

Abstract
Efforts to discover new drugs for Alzheimer’s disease emphasizing multiple targets was conducted seeking to inhibit amyloid oligomer formation and to prevent radical formation. The tryptoline and tryptamine cores of BACE1 inhibitors previously identified by virtual screening were modified in silico for additional modes of action. These core structures were readily linked to different side chains using 1,2,3-triazole rings as bridges by copper catalyzed azide-alkyne cycloaddition reactions. Three compounds among the sixteen designed compounds exerted multifunctional activities including β-secretase inhibitory action, anti-amyloid aggregation, metal chelating and antioxidant effects at micromolar levels. The neuroprotective effects of the multifunctional compounds 6h, 12c and 12h on Aβ1–42 induced neuronal cell death at 1 μM were significantly greater than those of the potent single target compound, BACE1 inhibitor IV and were comparable to curcumin. The observed synergistic effect resulting from the reduction of the Aβ1–42 neurotoxicity cascade substantiates the validity of our multifunctional strategy in drug discovery for Alzheimer’s disease.
Keywords: multifunction drugs, BACE1 inhibitor, anti-amyloid aggregation, chelator, antioxidant, neuroprotection
1. Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder with a multifactorial etiopathology involving β-amyloid peptide (Aβ40, Aβ42) accumulation, iron deregulation, oxidative damage and decreased acetylcholine levels [1–3]. β-Amyloid plaque pathogenesis has been a prime target in the search for new drugs for AD etiology treatment. In recent years, it has been evidenced that the Aβ oligomers are more toxic than the deposition of amyloid fibrils or plaques. These Aβ oligomers show a number of toxicity effects including synaptic damage, chondrial dysfunction, glutamate receptor remodeling and alteration of neurogenesis signaling pathways [4]. Therefore, Aβ obstruction and anti-Aβ aggregation are currently the main targets of interest for AD drug development. Aβ peptides are generated from amyloid precursor protein (APP) by β-secretase and γ-secretase cleaving enzymes. An Aβ peptide monomer can aggregate to form oligomers and finally plaques. Inhibition of β-secretase (BACE1), the key enzyme in Aβ peptide generation, and anti-Aβ aggregation are the most attractive targets to prevent Aβ oligomer formation. Metals are also found to play an important role in the pathophysiology of AD by inducing Aβ aggregation and producing harmful reactive oxygen species (ROS). Oxidative stress not only leads to metabolic dysfunction and apoptosis of neurons in AD but also enhances BACE1 expression and activity [5,6]. The bound transition metal ions (Cu(I) or Fe(II)) on Aβ oligomers are able to reduce molecular oxygen to hydrogen peroxide resulting in generation of ROS. Thus, metal chelation and radical scavenging are other attractive approaches to reduce neurotoxicity from amyloid aggregation and free radical generation [5,6].
According to the multi-pathogenesis of AD and the failure in clinical trials of many single target drugs, a multi-target-directed-ligand (MTDL) such as memoquin has been examined in current drug discovery. Memoquin exhibited multifunctional properties, acting as AChE inhibitor, free-radical scavenger and inhibitor of Aβ aggregation [3,7]. In the present study, we concentrated on MTDL development to increase drug efficacy for moderation of amyloid β peptide toxicity. Our multifunctional strategy aimed at inhibition of Aβ oligomer formation, moderation of metal levels and prevention of free radical formation, in addition to inhibition of BACE1 to enhance drug efficacy. From this strategy, we have modified our core BACE1 inhibitor structure by adding moieties to exert multifunctional properties in opposition to the AD etiology.
In a previous report, we discovered the core BACE1 inhibitor structure (tryptoline) from virtual screening of Thai medicinal plants [8]. To increase the efficacy, modification of a core structure and multifunctional design were performed. A new core structure (tryptamine) was introduced as a bioisostere of tryptoline in order to increase the hydrogen bond interaction and flexibility. In silico, tryptamine showed similar binding as tryptoline. Not only did the indole group of tryptamine fit with the hydrophobic S1 pocket (Leu30, Tyr71, Phe108, and Trp115) but also two hydrogen bonds were formed with catalytic residues Asp32 and Asp228 (Figure 1a). Based on the premise that more hydrogen bonding might yield higher binding affinity, the modification of new tryptamine core was carried out in parallel with the tryptoline core by adding moieties to exert anti-amyloid aggregation, metal chelating and antioxidant effects.
Figure 1.
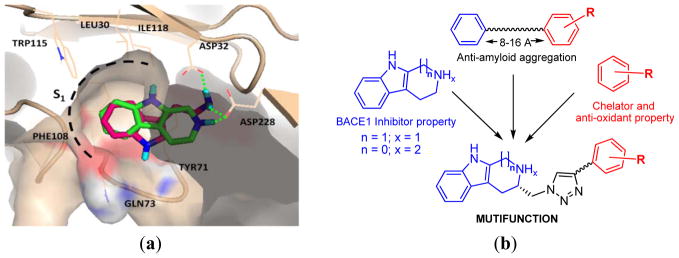
(a)The binding mode of the core structures with BACE1 enzyme, the former tryptoline core (green) and new tryptamine core (magenta) and (b) the design strategy for multifunctional compounds.
In order to gain the desired effects, an aromatic nucleus substituted with electron donating groups such as hydroxyl and halogen as well as conjugated phenolic moieties was added to the core structures using triazole as a linker (Figure 1b). The addition of aromatic nucleus was projected to produce – an anti-Aβ aggregation effect based on the pharmacophore reported by Reinke and Gestwicki [9]. The important anti-Aβ aggregation feature can be achieved with aromatic end groups separated by an optimum length of linker. Moreover, we have introduced active antioxidant and metal chelator functional groups on the added aromatic nucleus [10,11]. The purpose of these moieties was to achieve a multifunctional approach involving anti-Aβ aggregation, metal complexation and radical scavenging action.
2. Results and Discussion
2.1. Synthesis
The tryptoline azide (S)-3-(azidomethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (5) was synthesized as described previously [8]. The synthetic pathway to the tryptamine core, (S)-3-(-2-amino-3-(1H-1,2,3-triazol-1-yl)propyl)indole, is shown in Scheme 1.
Scheme 1.
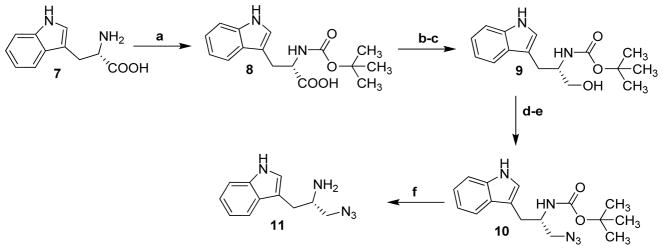
Preparation of (S)-3-(-2-amino-3-(1H-1,2,3-triazol-1-yl)propyl)indole (11).
Reagents and conditions: (a) Boc2O, NaOH, THF/H2O (1:1), rt, 18 h; (b) NMM, isobutyl chloroformate, 0 °C, N2, dry THF, 30 min; (c) NaBH4, MeOH, 0 °C, THF; (d) DPPA, DBU, 0 °C, DMF, 30 min; (e) NaN3, 70 °C, DMF, 2 h; (f) TFA, CH2Cl2, 0 °C to rt, 30 min.
The amino group of tryptophan (7) was protected by a Boc group to yield compound 8 [12]. Then, the carboxylic group of 8 was reduced to hydroxyl with NaBH4 [13]. The hydroxyl group of 9 was converted to azide 10 by a substitution reaction with NaN3 [14]. Finally, the protecting group was removed to yield tryptamine azide 11 [15].
The azido groups of tryptoline 5 and tryptamine 11 were reacted with different alkynes by copper catalyzed azide-alkyne cycloaddition reactions [16]. Commercial available alkynes a–c were used in the reaction to afford tryptolines 6a–c and tryptamines 12a–c (Scheme 2).
Scheme 2.
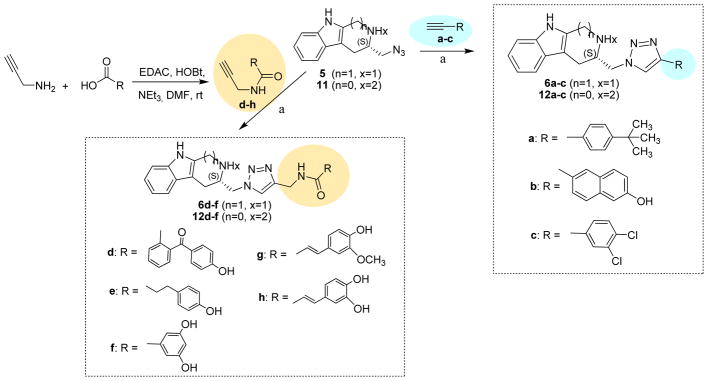
Synthesis of triazolyl methyltryptolines 6a–h and triazolyl methyltryptamines 12a–h.
Reagents and conditions: (a) CuSO4·5H2O, sodium ascorbate, H2O/t-BuOH/EtOH (2:2:1), rtto 50 °C, 18–24 h.
Alkynes d–h were prepared from propargyl amine and carboxylic acid compounds by using EDAC as coupling reagent [17]. The synthesized alkynes were used in the reaction to produce compounds 6d–h and 12d–h (Scheme 2). The chemical and biological properties of synthesized compounds are shown in Table 1.
Table 1.
Multifunctionality of triazole based compounds.
| Cpd | Log P | BACE1 | Anti-Aβ aggregation | Fe chelation | DPPH | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| % Inhibition (at 25 μM) (±SEM) | IC50 (μM) | % Inhibition (at 100 μM) (±SEM) | IC50 (μM) | % Capacity (at 100 μM) (±SEM) | Stoichiometric ratio (Fe:cpd) | % Inhibition (at 100 μM) (±SEM) | IC50 (μM) | ||
| 6a | 4.69 | 67.95 (±0.35) | 19.82 | NA | - | 9.67 (±0.32) | - | NA | - |
| 6ba | 3.86 | 73.63 (±1.47) | 18.86 | 7.19 (±1.19) | - | 12.35 (±0.68) | - | NA | - |
| 6c | 4.10 | 78.91 (±2.83) | 18.03 | 10.12 (±0.86) | - | 15.88 (±0.21) | - | NA | - |
| 6d | 3.01 | 14.10 (±1.79) | - | NA | - | 32.86 (±0.19) | - | 2.81 (±0.37) | - |
| 6e | 2.16 | 12.57 (±1.10) | - | NA | - | 20.63 (±0.35) | - | 5.45 (±0.33) | - |
| 6f | 1.41 | 21.28 (±1.92) | - | NA | - | 5.80 (±0.04) | - | NA | - |
| 6g | 2.01 | 21.14 (±1.13) | - | 66.36 (±2.02) | 82.90 | 8.88 (±0.25) | - | 47.03 (±0.12) | 106.41 |
|
| |||||||||
| 6h | 1.75 | 21.72 (±0.33) | - | 84.13 (±2.49) | 29.86 | 42.74 (±0.30) | 1:3 | 92.29 (±0.02) | 42.91 |
|
| |||||||||
| 12a | 4.42 | 18.91 (±0.40) | - | 17.81 (±0.86) | - | 42.65 (±1.07) | - | NA | - |
| 12b | 3.58 | 30.42 (±3.54) | - | 34.02 (±10.13) | - | 10.54 (±0.22) | - | NA | - |
|
| |||||||||
| 12c | 3.83 | 61.46 (±1.83) | 20.75 | 67.56 (±0.72) | 83.23 | 60.90 (±0.51) | 1:3 | NA | - |
|
| |||||||||
| 12d | 2.73 | 32.97 (±0.37) | - | 82.52 (±1.26) | 47.51 | 13.87 (±0.59) | - | 1.56 (±0.51) | - |
| 12e | 1.88 | 7.53 (±1.79) | - | 36.03 (±1.92) | - | 41.42 (±1.13) | - | NA | - |
| 12f | 1.13 | 16.57 (±2.91) | - | 3.80 (±1.56) | - | 13.06 (±0.16) | - | 1.32 (±0.78) | - |
| 12g | 1.74 | 16.23 (±0.45) | - | 46.06 (±1.63) | 109.83 | 77.70 (±0.67) | 1:3 | 40.79 (±0.31) | 130.44 |
|
| |||||||||
| 12h | 1.47 | 40.03 (±0.95) | - | 81.48 (±2.54) | 56.39 | 66.45 (±0.37) | 1:3 | 50.58 (±0.17) | 92.70 |
|
| |||||||||
| Inh IV (Merck®) | 1.23 | 96.51 (±1.33) | 0.015b | - | - | - | - | - | - |
| Curcumin | 2.56 | - | - | 82.90 (±0.82) | 0.63c | - | - | - | - |
| EDTA | −2.69 | - | - | - | - | 98.00 (±0.34) | - | - | |
| Ascorbic acid | −3.36 | - | - | - | - | - | - | 53.64 (±0.11) | 94.92 |
2.2. Chemical and Biological Assays
The triazole based compounds of both core structures were evaluated for inhibitory action against BACE1 and for additional activities such as anti-Aβ aggregation, metal chelating and antioxidant (Table 1). The BACE1 inhibitor IV (Calbiochem®) was used as a positive control. The BACE1 activity inhibitions were found to be 7.53% and 78.91% at 25 μM. Tryptolines 6a–c and tryptamine 12c showed good inhibition, with IC50 of 18–20 μM, as they all accommodated in the substrate binding site. The binding modes of these four compounds were apparently in a similar manner as shown in Figure 2. The core structures of compounds, tryptoline and tryptamine, fitted with the hydrophobic S1 and S1′ pockets and interacted with residues Leu30, Asp32, Tyr71, Thr72, Gln73, Phe108, Trp115, Ile118, Asp228, Gly230 and Thr231. The NH2 group and NH hydrogens in the core structures were involved in hydrogen bonding interactions with Asp32, Asp228 and/or Gln73. The triazole-bearing aromatic side chain accessed the S2–S4 sites and provided interactions with residues Tyr71, Thr72, Gln73, Gly230, Thr231, Thr232, Asn233, Arg235, Lys321 and Ser325 by hydrophobic, dipole induced dipole, dipole-dipole or hydrogen bond interactions. The effect of these compounds against cathepsin-D was determined for selectivity and no inhibition was observed at 100 μM. The interactions of these compounds with the Arg235 residue in S2 possibly contributed to the loss of inhibitory action against cathepsin-D because Asp235 is a unique residue in BACE1 compared with cat-D (Val233) and rennin (Ser222) [18].
Figure 2.
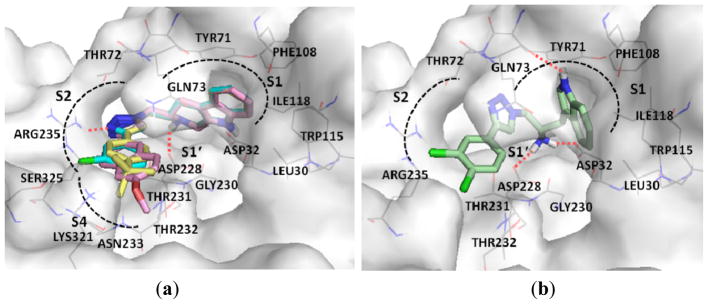
The binding modes of compounds with BACE1 enzyme; (a) tryptoline 6a (yellow), 6b (pink) and 6c (cyan); (b) tryptamine 12c (green).
The anti-Aβ aggregation activity of compounds bearing the new tryptamine core was generally higher than those of corresponding compounds with the former tryptoline core with the exception of 12h (Table 1). The distance between aromatic terminals in the 3D structure after energy minimization was determined to define the relationship between structure and anti-amyloid aggregation activity. Compounds having a length between aromatic terminals of 8–9 Å (compounds 12c, 12d and 12h) and 13–14 Å (compounds 6h and 6g) showed anti-Aβ aggregation activity over 50% (Figure 3). The optimal length between aromatic terminals of 8–9 Å is in agreement with Reinke and Gestwicki criteria [9], we also found the new optimal length of 13–14 Å.
Figure 3.
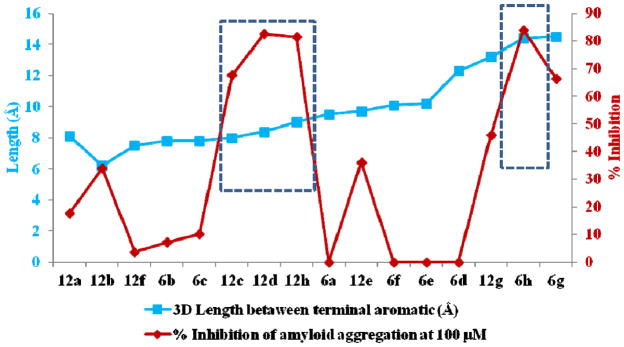
The relationship of bond length between aromatic terminal in the 3D structure and anti-amyloid aggregation activity.
The IC50 values for anti-Aβ aggregation of tryptolines 6g and 6h were 82.90 μM and 29.86 μM, respectively while those of tryptamines 12c, 12d and 12h were 83.23 μM, 47.51 μM, 56.39 μM, respectively. The substantial activity of these compounds possibly resulted from the capability to wrap the Aβ motif which is crucial for aggregation. The formation of H-bonds or hydrophobic interactions of the core structures such as those of tryptamines 12c, 12d and 12h with the key residue Asp23 possibly prevented Aβ from self-aggregation as Asp23 plays an important role in intermolecular salt bridge formation of Aβ peptides in protein aggregation (Figure 4a–c).
Figure 4.
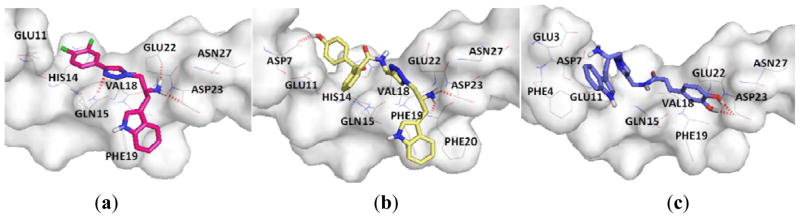
The binding mode of compounds with amyloid-β (1–42); (a) 12c (pink); (b) 12d (yellow); (c) 12h (blue).
Moreover, apart from the tryptamine and tryptotoline cores, the added moieties a–h also enhanced the Aβ binding. The m-OH substitution on the added aromatic nucleus of 6h provided a H-bond interaction with Asp7 in addition to the H-bond at Asp23. The interactions at both terminals secured the triazole linker to form hydrophobic and dipole-dipole interactions with Gln15, Val18 and Phe19 residues, these amino acid residues are self-recognition residues in the aggregation process (Figure 5a). Thus, 6h was two times more potent than 6g, which provided an H-bond only at one terminal (IC50 29.86 μM vs. 82.90 μM), the interactions at the terminal ends appeared to strengthen the Aβ wrapping. In case of 12g vs. 12h (Figure 5b), tryptamine 12h provided H-bond interactions at the terminal ends in the same scenario as 6h but flipped vertically, and the IC50 value of 12h is two times better than that of 12g (56.39 μM vs. 109.89 μM).
Figure 5.
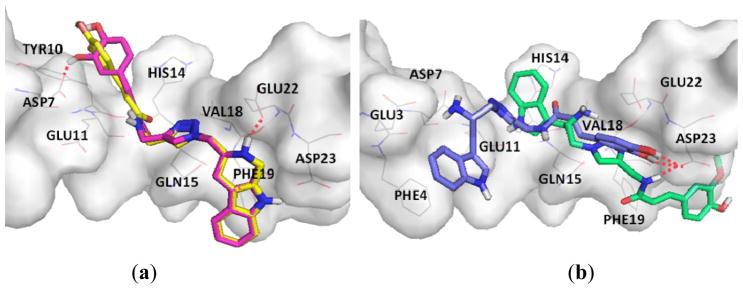
The binding modes of compounds with amyloid-β (1–42); (a) 6g (yellow) and 6h (pink); (b) 12g (green) and12h (blue).
In metal chelating capability, the tryptoline and tryptamine derivatives had chelating capacity between 5.80–77.70% at 100 μM. Generally, compounds containing the tryptoline core formed complexes with Fe2+ with less capacity than those with the tryptamine core due to the restriction ability of the NH in the tryptoline core to chelate with metal. The lone pair of electrons on the nitrogen atom in the core structure as well as the nitrogen atom in the triazole ring were the chelating functions. Compounds 12c, 12g and 12h exhibiting chelating capacities higher than 50% at 100 μM were selected for the determination of stoichiometric ratio. The stoichiometric ratio of these compounds 12c, 12g and 12h per metal were 3:1 (Figure 6).
Figure 6.

The chelating model of compound 12c with Fe2+.
For free radical scavenging activity, compounds containing conjugated phenolic moieties showed good activity, as anticipated. Compound 6h had high activity, with an IC50 value of 42.91 μM while compounds 6g, 12g and 12h showed moderate antioxidant properties, with IC50 values of 106.41 μM, 130.44 μM and 92.70 μM, respectively. Moreover, di-substitution of hydroxyl groups at m- and p-position helped to enhance the antioxidant activity as in 6h and 12h compared with 6g and 12g.
Compounds 6h, 12c and 12h were found to possess multifunctional activity as shown in Table 1. Compound 12c demonstrated substantial anti-Aβ aggregation and chelating effect in addition to the BACE1 inhibitory action (IC50 20.75 μM). The major effects of compounds 6h and 12h were anti-Aβ aggregation and antioxidant activity resulting from the conjugated phenolic side chain while the BACE1 inhibitory action appeared to be moderate. Their IC50 values for anti-Aβ aggregation were 29.86 μM and 56.39 μM, and those of antioxidant activity were 42.91 μM and 92.70 μM, respectively. The chelating capabilities of these three compounds were found to be moderate, 40–70% at 100 μM. Compounds 6h, 12c and 12h were selected for further investigation to demonstrate the synergistic effect on neuroprotection against β-amyloid toxicity in SH-SY5Y cells. The effect of these compounds on neurotoxicity induced by Aβ1–42 was determined at a non-toxic concentration (1 μM) using a colorimetric MTT method [21]. Curcumin, which is a potent antioxidant, anti-Aβ aggregation and chelating agent was included in the assay together with BACE1 Inhibitor IV. All test compounds significantly inhibited neuronal death induced by Aβ1–42 (Figure 7). As anticipated, the neuroprotective effect of the designed compounds using multi-target approach was superior to the single target compound. The multifunctional compounds 6h, 12c and 12h were able to decrease cell death to a greater extent than BACE1 inhibitor IV which is a potent single target compound. BACE1 inhibitor IV also improved cell viability as it reduces the amyloid beta level produced by the increase of BACE1 expression and activity under oxidative stress condition. The activation of the PKR (double-stranded RNA dependant protein kinase) pathway and eIF2α (eukaryotic translation initiation factor-2α) translational control [22] as well as a transcriptional regulation mediated by c-jun N-terminal kinase (JNK) pathway [23–25] are the causes of oxidative stress-induced BACE1 elevation. Increased levels of PKR mRNA, PKR protein and BACE1 were observed in SH-SY5Y after oxidative exposure [22]. Thus, inhibition of BACE1 enzyme helps in reducing the production of new amyloid beta peptide that causes neurotoxicity. Although the potency of multi-target compounds was apparently low in THE micromolar level against each individual target, the neuroprotection in SH-SY5Y cells was comparable to that provided by curcumin. The inclusion of BACE1 inhibitory action synergistically enhanced the neuroprotective effects of compounds 6h, 12c and 12h to the same level as curcumin, a potent nanomolar multi-target compound.
Figure 7.
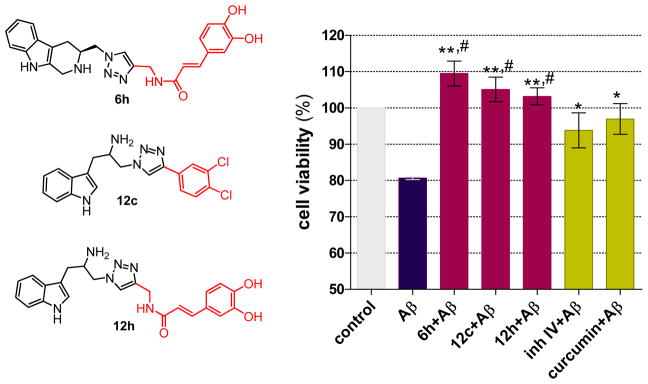
The protective effect of multifunctional compounds against Aβ-induced neuronal cell death by MTT viability assay at 1 μM, t-test * p < 0.05, ** p < 0.01 vs. Aβ treated cells and # p < 0.05 vs. BACE1 inhibitor IV.
3. Experimental
3.1. General
All ligands were generated and optimized with ChemDraw Ultra 9.0 and Chem3D Ultra 9.0. AutoDock program suit version 4.2on Garibaldi platform at The Scripps Research Institute was employed to perform the docking calculation. All chemical reagents were purchased from Aldrich or AK Science. 1H-NMR and 13C-NMR spectra were acquired on Bruker Avance 300 or 400 MHz instruments. Mass spectra were recorded on either a Thermo Finnigan or LCMS Bruker MicroTof. IR spectra were recorded on Nicolet FTIR 550. BACE1 enzyme and BACE1 substrate were purchased from Sino Biological® and Calbiochem®, respectively. Amyloid-β (1–42) from Anaspec® was used in ThT and MTT assay.
3.2. Docking Study of β-Secretase (BACE1)
The BACE1 template 2IRZ-F was constructed from two crystal structures of β-secretase (BACE1) bound to inhibitors (Protein Data Bank code: 2IRZ [26] and 1FKN [27]) as previously described [8]. Docking parameters in the docking studies were as follows: the number of genetic algorithm (GA) runs was 100; the population size was 150; the maximum number of energy evaluations was increased to 15,000,000 per run; and the maximum number of generations was 27,000.
3.3. Docking Study of Amyloid β (Aβ)
Amyloid β peptide (residues 1–42) template was prepared from crystal structure of Aβ monomer (PDB entry code: 1Z0Q [28]). The dimensions of grid were centered on the coordinates −1.733, 3.591 and −6.759 with 120 × 80 × 80 Å and 0.5 Å spacing between grids points. The docking parameters were as follows: the number of GA runs was 100; the population size was 150; the maximum number of energy evaluations was increased to 5,000,000 per run; and the maximum number of generations was 27,000.
3.4. Preparation of Azidomethyl Tryptamine Intermediates
(S)-2-(tert-Butoxycarbonylamino)-3-(1H-indol-3-yl)propanoic acid (8)
L-Tryptophan (20.45 g, 0.10 mol) in THF/H2O (1:1, 100 mL) was added with sodium hydroxide (8.80 g, 0.22 mol) and di-tert-butyl dicarbonate (24.01 g, 0.11 mol). The reaction was stirred at room temperature for 18 h. After the reaction was complete, water was added to dissolve the precipitate, then the THF was removed under reduced pressure and the aqueous layer was extracted with dichloromethane. The aqueous layer was acidified by 1 N HCl to pH 4 and extracted with dichloromethane and ethyl acetate (30 mL × 3). The organic phase was dried over sodium sulphate and concentrated to yield compound 8 as a white powder (24.74 g, 81%); m.p.: 141–143 °C; FTIR (KBr): 3373, 3338, 2524, 1717, 1647, 1505, 1400, 1251, 1163 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 10.82 (s, 1H, H1), 7.50 (d, J = 8.00 Hz, 1H, H4), 7.32 (d, J = 8.00 Hz, 1H, H7), 7.13 (d, J = 1.60 Hz, 1H, H2), 7.05 (t, J = 7.40 Hz, 1H, H6), 6.96 (t, J = 8.60 Hz, 1H, H5), 4.15–4.10 (m, 1H, H9), 3.11 (dd, J = 14.80, 4.80 Hz, 1H, H8b), 2.95 (dd, J = 14.40, 9.20 Hz, 1H, H8a), 1.31 (s, 9H, CH3); LRMS (API-ES) m/z 631.3 [2M+Na], 327.3 [M+Na].
(S)-tert-Butyl-1-hydroxy-3-(1H-indol-3-yl)propan-2-yl carba-mate (9)
To compound 8 (4.69 g, 15.42 mmol) in dry THF (30 mL) was added N-methylmorpholine (2.0 mL, 18.50 mmol) and isobutyl chloroformate (2.4 mL, 18.50 mmol) at 0 °C under nitrogen and the mixture was stirred for 30 min. The precipitate formed was immediately filtered and to the filtrate was added sodium borohydride (1.17 g, 30.84 mmol) at 0 °C. After 15 min, methanol (5 mL) was added dropwise at 0 °C. After the reaction was complete, THF was evaporated off and the residue was diluted with ethyl acetate (30 mL). The organic phase was washed with saturated sodium bicarbonate, 5% potassium bisulphate, and brine and dried over sodium sulphate. The concentrated residue was purified by column chromatography (hex/EtOAc; 7:3) to give a white powder of compound 9 (2.15 g, 48%); m.p.: 122–123 °C; FTIR (KBr): 3420, 3404, 3360, 1686, 1526, 1368, 1247, 1173, 999 cm−1; 1H-NMR (300 MHz, CDCl3): δ 8.13 (s, 1H, H1), 7.63 (d, J = 7.81 Hz, 1H, H4), 7.34 (d, J = 8.03 Hz, 1H, H7), 7.18 (t, J = 7.52 Hz, 1H, H6), 7.11 (t, J = 7.41 Hz, 1H, H5), 7.02 (d, J = 2.19 Hz, 1H, H2), 4.81 (br, 1H, carbamate NH), 4.02–3.92 (m, 1H, H9), 3.70–3.54 (m, 2H, H10), 2.97 (d, J = 6.84 Hz, 2H, H8), 2.52 (s, 1H, OH), 1.40 (s, 9H, CH3); LRMS (API-ES) m/z 603.3 [2M+Na], 313.2 [M+Na].
(S)-tert-Butyl-1-azido-3-(1H-indol-3-yl)propan-2-ylcarbamate (10)
Under nitrogen, diphenyl-phosphoryl azide (1.2 mL, 5.36 mmol) and DBU (0.8 mL, 5.36 mmol) were added dropwise to a cooled (0 °C) solution of compound 9 (1.04 g, 3.57 mmol) in DMF (5 mL). After the reaction was complete, sodium azide (1.16 g, 17.85 mmol) was added to the reaction at 0 °C and the reaction temperature was raised to 80 °C. The reaction was diluted with ethyl acetate (30 mL) and washed with water (30 mL) twice. The aqueous layer was washed with ethyl acetate (30 mL). Then the organic phases were combined, washed with saturated sodium bicarbonate, brine and dried over sodium sulphate. The concentrated residue was purified by column chromatography (hex/EtOAc; 9:1) to yield a white powder of compound 10 (0.5641 g, 50%); m.p.: 98–99 °C; FTIR (KBr): 3418, 3399, 3354, 2110, 1690, 1518, 1170 cm−1; 1H-NMR (300 MHz, CDCl3): δ 8.07 (s, 1H, H1), 7.62 (d, J = 7.63 Hz, 1H, H4), 7.34 (d, J = 7.87 Hz, 1H, H7), 7.18 (t, J = 7.52 Hz, 1H, H6), 7.11 (t, J = 7.41 Hz, 1H, H5), 7.02 (d, J = 2.21 Hz, 1H, H2), 4.69–4.67 (br, 1H, NH), 4.06–4.05 (br, 1H, H9), 3.41–3.28 (m, 2H, H10), 3.04–2.88 (m, 2H, H4), 1.40 (s, 9H, CH3); LRMS (API-ES) m/z 338.3 [M+Na].
(S)-1-Azido-3-(1H-indol-3-yl)propan-2-amine (11)
To compound 10 (0.52 g, 1.65 mmol) in dichloromethane (10 mL) was treated dropwise with trifluoroacetic acid (2.50 mL, 33.04 mmol) at 0 °C and stirred at room temperature for 30 min. Saturated sodium bicarbonate was added to the reaction for adjust pH = 8. The resulting solution was extracted with ethyl acetate 30 mL. The organic phase was dried over sodium sulphate. The concentrated residue was purified with column chromatography (CHCl3/MeOH; 20:1) to yield 11 as a brown oil (0.14 g, 40%): FTIR (ATR): 3407, 3345, 3283, 2097, 1660, 1577, 1091cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.84 (s, 1H, H1), 7.51 (d, J = 7.78 Hz, 1H, H4), 7.33 (d, J = 8.03 Hz, 1H, H7), 7.15 (s, 1H, H2), 7.05 (t, J = 7.08 Hz, 1H, H6), 6.96 (t, J = 7.05 Hz, 1H, H5), 3.27–3.24 (m, 1H, H10b), 3.17–3.12 (m, 2H, H10a, H9), 2.77 (dd, J = 14,10, 5.47 Hz, 1H, H8b), 2.65 (dd, J = 14.20, 6.51 Hz, 1H, H8a); LRMS (API-ES) m/z 216.3 [M+H].
3.5. Preparation of Alkynes d–h
A mixture of carboxylic acid (2 mmol) and N-hydroxybenzotriazole (1 mmol) were dissolved in DMF (5 mL). Propargylamine (2 mmol) and triethylamine (2 mmol) were added to the reaction and stirred at room temperature for 5 min. Then 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide·HCl (EDAC, 2.4 mmol) was added to the reaction. After 18 h, water (20 mL) was added to stop the reaction. The aqueous solution was extracted with ethyl acetate (10 mL × 3). The resulting solution was washed with 1N HCl, saturated NaHCO3, brine, dried with anhydrous NaSO4 and concentrated.
2-(4-Hydroxybenzoyl)-N-(prop-2-ynyl)benzamide (d)
Compound d was obtained from 2-(4-hydroxybenzoyl) benzoic acid as described above. The concentrated residue was washed with diethyl ether to yield a white powder (0.4239 g, 76%); m.p.: 165–167 °C; FTIR (KBr): 3319, 3308, 3240, 3240, 1654, 1612, 1518, 1346, 1201, 1166 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 9.48 (s, 1H, OH), 7.70 (d, J = 6.78 Hz, 1H, H6′), 7.57–7.46 (m, 2H, H4′, H5′), 7.23 (d, J = 7.4 Hz, 1H, H3′), 7.14 (d, J = 8.66 Hz, 2H, H2″, H6″), 7.01 (s, 1H, NH-amide), 6.69 (d, J = 8.70 Hz, 2H, H3″, H5″), 3.99 (dd, J = 17.75, 2.41 Hz, 1H, H1b), 3.83 (dd, J = 17.72, 2.42 Hz, 1H, H1a), 2.88–2.86 (m, 1H, H3); LRMS (API-ES) m/z 581.2 [2M+Na], 302.2 [M+Na].
3-(4-Hydroxyphenyl)-N-(prop-2-ynyl)propanamide (e)
Compound e was obtained from 3-(4-hydro xyphenyl) propinoic acid as described above as a white powder (0.2784 g, 69%); m.p.: 102–104 °C; FTIR (KBr): 3332, 3272, 3186, 1656, 1543, 1518, 1356, 1229, 1172 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 9.12 (s, 1H, OH), 8.24 (t, J = 5.24 Hz, 1H, NH), 6.96 (d, J = 8.34 Hz, 2H, H2″, H6″), 6.63 (d, J = 8.35 Hz, 2H, H3″, H5″), 3.84–3.81 (m, 2H, H1), 3.07 (t, J = 2.33 Hz, 1H, H3), 2.67 (t, J = 7.79 Hz, 2H, H2′), 2.30 (t, J = 7.81 Hz, 2H, H3′); LRMS (API-ES) m/z 429.3 [2M+Na], 226.2 [M+Na], 204.3 [M+H].
3,5-Dihydroxy-N-(prop-2-ynyl)benzamide (f)
Compound f was obtained from 3,5-dihydroxybenzoic acid as described above. The concentrated residue was washed with diethyl ether to yield a light brown powder (0.3242 g, 85%); m.p.: 82–83 °C; FTIR (KBr): 3518, 3397, 3345, 3271, 1624, 1591, 1524, 1371, 1214, 1160 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 9.49 (s, 2H, OH), 8.66 (t, J = 5.52 Hz, 1H, NH), 6.66 (d, J = 2.09 Hz, 2H, H2′, H6′), 6.35 (t, J = 2.05 Hz, 1H, H4′), 3.97–3.95 (m, 2H, H1), 3.05 (s, 1H, H3); LRMS (API-ES) m/z 405.3 [2M+Na], 214.2 [M+Na], 192.3 [M+H].
(E)-3-(4-Hydroxy-3-methoxyphenyl)-N-(prop-2-ynyl)acrylamide(g)
Compound g was obtained from ferulic acid as described above. The concentrated residue was purified by column chromatography on silica gel (CHCl3/EtOAc 7:3). A colorless oil was obtained (0.2295 g, 50%); FTIR (ATR): 3284, 2115, 1655, 1582, 1511, 1382, 1261, 1202, 1119, 1028 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.53 (d, J = 15.56 Hz, 1H, H3′), 6.99 (d, J = 7.96 Hz, 1H, H6″), 6.91 (s, 1H, H2″), 6.84 (d, J = 8.14 Hz, 1H, H5″), 6.36 (s, 2H, OH, NH), 6.29 (d, J = 15.54 Hz, 1H, H2′), 4.14 (s, 2H, H1), 3.81 (s, 3H, CH3); LRMS (ESI) m/z 484.63 [2M+Na], 254.14 [M+Na], 232.19 [M+H].
(E)-3-(3,4-Dihydroxyphenyl)-N-(prop-2-ynyl)acrylamide (h)
Compound h was obtained from caffeic acid as described above. The concentrated residue was purified by column chromatography on silica gel (CHCl3/EtOH 10:0.5). A white powder was obtained (0.0880 g, 20%); m.p.: 169–170 °C; FTIR (KBr): 3477, 3366, 3266, 1646, 1587, 1536, 1343, 1264, 1191, 973 cm−1; 1H-NMR (300 MHz, CD3OD-d4): δ 7.40 (d, J = 15.7 Hz, 1H, H3′), 7.00 (d, J = 2.02 Hz, 1H, H2″), 6.90 (dd, J = 8.21, 2.01 Hz, 1H, H6″), 6.75 (d, J = 8.16 Hz, 1H, H5″), 6.34 (d, J = 15.68 Hz, 1H, H2′), 4.05 (d, J = 2.54 Hz, 2H, H1), 2.59 (t, J = 2.54 Hz, 1H, H3); LRMS (ESI) m/z 240.06 [M+Na].
3.6. Preparation of Triazolylmethyl Tryptolines 6a–i and Triazolylmethyl Tryptamines 12a–h
A mixture of compound 5 or 11 (1.0 mmol), alkynes a–i (1.2 mmol), 5% mol CuSO4 and 20% mol sodium ascorbate in t-BuOH/H2O/EtOH (2:2:1, 10 mL) was stirred at room temperature to 50 °C for 24 h. After ethanol was evaporated out, water (10 mL) was added to the reaction. The aqueous solution was extracted with ethyl acetate (10 mL × 3). The organic solution was washed with brine, dried, concentrated.
(S)-3-((4-(4-tert-Butylphenyl)-1H-1,2,3-triazol-1-yl)methyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b] indole (6a)
Compound 6a was obtained from compound 5 and 1-tert-butyl-4-ethynylbenzene (a) as described above. The concentrated residue was purified by column chromatography (CHCl3/ EtOAc/NH4OH; 5:5:0.1). A light yellow powder of compound 6a was obtained (0.1261 g, 32%); m.p.: 218–220 °C; FTIR (KBr): 3407, 3293, 1625, 1492, 1226cm−1; 1H-NMR (300 MHz, acetone-d6): δ 9.84 (s, 1H, H9), 8.40 (s, 1H, H5′), 7.83 (d, J = 8.50 Hz, 2H, H2″, H6″), 7.47 (d, J = 8.51 Hz, 2H, H3″, H5″), 7.38 (d, J = 7.62 Hz, 1H, H5), 7.30 (d, J = 7.65 Hz, 1H, H8), 7.05–6.93 (m, 2H, H7, H6), 4.67 (dd, J = 13.81, 5.26 Hz, 1H, H10b), 4.58 (dd, J = 13.83, 7.82 Hz, 1H, H10a), 4.06 (d, J = 16.34 Hz, 1H, H1b), 3.98 (d, J = 16.34 Hz, 1H, H1a), 3.52–3.43 (m, 1H, H3), 2.70 (dd, J = 15.16, 3.20 Hz, 1H, H4b), 2.52 (dd, J = 15.11, 9.80 Hz, 1H, H4a), 1.33 (s, 9H, CH3); 13C-NMR (300 MHz, acetone-d6): δ 151.394, 147.682, 137.343, 134.567, 129.701, 128.526, 126.420, 126.030, 121.882, 121.565, 119.457, 118.112, 111.647, 107.588, 55.343, 55.034, 43.211, 35.120, 31.575, 26.611; LRMS (API-ES) m/z 793.4 [2M+Na], 771.5 [2M+H], 408.4 [M+Na], 386.4 203.2 [M+H]; HRMS (ESI) m/z calcd for [M+] 385.5047, Found 408.1892 [M+Na], 386.2073 [M+H].
(S)-3-((4-(6-Methoxynaphthalen-2-yl)-1H-1,2,3-triazol-1-yl)me-thyl)-2,3,4,9-tetrahydro-1H-pyrido- [3,4-b]indole (6b)
Compound 6b was obtained from compound 5 and 2-ethynyl-6-methoxynaphthalene (b) as described above. After water was added to the reaction mixture, the precipitate was filtered through sintered glass and washed with water and ethyl acetate to yield a light brown solid of 6b (0.1548 g, 38%); m.p.: 232–234 °C; FTIR (KBr): 3421, 3302, 3243, 1613, 1501, 1266, 1215, 1029 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.70 (s, 1H, H9), 8.67 (s, 1H, H5′), 8.33 (s, 1H, H5″), 7.96 (d, J = 8.55 Hz, 1H, H7″), 7.88 (d, J = 8.95 Hz, 2H, H3″, H8″), 7.34–7.32 (m, 2H, H1″, H5), 7.26 (d, J = 7.85 Hz, 1H, H8), 7.18 (dd, J = 8.94, 2.44 Hz, 1H, H3″), 6.99 (t, J = 7.49, 7.49 Hz, 1H, H7), 6.91 (t, J = 6.94, 6.94 Hz, 1H, H6), 4.58 (s, 2H, H10), 3.94 (s, 2H, H1), 3.88 (s, 3H, CH3), 2.67 (d, J = 12.69 Hz, 1H, H4b), 2.49 (m, 1H, H4a); 13C-NMR (300 MHz, DMSO-d6): δ 157.530, 146.446, 135.899, 133.942, 129.594, 128.655, 127.447, 127.097, 126.202, 124.242, 123.485, 122.075, 120.479, 119.159, 118.358, 117.193, 110.949, 106.168, 106.014, 55.310, 54.164, 53.498, 41.959, 25.484; LRMS (API-ES) m/z 841.4 [2M+Na], 432.1 [M+Na], 410.3 [M+H]; HRMS (ESI) m/z calcd for [M+] 409.4830, Found 432.1699 [M+Na], 401.1896 [M+H].
(S)-3-((4-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-1-yl)methyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]-indole (6c)
Compound 6c was obtained from compound 5 and 1,2-dichloro-4-ethynylbenzene (c) as described above. The concentrated residue was purified by column chromatography (CHCl3/ EtOAc/NH4OH; 5:5:0.1). A light orange powder of compound 6c was obtained (0.2142 g, 52%); m.p.: 220–221 °C; FTIR (KBr): 3292, 1554, 1130, 803 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.71 (s, 1H, H9), 8.75 (s, 1H, H5′), 8.11 (d, J = 1.93 Hz, 1H, H2″), 7.86 (dd, J = 8.39, 1.98 Hz, 1H, H6″), 7.70 (dd, J = 8.41, 1.90 Hz, 1H, H5″), 7.33 (d, J = 7.56 Hz, 1H, H5), 7.26 (d, J = 7.8 Hz, 1H, H8), 6.99 (t, J = 7.49 Hz, 1H, H7), 6.91 (t, J = 7.64 Hz, 1H, H6), 4.63–4.49 (m, 2H, H10), 3.94 (d, J = 16.66 Hz, 1H, H1b), 3.87 (d, J = 16.73 Hz, 1H, H1a), 3.32 (m, 1H, H3), 2.68 (dd, J = 14.85, 3.63 Hz, 1H, H4b), 2.50–2.41 (m, 1H, H4a); 13C-NMR (300 MHz, DMSO-d6): δ 143.870, 135.752, 133.878, 131.684, 131.574, 131.115, 129.945, 126.969, 126.679, 125.099, 123.058, 120.320, 118.190, 117.059, 110.806, 105.850, 54.129, 53.325, 41.821, 25.357; LRMS (API-ES) m/z 420.2 [M+Na], 398.2 [M+]; HRMS (ESI) m/z calcd for [M+] 398.2885, Found 420.0629 [M+Na].
(S)-3-((4((N-(3-(4-Hydroxybenzoyl)benzoyl)azyl)methyl)-1H-1,2,3-triazol-1-yl)methyl)-2,3,4,9-tetra-hydro-1H-pyrido[3,4b]indole (6d)
Compound 6d was obtained from compound 5 and 2-(4-hydroxybenzoyl)-N-(prop-2-ynyl)benzamide (d) as described above. The concentrated residue was purified by column chromatography (EtOAc/EtOH/NH4OH; 9:1:0.1). A light brown powder of compound 6d was obtained (0.1773 g, 35%); m.p.: 213–215 °C; FTIR (KBr): 3409, 1682, 1613, 1513, 1372, 1277, 1169 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 10.67 (s, 1H, H1), 9.47 (s, 1H, OH), 7.75 (d, J = 5.2 Hz, 1H, H6″), 7.71 (d, J = 7.2 Hz, 1H, H5″), 7.56–7.47 (m, 2H, H4″, H5″), 7.30 (t, J = 6.80 Hz, 1H, H7′), 7.24 (d, J = 7.60 Hz, 2H, H5, H8), 7.09 (d, J = 8.4 Hz, 2H, H2‴, H6‴), 7.00–6.89 (m, 3H, H7, H5′, H6), 6.65 (dd, J = 8.60, 2.60 Hz, 2H, H3‴, H5‴), 4.59 (d, J = 15.2 Hz, H6′b), 4.41–4.34 (m, 2H, H10), 4.22 (dd, J = 15.8, 3.4 Hz, 1H, H6′a), 3.91 (d, J = 16.0 Hz, 1H, H1b), 3.84 (d, J = 16.4 Hz, 1H, H1a), 3.25–3.23 (m, 1H, H3), 2.60–2.55 (m, 1H, H4a), 2.38–2.31 (m, 1H, H4b); 13C-NMR (300 MHz, DMSO-d6): δ 166.605, 157.196, 150.040, 135.937, 132.581, 130.104, 129.597, 129.032, 127.317, 126.899, 122.803, 122.470, 120.561, 118.344, 117.186, 115.036, 110.939, 90.755, 59.783, 34.500; LRMS (ESI) m/z 529.27 [M+Na], 489.12 [M+-17]; HRMS (ESI) m/z calcd for [M+] 506.5551, Found 529.1922[M+Na], 489.1922 [M+-17].
(S)-3-((4-(N-(((4-Hydroxyphenyl)propanoyl)azyl)methyl)-1H-1,2,3-triazol-1-yl)methyl)-2,3,4,9-tetra-hydro-1H-pyrido[3,4-b]indole (6e)
Compound 6e was obtained from compound 5 and 3-(4-hydroxyphenyl)-N-(prop-2-ynyl)propanamide (e) as described above. The concentrated residue was washed with chloroform, ethyl acetate (20 mL) for each solvent. A yellow brown powder of compound 6e was obtained (0.1685 g, 39%); m.p.: 218–220 °C; FTIR (KBr): 3407, 3303, 3262, 1641, 1514, 1381, 1261, 1153 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.77 (s,,1H, H9), 9.21 (s, 1H, OH), 8.35 (t, J = 5.30 Hz, 1H, H7′), 7.78 (s, 1H, H5′), 7.31 (d, J = 7.65 Hz, 1H, H5), 7.26 (d, J = 7.90 Hz, 1H, H8), 7.02–6.89 (m, 4H, H7, H2″, H6″, H6), 6.65 (d, J = 8.29 Hz, 2H, H3″, H5), 4.52 (s, 1H, H10), 4.28 (d, J = 5.39 Hz, 2H, H1), 3.96 (s, 2H, H6′), 2.70 (t, J = 7.63 Hz, 2H, H9′), 2.63 (br, 1H, H4b), 2.43 (br, 1H, H4a) 2.34 (t, J = 7.71 Hz, 2H, H10′); 13C-NMR (300 MHz, DMSO-d6): δ 171.55, 155.479, 135.881, 131.295, 129.125, 126.844, 120.572, 118.377, 117.179, 115.071, 110.980, 105.700, 53.304, 37.338, 34.233, 30.332; LRMS (ESI) m/z 453.60 [M+Na]; HRMS (ESI) m/z calcd for [M+] 430.5022, Found 453.1911 [M+Na].
(S)-3-((4-(((N-(3,5-Dihydroxy)benzoyl)azyl)methyl)-1H-1,2,3-triazol-1-yl)methyl)-2,3,4,9-tetra hydro-1H-pyrido[3,4-b]indole(6f)
Compound 6f was obtained from compound 5 and 3,5-dihydroxy-N-(prop-2-ynyl)benzamide (f) as described above. The concentrated residue was purified by column chromatography (EtOAc/EtOH/NH4OH; 10:1:0.1). A light pink powder of 6f was obtained (0.0937 g, 22%); m.p.: 180–182 °C; FTIR (KBr): 3395, 3284, 1644, 1596, 1543, 1353, 1163, 1004 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.71 (s, 1H, H9), 9.48 (s, 2H, OH), 8.79 (t, J = 5.61 Hz, 1H, H7′), 7.98 (s, 1H, H5′), 7.30 (d, J = 7.58 Hz, 1H, H5), 7.25 (d, J = 7.85 Hz, 1H, H8), 6.99 (t, J = 6.94 Hz, 1H, H7), 6.92 (t, J = 7.34 Hz, 1H, H6), 6.71 (d, J = 2.13 Hz, 2H, H2″, H6″), 6.37 (d, J = 2.10 Hz, 1H, H4″), 4.56–4.43 (m, 4H, H10, H1), 4.05–3.85 (m, 2H, H6′), 3.34 (m, 1H, H3), 2.62 (dd, J = 15.02, 3.46 Hz, 1H, H4b), 2.39 (dd, J = 14.62, 9.51 Hz, 1H, H4a); 13C-NMR (300 MHz, DMSO-d6): δ 166.446, 158.288, 145.029, 136.437, 135.830, 133.438, 126.934, 123.752, 120.481, 118.345, 117.152, 110.921, 105.846, 105.568, 105.219, 53.574, 53.513, 41.831, 34.923, 25.176; LRMS (ESI) m/z 441.27 [M+Na], 419.14 [M+H]; HRMS (ESI) m/z calcd for [M+] 418.4485, Found 419.2116 [M+H].
(S)-3-((4-(N-(((E)-(3-Methoxy-4-hydroxyphenyl)propenoyl)azyl)methyl)-1H-1,2,3-triazol-1-yl)- methyl)-2,3,4,9-tetrahydro-1H-pyri do[3,4-b]indole (6g)
Compound 6g was obtained from compound 5 and(E)-3-(4-hydroxy-3-methoxyphenyl)-N-(prop-2-ynyl)acrylamide (g) as described above. The concentrated residue was purified by column chromatography (EtOAc/EtOH/NH4OH; 9:1:0.1). A light yellow powder of 6g was obtained (0.1356 g, 30%); m.p.: 199–200 °C; FTIR (KBr): 3373, 3262, 1650, 1584, 1517, 1327, 1280, 1204, 1169, 1030 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.69 (s, 1H, H9), 9.45 (s, 1H, OH), 8.46 (s, 1H, H7′), 8.01 (s, 1H, H5′), 7.40–7.24 (m, 3H, H10′, H5, H8), 7.12 (s, 1H, H2″), 7.01–6.98 (m, 2H, H7, H6″), 6.91 (t, J = 7.34 Hz, 1H, H6), 6.79 (d, J = 8.14 Hz, 1H, H5″), 6.50 (d, J = 15.70 Hz, 1H, H9′), 4.49–4.44 (m, 4H, H10, H1), 3.93–3.87 (m, 2H, H6′), 3.79 (s, 3H, CH3), 3.29 (s, 1H, H3), 2.63–2.34 (m, 2H, H4); 13C-NMR (300 MHz, DMSO-d6): δ 165.330, 148.322, 147.807, 144.567, 139.392, 135.766, 133.904, 126.981, 126.351, 123.649, 121.507, 120.370, 118.675, 118.246, 117.081, 115.674, 110.859, 105.916, 55.512, 53.806, 53.506, 41.891, 34.383, 25.370; LRMS (ESI) m/z 459.42 [M+H]; HRMS (ESI) m/z calcd for [M+] 458.5123, Found 481.1977 [M+Na], 459.2158 [M+H].
(S)-3-((4-(N-(((E)-(3,4-Dihydroxyphenyl)propenoyl)azyl)methyl)-1H-1,2,3-triazol-1-yl)methyl)-2,3,4,9- tetrahydro-1H-pyrido[3,4-b]indole (6h)
Compound 6h was obtained from compound 5 and((E)-3-(3,4-dihydroxyphenyl)-N-(prop-2-ynyl)acrylamide (h) as described above. The concentrated residue was purified by column chromatography (EtOAc/EtOH/NH4OH; 9:1:0.1). A light yellow powder of 6h was obtained (0.0710 g, 18%); m.p.: 176–178 °C; FTIR (KBr): 3392, 3271, 1650, 1600, 1527, 1375, 1283, 1112 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.76 (s, 1H, H9), 9.20 (m, 2H, OH), 8.54 (s, 1H, H7′), 8.04 (s, 1H, H5′), 7.35–7.27 (m, 3H, H5, H8, H10′), 7.04–6.91 (m, 3H, H7, H6, H2″), 6.85 (d, J = 8.24 Hz, 1H, H6″), 6.76 (d, J = 8.03 Hz, 1H, H5″), 6.40 (d, J = 15.72 Hz, 1H, H9′), 4.55–4.44 (m, 4H, H10, H1), 4.03–3.42 (m, 2H, H6′), 3.48–3.41 (m, 1H, H3), 2.67 (d, J = 11.88 Hz, 1H, H4b), 2.51–2.45 (m, 1H, H4a); 13C-NMR (300 MHz, DMSO-d6): δ 165.369, 147.396, 145.552, 144.686, 139.542, 135.830, 132.971, 126.831, 126.305, 123.783, 120.523, 118.354, 118.152, 117.158, 115.770, 113.801, 110.933, 105.716, 53.439, 53.342, 41.738, 34.334, 24.949; LRMS (ESI) m/z 445.50 [M+H]; HRMS (ESI) m/z calcd for [M+] 444.4858, Found 467.1815 [M+Na], 445.2002 [M+H].
(S)-1-(4-(4-tert-Butylphenyl)-1H-1,2,3-triazol-1-yl)-3-(1H-indol-3-yl)propan-2-amine (12a)
Compound 12a was obtained from compound 11 and 1-tert-butyl-4-ethynylbenzene (a) as described above. The concentrated residue was purified by column chromatography (CHCl3/EtOAc/NH4OH; 9:1:0.1) to yield a brown semisolid of compound 12a (0.1530 g, 41%); FTIR (ATR): 3407, 3358, 3287, 1665, 1583, 1265, 1051 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.89 (s, 1H, H1), 8.49 (s, 1H, H5′), 7.75 (d, J = 8.36 Hz, 2H, H2″, H6″), 7.52 (d, J = 7.80 Hz, 1H, H4), 7.44 (d, J = 8.40 Hz, 2H, H3″, H5″), 7.35 (d, J = 8.02 Hz, 1H, H7), 7.23 (d, J = 2.10 Hz, 1H, H2), 7.06 (t, J = 7.09 Hz, 1H, H6), 6.97 (t, J = 7.39 Hz, 1H, H5), 4.41 (dd, J = 13.46, 4.56 Hz, 1H, H10b), 4.23 (dd, J = 13.54, 7.76 Hz, 1H, H10a), 3.40 (m, 1H, H9), 2.81 (dd, J = 14.22, 5.80 Hz, 1H, H8b), 2.69 (dd, J = 14.22, 7.13 Hz, 1H, H8a), 1.29 (s, 9H, CH3); 13C-NMR (300 MHz, DMSO-d6): δ 150.171, 145.965, 136.265, 128.177, 127.446, 125.561, 124.900, 123.718, 121.816, 120.917, 118.360, 118.304, 111.409, 110.646, 55.733, 52.042, 34.321, 31.060; LRMS (API-ES) m/z 374.4 [M+H]; HRMS (ESI) m/z calcd for [M+] 373.4940, Found 374.2425 [M+H].
(S)-1-(1H-Indol-3-yl)-3-(4-(6-methoxynaphthalen-2-yl)-1H-1,2,3-triazol-1-yl)propan-2-amine (12b)
Compound 12b was obtained from compound 11 and 2-ethynyl-6-methoxynaphthalene (b) as described above. The concentrated residue was purified with column chromatography (CHCl3/EtOAc/NH4OH; 8:2:0.1) to yield a white solid of 12b (0.1078 g, 27%); m.p.: 85–87 °C; FTIR (KBr): 3413, 3274, 1612, 1549, 1260, 1163, 1030 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 10.89 (s, 1H, H9), 8.60 (s, 1H, H5′), 8.30 (s, 1H, H5″), 7.93 (dd, J = 8.52, 1.55 Hz, 1H, H7″), 7.86 (d, J = 9.29 Hz, 2H, H4″, H8″), 7.53 (d, J = 7.86 Hz, 1H, H4), 7.34 (d, J = 8.07 Hz, 1H, H7), 7.31 (d, J = 2.40 Hz, 1H, H1″), 7.23 (d, J = 2.17 Hz, 1H, H5″), 7.16 (dd, J = 8.95, 2.51 Hz, 1H, H3″), 7.05 (t, J = 7.52 Hz, 1H, H6), 6.96 (t, J = 7.44 Hz, 1H, H5), 4.43 (dd, J = 13.53, 4.53 Hz, 1H, H10b), 4.25 (dd, J = 13.52, 7.83 Hz, 1H, H10a), 3.47–3.41 (m, 1H, H9), 2.82 (dd, J = 14.22, 5.89 Hz, 1H, H8a), 2.70 (dd, 14.24, 7.13 Hz, 1H, H8b), 1.56 (s, 2H, NH2); 13C-NMR (300 MHz, DMSO-d6): δ 157.407, 146.277, 136.297, 133.824, 129.491, 128.547, 127.312, 126.121, 124.137, 123.925, 123.352, 122.189, 120.989, 119.091, 118.375, 111.461, 110.084, 106.019, 55.210, 54.893, 51.857, 30.201; LRMS (API-ES) m/z 817.5 [2M+Na], 398.4 [M+H]; HRMS (ESI) m/z calcd for [M+] 397.4723, Found 398.2066 [M+H].
(S)-1-(4-(3,4-Dichlorophenyl)-1H-1,2,3-triazol-1-yl)-3-(1H-indol-3-yl)propan-2-amine (12c)
Compound 12c was obtained from compound 11 and 1,2-dichloro-4-ethynylbenzene (c) as described above. The concentrated residue was purified with column chromatography (CHCl3/EtOAc/NH4OH; 9:1:0.1) to yield a light pink powder of compound 12c (0.2500 g, 65%); m.p.: 149–151 °C; FTIR (KBr): 3446, 3357, 3264, 1608, 1555, 1132, 800 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.89 (s, 1H, H1), 8.68 (s, 1H, H5′), 8.08 (d, J = 1.86 Hz, 1H, H2″), 7.84 (dd, J = 8.39, 1.94 Hz, 1H, H6″), 7.69 (d, J = 8.4 Hz, 1H, H5″), 7.53 (d, J = 7.78 Hz, 1H, H4), 7.35 (d, J = 7.97 Hz, 1H, H7), 7.23 (d, J = 1.97 Hz, 1H, H2), 7.06 (t, J = 7.48 Hz, 1H, H6), 6.97 (t, J = 7.38 Hz, 1H, H5), 4.43 (dd, J = 13.52, 4.38 Hz, 1H, H10b), 4.24 (dd, J = 13.49, 7.88 Hz, 1H, H10a), 3.42 (m, 1H, H9), 2.82 (dd, J = 14.26, 5.92 Hz, 1H, H8b), 2.70 (dd, J = 14.25, 6.99 Hz, 1H, H8a), 1.54 (s, 2H, NH2); 13C-NMR (300 MHz, DMSO-d6): δ 143.770, 136.265, 131.680, 131.136, 129.909, 127.427, 126.682, 125.096, 123.713, 123.186, 120.924, 118.363, 118.310, 111.412, 110.631, 55.942, 52.023, 31.081; LRMS (API-ES) m/z 386.2 [M+]; HRMS (ESI) m/z calcd for [M+] 386.2778, Found 386.1003 [M+].
(S)-N-((1-(2-Amino-3-(1H-indol-3-yl)propyl)-1H-1,2,3-triazol-4-yl)methyl)-2-(4-hydroxybenzoyl)- benzamide (12d)
Compound 12d was obtained from compound 11 and 2-(4-hydroxybenzoyl)-N-(prop-2-ynyl)benzamide (d) as described above. The concentrated residue was purified with column chromatography (EtOAc/EtOH/NH4OH; 9:1:0.1) to yield a light yellow powder of compound 12d (0.1086 g, 22%); m.p.: 177–179 °C; FTIR (KBr): 3404, 3347,1684, 1612, 1511, 1394, 1202, 1052 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 10.98 (s, 1H, H1), 7.78 (s, 1H, H5′), 7.77 (s, 1H, H6″), 7.62–7.53 (m, 3H, H4, H4″, H5″), 7.40 (d, J = 8.00 Hz, 1H, H7), 7.30 (d, J = 7.20 Hz, 1H, H3″), 7.25 (d, J = 1.60 Hz, 1H, H2), 7.13 (d, J = 8.40 Hz, 2H, H2‴, H6‴), 7.10 (d, J = 7.6 Hz, 1H, H6), 7.01 (t, J = 7.40 Hz, 1H, H5), 6.71 (d, J = 8.4 Hz, 2H, H3‴, H5‴), 4.64 (d, J = 15.60 Hz, 1H, H6′b), 4.34–4.29 (m, 2H, H10), 4.26 (d, J = 15.6 Hz, H6′a) 4.18–4.22 (m, 1H, H10), 2.81–2.76 (m, 1H, H8a), 2.60 (dd, J = 14.00, 7.20 Hz, 1H, H8b); 13C-NMR (400 MHz, DMSO-d6): δ 166.999, 157.809, 150.502, 144.332, 136.705, 132.968, 130.543, 129.864, 129.422, 127.881, 127.675, 124.327, 124.121, 123.221, 122.885, 121.337, 118.836, 118.736, 115.503, 118.842, 111.125, 91.188, 55.899, 52.452, 34.719, 31.257; LRMS (API-ES) m/z 1011.3 [2M+Na], 477.2 [M+-17]; HRMS (ESI) m/z calcd for [M+] 494.5444, Found 477.1888 [M+-17].
(S)-N-((1-(2-Amino-3-(1H-indol-3-yl)propyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-hydroxyphenyl) propanamide (12e)
Compound 12e was obtained from compound 11 and 3-(4-hydroxyphenyl)-N-(prop-2-ynyl)propanamide (e) as described above. The concentrated residue was purified with column chromatography (CHCl3/MeOH/NH4OH; 10:0.2:0.1) to yield brown semisolid of compound 12e (0.2007 g, 49%): FTIR (ATR): 3398, 3338, 3281, 1650, 1613, 1515, 1340, 1232, 1052 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.90 (s, 1H, H1), 8.31 (t, J = 5.50 Hz, 1H, H7′), 7.74 (s, 1H, H5′), 7.52 (d, J = 7.79 Hz, 1H, H4), 7.36 (d, J = 8.00 Hz, 1H, H7), 7.23 (s, 1H, H2), 7.08 (t, J = 7.13 Hz, 1H, H6), 7.01–6.96 (m, 3H, H5, H2″, H6″), 6.66 (d, J = 8.37 Hz, 2H, H3″, H5″), 4.38–4.14 (m, 4H, H10, H6′), 3.34 (s, 1H, H9), 2.81–2.62 (m, 4H, H8, H9′), 2.35 (t, J = 7.75 Hz, 2H, H10′); 13C-NMR (300 MHz, DMSO-d6): δ 171.437, 155.429, 144.662, 136.286, 131.321, 129.111, 127.425, 123.704, 123.565, 120.951, 118.376, 118.334, 115.040, 111.433, 110.645, 55.350, 52.098, 37.310, 34.205, 30.923, 30.287; LRMS (API-ES) m/z 859.5 [2M+Na], 441.4 [M+Na], 419.3 [M+H]; HRMS (ESI) m/z calcd. for [M+] 418.4915, Found 441.1939 [M+Na].
(S)-N-((1-(2-Amino-3-(1H-indol-3-yl)propyl)-1H-1,2,3-triazol-4-yl)methyl)-3,5-dihydroxybenzamide (12f)
Compound 12f was obtained from compound 11 and 3,5-dihydroxy-N-(prop-2-ynyl) benzamide (f) as described above. The concentrated residue was purified with column chromatography (CHCl3/MeOH/NH4OH; 10:0.4:0.1) to yield compound 12f as a light orange solid (0.1304 g, 32%); m.p.: 68–70 °C; FTIR (KBr): 3415, 1679, 1595, 1536, 1340, 1206, 1165 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.91 (s, 1H, H1), 9.51 (s, 2H, OH), 8.76 (t, J = 5.58 Hz, 1H, H7′), 7.95 (s, 1H, H5′), 7.50 (d, J = 7.74 Hz, 1H, H4), 7.35 (d, J = 8.02 Hz, 1H, H7), 7.22 (d, J = 2.00 Hz, 1H, H2), 7.07 (t, J = 7.09 Hz, 1H, H6), 6.96 (t, J = 7.05 Hz, 1H, H5), 6.71 (d, J = 2.08 Hz, 2H, H2″, H6″), 6.73 (s, 1H, H4″), 4.46 (d, J = 5.55 Hz, 2H, H6′), 4.37 (dd, J = 13.55, 4.41 Hz, 1H, H10b), 4.20 (dd, J = 13.48, 7.66 Hz, 1H, H10a), 3.34 (m, 1H, H9), 2.77 (dd, J = 14.41, 5.94 Hz, 1H, H8b), 2.66 (dd, J = 14.15, 7.30 Hz, 1H, H8a); 13C-NMR (300 MHz, DMSO-d6): δ 166.409, 158.268, 144.853, 136.426, 136.289, 127.411, 123.799, 123.733, 120.948, 118.350, 111.439, 110.568, 105.537, 105.179, 55.221, 52.074, 34.907, 30.829; LRMS (API-ES) m/z 429.3 [M+Na], 407.5 [M+H]; HRMS (ESI) m/z calcd for [M+] 406.4378, Found 429.1615 [M+Na], 407.1798 [M+H].
(S,E)-N-((1-(2-Amino-3-(1H-indol-3-yl)propyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(4-hydroxy-3-methoxyphenyl) acrylamide (12g)
Compound 12g was obtained from compound 11 and (E)-3-(4-hydroxy-3-methoxyphenyl)-N-(prop-2-ynyl)acrylamide (g) as described above. The concentrated residue was purified by column chromatography (CH2Cl2/EtOH/NH4OH; 9.5:0.5:0.1) to yield compound 12g as a yellow solid (0.1020 g, 23%); m.p.: 118–119 °C; FTIR (KBr): 3395, 3344, 3262, 1656, 1593, 1511, 1336, 1277, 1229, 1121, 1030 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 10.88 (s, 1H, H1), 8.43 (t, J = 5.68 Hz, 1H, H7′), 7.98 (s, 1H, H5′), 7.51 (d, J = 9.73 Hz, 1H, H4), 7.39–7.34 (m, 2H, H10′, H7), 7.22 (d, J = 2.13 Hz, 1H, H2), 7.12 (d, J = 1.69 Hz, H2″), 7.06 (t, J = 7.49 Hz, 1H, H6), 7.02–6.94 (m, 2H, H6″, H5), 6.79 (d, J = 8.11 Hz, 1H, H5″), 6.49 (d, J = 15.69 Hz, 1H, H9′), 4.43 (d, J = 5.47 Hz, 2H, H6′), 4.36 (dd, J = 13.44, 4.60 Hz, 1H, H10b), 4.19 (dd, J = 13.42, 7.56 Hz, 1H, H10a), 3.80 (s, 3H, CH3), 3.36–3.32 (m, 1H, H9), 2.77 (dd, J = 14.27, 5.81 Hz, 1H, H8b), 2.65 (dd, J = 14.18, 7.23 Hz, 1H, H8a); 13C-NMR (300 MHz, DMSO-d6): δ 165.283, 148.304, 147.797, 144.437, 139.345, 136.257, 127.405, 126.336, 123.705, 123.664, 121.496, 120.910, 118.661, 118.352, 118.308, 115.653, 111.399, 110.838, 110.680, 55.509, 55.409, 52.102, 34.372, 31.018; LRMS (ESI) m/z 469.58 [M+Na], 447.58 [M+H]; HRMS (ESI) m/z calcd for [M+] 446.5016, Found 469.1923 [M+Na], 447.2119 [M+H].
(S,E)-N-((1-(2-Amino-3-(1H-indol-3-yl)propyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(3,4-dihydroxy- phenyl) acrylamide (12h)
Compound 12h was obtained from compound 11 and ((E)-3-(3,4-dihydroxyphenyl)-N-(prop-2-ynyl)acrylamide (h) as described above. The concentrated residue was purified by column chromatography (CHCl3/EtOH; 8:2) to compound 12h as a yellow brown semisolid (0.0562 g, 13%): FTIR (ATR): 3404, 3284, 1656, 1593, 1524, 1340, 1270, 1118 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 10.92 (s, 1H, H1), 8.51–8.46 (m, 1H, H7′), 7.96 (s, 1H, H5′), 7.49 (d, J = 7.60 Hz, 1H, H4), 7.33 (d, J = 8.40 Hz, 1H, H10′), 7.27 (s, 1H, H2), 7.23–7.19 (m, 2H, H7, H5″), 7.05 (t, J = 7.60 Hz, 1H, H6), 6.96 (t, J = 7.40 Hz, 1H, H5), 6.92 (d, J = 2.00 Hz, 1H, H2″), 6.81 (dd, J = 8.40, 2.00 Hz, 1H, H6″), 6.72 (d, J = 8.40 Hz, 1H, H9′), 4.40–4.36 (m, 3H, H6′, H10b), 4.24 (dd, J = 13.80, 7.40 Hz, 1H, H10a), 2.78 (dd, J = 14.40, 6.40 Hz, 1H, H8b), 2.69 (dd, J = 14.20, 7.00 Hz, 1H, H8a); 13C-NMR (400 MHz, DMSO-d6): δ 171.956, 165.794, 147.848, 146.002, 145.034, 139.970, 136.721, 129.338, 127.782, 126.729, 124.289, 121.413, 120.903, 118.813, 118.584, 116.204, 114.267, 111.880, 52.315, 34.765, 22.928, 21.632; LRMS (ESI) m/z 455.67 [M+Na]; HRMS (ESI) m/z calcd for [M+] 432.4751, Found 455.1807 [M+Na], 447.2119 [M+H].
3.7. β-Secretase Inhibition Assay
β-Secretase enzyme was reconstituted with 50 mM Tris (pH 7.5), 10% glycerol to yield concentration of 0.5 unit/μL. The enzyme stock solution was diluted with 100 mM sodium acetate buffer (pH 4.5) to 0.01 unit/μL. Thirty microliters of enzyme solution were added to each well that contained 20 μL of 5% DMSO of test compound. Then, fifty microliters of 50 μM β-secretase substrate IV from Calbiochem were added to the reaction plate. The fluorescence was measured at Ex = 380 nm and Em = 510 nm by using SpectraMax Gemini EM™ [29]. β-Secretase inhibitor IV was used as positive control. The assays were run in triplicate. The results of test compound 25 μM were analyzed. Compounds having inhibitory activity more than 50% were further evaluated for IC50 and data were analyzed via GrahPad Prism 4 in nonlinear regression curve fit.
3.8. Cathepsin D Assay
Sensolyte®520 Cathepsin D assay kit was purchased from Anaspec. Forty microliters of cathepsin D (0.25 μg/mL in 5 mM DTT assay buffer) was added to a greiner black 96 well plate. Ten microliters of each test compound (1000 μM in 10% DMSO) were added to each well. After 10 min incubation at 37 °C, 50 μL of 0.01 mM cathepsin D substrate was added to each well. The assay reactions were incubated for 30 min at 37 °C. The fluorescence was measured at Ex = 490 nm and Em = 520 nm by using SpectraMax Gemini EM™. Pepstatin A was included as a positive control. The assays were run in triplicate. The data were analyzed.
3.9. Amyloid β Preparation and Aggregation Assay
Stock solution of amyloid-β (1-42) peptide (Anaspec; 1384 μM in 1% NH4OH) was diluted with 50 mM Tris buffer (pH 7.4) to yield 25 μM working solution. Nine microliters amyloid-β solution was added to a greiner transparent 96 well plate. One microliter of each test compounds (1,000 μM in DMSO) were added to each well and mixed gently by tapping. The final concentrations of each compound were 100 μM in 10% DMSO. The reaction plate was incubated in dark at 37 °C for 48 h. After incubation, 200 μL of 5 μM ThT (Sigma) in 50 mM Tris buffer (pH 7.4) was added to each well. The fluorescence was measured at Ex = 446 nm and Em = 490 nm by using SpectraMax Gemini EM™ [30]. The assays were run in triplicate. Curcumin was used as a positive control. The result data were analyzed and compounds having inhibition over 50% were evaluated for IC50 values and data were analyzed via GraphPad Prism 4 by nonlinear regression curve fit.
3.10. Fe (II) Chelation Capcity Assay
An aqueous solution of 0.2 mM ferrous sulphate was prepared. Fifty microliters of this solution was added to 96 well plate, then 40 μL of 500 μM of test compounds in 50% DMSO were added to each well followed by DI water to adjust the volume to 150 μL. The mixtures were incubated at room temperature for 10 min. After incubation, 50 μL of 1 mM ferrozine (Sigma) was added to the reaction mixture. The absorbance was measured at 562 nm by using infinite 200 pro™, Tecan [31,32]. EDTA was used as a positive control. Chelation capacity of each compounds were calculated. Compounds having chelating capacity over than 50% were evaluated for stoichiometric ratio of compound: metal.
3.11. Free Radical Scavenging Assay
The test samples were prepared in 50% DMSO at 500 μM. Seventy microliters of DPPH solution (500 μM in methanol) was added to 96 well plate. The assay mixture was adjusted to 80 μL volume by methanol. After adjusting the volume, 20 μL of test compounds was added to each well. The reaction plate was incubated at room temperature in dark for 30 min. The absorbance was measured at 517 nm by using infinite 200 pro™, Tecan [33]. Ascorbic acid was used as a positive control. Percent inhibition was calculated and compounds showing activity were evaluated for the IC50 values.
3.12. Cell Culture and Cell Viability Assay by MTT Method
The SH-SY5Y cells were cultured in a medium containing minimum essential medium (MEM): F-12 (1:1), 10% fetal bovine serum, MEM non-essential amino acids (0.5×), 0.5 mM sodium pyruvate and 100 units/ml of penicillin and 100 μg/mL of streptomycin. The cells were maintained under a humidified incubator with 5% CO2 in air at 37 °C.
SH-SY5Y cells were seeded in 96-well plates (2 × 104 cells per well) for 24 h. After 24 h, cells were treated with 10 μM of test compounds for 2 h prior to exposure to 1 μM aggregated β-amyloid (1-42). The aggregated β-amyloid was prepared by incubating in a medium without serum, penicillin and streptomycin at 37 °C for 72 h. The cells treated with aggregated β-amyloid were incubated for 24 h, 15 μL of MTT reagent (5 mg/mL MTT in serum free medium containing 10 μM HEPES) was added to each well and incubated at 37 °C for 3 h. The medium was removed from each well. One hundred microlitres of 0.04 N HCl in isopropanol was added to each well to dissolve blue formazan crystals, which is the product of MTT catalyzed by mitochondrial dehydrogenase. The absorbance was measured at 570/630 nm using a Synergy™ HT multi-detection microplate reader (Bio-Tek Instruments, Winooski, VT, USA) [34].
4. Conclusions
In this research, the tryptoline core compound previously reported as BACE1 inhibitor was modified in silico to possess multi-modes of action for the treatment of Alzheimer’s disease i.e., anti-amyloid aggregation, metal chelating and radical scavenging action. The in silico designed compounds are tryptoline- and tryptamine-based BACE1 inhibitors containing additional moieties to exert multi-functionality. Among sixteen new compounds, the major action of compound 6h were anti-Aβ aggregation and antioxidative action. Two compounds, 12c and 12h, were multifunctional compounds with three actions. Compound 12c acted as a BACE1 inhibitor, anti-amyloid aggregation and metal chelator, while compound 12h was an Aβ aggregation blocker, chelator and antioxidant. The IC50 values of compound 12c against BACE1 and amyloid-β aggregation were 20.75 μM and 83.23 μM, while the IC50 values of compound 12h against amyloid-β aggregation and antioxidant were 56.39 μM and 92.70 μM. Furthermore, these compounds acted as metal chelators with a stoichiometric ratio of ligand per metal 3:1. Despite the fact that the individual activities at each targets of compounds 6h, 12c and 12h were rather weak (in the micromolar range), the neuroprotective effect against Aβ1-42 insult in SH-SY5Y cells of these multifunctional ligands were better than that of single targeted ligands i.e., BACE1 inhibitor IV and comparable to the potent nanomolar curcumin. The results indicated the success of the multifunction strategy which suits the multi-pathogenesis of AD by reducing the neurotoxicity cascade from Aβ1-42.
Acknowledgments
This work was supported by Thailand Research Fund (TRF) through the Royal Golden Jubilee Ph.D. Program (Grant No. PHD/0209/2548), the Office of the Higher Education Commission, Ministry of Education, Thailand under Grant No. CHE-RES-RG 2551, Mahidol University and The Scripps Research Institute.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
References
- 1.Jakob-Roetne R, Jacobsen H. Alzheimer’s disease: From pathology to therapeutic approaches. Angew Chem Int Ed Engl. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 2.Mandel S, Amit T, Bar-Am O, Youdim MB. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog Neurobiol. 2007;82:348–360. doi: 10.1016/j.pneurobio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 4.Galimberti D, Scarpini E. Progress in Alzheimer’s disease. J Neurol. 2012;259:201–211. doi: 10.1007/s00415-011-6145-3. [DOI] [PubMed] [Google Scholar]
- 5.Budimir A. Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm. 2011;61:1–14. doi: 10.2478/v10007-011-0006-6. [DOI] [PubMed] [Google Scholar]
- 6.Tabner BJ, Mayes J, Allsop D. Hypothesis: Soluble Aβ oligomers in association with redox-active metal ions are the optimal generators of reactive oxygen species in Alzheimer’s disease. Int J Alzheimer’s Dis. 2010;2011 doi: 10.4061/2011/546380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolognesi ML, Cavalli A, Melchiorre C. Memoquin: A Multi-target-directed ligand as an innovative therapeutic opportunity for Alzhemer’s disease. Neurotherapeutics. 2009;6:152–162. doi: 10.1016/j.nurt.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiaranaikulwanitch J, Boonyarat C, Fokin VV, Vajragupta O. Triazolyltryptoline derivatives as β-secretase inhibitors. Bioorg Med Chem Lett. 2010;10:6572–6576. doi: 10.1016/j.bmcl.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinke AA, Gestwicki JE. Structure-activity relationships of amyloid beta-aggregation inhibitiors based on curcumin: Influence of linker length and flexibility. Chem Biol Drug Des. 2007;70:206–215. doi: 10.1111/j.1747-0285.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- 10.Zheng W, Wang SY. Antioxidant activity and phenolic compounds in selected herbs. J Agric Food Chem. 2001;49:5165–5170. doi: 10.1021/jf010697n. [DOI] [PubMed] [Google Scholar]
- 11.Ebrahimzadeh MA, Pourmorad F, Bekhradnia AR. Iron chelating activity, phenol and flavonoid content of some medicinal plants from Iran. Afr J Biotechnol. 2008;7:3188–3192. [Google Scholar]
- 12.Xiao J, Xu J, Cui S, Liu H, Wang S, Li Y. Supramolecular helix of an amphiphilic pyrene derivative induced by chiral tryptophan through electrostatic interactions. Org Lett. 2008;10:645–648. doi: 10.1021/ol7030087. [DOI] [PubMed] [Google Scholar]
- 13.Page D, Naismith A, Schmidt R, Coupal M, Labarre M, Gosselin M, Bellemare D, Payza K, Brown W. Novel C-terminus modifications of the Dmt-Tic Motif: A new class of dipeptide analogues showing altered pharmacological profiles toward the opioid receptors. J Med Chem. 2001;44:2387–2390. doi: 10.1021/jm015532k. [DOI] [PubMed] [Google Scholar]
- 14.Galli U, Ercolano E, Carraro L, Blasi Roman CR, Sorba G, Canonico PL, Genazzani AA, Tron GC, Billington RA. Synthesis and biological evaluation of isosteric analogues of FK866, an inhibitor of NAD salvage. ChemMedChem. 2008;3:771–779. doi: 10.1002/cmdc.200700311. [DOI] [PubMed] [Google Scholar]
- 15.Xie J, Seto CT. A two stage click-based library of protein tyrosine phosphatase inhibitors. Bioorg Med Chem. 2007;15:458–473. doi: 10.1016/j.bmc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: Copper (I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Kumagai N, Muncipinto G, Schreiber SL. Short synthesis of skeletally and stereochemically diverse small molecules by coupling petasis condensation reactions to cyclization reactions. Angew Chem Int Ed Engl. 2006;45:3635–3638. doi: 10.1002/anie.200600497. [DOI] [PubMed] [Google Scholar]
- 18.Brady SF, Singh S, Crouthamel MC, Holloway MK, Coburn CA, Garsky VM, Boqusky M, Pennington MW, Vacca JP, Hazuda D, et al. Rational design and synthesis of selective BACE-1 inhibitors. Bioorg Med Chem Lett. 2004;14:601–604. doi: 10.1016/j.bmcl.2003.11.061. [DOI] [PubMed] [Google Scholar]
- 19.Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR. Structure-based design of potent and selective cell-permeable inhibitors of human β-secretase (BACE1) J Med Chem. 2004;47:6447–6450. doi: 10.1021/jm049379g. [DOI] [PubMed] [Google Scholar]
- 20.Ono K, Haseqawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J Neurosci Res. 2004;75:742–750. doi: 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- 21.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 22.Mouton-Liger F, Paquet C, Dumurgier J, Bouras C, Pradier L, Gray F, Hugon J. Oxidative stress increase BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim Biophys Acta. 2012;1822:885–896. doi: 10.1016/j.bbadis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, et al. Oxidative stress activates a positive feedback between the gamma- and betasecretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quiroz-Baez R, Rojas E, Arias C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem Int. 2009;55:662–670. doi: 10.1016/j.neuint.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 25.Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, Liao FF. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener. 2011;6 doi: 10.1186/1750-1326-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajapakse HA, Nantermet PG, Selnick HG, Munshi S, McGaughey GB, Lindsley SR, Young MB, Lai MT, Espeseth AS, Shi XP, et al. Discovery of oxadiazoyl tertiary carbinamine inhibitors of β-secretase (BACE-1) J Med Chem. 2006;49:7270–7273. doi: 10.1021/jm061046r. [DOI] [PubMed] [Google Scholar]
- 27.Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science. 2000;290:150–153. doi: 10.1126/science.290.5489.150. [DOI] [PubMed] [Google Scholar]
- 28.Tomaselli S, Esposito V, Vangone P, van Nuland NA, Bonvin AM, Guerrini R, Tancredi T, Temussi PA, Picone D. The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1-42) peptide in aqueous media is reversible: A step by step conformational analysis suggests the location of beta conformation seeding. ChemBioChem. 2006;2:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 29.Ermolieff J, Loy JA, Koelsch G, Tang J. Proteolytic activation of recombinant pro-memapsin 2 (pro-β-secretase) studied with new fluorogenic substrates. Biochemistry. 2000;39:12450–12456. doi: 10.1021/bi001494f. [DOI] [PubMed] [Google Scholar]
- 30.Levine H., III Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Prot Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stookey LL. Ferrozine-a new spectrophotometric reagent for Iron. Anal Chem. 1970;42:779–781. [Google Scholar]
- 32.Karamac M. Chelation of Cu(II), Zn(II) and Fe(II) by tannin constituents of selected edible nuts. Int J Mol Sci. 2009;10:5485–5497. doi: 10.3390/ijms10125485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma OP, Bhat TK. DPPH antioxidant assay revisited. Food Chem. 2009;113:1202–1205. [Google Scholar]
- 34.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]