechanisms underlying the distinctive features of this syndrome: HPC formation, calcification of the interosseous membrane and dislocation of the head of the radius. However, it is evident that OI type V involves aberrant mineralization that is not seen in other OI types associated with abnormal synthesis of type I collagen. IFITM5 is involved in both mineralization and bone growth, but very little is known about its function (9–11). The mechanism by which the IFITM5 mutation cause the OI type V phenotype is unknown. We speculate that it might be related to an acquired molecular function due to the elongation of the protein, such as sustained signaling through an important osteogenesis signaling cascade.
The identification of the same mutation in 34 previous OI type V families (7,8,31), and in these 12 families contrasts markedly with the wide distribution of type I collagen mutations in other OI phenotypes. IFITM5 is a 14.8 kDa protein that has two transmembrane domains with both the amino and carboxy terminus present extracellularly. These termini exhibit the least conservation within the IFITM family of proteins, and are the regions that most likely interact with other proteins (11,32). The first functional studies of IFITM5, also termed Bril, included expression studies by northern blotting and in situ hybridization, both suggesting a bone specific expression pattern (10,11). In vitro studies suggest a role in mineralization, including expression during osteoblast differentiation as well as increased mineralization in an overexpression model (10,11). Knockout mice show a possible defect in prenatal bone development, since newborn mice display shorter bones that are sometimes bent in appearance. However, these skeletal phenotypes were not present in adult mice (10). Furthermore, IFITM5 has only one known binding partner, FKBP11. It has been shown that the binding of IFITM5 to FKBP11 disrupts binding of CD9 with the FKBP11-CD81-CD9/FPRP complex and increases expression of interferon-induced genes (9,10). Thus, there may be an immune component to OI type V clinical manifestations.
We suspect that IFITM5 not only has a role in bone development but also may play a role in the immune response in bone. This mutation extends the amino terminal extracellular domain by five amino acids and it is likely that this mediates protein interactions with IFITM5. It is possible that not only binding of FKBP11 may be altered, but also the downstream interferon response. It is also likely that IFITM5 interacts with other proteins in vivo and the mutation might increase, decrease or change this binding pattern. Through this process, it is likely that other components of osteogenesis are affected by the mutation and would lead to the low bone mass phenotype. Conditions under fracture repair may be different owing to the immune response and thus have an opposite phenotype, thus increasing bone formation without proper feedback on fracture repair. Introducing the mutation in transgenic mice should allow the dissection of the mechanisms leading to osteogenesis imperfecta and hyperplastic callous formation.
Acknowledgements
We thank Alyssa Tran for patient enrollment and Yuqing Chen for technical assistance. Research funding includes NIH grants PO1 HD22657 and PO1 HD070394. PC is supported by a CIHR clinician-scientist training award and the O'Malley Foundation. SNSC is supported by fellowship grants from the Osteogenesis Imperfecta Foundation and the National Urea Cycle Disorders Foundation. This project was sponsored in part by the Charitable and Research Foundation, Punta Gorda, FL. Author roles: JRS, SCSN, MB, TTR, and BHL performed clinical assessments. PM, JRS, PMC, MG, CL, SCSN, DHC, YW and FSB compiled the clinical data. JTL, DMB, MNB, BCD, MG and PMC performed the analysis of the next-generation sequencing data. CL and MG performed the Sanger sequencing. RAG, JRS and BHL coordinated the study. PMC, JRS and BHL take responsibility for the integrity of the data.
References
- 1.Shapiro JR, Sponsellor PD. Osteogenesis imperfecta: questions and answers. Curr Opin Pediatr. 2009;21(6):709–716. doi: 10.1097/MOP.0b013e328332c68f. [DOI] [PubMed] [Google Scholar]
- 2.Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 4.Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ. Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res. 2000;15(9):1650–1658. doi: 10.1359/jbmr.2000.15.9.1650. [DOI] [PubMed] [Google Scholar]
- 5.Brenner RE, Schiller B, Pontz BF, Lehmann H, Teller WM, Spranger J, Vetter U. [Osteogenesis imperfecta in childhood and adolescence] Monatsschr Kinderheilkd. 1993;141(12):940–945. [PubMed] [Google Scholar]
- 6.Wenstrup RJ, Willing MC, Starman BJ, Byers PH. Distinct biochemical phenotypes predict clinical severity in nonlethal variants of osteogenesis imperfecta. Am J Hum Genet. 1990;46(5):975–982. [PMC free article] [PubMed] [Google Scholar]
- 7.Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, Park WY, Park SS, Ikegawa S, Yoo WJ, Choi IH, Kim JW. A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91(2):343–348. doi: 10.1016/j.ajhg.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, Wollnik B, Netzer C. A mutation in the 5'-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91(2):349–357. doi: 10.1016/j.ajhg.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanagata N, Li X. Osteoblast-enriched membrane protein IFITM5 regulates the association of CD9 with an FKBP11-CD81-FPRP complex and stimulates expression of interferon-induced genes. Biochem Biophys Res Commun. 2011;409(3):378–384. doi: 10.1016/j.bbrc.2011.04.136. [DOI] [PubMed] [Google Scholar]
- 10.Hanagata N, Li X, Morita H, Takemura T, Li J, Minowa T. Characterization of the osteoblast-specific transmembrane protein IFITM5 and analysis of IFITM5-deficient mice. J Bone Miner Metab. 2011;29(3):279–290. doi: 10.1007/s00774-010-0221-0. [DOI] [PubMed] [Google Scholar]
- 11.Moffatt P, Gaumond MH, Salois P, Sellin K, Bessette MC, Godin E, de Oliveira PT, Atkins GJ, Nanci A, Thomas G. Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res. 2008;23(9):1497–1508. doi: 10.1359/jbmr.080412. [DOI] [PubMed] [Google Scholar]
- 12.Dobrocky I, Seidl G, Grill F. MRI and CT features of hyperplastic callus in osteogenesis imperfecta tarda. Eur Radiol. 1999;9(4):665–668. doi: 10.1007/s003300050729. [DOI] [PubMed] [Google Scholar]
- 13.Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, Flegal KM, Guo SS, Wei R, Mei Z, Curtin LR, Roche AF, Johnson CL. CDC growth charts: United States. Adv Data. 2000;(314):1–27. [PubMed] [Google Scholar]
- 14.Campeau PM, Kim JC, Lu JT, Schwartzentruber JA, Abdul-Rahman OA, Schlaubitz S, Murdock DM, Jiang MM, Lammer EJ, Enns GM, Rhead WJ, Rowland J, Robertson SP, Cormier-Daire V, Bainbridge MN, Yang XJ, Gingras MC, Gibbs RA, Rosenblatt DS, Majewski J, Lee BH. Mutations in KAT6B, encoding a histone acetyltransferase, cause Genitopatellar syndrome. Am J Hum Genet. 2012;90(2):282–289. doi: 10.1016/j.ajhg.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Havlak P, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Weinstock GM, Gibbs RA. The Atlas genome assembly system. Genome Res. 2004;14(4):721–732. doi: 10.1101/gr.2264004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 20.Lee DY, Cho TJ, Choi IH, Chung CY, Yoo WJ, Kim JH, Park YK. Clinical and radiological manifestations of osteogenesis imperfecta type V. J Korean Med Sci. 2006;21(4):709–714. doi: 10.3346/jkms.2006.21.4.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeitlin L, Rauch F, Travers R, Munns C, Glorieux FH. The effect of cyclical intravenous pamidronate in children and adolescents with osteogenesis imperfecta type V. Bone. 2006;38(1):13–20. doi: 10.1016/j.bone.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 22.Ramirez N, Vilella FE, Colon M, Flynn JM. Osteogenesis imperfecta and hyperplastic callus formation in a family: a report of three cases and a review of the literature. J Pediatr Orthop B. 2003;12(2):88–96. doi: 10.1097/01.bpb.0000043724.21564.32. [DOI] [PubMed] [Google Scholar]
- 23.Garretsen AJ, Cremers CW, Huygen PL. Hearing loss (in nonoperated ears) in relation to age in osteogenesis imperfecta type I. Ann Otol Rhinol Laryngol. 1997;106(7 Pt 1):575–582. doi: 10.1177/000348949710600709. [DOI] [PubMed] [Google Scholar]
- 24.Pillion JP, Vernick D, Shapiro J. Hearing loss in osteogenesis imperfecta: characteristics and treatment considerations. Genet Res Int. 2011;2011:983942. doi: 10.4061/2011/983942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheung MS, Azouz EM, Glorieux FH, Rauch F. Hyperplastic callus formation in osteogenesis imperfecta type V: follow-up of three generations over ten years. Skeletal Radiol. 2008;37(5):465–467. doi: 10.1007/s00256-007-0441-0. [DOI] [PubMed] [Google Scholar]
- 26.Reichenbach H, Hormann D, Theile H. Hereditary congenital posterior dislocation of radial heads. Am J Med Genet. 1995;55(1):101–104. doi: 10.1002/ajmg.1320550124. [DOI] [PubMed] [Google Scholar]
- 27.Fassier AM, Rauch F, Aarabi M, Janelle C, Fassier F. Radial head dislocation and subluxation in osteogenesis imperfecta. J Bone Joint Surg Am. 2007;89(12):2694–2704. doi: 10.2106/JBJS.F.01287. [DOI] [PubMed] [Google Scholar]
- 28.Castillo H, Samson-Fang L. Effects of bisphosphonates in children with osteogenesis imperfecta: an AACPDM systematic review. Dev Med Child Neurol. 2009;51(1):17–29. doi: 10.1111/j.1469-8749.2008.03222.x. [DOI] [PubMed] [Google Scholar]
- 29.Cheung MS, Glorieux FH. Osteogenesis Imperfecta: update on presentation and management. Rev Endocr Metab Disord. 2008;9(2):153–160. doi: 10.1007/s11154-008-9074-4. [DOI] [PubMed] [Google Scholar]
- 30.Byers PH, Pyott SM. Recessively inherited forms of osteogenesis imperfecta. Annu Rev Genet. 2012;46:475–497. doi: 10.1146/annurev-genet-110711-155608. [DOI] [PubMed] [Google Scholar]
- 31.Rauch F, Moffatt P, Cheung M, Roughley P, Lalic L, Lund AM, Ramirez N, Fahiminiya S, Majewski J, Glorieux FH. Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c.-14C>T mutation in all patients. J Med Genet. 2013;50(1):21–24. doi: 10.1136/jmedgenet-2012-101307. [DOI] [PubMed] [Google Scholar]
- 32.Hickford D, Frankenberg S, Shaw G, Renfree MB. Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genomics. 2012;13:155. doi: 10.1186/1471-2164-13-155. [DOI] [PMC free article] [PubMed] [Google Scholar]