

Abstract
In this article the calcium/calmodulin-dependent protein kinases are reviewed. The primary focus is on the structure and function of this diverse family of enzymes, and the elegant regulation of their activity. Structures are compared in order to highlight the conserved architecture of their catalytic domains with respect to each other as well as protein kinase A, a prototype for kinase structure. In addition to reviewing structure and function in these enzymes, the variety of biological processes for which they play a mediating role are also examined. Finally, how the enzymes become activated in the intracellular setting is considered by exploring the reciprocal interactions that exist between calcium binding to calmodulin when interacting with the CaM-kinases.
Keywords: Calmodulin, protein kinase, Ca2+-signaling, 3-D structure, phosphotransferase, autophosphorylation
Introduction
The divalent cation calcium (Ca2+) is one of the most widely utilized second messengers in cellular signaling. In fact, Ca2+ is so integral to cellular physiology that it would be difficult to find a process in mammalian cells that is not influenced by its action in some way. Interestingly, despite its critical nature in the cell, Ca2+ is highly reactive, and therefore toxic to cells upon prolonged exposure to high levels. Because of this toxicity, it is essential that cells preserve a low intracellular concentration of Ca2+ (50–100 nM) in their basal state. On the other hand, it is this highly reactive nature of Ca2+ and the low intracellular concentration that make it such a potent molecule for use in cellular signaling. The myriad of pumps, exchangers and buffers that work to maintain the low intracellular concentration of this ion ensure that a large chemical gradient for Ca2+ will always exist across the plasma membrane of the cell. So, when integral membrane proteins like Ca2+-permeable receptors or voltage-gated Ca2+ channels are opened, there is a rapid influx of Ca2+ into the cell that raises the concentration an order of magnitude or more. Due to the spatial distribution and temporal kinetics of different Ca2+ permeable receptors and channels, the actual elevation of intracellular Ca2+ can vary widely between different cell types and cellular sub-compartments, and local concentrations have been estimated to reach levels as high as 10–100 μM during an induced Ca2+ influx. Extracellular Ca2+ is not the only source for the signaling ion however. Ca2+ can also be released from intracellular stores via activation of inositol trisphosphate (IP3) receptors or ryanodine receptors located in the membrane of both the endoplasmic and sarcoplasmic reticulum. These intra-cellular stores are typically stimulated to release by either a chemical or mechanical communication between receptors/ion channels in the plasma membrane and the organelles that maintain Ca2+ stores. Often, this communication will initiate a positive feedback loop that can send waves of Ca2+ release outward from the starting point through the entire cell, and sometimes into neighboring cells in the same tissue. The resulting spatial and temporal profile of the Ca2+ signal can thus be quite complex, and is dictated by the specific cell type’s expression pattern of Ca2+ release molecules [1]. These signals are decoded by the intracellular biochemical machinery to regulate biological effects such as contraction, secretion, cell proliferation, and induction of synaptic plasticity, to name a few. For a more complete review of Ca2+ signaling see references [1 and 2].
Many of the second messenger effects of Ca2+ are mediated through the ubiquitous Ca2+ sensing protein, calmodulin (CaM, Figure 1). CaM is a 17 kDa Ca2+-binding molecule that has been highly conserved throughout biological evolution. It is composed of an N- and C-terminal lobe tethered by a highly flexible helical linker region that allows CaM to adopt a variety of conformations when bound to different targets [3]. Each lobe of CaM contains a pair of EF-hand motifs allowing it to bind four Ca2+ ions, and saturation of CaM with Ca2+induces a conformational change that permits the protein to interact with and activate a surprisingly diverse set of target enzymes. As such, CaM has no enzymatic activity, and its function is to integrate the Ca2+signal and transduce it to other downstream enzymes. With this in mind, it is intriguing to note that, by itself, CaM’s Ca2+ binding kinetics (Kd of ~10 μM) would make it a relatively poor Ca2+ sensor in a cellular context where global Ca2+ concentrations tend to peak around 1 μM [4]. Of course this would not hold true in Ca2+ microdomains where the concentration is expected to be much higher (up to 100 μM), but it does beg the question of why such an integral Ca2+-signal transduction molecule has a relatively low affinity for the ion? The answer to this question likely lies in the difference between CaM’s affinity for Ca2+ by itself versus its affinity for Ca2+ in the presence of target enzyme. Research by several labs over the last decade has illustrated thermodynamic coupling between CaM, Ca2+, and its downstream targets [5–12]. The increases in CaM’s Ca2+-binding affinity can be substantial, suggesting that, in a cellular context, the constellation of different targets tunes CaM’s Ca2+-binding affinity accordingly. Of the many downstream targets of CaM, a family of enzymes known as the calmodulin-dependent kinases (CaM-kinases) is one of the best characterized and will be the focus of this review. CaM-kinases are critically important for proper cellular function as their range of influence spans processes as diverse as gene transcription, cell survival/cell death (apoptosis), cytoskeletal reorganization, and learning and memory. In addition to reviewing the structure of these kinases and how this structure dictates their enzymatic activity and regulation, we will also touch on the cellular processes regulated by the distinct members of the CaM-kinase family.
Figure 1.
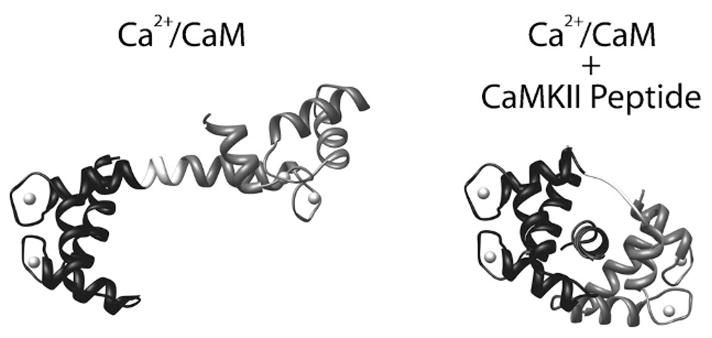
Structure of CaM and its interaction with a CaMKII binding peptide. (Left) Ribbon diagram of CaM in the Ca2+-saturated state (Ca2+/CaM) depicting its extended “barbell” shape (PDB# 1CLL). The N-terminal lobe (residues 1–75) is shown in black, and the C-terminal lobe (residues 81–148) is shown in grey. Between the two lobes is the flexible linker region (residues 76–80) which is shown in white. Calcium ions bound by the two pairs of EF-hands are shown as spheres. (Right) Ribbon diagram of Ca2+/CaM bound to a peptide corresponding to the CaM binding domain of CaMKII (PDB# 1CM1). N- and C-terminal lobes are shown in black and gray respectively, and the linker region is white. Note that the linker region has uncoiled from the helical conformation seen in the structure on the left, allowing Ca2+/CaM to wrap around the peptide when bound.
Kinase Structure and the Phosphotransferase Reaction
The term kinase refers to a large category of enzymes that catalyze the transfer of phosphate from the gamma position of ATP to the hydroxyl group of Ser, Thr, or Tyr within protein substrates. The first direct evidence of kinase activity was observed in 1954 by Burnett and Kennedy, as the phosphorylation of the protein casein by a partially purified protein sample [13]. Since then, the importance of protein kinases in the regulation of protein function has become an integral part of our understanding of biology (for a historical review on the subject see references [14] and [15]). In 1991, Knighton et al. solved the crystal structure of the catalytic domain of the cAMP-dependent protein kinase (PKA) [16]. This was the first protein kinase structure solved, and provided the earliest glimpse into the mechanics of kinase catalytic function. Over the next few years the X-Ray crystallographic structures of cyclin-dependent kinase 2 (cdk2) and the extracellular-regulated kinase 2 (ERK2) were solved [17,18], and it became evident that all protein kinases likely shared a common structural motif [19]. To this day the most well studied protein kinase is PKA, and it thus serves as a model for kinase structure and function.
According to sequence comparisons of various protein kinases they are known to share a conserved region of ~250 amino acids that are required for activity (Figure 2). Among these amino acids there are several homologous regions and highly conserved residues. This conserved region is responsible for a structural core that is present in all known kinase molecules; a bi-lobed catalytic core comprised of a small ATP binding domain, made primarily of β-strands, (orange domains in Figure 2) and a larger substrate binding domain, made primarily of α-helices, (green domains in Figure 2) connected by a small linker region [20]. This linker region acts as a hinge that allows the two globular domains of the catalytic region to open and close through the different stages of the catalytic cycle [21]. On top of ATP and substrate, the phosphotransferase reaction catalyzed by kinases requires a divalent metal ion (typically Mg2+). This ion functions to both facilitate phosphoryl transfer as well as stabilize ATP binding [20].
Figure 2.

Conservation of bi-lobed structure and functional regions within the catalytic domains of PKA, CaMKI and CaMKII. (A) Depicted is a surface rendering of crystal structures for PKA (PDB# 1J3H), CaMKI (PDB# 1A06) and CaMKII (PDB# 2BDW) catalytic domains. Conserved functional regions are colored orange (ATP-binding lobe), green (substrate-binding lobe) and yellow (glycine-rich loop). The autoinhibitory domains of CaMKI and CaMKII are colored red. The white regions on PKA are N- and C-terminal residues not conserved in the CaM-kinases. Note the conserved bi-lobed structure with a catalytic cleft at the interface between the lobes. (B) Multi-sequence alignment of amino acids spanning the catalytic domains of PKA and all the CaM-kinases (except CaMKIII due to its lack of homology). Colored bars above the sequences correspond to the amino acids which make up the functional regions of the structures above displayed in the same color. The residues C-terminal to the catalytic domains of CaMKI and CaMKII are shown in order to include their autoinhibitory domains. Histograms below the aligned residues show the high level of conserved amino acids in the catalytic domains of all the CaM-kinases with respect to each other and PKA. Amino acid positions marked by the asterisks (*) and the arrowhead correspond to the four amino acids in CaMKI involved in substrate binding. The amino acid marked by the arrowhead is of interest as it is conserved within the multifunctional but not the substrate-specific CaM-kinases. Sequences were aligned using ClustalX (http://bips.u-strasbg.fr/fr/Documentation/ClustalX/).
Nestled between the lobes described above is the catalytic cleft where the phosphotransferase reaction takes place. Surrounding this site is the glycine-rich loop which helps to bind ATP [22,23], and three highly conserved amino acid residues that play important roles in facilitating the transfer of the γ phosphate from ATP to the hydroxyl group of the phosphorylation site on protein substrates (Figure 3). Asp184 in PKA is a strictly conserved residue in all kinases that interacts with the critical magnesium (Mg2+) ion. This Mg2+ ion chelates both the β and γ phosphate of ATP and, in doing so, is thought to play two essential roles in the mechanism of phosphate transfer: 1) correctly position the γ phosphate for direct transfer to the substrate and 2) masking the charge of the γ phosphate, thus reducing electrostatic repulsion on the incoming hydroxyl group of the substrate. It is not entirely clear which of the two is the most prevalent role of Asp184; however, yeast expressing a mutant form of PKA in which Asp184 is replaced with Ala are no longer viable, supporting its critical role in catalytic function [24]. Lys72 is a second conserved amino acid in the protein kinase family. It contributes to further stabilization of the phosphates through interactions with the α and β phosphate groups of ATP. The main function of Lys72 is to facilitate the transfer of phosphate from ATP to substrate, while having little effect on the kinase’s affinity for the nucleotide. Mutational studies in Lck and ERK2 [25,26], where Lys72 was replaced with Arg, show lowered catalytic function with no effect on affinity for ATP. Asp166, the third highly conserved residue in the PKA catalytic site, has been shown to form a hydrogen bond with the hydroxyl group of substrates, and due to its proximity with respect to the γ phosphate of ATP is thought to target the hydroxyl group for nucleophilic attack on the terminal phosphate [20,27]. Note that this class of kinase molecules catalyzes the direct transfer of phosphate onto the substrate protein as opposed to other kinases that are themselves a phosphorylated intermediate in the reaction pathway. Although subtle differences between the structure of PKA and other kinases have been observed, it is believed that the same basic mechanisms of phosphoryl transfer are shared by the family of CaM-dependent protein kinases reviewed in this chapter.
Figure 3.
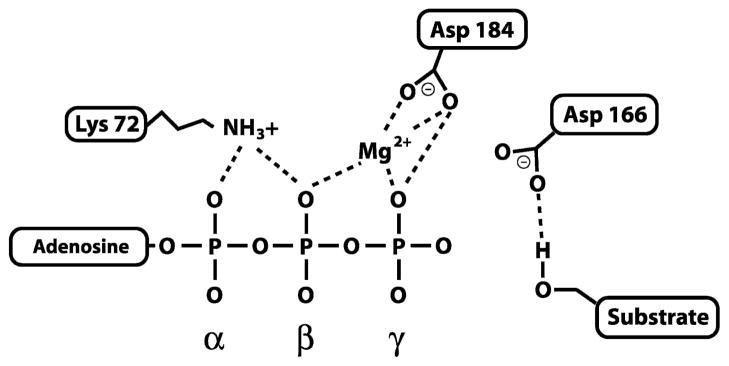
Diagram of amino acids involved in mediating the phosphotransferase reaction in PKA. The three amino acids depicted are those found in PKA, but they are highly conserved in all protein kinases. Lys72 and Asp184 facilitate phosphoryl transfer and stabilize interactions with the nucleotide and the essential Mg2+ ion, while Asp166 helps position the hydroxyl group of the protein substrate for nucleophilic attack on the γ phosphate of ATP. Dashed lines represent interactions between atoms in the conserved residues with ATP, Mg2+ and protein substrate.
In order for the phosphotransferase reaction to proceed efficiently, the phospho-accepting substrate must also be bound to the catalytic domain in the proper orientation. Along with Asp166 described above, there are four other amino acids in the substrate-binding lobe thought to be important for substrate binding (Glu127, Phe129, Glu230 and Pro236). In Figure 2 their positions within the sequence are labeled with asterisks and an arrowhead. From the crystal structure of CaMKI, Glu102, Phe104, Ile210 and Pro216 were found to be structurally analogous to these amino acids in PKA (Panel B, Figure 2) [28]. Glu102 in CaMKI interacts electrostatically with basic residues in the substrate recognition sequence while Ile210, Pro216 and Phe104 form a hydrophobic pocket that recognizes hydrophobic residues in protein substrates. There is a high degree of conservation of these amino acids through the CaM-kinase family (see Figure 2).
General Characteristics of Ca2+/Calmodulin-activated Protein Kinases (CaM-kinases)
Members of the CaM-kinase family are all classified as Ser/Thr kinases whose substrate P-sites (the targeted site of phosphorylation) all contain either a Ser or a Thr. As their name implies, activation is initially dependent on the binding of Ca2+/CaM but, as will become apparent, some of them are capable of becoming independent of Ca2+/CaM following activation, or require additional modification (i.e. phosphorylation) by other regulators in order to achieve full activation. These differences in regulation are what allow a relatively small family of proteins to display such elegant control over so many disparate cellular functions. The overall domain structure of the CaM-kinases is very similar to that described for PKA (Figure 2 and 4), where a bi-lobed catalytic domain is followed by a regulatory domain containing both an autoinhibitory domain as well as a CaM-binding domain. The autoinhibitory and CaM-binding domains overlap slightly and binding of Ca2+/CaM regulates functioning of the autoinhibitory domain. At basal Ca2+ levels, CaM-kinases are maintained in a dormant state through an autoinhibitory mechanism involving the regulatory domain just downstream of the catalytic domain. For autoinhibition to occur the autoinhibitory domain must interact in a way that blocks the binding of substrate, or distorts the catalytic site in order to render it nonfunctional. Current data suggest that both of these strategies, and sometimes a combination of both, are used by CaM-kinases. Regardless of the mechanism of autoinhibition, as intracellular Ca2+ concentrations rise (through the opening of receptor/ion channels or release from intracellular stores) CaM becomes saturated with four Ca2+ ions and undergoes a conformational change allowing it to bind to its target site on CaM-kinases.
Figure 4.
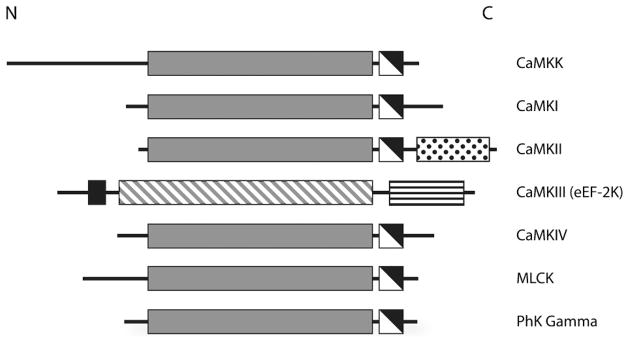
General domain structure of the CaM-kinases. The conserved CaM-kinase domains are shown in gray (catalytic domains), white (autoinhibitory domains) and black (CaM-binding domains). The catalytic domain of CaMKIII is diagonally hatched, because it bears little resemblance to the other CaM-kinase catalytic domains. The C-terminal association domain of CaMKII and the eEF-2 recognition domain in CaMKIII are shown as white with black dots and white with horizontal black lines, respectively. Note the general domain organization with an N-terminal catalytic domain followed by a regulatory region that contains overlapping autoinhibitory and CaM-binding domains.
This binding relieves autoinhibition by disrupting the interactions between the autoinhibitory and the catalytic domains. Once a CaM-kinase becomes activated, ATP and the protein substrate must be bound simultaneously to a competent catalytic site for phosphoryl transfer to occur.
The crystal structures of the catalytic domains of two CaM-kinases (CaMKI and CaMKII) have been solved, and available data support the idea that all CaM-kinases share similar catalytic domain structures (Figure 2). It is also apparent that their architecture is comparable to the bi-lobed structure of PKA, showing two globular domains (one that binds ATP and another that binds substrate) connected by a linker to form a catalytic cleft (Figure 2).
When describing the family of CaM-kinases (Figure 4) it is helpful to break them down in to two separate classes. There are multifunctional CaM-kinases (CaMKK, CaMKI, CaMKII and CaMKIV) which each have multiple downstream targets and substrate-specific CaM-kinases (CaMKIII, phosphorylase kinase, and the myosin light chain kinases) that have only one known downstream target. The activation of multifunctional kinases can lead to signaling that affects many downstream pathways controlling a variety of cellular functions, while substrate-specific kinases tend to serve a dedicated function within the cell or tissue where they are expressed. It is not known exactly how the substrate-specific CaM-kinases exhibit such high specificity with respect to the multi-functional kinases, as we are lacking structural information for a CaM-kinase with a substrate bound to its catalytic domain, but several factors are likely to play a role. Among CaM-kinase substrates there is a generalized consensus sequence (Arg-X-X-Ser/Thr) that is used to predict potential phosphorylation sites within proteins; however, it is only a general rule and differences within specific substrates do exist (for review see references [29,30]). In addition to differences in substrate recognition sequences, conformational distinctions between various substrates is also known to play a role in substrate recognition [29]. It is also interesting to note that Ile210 (one of the substrate binding residues described above) is conserved through the multifunctional, but not the substrate-specific CaM-kinases (Figure 2). Perhaps this difference is involved in conveying substrate specificity to these enzymes. Some CaM-kinases are themselves substrates for other CaM-kinases or even themselves (an event known as autophosphorylation). It is obvious that this family of enzymes has evolved to fill an important role in integrating intracellular Ca2+ signals and a closer look at each member of the family will provide an even clearer picture of just how well suited each member is for serving their individual cellular roles. A summary of various kinases, their structural features, mechanisms of activation, and physiological roles can be found in Table 1.
Table 1.
General Characteristics of the CaM-dependent Protein Kinases.
| Kinase | Type | Subunit Composition | Mechanism of Activation | Targets | Physiological Role |
|---|---|---|---|---|---|
| CaMKK | Multi-functional | Monomer | Ca2+/CaM | CaMKI, CaMKIV | Gene Transcription, Apoptosis |
| CaMKI | Multi-functional | Monomer | Ca2+/CaM and CaMKK | Synapsin 1, CREB | Gene Transcription, Vesicle Mobilization |
| CaMKII | Multi-functional | Dodecamer | Ca2+/CaM Autophosphorylation | CaMKII, AMPA/NMDA receptors, L-type Ca2+ channels | Synaptic Plasticity, Regulation of Ion Channels, Gene Transcription |
| CaMKIV | Multi-functional | Monomer | Ca2+/CaM,CaMKK and Autophosphorylation | CaMKIV, CREB, CBP, SRF, HDAC4, Oncoprotein 18 | Gene Transcription |
| CaMKIII | Substrate Specific | Monomer | Ca2+/CaM | Elongation Factor 2 | Facilitate Protein Translation |
| MLCK | Substrate Specific | Monomer | Ca2+/CaM | RLC of Myosin | Muscle Contraction, Intracellular Transport |
| Phosphorylase Kinase | Substrate Specific | Tetramer of Tetramers | Ca2+/CaM PKA | Glycogen Phosphorylase | Glycogen Metabolism |
Multifunctional CaM-Kinases
CaM-kinase kinase (CaMKK)
As mentioned above, some members of the CaM-kinase family are substrates for other kinases that are themselves dependent on Ca2+/CaM. This cascade was first discovered in 1994 when both Lee & Edelmen and Tokumitsu et al. independently purified a Ca2+/CaM-dependent protein activator of CaMKI and CaMKIV, respectively [31,32]. It was soon discovered that these activators were the same molecule, now known as calmodulin-dependent protein kinase kinase (CaMKK), and it is the most upstream element of a CaM-kinase cascade involving the three mentioned CaM-kinases [33]. The exact benefit of having independent enzymes in a signaling cascade activated by the same allosteric regulator (Ca2+/CaM) is still not entirely clear, but the CaM-kinase cascade is not the only example of such a system. One potential benefit of such an arrangement is a kinetic proof-reading effect, whereby the diffusion limited steps of the coupled reactions ensure that the downstream enzymes are not fully activated unless the duration of the Ca2+ signal persists long enough for all the components to bind Ca2+/CaM and then find one another. This would make sense seeing as how the enzymes of the CaM-kinase cascade regulate gene transcription events which induce long-term biological changes that should only be set in motion by an error-proofed mechanism.
There are two isoforms of CaMKK, a 505 amino acid α isoform and a slightly larger β isoform composed of 587 amino acids. Each is expressed from separate genes, and they are both expressed in cells as a monomer. CaMKK is most highly expressed in brain tissue, but the α isoform is also expressed in the thymus and spleen [34]. In the cell, CaMKK resides in the cytosol and the nucleus where it is poised to respond to both cytosolic and nuclear changes in Ca2+ concentration and act on its two known substrates in the CaM-kinase cascade. The domain structure of both CaMKK isoforms is similar to other CaM-kinases with a catalytic, autoinhibitory, and CaM-binding domain extending through the sequence from the N- to C-terminus (Figure 5) [35]. While the crystal structure of CaMKK is yet to be solved, it would presumably bear a close resemblance to other CaM-kinases that possess features very similar to those of PKA (Figure 2). Like all CaM-dependent kinases, CaMKK is held in an inactive state by its autoinhibitory domain, which interacts with the catalytic domain to prevent kinase activity. Binding of Ca2+/CaM relieves this inhibition by displacing the autoinhibitory domain and exposing a competent catalytic site. This leads to the activation of CaMKK and the subsequent phosphorylation of its downstream substrates CaMKI and CaMKIV, an event which is necessary, but not sufficient, for their activation.
Figure 5.
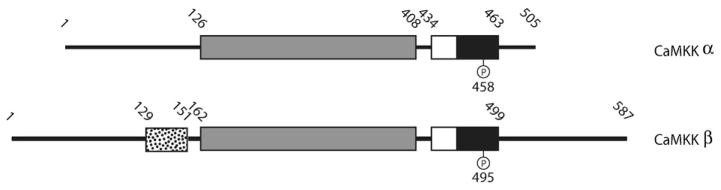
Domain structure of CaMKK isoforms. The conserved CaM-kinase domains are shown in gray (catalytic domains), white (autoinhibitory domains) and black (CaM-binding domains). The 23 amino acid stretch (residues 129–151) involved in regulating autonomous activity in the β isoform is represented as white with black dots. The inhibitory phosphorylation site in the α isoform (Ser458), which is phosphorylated by PKA, can be seen in the CaM-binding domain as well as the analogous site, based on sequence similarity between the isoforms, in the β isoform (Ser495). Numbers above the diagrams represent residue numbers at experimentally defined boundaries of various domains.
CaMKK is itself a substrate for phosphorylation. Activated PKA phosphorylates CaMKK at Ser458, a residue within the kinase’s CaM-binding domain, which prevents CaM binding and thus blocks the ability of CaMKK to respond to increased Ca2+ [36]. Because of the location for PKA phosphorylation on CaMKK and its ability to block Ca2+/CaM binding, the temporal sequence of these two distinct signaling events plays an important role in directing signaling through CaMKK. For example, if cAMP levels are raised by forskolin stimulation (an activator of adenylyl cyclases that makes cAMP and activates PKA) prior to an increase in Ca2+ then PKA phosphorylation of CaMKK inhibits its activation by Ca2+/CaM, thus blocking full activation of CaMKI or CaMKIV [36]. However, if the Ca2+ increase occurs before forskolin stimulation, CaM binds to CaMKK, blocking Ser458 within the CaM-binding domain from being phosphorylated and thus preventing PKA-mediated inhibition of the CaMKK pathway [35]. Because CaMKK is the first enzyme in the CaM-kinase cascade its phosphorylation by PKA not only inhibits its activation, but it also suppresses signaling through its substrates CaMKI and CaMKIV as well. Recently it was discovered that CaMKK also mediates cell death as a substrate for the death associated protein kinase (DAPK), thus extending its known cellular role [37].
The catalytic domain of CaMKK contains two distinct features not seen in the other CaM-kinases. The first is a lack of conserved acidic residues that normally recognize basic residues in protein substrates, and the second is an Arg-Pro-rich insert that is critical for recognition of CaMKI and CaMKIV as substrates [38]. Deletion of this insert from the catalytic domain of CaMKK occludes its ability to phosphorylate CaMKI and CaMKIV, but not the low affinity substrate protein kinase B (PKB) [39]. The α and β isoforms of CaMKK show unique differences in their mechanisms of regulation despite sharing the same family of substrates. The β isoform was discovered to exhibit Ca2+/CaM-independent activity following activation by Ca2+/CaM, and mutational studies revealed that a 23 amino acid sequence (residues 129–151 in CaMKKβ) participates in the release of its autoinhibitory domain from the catalytic core, resulting in autonomous activity [40]. This difference in regulation between the two isoforms of CaMKK means those cell types or compartments that contain the β isoform will sustain CaMKK activity beyond the lifetime of the Ca2+ signal that initiated its activation.
CaM-kinase I (CaMKI)
As noted earlier, CaMKI is one of two downstream targets in the CaM-kinase cascade, and therefore its activation requires both binding by Ca2+/CaM as well as subsequent phosphorylation by CaMKK. It is a monomeric kinase of approximately 42 kDa that is expressed from three genes encoding α, β, and γ isoforms, that are broadly distributed throughout most mammalian cells [38]. CaMKI is a cytosolic protein and presumably serves a diverse set of functions despite the fact that little is known about the extent of its protein substrates. To date its best characterized substrates are synapsin 1, a presynaptic vesicle-associated protein that tethers neurotransmitter-filled vesicles to the presynaptic actin cytoskeleton, and the cAMP response element binding protein (CREB), a protein intimately linked to regulation of gene transcription [41,42]. Upon phosphorylation of synapsin 1 by CaMKI its affinity for the vesicle is decreased, thus increasing the mobility of the vesicle within the presynaptic terminal. Phosphorylation of CREB facilitates binding of the transcription factor to the cAMP response element (CRE) in CREB regulated genes and initiates formation of transcriptional machinery.
CaMKIα is composed of 374 amino acids and, as one would expect, it displays the prototypical CaM-kinase domain structure (Figure 6). Amino acids 10–275 make up the catalytic domain which is composed of the ATP- and substrate-binding domains, and amino acids 276–316 contain the regulatory domain which includes the CaM-binding and the autoinhibitory domain. During activation, Ca2+/CaM binds to a region adjacent to the autoinhibitory domain and in doing so removes the autoinhibitory domain from the catalytic site, allowing the binding of both ATP and protein substrate. Binding of Ca2+/CaM is also necessary to expose Thr177 in the activation loop of CaMKI that serves as the substrate site for CaMKK phosphorylation. When CaMKI is phosphorylated on Thr177, its activity rises by ten- to twenty-fold, but unlike CaMKK, CaMKI remains entirely Ca2+/calmodulin-dependent. Therefore, as the Ca2+ level returns to the basal state, the activity of CaMKI will decrease regardless of the phosphorylation state of the regulatory amino acid Thr177.
Figure 6.
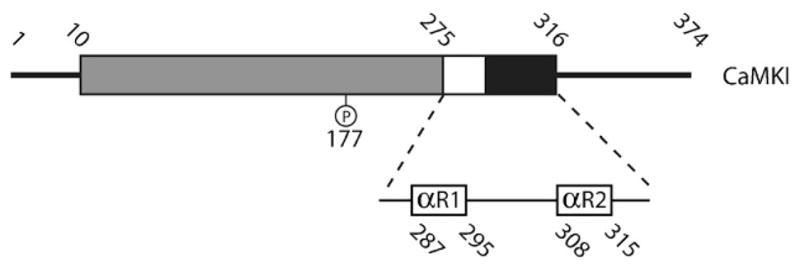
Detailed domain structure of CaMKI. The conserved CaM-kinase domains are shown in gray (catalytic domain), white (autoinhibitory domain) and black (CaM-binding domain). Thr177, the regulatory site phosphorylated by CaMKK, is shown in the catalytic domain. The regulatory region of CaMKI, which contains the autoinhibitory domain and CaM-binding domain, is blown up to show the amino acid sequences that correspond to the regulatory helices within the regulatory domain that are shown in 3-D in Figure 6. Numbers above the diagram represents residue numbers at experimentally defined boundaries of various domains.
In 1996, CaMKI became the first member of the CaM-kinase family to have its chemical structure solved via X-ray diffraction (see Figure 2). Because the kinase was in an inactive state the structure allowed for a detailed understanding of how interactions between the autoinhibitory and the catalytic domains lead to suppression of kinase activity [28]. What was immediately clear was that the bi-lobed structure of the catalytic domain of PKA was conserved for CaMKI, and that the intersection between these two lobes acts as the catalytic cleft. In a bit of a surprise, the mechanism of autoinhibition turned out to be more complex then originally expected. Autoinhibition was thought to occur through a pseudosubstrate mechanism where the autoinhibitory domain mimics the natural protein substrates, thus blocking access to the catalytic site through nothing more than steric hindrance. However, this is not entirely the case. The autoinhibitory domain of CaMKI is composed of two α-helices (αR1 and αR2) connected by a loop, and αR1 and αR2 make contact with hydrophobic amino acid residues in the substrate- and ATP-binding lobes respectively (Figure 7). While αR1 does appear to act as a pseudosubstrate to block interactions with target proteins, it is clear that helix αR2 interacts with the ATP-binding site, altering its conformation so as to interfere with its ability to bind nucleotide [28]. Based on the 3-D structure, it is hypothesized that early interaction of Ca2+/CaM with αR2 disrupts the regulatory helix’s contact with the upper catalytic lobe, and allows the lobe to assume a new conformation readily capable of binding ATP. Next, an ensuing change in αR1 conformation, or perhaps interaction between Ca2+/CaM and αR1, leads to removal of the pseudosubstrate block on the lower protein substrate-binding lobe. Once the catalytic domain of CaMKI is relieved of inhibition by both regulatory helices it is free to bind both ATP and target protein, and to engage in the phosphotransferase reaction. As mentioned above, phosphorylation of Thr177 leads to a 10–20 fold increase in CaMKI activity. In the crystal structure, the activation loop (containing Thr177) was not resolved, suggesting that in the inactive state the residues forming the loop are mobile and unstructured. This mobility most likely contributes to the kinase’s lower activity prior to phosphorylation at Thr177, and phosphorylation at this site by CaMKK presumably stabilizes the activation loop, providing a more competent catalytic site. This seems reasonable based on the crystal structure of a phosphorylated form of PKA, where the phosphorylation of an analogous residue in the activation loop stabilizes interactions with residues near the catalytic site [16].
Figure 7.

Interactions of the CaMKI regulatory domain with the catalytic core. Depicted above is a surface rendering of CaMKI with a ribbon diagram of the regulatory domain lying across the face of the molecule. The ATP-binding lobe of the catalytic core is colored dark gray and the substrate-binding domain is light gray. The regulatory domain, which contains both the autoinhibitory and CaM-binding domains, is black. The structure of the regulatory domain is composed of two regulatory helices (αR1 and αR2) connected by a loop. Regulatory helix αR1 makes contacts with the substrate binding domain, blocking interactions with substrate, and αR2 inhibits ATP binding through interactions with residues in the ATP-binding domain.
CaM-kinase IV (CaMKIV)
CaMKIV is the third enzyme in the CaM-kinase cascade, and there are two isoforms (α and β) that are both expressed as a monomer of approximately 65 kDa from a single gene (Figure 8). CaMKIVα is found primarily in neuronal tissue, T-cells, and testis, while the β splice variant, which contains 28 additional N-terminal residues, is expressed differentially throughout development in cerebellar granule cells [43,44]. Like CaMKI, in order to become active, CaMKIV must undergo a multi-step process where first Ca2+/CaM binds, then CaMKK phosphorylates the kinase in its activation loop on Thr196. Unlike CaMKI, however, once phosphorylated by CaMKK, CaMKIV may undergo an intrasubunit autophosphorylation of its Ser/Thr-rich N-terminus. This autophosphorylation event not only maximally activates CaMKIV, but it also generates Ca2+/CaM-independent activity that allows the kinase to retain functionality beyond the duration of a transient elevation in Ca2+. An additional level of self-regulation occurs when CaMKIV autophosphorylates Ser332 in its CaM-binding domain. This prevents subsequent binding by Ca2+/CaM until Ser332-P is dephosphorylated by an available phosphatase. The protein phosphatase PP2a has been shown to stably associate with CaMKIV to form a signaling complex that allows for tightly controlled phosphorylation/dephosphorylation kinetics of CaMKIV’s activation loop [45].
Figure 8.
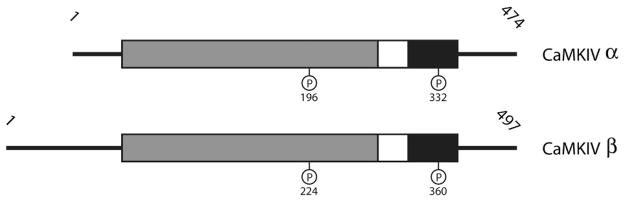
Domain structure of CaMKIV isoforms. The conserved CaM-kinase domains are shown in gray (catalytic domains), white (autoinhibitory domains) and black (CaM-binding domains). Thr196, the regulatory site phosphorylated by CaMKK in the α isoform, is shown in the catalytic domain, and the site of autophosphorylation that blocks CaM-binding (Ser332) is shown in the CaM-binding domain. Based on the fact that the β isoform only differs by 28 additional N-terminal residues Thr224 and Ser360 represent phosphorylation sites analogous to those in CaMKKα. Numbers above the diagrams represent the length of each isoform in amino acid residues.
CaMKIV can be found in both the cytosol and the nucleus, where it is poised to regulate Ca2+-dependent gene transcription via phosphorylation of several transcription factors like CREB, the serum response factor (SRF), and histone deacetylase 4 (HDAC4) [46–48]. The most well characterized nuclear event regulated by CaMKIV is the phosphorylation of CREB at Ser133, a site well known for activating the transcriptional properties of CREB. Activation of gene transcription by CREB can also be initiated by CaMKIV through the phosphorylation of the CREB binding protein (CBP), a co-activator of CREB that facilitates stabilizing interactions with the general transcription machinery [49]. Recent evidence suggests that initiation of gene transcription by CaMKIV requires that it achieve an autonomous (Ca2+-independent) state [50]. As mentioned above CaMKIV can form a stable signaling complex with PP2a, and this interaction has now been shown to negatively regulate CaMKIV’s ability to activate gene transcription by actively dephosphorylating the kinase and preventing autonomous activity [51]. In the cytosol, CaMKIV’s role is not as well understood, but it does phosphorylate oncoprotein 18 at Ser16 which prevents its association with tubulin [52].
Not surprisingly, the domain structure of CaMKIV is nearly the same as for all other CaM-kinases with an N-terminal catalytic domain just upstream of the autoinhibitory domain and CaM-binding domain (Figure 8). To date, the structure of CaMKIV has still not been solved, but due to its similarity to CaMKI, both in its domain structure and its role as a substrate for CaMKK, it is likely to share a very similar 3-D structure as well.
CaM-kinase II (CaMKII)
CaMKII is not a component of the traditional three-part CaM-kinase cascade involving the molecules described above, but it plays an equally integral role in translating intracellular Ca2+ signals to a variety of downstream targets. In fact, because of its unique macromolecular structure and its critical role in regulating processes such as synaptic plasticity and spatial memory [53], CaMKII is one of the most well studied members of the CaM-kinase family. There are four isoforms of CaMKII (α, β, γ and δ) in mammals and each is expressed from a separate gene [54]. Because of slight differences in the isoforms’ primary structure as well as alternative splicing, the four proteins can range in size from 50–68 kDa. The α and β isoforms are brain specific and together make up approximately 1% of total brain protein and up to 2% of total protein in the hippocampus (a brain structure well established in processing and encoding new memories; [55]). The γ and δ isoforms are expressed in much lower concentration throughout the rest of the body [56].
One striking difference between CaMKII and the other multifunctional CaM-kinases is that it is the only one that does not exist as a monomer in cells. All isoforms of CaMKII share the same domain structure, which contains an N-terminal portion similar to other CaM-kinases plus a C-terminal association domain (Figure 9) that is required for assembly of the CaMKII holoenzyme into a twelve subunit complex [54]. This dodecameric structure displays a very unique macro-molecular architecture. In 2000, the structure of the active form of αCaMKII was solved by our lab to approximately 25Å resolution using single particle reconstruction from cryo-electron micrographs [57]. The structure revealed a distinctive multi-subunit assembly in which the association domains of each subunit combine to form a barrel-shaped core with six catalytic domains extending radially from both ends of the core (Figure 10A). Subsequently, using the same technique, the structures of the β, γ and δ isoforms were solved and, despite some minor differences, they share the same overall macromolecular architecture at the resolution obtained [58]. In 2003, the crystal structure of the truncated association domain of CaMKII was solved [59], and it surprisingly revealed a tetradecameric (14–mer) assembly. Because there is no evidence for a tetradecameric assembly in the holoenzyme, it is likely due to the absence of the N-terminal catalytic domain or perhaps the bacterial expression system used to make the protein for crystallization in these experiments [60]. However, it is worth noting that a similar fragment of the association domain of the γ isoform of CaMKII expressed in bacteria was also crystallized (see PDB file 2UX0), and this structure was a dodecamer [61]. While the structure and function of purified holoenzymes composed of individual subunits is often studied, it is known that inside of cells the subunits can mix to form heteromeric CaMKII holoenzymes composed of varying ratios of the different isoforms [62–64]. Subunit mixing adds significantly to the complexity of CaMKII’s potential structural configurations as well as to the diversity of its biological function. For example, βCaMKII contains an actin binding site whose affinity for actin is negatively regulated in a Ca2+-dependent manner [65–68]. Therefore, a CaMKII holoenzyme composed of both α and β subunits will be targeted to the actin cytoskeleton at basal Ca2+ concentrations, thus potentially restricting its ability to be activated and restricting the phosphorylation of substrates by limiting diffusion. As intracellular Ca2+ concentrations rise, the β subunit containing holoenzymes will leave the cytoskeletal fraction and enter the cytosol, where they are free to phosphorylate other protein substrates. Due to the structural arrangement of its subunits, CaMKII exhibits an intriguingly complex form of regulation by Ca2+/CaM (Figure 10B). Like other CaM-kinases, CaMKII initially exists in a quiescent state as its autoinhibitory domain blocks catalytic activity. Upon elevation of intracellular Ca2+, Ca2+-saturated CaM can bind to and fully activate each subunit of the CaMKII holoenzyme. Once Ca2+/CaM-bound, one subunit can then be autophosphorylated at Thr286 (in αCaMKII; Thr287 in the other isoforms) by a neighboring activated CaMKII subunit, generating Ca2+-independent activity [69]. This set of reactions is conceptually very similar to those in the CaM-kinase cascade involving CaMKK and CaMKI or CaMKIV. In either case, it is imperative that both the kinase catalyzing the phosphotransferase reaction as well as the kinase acting as the substrate be bound to Ca2+/CaM. In the case of CaMKII, however, the assembly of catalytic subunits into the holoenzyme greatly increases the probability of phosphorylation occurring, whereas in the CaM-kinase cascade the rate of activation by phosphorylation is dependent upon several diffusion-limited reaction steps. Elegant muta-genesis and biochemical studies showed that this autophosphorylation is an intra-holoenzyme reaction occurring between two adjacent, Ca2+/CaM-bound CaMKII subunits and that it does not occur between activated subunits in separate holoenzymes [70,71]. As mentioned above, phosphorylation at Thr286 generates Ca2+/CaM-independent or autonomous activity. In addition, autophosphorylation of Thr286 also increases the affinity of CaMKII for Ca2+/CaM by more than 1000-fold in an event known as “CaM-trapping” [72]. Together, CaM-trapping and autonomous activity allow the kinase to remain activated beyond the timeframe of the Ca2+ transient that initially switched on the molecule. Because of this property, CaMKII is often referred to as a “memory molecule,” [73–75] and autophosphorylation of Thr286 in CaMKII is necessary for certain forms of synaptic plasticity and certain forms of learning and memory [53]. Adding to the complexity of CaMKII regulation, Thr286 is not the only site of autophosphorylation. Two neighboring residues in the CaM-binding domain (Thr305 and Thr306) are also targets for autophosphorylation, and phosphorylation at these sites prevents binding of Ca2+/CaM [76]. This regulatory mechanism is analogous to the phosphorylation of Ser332 in the CaM-binding domain of CaMKIV, and is referred to as inhibitory autophosphorylation. Although inhibitory autophosphorylation blocks Ca2+/CaM binding, it does not block autonomous activity if Thr286 is phosphorylated prior to Thr305/306 [77], making the temporal order of each phosphorylation event critical in regulating CaMKII activity. Autophosphorylation of Thr305/306 does however inhibit further autophosphorylation as Ca2+/CaM must be bound to the catalytic subunit in order for it to act as either a kinase toward other subunits or as a substrate for autophosphorylation. Experiments using knock-in mice expressing mutants of CaMKII either incapable of phosphorylating Thr305/306 or mimicking persistent phosphorylation at this site have illustrated its importance in regulating synaptic plasticity by altering the kinase’s ability to incorporate into the postsynaptic density [78]. The postsynaptic density is a proteindense organelle lining the intracellular side of the postsynaptic membrane that organizes synaptic signal transduction molecules [79,80]. These results highlight the importance of subcellular targeting of CaMKII to properly serve its functions, and that targeting can be impacted by autophosphorylation of the kinase.
Figure 9.

Domain structure of CaMKII isoforms. The conserved CaM-kinase domains are shown in gray (catalytic domains), white (autoinhibitory domains) and black (CaM-binding domains), and the C-terminal association domain is shown as white with black dots. The site of autophosphorylation (Thr286 in the α isoform and Thr287 in the β, γ and δ isoforms) that generates CaM-trapping and autonomous activity is shown in the autoinhibitory domains. The other sites of autophosphorylation (Thr305/306 in the α isoform and Thr306/307 in the β, γ and δ isoforms) are shown in the CaM-binding domain. The aligned amino acid sequences from the region between the regulatory and association domain of each isoform is shown in the blow-up beneath the diagrams. This is the region of the isoforms that shows the highest amount of variability due to alternative splicing. The stretch of amino acids (residues 354–392) highlighted in the variable region of CaMKIIβ plays a role in targeting holoenzymes containing this isoform and likely is responsible for actin binding. Numbers above the diagrams represent the length of each isoform in amino acid residues.
Figure 10.

Structure and regulation of CaMKII. (A) Side and top view of CaMKII holoenzyme solved by single particle reconstruction from cryo-electron micrographs. The image is composed of a surface rendering of the entire holoenzyme (red mesh) superimposed on the core formed by the truncated C-terminal association domains (yellow). The catalytic domains can be seen as feet attached to the core via a stalk. Note the two-fold symmetry seen in the side view and the six-fold symmetry visible in the top view. (B) Diagram depicting the sequence of events leading to the activation and autophosphorylation of CaMKII. 1) CaMKII initially resides in an inactive state due to autoinhibition. 2) As Ca2+ levels rise CaM becomes saturated with the ion and binds individual subunits of the holoenzyme, thus inducing Ca2+/CaM-dependent activity. 3) If adjacent subunits within a holoenzyme are activated by Ca2+/CaM then autophosphorylation at Thr286 may occur between them, which leads to CaM-Trapping. 4) When Ca2+/CaM dissociates from CaMKII the subunits phosphorylated at Thr286 remain autonomously active beyond the duration of the Ca2+ signal that caused its activation.
The induction of autonomous activity and CaM-trapping by autophosphorylation of Thr286 coupled with CaMKII’s ability to detect changes in the frequency of Ca2+ oscillations [81] has led to a theory of how CaMKII can act as a trigger for synaptic plasticity in neurons. Because autophosphorylation of Thr286 requires that two adjacent subunits be bound by Ca2+/CaM simultaneously, it is unlikely that this configuration will occur during a brief Ca2+ spike, but as the frequency of oscillations increases, the probability of such an occurrence increases substantially. This is due to the dramatically decreased off-rate for Ca2+/CaM when it becomes trapped by CaMKII [72,82,83]. Eventually a subpopulation of CaMKII subunits will exist in a persistently activated, CaM-trapped state, allowing kinase activity induced by the Ca2+transients to persist long after intracellular concentrations of the ion have returned to their basal state. This prolonged activity presumably allows CaMKII to continue modifying substrates near the synapse that lead to increased efficiency of synaptic transmission. One target for such regulation is the AMPA subtype of the glutamate receptor whose phosphorylation by CaMKII leads to an increased single channel conductance, thus increasing the current generated via glutamate-mediated transmission [84,85]. Another mechanism for increasing synaptic currents is to increase the number of AMPA receptors, and CaMKII mediates this process by facilitating incorporation of new AMPA receptors into the post-synaptic membrane [86].
In the heart, CaMKII functions to regulate Ca2+-dependent facilitation (CDF) of L-type Ca2+ channels by potentiating Ca2+ influx during repetitive activity. Frequency-dependent alterations in Ca2+ influx plays a role in the force-frequency relationship of cardiac contraction, and this increase in contraction strength, along with faster heart rates, drives the positive inotropic response that occurs during exercise [87–89]. In order to regulate CDF, CaMKII was discovered to tether itself to the C-terminal portion of α1C (an L-type Ca2+ channel regulatory protein) where it can phosphorylate the channel to increase Ca2+ conductance. It is thought that by tethering itself to α1C the CaMKII holoenzyme is in a prime location to act as a dedicated frequency decoder of voltage-driven Ca2+ spikes as well as a local kinase effector that regulates Ca2+ channel activity [90].
Due to the extraordinarily high concentration of CaMKII in neurons and its highly symmetric structure, many have proposed a potential structural role for CaMKII. A picture is emerging in which CaMKII acts as a scaffolding protein at postsynaptic specializations to arrange synaptic proteins into organized signaling complexes. NMDA receptors, densin-180 (a trans-synaptic protein), and α-actinin (an actin-binding protein) are among the most well characterized binding partners of CaMKII at the synapse [91,92], and because of the kinase’s six-fold symmetry it is possible that each holoenzyme could bind multiple binding partners at the same time. CaMKII is known to translocate to synapses in an activity dependent manner [90,93,94] where it can combine with any of the binding partners mentioned above. CaMKII’s interaction with NMDA receptors is mediated through the C-terminal tail of the NR2b subunit [95,96], which intriguingly leads to sustained autonomous activity once bound. The initial binding is Ca2+/CaM dependent, but once bound CaMKII’s activity remains autonomous in a Ca2+/CaM and phosphorylation-independent manner [97]. This finding is particularly interesting since CaMKII’s interaction with NR2b has been shown to regulate synaptic plasticity [98].
In 2005, the crystal structure of an inactive, auto-inhibited catalytic domain of CaMKII was solved and revealed the prototypical kinase catalytic core [99]. The bi-lobed structure contains both a small ATP-binding domain and a larger substrate binding domain (see Figure 2) with a catalytic cleft at the interface of the two. Surprisingly, the crystal structure revealed a dimeric complex with the two autoinhibitory domains forming a coiled coil which blocks access to the ATP-and substrate-binding lobe of the catalytic domains. It is hypothesized that this interaction may function to decrease the probability of random autophosphorylation in an environment (the CaMKII holoenzyme) where the concentration of catalytic domains is extremely high. However, the existence and biological significance of this interaction between potential dimer pairs of the catalytic domains is yet to be established in an intact holoenzyme. Further, crystal structures of the catalytic domains of the active but autoinhibited forms of the β, γ and δ isoforms have become available (see PDB files 3BHH, 2V7O, and 2VN9). Interestingly, in none of these structures was this coiled coil structure between the autoinhibitory domains observed [100–102]. The reasons for such discrepancies are not apparent, but clearly there are important issues that need to be resolved concerning the structure and regulation of CaMKII.
Substrate-Specific CaM-Kinases
CaM-kinase III (CaMKIII) or Elongation Factor 2 Kinase (EF2K)
In 1983, Dr. Clive Palfrey published a paper describing a Ca2+/CaM-dependent kinase that phosphorylates an identical 100 kDa substrate in a variety of tissues. Two years later that same 100 kDa substrate was used to partially purify and characterize the CaM-kinase for which it is a target, and Nairn et al. [103] showed that the kinase, which they named CaMKIII, was the only CaM-kinase capable of phosphorylating the 100 kDa substrate. They also showed that synapsin 1, phosphorylase b, myosin light chain and histone were poor substrates for the kinase, making CaMKIII a dedicated kinase with only one known substrate. The substrate for CaMKIII was soon after revealed to be elongation factor 2 (EF2), a translational regulator that aids in the growth of polypeptide chains during protein translation [104]. Because of this discovery CaMKIII is now typically referred to as elongation factor 2 kinase (EF2K) since there are no other known substrates for the enzyme. Phosphorylation of Thr56 and Thr58 in EF2 by CaMKIII leads to the suppression of protein translation by dissociating EF2 from the ribosome. CaMKIII is expressed in cells as a monomer of approximately 100 kDa, and it is found in the cytosol, thus coupling Ca2+ increases in the cytosol to the suppression of protein synthesis.
The domain structure of CaMKIII bears the least resemblance to the other CaM-kinase family members. The CaM-binding domain resides in the N-terminus and the EF2 recognition domain resides in the C-terminus that flanks the catalytic domain. This domain organization displays little likeness to that of all other CaM-kinases. Like CaMKII and CaMKIV, once Ca2+/CaM binds CaMKIII it undergoes auto-phosphorylation which generates Ca2+/CaM-independent activity. This same autonomous activity can also be achieved from phosphorylation of CaMKIII by PKA. In addition, recent experiments have shown CaMKIII is activated by an undetermined member of the mammalian target of the rapamycin (mTOR) pathway as well as slight decreases in intracellular pH, linking its activity to pathways regulating cell growth as well as tissue acidosis like that experienced during ischemic and hypoxic conditions [105,106]. In light of all the different modes of regulation of CaMKIII, the traditional view that the enzyme provides a simple link between increased Ca2+ and decreased protein synthesis has been modified to one in which CaMKIII integrates a variety of cellular signals that all function to regulate the rate of translation.
Myosin Light Chain Kinase (MLCK)
Myosin light chain kinase is expressed from two separate genes that encode the isoforms known as skeletal muscle MLCK (skMLCK) and smooth muscle MCLK (smMLCK). It is obvious from there names that the two isoforms exist in striated and smooth muscle respectively, but the smMLCK isoform is also expressed in many other tissues including the brain [43]. Both isoforms are expressed in cells as monomers where skMLCK ranges in size from 69–150 kDa and smMLCK ranges in size from 130–150 kDa. This disparity in protein size is the consequence of mRNA splicing as well as the use of alternative promoters in each gene [107]. The primary function of MLCK is to stimulate muscle contraction through the phosphorylation of the regulatory light chain (RLC) of myosin II, a eukaryotic motor protein that interacts with filamentous actin. Smooth muscle contraction is initiated through phosphorylation of the RLC via its stimulation of actin-activated myosin ATPase activity [108], whereas contraction in striated muscle is potentiated through the formation of force-bearing cross-bridges by myosin [109]. In the brain, MLCK regulates myosin based transport in axons and nerve terminals as well as Ca2+-mediated recruitment of vesicles in presynaptic terminals [110]. It is interesting to note that, although MLCK is a restricted CaM-kinase with only one known substrate (RLC), it is linked to a variety of cellular functions due to the diverse biological function of myosin II.
Both isoforms of MLCK contain the prototypical domain structure of the CaM-kinase family with a catalytic domain followed by a regulatory sequence containing an autoinhibitory domain and a CaM-binding domain; however, smMLCK contains additional N-terminal amino acids containing several distinct protein motifs that regulate its localization (Figure 11). There is a short and a long form of smMLCK, and the short form (S-smMLCK) contains a fibronectin module, two immunoglobulin (IG) modules, PEVK repeats, and an actin-binding domain upstream of the catalytic domain. The long form (L-smMLCK) shares the exact same structure, but includes an additional six IG modules upstream of the actin-binding domain. Although no 3-D structure exists for MLCK, it is expected to share the same bilobed structure, because of the reasonably high degree of sequence similarity in its catalytic domain compared to the other CaM-kinases as well as PKA.
Figure 11.

Domain structure of MLCK isoforms. The conserved CaM-kinase domains are shown in gray (catalytic domains), white (autoinhibitory domains) and black (CaM-binding domains). The short isoform of smMLCK (S-smMLCK) contains an actin-binding domain (A), PEVK repeats (P), two immunoglobulin modules (IG) and a fibronectin module (F), while the long isoform (L-smMLCK) contains an additional six immunoglobulin modules N-terminal to the actin binding domain. These regions help to target the two isoforms to the appropriate intracellular location. Numbers above the diagrams represent the length of each isoform in amino acid residues.
Both types of MLCK are maintained in an inactive state at basal Ca2+ levels through binding of the autoinhibitory domain to the catalytic core. The autoinhibitory domain binds to the substrate binding lobe of the catalytic core, blocking substrate binding, but does not inhibit binding of ATP. Initially, due to sequence similarities between the regulatory domain of MLCK and the area surrounding Ser19, the phosphorylation site for MLCK in the myosin RLC, it was proposed that this region of the regulatory domain bound to the catalytic domain via a pseudosubstrate mechanism. This sequence overlaps with the N-terminal portion of the CaM-binding domain, and indeed a synthetic peptide corresponding to this region of the MLCK regulatory domain competitively inhibited the kinase’s ability to phosphorylate the RLC [111,112]. However, further experimentation has led to the hypothesis that it is not a pseudosubstrate model of autoinhibition, but rather intrasteric regulation dependent on amino acid residues which are spatially distinct from those proposed to act as a pseudosubstrate [113–115]. In this new model of intrasteric regulation it is thought that the autoinhibitory domain makes contacts with amino acids in the catalytic core of the kinase and blocks the phosphotransferase reaction without directly interacting with residues responsible for substrate binding. smMLCK is also a known substrate for phosphorylation by CaMKII in vivo [116] where it phosphorylates the kinase in the C-terminal portion of the CaM-binding domain. This phosphorylation inhibits binding of Ca2+/CaM and leads to a decrease in the phosphorylation of RLC and a relaxation of smooth muscle cells [117].
Phosphorylase Kinase
Although protein kinase activity was first observed by Burnett and Kennedy in 1954 [13], phosphorylase kinase was the first protein kinase to be purified, characterized and have its precise function determined [118,119]. To date, its only known biological function is the phosphorylation of glycogen phosphorylase, despite being one of the largest and most structurally complex CaM-kinases known. Tissue distribution of phosphorylase kinase is widespread, with the majority being present in skeletal muscle and liver, where the kinase modulates the energy source for muscle contraction and contributes to blood-glucose homeostasis, respectively [43]. In fact, phosphorylase kinase comprises 0.5 to 1% of total soluble protein found in skeletal muscle. Phosphorylase kinase is composed of four different subunits termed α, β, γ and δ which assemble to form a heterotetramer (Figure 12). Four of these heterotetramers then associate to form the phosphorylase kinase holoenzyme, which has a molecular weight of approximately 1,300 kDa. Due to alternative splicing and expression of different isoforms of the individual subunits, a variety of holoenzymes can be constructed depending on the cell type in which it is expressed.
Figure 12.

Schematic representation of the phosphorylase kinase holoenzyme structure and regulation. The phosphorylase kinase holoenzyme is a tetramer of heterotetramers. Each heterotetramer is composed of one γ (catalytic), one δ (CaM) and one α and β (both regulatory) subunit. Four heterotetramers are then combined to form the holoenzyme. Although the exact structural arrangement of holoenzyme subunits is not known, the “butterfly” shape was chosen because it is a common view seen in electron micrographs of purified holoenzymes. The δ subunit (or CaM) is stably bound to the holoenzyme through interactions with the γ and at least the α subunit as well, where it regulates catalytic activity in a Ca2+-dependent manner. Phosphorylation of the β subunit at Ser26 by PKA leads directly to increased catalytic activity, and phosphorylation of the α subunit at Ser1018 amplifies this increased activity. Phosphorylation of Ser1018 by itself does not alter phosphorylase kinase activity. Further Ca2+-dependent regulation is imparted through the reversible binding of exogenous Ca2+/CaM to the α and β subunits, which functions as an alternate form of regulation in the absence of PKA phosphorylation.
The γ subunit is the 45 kDa catalytic subunit of phosphorylase kinase, and it bears resemblance to all other CaM-kinases with an N-terminal catalytic domain followed by a regulatory domain consisting of an autoinhibitory domain and a CaM-binding domain. The α, β and δ subunits are all regulatory subunits, but intriguingly the δ subunit is CaM that is stably bound as a structural component of the phosphorylase kinase holoenzyme and remains attached even at nanomolar concentrations of intra-cellular Ca2+. It is not entirely clear as to how this tight binding is maintained, but the CaM-binding domain in the γ subunit shows high sequence similarity to the troponin-C binding domain (a protein highly homologous to CaM) of troponin-I, a complex which also does not dissociate at basal levels of Ca2+. apo-CaM and Ca2+/CaM can reside in an extended or compact conformation, and like in the troponin I-C complex, CaM bound to the γ subunit of phosphorylase kinase appears to be in an extended conformation, perhaps allowing for more intermolecular contacts to occur that stabilize the interactions. Further contacts appear to be formed with the addition of the remaining regulatory subunits. For example, the mild denaturant urea is able to dissociate the γ-δ complex in the presence or absence of Ca2+, whereas it is unable to dissociate the ternary γ-δ-α complex in the presence of Ca2+ [120].
In the presence of low intracellular Ca2+ phosphorylase kinase resides in an inhibited state, in which the autoinhibitory domain binds the catalytic core and blocks binding of glycogen phosphorylase. Upon elevation of Ca2+ the δ subunit becomes saturated, inducing a conformational change that displaces the autoinhibitory domain and allows the phosphotransferase reaction to occur. The α and β regulatory subunits are relatively large proteins, and constitute approximately 80% of the mass of the phosphorylase kinase holoenzyme [121,122]. While both subunits function to inhibit phosphorylase kinase activity, the exact mechanism of their action is yet to be determined [123,124]. Although both subunits contain many phosphorylation sites in vitro, Ser1018 and Ser26 are the major sites of regulation within the α and β subunits, respectively, and both sites are phosphorylated by PKA in vivo [121]. Phosphorylation of Ser26 in the β subunit leads directly to an increase in activity of the γ subunit, and phosphorylation of Ser1018 in the α subunit further amplifies activity. Phosphorylation of Ser1018 alone, however, does not lead to activation of the kinase [125,126]. PKA mediated phosphorylation facilitates activity via an increase in the Vmax of phosphorylase kinase as opposed to a decrease in Km, which was originally thought to be the case. This makes sense, seeing as how concentrations of its substrate (glycogen phosphorylase) are well in excess of the enzyme’s Km for the protein target [127]. In addition to Ca2+-dependent regulation by the stably associated δ subunit (CaM), further regulation is imparted by what is known as the δ’ subunit or exogenous Ca2+/CaM. Unlike the δ subunit which binds more-or-less permanently to the γ subunit to regulate activity, the δ’ subunit associates and dissociates in a Ca2+-dependent manner to both the α and β subunits. Interestingly, binding of exogenous Ca2+/CaM to the α or β subunit does not activate the kinase further if it has been previously phosphorylated. Instead, it seems to be an alternate form of regulation that can function in the absence of phosphorylation to increase kinase activity [128]. On top of being regulated by Ca2+ via the δ and δ’ subunits and by cAMP through phosphorylation by PKA, phosphorylase kinase activity can also be stimulated through binding of ADP and by changes in pH, making it a central point for the regulation of glycogen metabolism by a wide variety of cellular conditions. So despite its seemingly simple biological function, phosphorylase kinase is one of the most intricately regulated kinases known to date.
The crystal structure of the catalytic subunit of phosphorylase kinase has been solved [129], and it bears striking resemblance to the catalytic core of PKA (as would be expected). It is composed of a smaller ATP-binding domain and a larger substrate binding domain joined to form the typical bi-lobed catalytic core. The C-terminal region of the protein, containing the regulatory domain, was truncated in order to produce a crystal that would diffract to high resolution, so it is not known how that autoinhibitory region specifically interacts with the catalytic domain to block the phosphorylation reaction.
Perspective and Summary
At this point it should be clear just how critical the impact of signaling through CaM-kinases is within the cell. Due to the widespread use of Ca2+ as a second messenger and the importance of phosphorylation as a regulator of protein function, the CaM-kinases represent a family of enzymes poised to mediate a vast array of cellular processes. With such a complex network of signaling pathways, all controlled by a single signaling ion (Ca2+) and the same allosteric regulator (CaM), a valid question is, how is the appropriate decision made for the activation of a particular CaM kinase pathway? In short, how does CaM “decide” which targets to activate in response to a variety of Ca2+ transients when the targets are all competing for the same molecule?
As mentioned in the Introduction, CaM has a Kd of ~10 μM for Ca2+ in vitro, which is a value well above the peak global Ca2+ concentration typically observed in cells (~1 μM) [4]. It is true that Ca2+-microdomains likely exist in different cellular compartments or at the site of Ca2+ influx through ion channels where concentrations can reach values up to 100 μM, but it is still counterintuitive for a ubiquitous Ca2+-sensor to express such relatively poor Ca2+-binding kinetics. However, it is now well documented that when CaM interacts with target proteins, such as the CaM-kinases, CaM’s Ca2+-binding affinity can increase dramatically. In this regard, the Ca2+-binding properties of free CaM are underestimates of the Ca2+-binding affinity of CaM when one considers the diversity and concentration of CaM-binding targets within the cell. This target-dependent tuning of Ca2+ binding kinetics is now thought to play a key role in adjusting the Ca2+-sensing properties of CaM to respond appropriately to what might appear to be modest increases in intracellular Ca2+. Depending on the magnitude of these reciprocal interactions between Ca2+, CaM and CaM-binding target, one could also envision an increase in the Ca2+-binding affinity of CaM, whereby it might remain Ca2+-saturated and therefore bound to a target even at resting Ca2+-levels. Some of the best systems in which these reciprocal effects have been characterized are with members of the CaM-kinase family. Both MLCK and CaMKII (even more so in its Thr286 phosphorylated form) have been shown to significantly increase CaM’s affinity for Ca2+ [6,10,11], as have peptides corresponding to the CaM-binding domains of phosphorylase kinase, CaM-KII, and skMLCK and smMLCK [10]. Interestingly, it appears that regulation of CaM’s Ca2+-binding properties can be tuned not only differentially between varying target proteins, but also in a lobe-specific manner, leading to an increased complexity of Ca2+-binding kinetics among various Ca2+/CaM-target protein complexes. This dynamic modification of binding-kinetics could underlie CaM’s ability to regulate so many targets in response to the same second messenger. In fact, the changes observed through thermodynamic coupling have led to the hypothesis that different target enzymes reside in different modes of interaction with CaM during basal Ca2+ states, which allows them to respond differentially to Ca2 signals with varying temporal profiles. For example, in the most extreme case, phosphorylase kinase actually incorporates apo-CaM into its holoenzyme structure as a stably bound subunit, even at low intracellular Ca2+ concentrations. This arrangement then alleviates the need for multiple diffusion limited steps to produce activation of the kinase, and is dependent only on Ca2+’s ability to find and bind to the delta subunit of the holoenzyme. This arrangement would increase the rate at which the kinase is able to respond to an incoming Ca2+ signal. MLCK and CaMKII are perhaps examples of an intermediate form of coupling where it is hypothesized that they could reside in a one lobe-bound form of CaM at resting Ca2+ levels (presumably the C-lobe due to its higher Ca2+-binding affinity[10]). This would mean that Ca2+-saturation of the N-terminal domain would be the only required event for their activation, once again limiting the number of diffusion restricted steps involved in the activation pathway. As mentioned earlier, CaMKII’s dodecameric structure also works to increase the probability of full, autophosphorylated activation by clustering subunits and thus increasing the probability of their interacting once bound to CaM. Lastly, at the other extreme, there are CaMKK, and CaMKI or CaMKIV, the components of the CaM-kinase cascade described above. Although the effect of these kinases on CaM’s affinity for Ca2+ has not been documented, it is highly probable that they induce some change in affinity. However, because they are monomeric soluble proteins, their activation will be dependent on several diffusion limited steps: binding of Ca2+ to CaM, followed by Ca2+/CaM-binding to CaMKK, and then finally phosphorylation of CaMKI and CaMKIV only if they, too, are Ca2+/CaM bound. This arrangement allows for multiple checkpoints prior to activation by requiring a Ca2+ signal of adequate amplitude and duration in order for all of these steps to take place before committing to this signaling pathway. The regulation of CaM’s Ca2+ binding properties by target enzymes is of fundamental importance to fully appreciate under which circumstances each of these CaM-kinases will be activated in the intracellular setting. Much work is still required before a complete understanding of these reciprocal effects on Ca2+/CaM signaling is fully realized. But, the available findings have already provided important insights into how a protein seemingly as simple as CaM can function to regulate such a diverse set of CaM-dependent targets, like those in the CaM-kinase family, to control their downstream biological effects.
Acknowledgments
The authors would like to acknowledge the creative group of people who have contributed to our structural work on CaMKII. These are Steve Kolodziej, Andy Hudmon, James Stoops and Pawel Penczek. We also thank Hugo Sanabria, Amelie Forest, Aron Goins, Yoshi Kubota and John Putkey for many stimulating discussions concerning the Ca2+/calmodulin signaling system. Dr. Stefan Knapp is also acknowledged for discussions concerning his group’s successes at determining crystal structures for the different CaMKII isoforms. Finally, we gratefully acknowledge the financial support for the work on CaMKII through NIH grant NS26086 (MNW) and MTS acknowledges support from NIH training grant T32 NS07467.
References
- 1.Berridge M, Lipp P, Bootman M. Calcium signaling. Curr Biol. 1999;9:R157–159. doi: 10.1016/s0960-9822(99)80101-8. [DOI] [PubMed] [Google Scholar]
- 2.Bootman MD, Lipp P, Berridge MJ. The organisation and functions of local Ca(2+) signals. J Cell Sci. 2001;114:2213–2222. doi: 10.1242/jcs.114.12.2213. [DOI] [PubMed] [Google Scholar]
- 3.Hoeflich KP, Ikura M. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 2002;108:739–742. doi: 10.1016/s0092-8674(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 4.Persechini A, Stemmer PM. Calmodulin is a limiting factor in the cell. Trends Cardiovasc Med. 2002;12:32–37. doi: 10.1016/s1050-1738(01)00144-x. [DOI] [PubMed] [Google Scholar]
- 5.Bayley PM, Findlay WA, Martin SR. Target recognition by calmodulin: dissecting the kinetics and affinity of interaction using short peptide sequences. Protein Sci. 1996;5:1215–228. doi: 10.1002/pro.5560050701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaertner TR, Putkey JA, Waxham MN. RC3/Neurogranin and Ca2+/calmodulin-dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium. J Biol Chem. 2004;279:39374–39382. doi: 10.1074/jbc.M405352200. [DOI] [PubMed] [Google Scholar]
- 7.Johnson JD, Snyder C, Walsh M, Flynn M. Effects of myosin light chain kinase and peptides on Ca2+ exchange with the N- and C-terminal Ca2+ binding sites of calmodulin. J Biol Chem. 1996;271:761–767. doi: 10.1074/jbc.271.2.761. [DOI] [PubMed] [Google Scholar]
- 8.Olwin BB, Edelman AM, Krebs EG, Storm DR. Quantitation of energy coupling between Ca2+, calmodulin, skeletal muscle myosin light chain kinase, and kinase substrates. J Biol Chem. 1984;259:10949–10955. [PubMed] [Google Scholar]
- 9.Olwin BB, Storm DR. Calcium binding to complexes of calmodulin and calmodulin binding proteins. Biochemistry. 1985;24:8081–8086. doi: 10.1021/bi00348a037. [DOI] [PubMed] [Google Scholar]
- 10.Peersen OB, Madsen TS, Falke JJ. Intermolecular tuning of calmodulin by target peptides and proteins: differential effects on Ca2+ binding and implications for kinase activation. Protein Sci. 1997;6:794–807. doi: 10.1002/pro.5560060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Persechini A, White HD, Gansz KJ. Different mechanisms for Ca2+ dissociation from complexes of calm-odulin with nitric oxide synthase or myosin light chain kinase. J Biol Chem. 1996;271:62–67. doi: 10.1074/jbc.271.1.62. [DOI] [PubMed] [Google Scholar]
- 12.Persechini A, Yano K, Stemmer PM. Ca(2+) binding and energy coupling in the calmodulin-myosin light chain kinase complex. J Biol Chem. 2000;275:4199–4204. doi: 10.1074/jbc.275.6.4199. [DOI] [PubMed] [Google Scholar]
- 13.Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. J Biol Chem. 1954;211:969–980. [PubMed] [Google Scholar]
- 14.Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 15.Pinna LA. A historical view of protein kinase CK2. Cell Mol Biol Res. 1994;40:383–390. [PubMed] [Google Scholar]
- 16.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 17.De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 19.Taylor SS, Radzio-Andzelm E. Three protein kinase structures define a common motif. Structure. 1994;2:345–355. doi: 10.1016/s0969-2126(00)00036-8. [DOI] [PubMed] [Google Scholar]
- 20.Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev. 2001;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- 21.Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Aimes RT, Hemmer W, Taylor SS. Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry. 2000;39:8325–8332. doi: 10.1021/bi992800w. [DOI] [PubMed] [Google Scholar]
- 23.Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- 24.Gibbs CS, Zoller MJ. Rational scanning muta-genesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. J Biol Chem. 1991;266:8923–8931. [PubMed] [Google Scholar]
- 25.Carrera AC, Alexandrov K, Roberts TM. The conserved lysine of the catalytic domain of protein kinases is actively involved in the phosphotransfer reaction and not required for anchoring ATP. Proc Natl Acad Sci U S A. 1993;90:442–446. doi: 10.1073/pnas.90.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MJ, Harkins PC, Zhang J, Baer R, Haycock JW, Cobb MH, Goldsmith EJ. Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5’-triphosphate. Biochemistry. 1996;35:5641–5646. doi: 10.1021/bi952723e. [DOI] [PubMed] [Google Scholar]
- 27.Madhusudan, Trafny EA, Xuong NH, Adams JA, Ten Eyck LF, Taylor SS, Sowadski JM. cAMP-dependent protein kinase: crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 1994;3:176–187. doi: 10.1002/pro.5560030203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldberg J, Nairn A, Kuriyan J. Structural Basis for the Autoinhibition of Calcium/Calmodulin-Dependent Protein Kinase I. Cell. 1996;84:875–887. doi: 10.1016/s0092-8674(00)81066-1. [DOI] [PubMed] [Google Scholar]
- 29.Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- 30.Pearson RB, Kemp BE. Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol. 1991;200:62–81. doi: 10.1016/0076-6879(91)00127-i. [DOI] [PubMed] [Google Scholar]
- 31.Lee JC, Edelman AM. A protein activator of Ca(2+)-calmodulin-dependent protein kinase Ia. J Biol Chem. 1994;269:2158–264. [PubMed] [Google Scholar]
- 32.Tokumitsu H, Brickey DA, Glod J, Hidaka H, Sikela J, Soderling TR. Activation mechanisms for Ca2+/calmodulin-dependent protein kinase IV. Identification of a brain CaM-kinase IV kinase. J Biol Chem. 1994;269:28640–28647. [PubMed] [Google Scholar]
- 33.Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- 34.Tokumitsu H, Enslen H, Soderling TR. Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J Biol Chem. 1995;270:19320–19324. doi: 10.1074/jbc.270.33.19320. [DOI] [PubMed] [Google Scholar]
- 35.Tokumitsu H, Wayman GA, Muramatsu M, Soderling TR. Calcium/calmodulin-dependent protein kinase kinase: identification of regulatory domains. Biochemistry. 1997;36:12823–12827. doi: 10.1021/bi971348i. [DOI] [PubMed] [Google Scholar]
- 36.Wayman GA, Tokumitsu H, Soderling TR. Inhibitory cross-talk by cAMP kinase on the calmodulin-dependent protein kinase cascade. J Biol Chem. 1997;272:16073–16076. doi: 10.1074/jbc.272.26.16073. [DOI] [PubMed] [Google Scholar]
- 37.Schumacher AM, Schavocky JP, Velentza AV, Mirzoeva S, Watterson DM. A calmodulin-regulated protein kinase linked to neuron survival is a substrate for the calmodulin-regulated death-associated protein kinase. Biochemistry. 2004;43:8116–8124. doi: 10.1021/bi049589v. [DOI] [PubMed] [Google Scholar]
- 38.Soderling TR, Stull JT. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev. 2001;101:2341–2352. doi: 10.1021/cr0002386. [DOI] [PubMed] [Google Scholar]
- 39.Tokumitsu H, Takahashi N, Eto K, Yano S, Soderling TR, Muramatsu M. Substrate recognition by Ca2+/Calmodulin-dependent protein kinase kinase. Role of the arg-prorich insert domain. J Biol Chem. 1999;274:15803–15810. doi: 10.1074/jbc.274.22.15803. [DOI] [PubMed] [Google Scholar]
- 40.Tokumitsu H, Iwabu M, Ishikawa Y, Kobayashi R. Differential regulatory mechanism of Ca2+/calm-odulin-dependent protein kinase kinase isoforms. Biochemistry. 2001;40:13925–13932. doi: 10.1021/bi010863k. [DOI] [PubMed] [Google Scholar]
- 41.Nairn AC, Greengard P. Purification and characterization of Ca2+/calmodulin-dependent protein kinase I from bovine brain. J Biol Chem. 1987;262:7273–72781. [PubMed] [Google Scholar]
- 42.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 43.Robison J, Colbran R. Calcium/Calmodulin-Dependent Kinases. Encyclopedia of Biological Chemistry. 2004;1:281–286. [Google Scholar]
- 44.Sakagami H, Kondo H. Cloning and sequencing of a gene encoding the beta polypeptide of Ca2+/calmodulin-dependent protein kinase IV and its expression confined to the mature cerebellar granule cells. Brain Res Mol Brain Res. 1993;19:215–218. doi: 10.1016/0169-328x(93)90029-o. [DOI] [PubMed] [Google Scholar]
- 45.Westphal RS, Anderson KA, Means AR, Wadzinski BE. A signaling complex of Ca2+-calmodulin-dependent protein kinase IV and protein phosphatase 2A. Science. 1998;280:1258–1261. doi: 10.1126/science.280.5367.1258. [DOI] [PubMed] [Google Scholar]
- 46.Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calm-odulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem. 1994;269:15520–15527. [PubMed] [Google Scholar]
- 47.Miranti CK, Ginty DD, Huang G, Chatila T, Greenberg ME. Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin-dependent kinase. Mol Cell Biol. 1995;15:3672–3684. doi: 10.1128/mcb.15.7.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miska EA, Langley E, Wolf D, Karlsson C, Pines J, Kouzarides T. Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res. 2001;29:3439–3447. doi: 10.1093/nar/29.16.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 50.Chow FA, Anderson KA, Noeldner PK, Means AR. The autonomous activity of calcium/calmodulin-dependent protein kinase IV is required for its role in transcription. J Biol Chem. 2005;280:20530–20538. doi: 10.1074/jbc.M500067200. [DOI] [PubMed] [Google Scholar]
- 51.Anderson KA, Noeldner PK, Reece K, Wadzinski BE, Means AR. Regulation and function of the calcium/calmodulin-dependent protein kinase IV/protein serine/threonine phosphatase 2A signaling complex. J Biol Chem. 2004;279:31708–31716. doi: 10.1074/jbc.M404523200. [DOI] [PubMed] [Google Scholar]
- 52.Marklund U, Larsson N, Brattsand G, Osterman O, Chatila TA, Gullberg M. Serine 16 of oncoprotein 18 is a major cytosolic target for the Ca2+/calmodulin-dependent kinase-Gr. Eur J Biochem. 1994;225:53–60. doi: 10.1111/j.1432-1033.1994.00053.x. [DOI] [PubMed] [Google Scholar]
- 53.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 54.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Erondu NE, Kennedy MB. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J Neurosci. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tobimatsu T, Fujisawa H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J Biol Chem. 1989;264:17907–17912. [PubMed] [Google Scholar]
- 57.Kolodziej SJ, Hudmon A, Waxham MN, Stoops JK. Three-dimensional reconstructions of calcium/calm-odulin-dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J Biol Chem. 2000;275:14354–14359. doi: 10.1074/jbc.275.19.14354. [DOI] [PubMed] [Google Scholar]
- 58.Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:12484–12494. doi: 10.1074/jbc.M313597200. [DOI] [PubMed] [Google Scholar]
- 59.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–1251. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 60.Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J. Oligo-merization states of the association domain and the holoe-nyzme of Ca2+/CaM kinase II. FEBS J. 2006;273:682–694. doi: 10.1111/j.1742-4658.2005.05088.x. [DOI] [PubMed] [Google Scholar]
- 61.Bunkoczi G, Debreczeni JE, Salah E, Gileadi O, Rellos P, Arrowsmith CH, Edwards A, Sundstrom M, Weigelt J, Von Delft F, Turnbull A, Pike ACW, Knapp S. Structure of the Oligomerisation Domain of Calcium-Calmodulin-Dependent Protein Kinase II Gamma. 2007 http://www.pdb.org, To be Published.
- 62.Vallano ML. Separation of isozymic forms of type II calcium/calmodulin-dependent protein kinase using cation-exchange chromatography. J Neurosci Methods. 1989;30:1–9. doi: 10.1016/0165-0270(89)90067-8. [DOI] [PubMed] [Google Scholar]
- 63.Bennett MK, Erondu NE, Kennedy MB. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J Biol Chem. 1983;258:12735–12744. [PubMed] [Google Scholar]
- 64.Miller SG, Kennedy MB. Distinct forebrain and cerebellar isozymes of type II Ca2+/calmodulin-dependent protein kinase associate differently with the postsynaptic density fraction. J Biol Chem. 1985;260:9039–9046. [PubMed] [Google Scholar]
- 65.O’Leary H, Lasda E, Bayer KU. CaMKIIbeta association with the actin cytoskeleton is regulated by alternative splicing. Mol Biol Cell. 2006;17:4656–4665. doi: 10.1091/mbc.E06-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 67.Ohta Y, Nishida E, Sakai H. Type II Ca2+/calmodulin-dependent protein kinase binds to actin filaments in a calmodulin-sensitive manner. FEBS Lett. 1986;208:423–426. doi: 10.1016/0014-5793(86)81061-4. [DOI] [PubMed] [Google Scholar]
- 68.Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron. 1998;21:593–606. doi: 10.1016/s0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- 69.Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by auto-phosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 70.Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multi-functional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 71.Bradshaw JM, Hudmon A, Schulman H. Chemical quenched flow kinetic studies indicate an intra-holoenzyme autophosphorylation mechanism for Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2002;277:20991–20998. doi: 10.1074/jbc.M202154200. [DOI] [PubMed] [Google Scholar]
- 72.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 73.Lisman JE. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci U S A. 1985;82:3055–3057. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mullasseril P, Dosemeci A, Lisman JE, Griffith LC. A structural mechanism for maintaining the ‘on-state’ of the CaMKII memory switch in the post-synaptic density. J Neurochem. 2007;103:357–364. doi: 10.1111/j.1471-4159.2007.04744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- 76.Hanson PI, Schulman H. Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J Biol Chem. 1992;267:17216–17224. [PubMed] [Google Scholar]
- 77.Hashimoto Y, Schworer CM, Colbran RJ, Soderling TR. Autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. Effects on total and Ca2+-independent activities and kinetic parameters. J Biol Chem. 1987;262:8051–8055. [PubMed] [Google Scholar]
- 78.Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron. 2002;36:493–505. doi: 10.1016/s0896-6273(02)01007-3. [DOI] [PubMed] [Google Scholar]
- 79.Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- 80.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 81.De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 82.Putkey JA, Waxham MN. A peptide model for calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 1996;271:29619–29623. doi: 10.1074/jbc.271.47.29619. [DOI] [PubMed] [Google Scholar]
- 83.Waxham MN, Tsai AL, Putkey JA. A mechanism for calmodulin (CaM) trapping by CaM-kinase II defined by a family of CaM-binding peptides. J Biol Chem. 1998;273:17579–17584. doi: 10.1074/jbc.273.28.17579. [DOI] [PubMed] [Google Scholar]
- 84.Benke TA, Luthi A, Isaac JT, Collingridge GL. Modulation of AMPA receptor unitary conductance by synaptic activity. Nature. 1998;393:793–797. doi: 10.1038/31709. [DOI] [PubMed] [Google Scholar]
- 85.Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 87.Koch-Weser J, Blinks JR. The Influence of the Interval between Beats on Myocardial Contractility. Pharmacol Rev. 1963;15:601–652. [PubMed] [Google Scholar]
- 88.Ross J, Jr, Miura T, Kambayashi M, Eising GP, Ryu KH. Adrenergic control of the force-frequency relation. Circulation. 1995;92:2327–2332. doi: 10.1161/01.cir.92.8.2327. [DOI] [PubMed] [Google Scholar]
- 89.Zygmunt AC, Maylie J. Stimulation-dependent facilitation of the high threshold calcium current in guinea-pig ventricular myocytes. J Physiol. 1990;428:653–671. doi: 10.1113/jphysiol.1990.sp018233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–6. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 92.Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, Kennedy MB. Densin-180 forms a ternary complex with the (alpha)-subunit of Ca2+/calmodu-lin-dependent protein kinase II and (alpha)-actinin. J Neuro-sci. 2001;21:423–433. doi: 10.1523/JNEUROSCI.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Otmakhov N, Tao-Cheng JH, Carpenter S, Asrican B, Dosemeci A, Reese TS, Lisman J. Persistent accumulation of calcium/calmodulin-dependent protein kinase II in dendritic spines after induction of NMDA receptor-dependent chemical long-term potentiation. J Neurosci. 2004;24:9324–9331. doi: 10.1523/JNEUROSCI.2350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Strack S, Choi S, Lovinger DM, Colbran RJ. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J Biol Chem. 1997;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- 95.Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- 97.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 98.Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 99.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 100.Filippakopoulos P, Rellos P, Niesen F, Burgess N, Bullock A, Berridge G, Pike ACW, Ugochukwu E, Pilka ES, Von Delft F, Arrowsmith CH, Edwards AM, Weigelt J, Knapp S. Crystal Structure of Human Calcium/Calmodulin-Dependent Protein Kinase IIB Isoform 1 (CAMK2B) 2007 http://www.pdb.org, To be Published.
- 101.Pike ACW, Rellos P, Fedorov O, Burgess-Brown N, Shrestha L, Ugochukwu E, Pilka ES, Von Delft F, Edwards A, Weigelt J, Arrowsmith CH, Sundstrom M, Knapp S. Crystal Structure of Human Calcium-Calmodulin-Dependent Protein Kinase II Gamma. 2007 http://www.pdb.org, To be Published.
- 102.Roos AK, Rellos P, Salah E, Pike ACW, Fedorov O, Pilka ES, Von Delft F, Arrowsmith CH, Weigelt J, Edwards A, Bountra C, Knapp S. Crystal Structure of Human Calcium Calmodulin Dependent Protein Kinase II Delta Isoform 1, Camkd. 2008 http://www.pdb.org, To be Published.
- 103.Nairn AC, Bhagat B, Palfrey HC. Identification of calmodulin-dependent protein kinase III and its major Mr 100,000 substrate in mammalian tissues. Proc Natl Acad Sci U S A. 1985;82:7939–7943. doi: 10.1073/pnas.82.23.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nairn AC, Palfrey HC. Identification of the major Mr 100,000 substrate for calmodulin-dependent protein kinase III in mammalian cells as elongation factor-2. J Biol Chem. 1987;262:17299–17303. [PubMed] [Google Scholar]
- 105.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dorovkov MV, Pavur KS, Petrov AN, Ryazanov AG. Regulation of elongation factor-2 kinase by pH. Biochemistry. 2002;41:13444–13450. doi: 10.1021/bi026494p. [DOI] [PubMed] [Google Scholar]
- 107.Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- 108.Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- 109.Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol. 1993;264:C1085–1095. doi: 10.1152/ajpcell.1993.264.5.C1085. [DOI] [PubMed] [Google Scholar]
- 110.Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–23. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kemp BE, Pearson RB, Guerriero V, Jr, Bagchi IC, Means AR. The calmodulin binding domain of chicken smooth muscle myosin light chain kinase contains a pseudosubstrate sequence. J Biol Chem. 1987;262:2542–2548. [PubMed] [Google Scholar]
- 112.Kemp BE, Pearson RB. Intrasteric regulation of protein kinases and phosphatases. Biochim Biophys Acta. 1991;1094:67–76. doi: 10.1016/0167-4889(91)90027-u. [DOI] [PubMed] [Google Scholar]
- 113.Fitzsimons DP, Herring BP, Stull JT, Gallagher PJ. Identification of basic residues involved in activation and calmodulin binding of rabbit smooth muscle myosin light chain kinase. J Biol Chem. 1992;267:23903–23909. [PMC free article] [PubMed] [Google Scholar]
- 114.Krueger JK, Padre RC, Stull JT. Intrasteric regulation of myosin light chain kinase. J Biol Chem. 1995;270:16848–16853. doi: 10.1074/jbc.270.28.16848. [DOI] [PubMed] [Google Scholar]
- 115.Tanaka M, Ikebe R, Matsuura M, Ikebe M. Pseudosubstrate sequence may not be critical for autoinhibition of smooth muscle myosin light chain kinase. EMBO J. 1995;14:2839–2846. doi: 10.1002/j.1460-2075.1995.tb07283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tansey MG, Word RA, Hidaka H, Singer HA, Schworer CM, Kamm KE, Stull JT. Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent protein kinase II in smooth muscle cells. J Biol Chem. 1992;267:12511–12516. [PubMed] [Google Scholar]
- 117.Word RA, Tang DC, Kamm KE. Activation properties of myosin light chain kinase during contraction/relaxation cycles of tonic and phasic smooth muscles. J Biol Chem. 1994;269:21596–21602. [PubMed] [Google Scholar]
- 118.Fischer EH, Krebs EG. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J Biol Chem. 1955;216:121–32. [PubMed] [Google Scholar]
- 119.Krebs EG, Fischer EH. The phosphorylase b to a converting enzyme of rabbit skeletal muscle. Biochim Biophys Acta. 1956;20:150–157. doi: 10.1016/0006-3002(56)90273-6. [DOI] [PubMed] [Google Scholar]
- 120.Chan KF, Graves DJ. Rabbit skeletal muscle phosphorylase kinase. Interactions between subunits and influence of calmodulin on different complexes. J Biol Chem. 1982;257:5956–5961. [PubMed] [Google Scholar]
- 121.Kilimann MW, Zander NF, Kuhn CC, Crabb JW, Meyer HE, Heilmeyer LM., Jr The alpha and beta subunits of phosphorylase kinase are homologous: cDNA cloning and primary structure of the beta subunit. Proc Natl Acad Sci U S A. 1988;85:9381–9385. doi: 10.1073/pnas.85.24.9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zander NF, Meyer HE, Hoffmann-Posorske E, Crabb JW, Heilmeyer LM, Jr, Kilimann MW. cDNA cloning and complete primary structure of skeletal muscle phosphorylase kinase (alpha subunit) Proc Natl Acad Sci U S A. 1988;85:2929–2933. doi: 10.1073/pnas.85.9.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Paudel HK, Carlson GM. Inhibition of the catalytic subunit of phosphorylase kinase by its alpha/beta subunits. J Biol Chem. 1987;262:11912–11915. [PubMed] [Google Scholar]
- 124.Sanchez VE, Carlson GM. Isolation of an autoinhibitory region from the regulatory beta-subunit of phosphorylase kinase. J Biol Chem. 1993;268:17889–17895. [PubMed] [Google Scholar]
- 125.Pickett-Gies CA, Walsh DA. Subunit phosphorylation and activation of skeletal muscle phosphorylase kinase by the cAMP-dependent protein kinase. Divalent metal ion, ATP, and protein concentration dependence. J Biol Chem. 1985;260:2046–2056. [PubMed] [Google Scholar]
- 126.Ramachandran C, Goris J, Waelkens E, Merlevede W, Walsh DA. The interrelationship between cAMP-dependent alpha and beta subunit phosphorylation in the regulation of phosphorylase kinase activity. Studies using subunit specific phosphatases. J Biol Chem. 1987;262:3210–3218. [PubMed] [Google Scholar]
- 127.Newsholme P, Walsh DA. A kinetic re-interpretation of the regulation of rabbit skeletal-muscle phosphorylase kinase activity by Ca2+ and phosphorylation. Biochem J. 1992;283 ( Pt 3):845–848. doi: 10.1042/bj2830845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Picton C, Klee CB, Cohen P. Phosphorylase kinase from rabbit skeletal muscle: identification of the calmodulin-binding subunits. Eur J Biochem. 1980;111:553–561. doi: 10.1111/j.1432-1033.1980.tb04971.x. [DOI] [PubMed] [Google Scholar]
- 129.Owen DJ, Noble ME, Garman EF, Papageorgiou AC, Johnson LN. Two structures of the catalytic domain of phosphorylase kinase: an active protein kinase complexed with substrate analogue and product. Structure. 1995;3:467–482. doi: 10.1016/s0969-2126(01)00180-0. [DOI] [PubMed] [Google Scholar]