

Abstract
During corticogenesis, pituitary adenylate cyclase-activating polypeptide (PACAP; ADCYAP1) may contribute to proliferation control by activating PAC1 receptors of neural precursors in the embryonic ventricular zone. PAC1 receptors, specifically the hop and short isoforms, couple differentially to and activate distinct pathways that produce pro- or anti-mitogenic actions. Previously we found that PACAP was an anti-mitogenic signal from embryonic day 13.5 (E13.5) onwards both in culture and in vivo, and activated cAMP signaling through the short isoform. However, we now find that mice deficient in PACAP exhibited a decrease in the BrdU labeling index in E9.5 cortex, suggesting PACAP normally promotes proliferation at this stage. To further define mechanisms, we established a novel culture model in which the viability of very early cortical precursors (E9.5 mouse and E10.5 rat) could be maintained. At this stage, we found that PACAP evoked intracellular calcium fluxes and increased phospho-PKC levels, as well as stimulated G1 cyclin mRNAs and proteins, S-phase entry and proliferation without affecting cell survival. Significantly, expression of hop receptor isoform was 24-fold greater than the short isoform at E10.5, a ratio that was reversed at E14.5 when short expression was 15-fold greater and PACAP inhibited mitogenesis. Enhanced hop isoform expression, elicited by in vitro treatment of E10.5 precursors with retinoic acid, correlated with sustained pro-mitogenic action of PACAP beyond the developmental switch. Conversely, depletion of hop receptor using shRNA abolished PACAP mitogenic stimulation at E10.5. These observations suggest PACAP elicits temporally specific effects on cortical proliferation via developmentally-regulated expression of specific receptor isoforms.
Introduction
In developing cerebral cortex, positive and negative regulation of neuronal precursor proliferation and differentiation by extracellular factors influences correct cell types and numbers (Vaccarino et al., 1999; Menard et al., 2002). The PACAP ligand/PAC1 receptor system is expressed widely in multiple regions of the embryonic nervous system. The actions of PACAP signaling are complex: the peptide functions in precursor cell cycle progression, differentiation and survival. While previous studies support this contention, it is apparent that PACAP is an anti-mitogenic signal in most contexts (Lu and DiCicco-Bloom, 1997; Waschek et al., 1998; Suh et al., 2001; Nicot et al., 2002; Vaudry et al., 2002b). Defining the role of PACAP in brain development may be important because recent studies suggest PACAP signaling abnormalities may contribute to schizophrenia (Hashimoto et al., 2007), post-traumatic stress disorder (PTSD) (Ressler et al., 2011) and possibly autism (Nijmeijer et al., 2010).
PACAP acts on heptahelical G protein-coupled receptors (GPCRs): PAC1, VPAC1 and VPAC2 (Harmar et al., 1998). PAC1 is the most abundant receptor especially in central nervous system (Spengler et al., 1993; Basille et al., 2000) and has multiple splice isoforms, which are characterized by the absence (short) or presence of a 28 amino acid insert (hop) in the third intracellular loop (Spengler et al., 1993). Significantly, the short isoform and the insert-containing, hop isoform couple to different transduction pathways (Spengler et al., 1993; Vaudry et al., 2002a) and exhibit anti- or pro-mitogenic effects respectively. In E13.5 or later cortical precursors, which predominantly express the short isoform that increases cAMP levels and activates PKA, PACAP elicits cell cycle exit and promotes differentiation (Lu and DiCicco-Bloom, 1997; Lu et al., 1998), a finding replicated in vivo (Suh et al., 2001). In sharp contrast, the hop isoform activates both adenylate cyclase (AC) and phospholipase C (PLC) pathways and mediates mitogenic stimulation (Lu et al., 1998; DiCicco-Bloom et al., 2000). Furthermore, ectopic over-expression of hop isoform in E14.5 precursors converted PACAP anti-mitogenic effects into pro-mitogenic activity (Nicot and DiCicco-Bloom, 2001). These results suggest the natural expression of different PAC1 isoforms is important for regulating precursor mitosis.
The presence of total PAC1 gene transcripts as well as both individual short and hop mRNA isoforms has been reported from primitive streak stage E9 to postnatal periods (Waschek et al., 1998; Basille et al., 2000; Zhou et al., 2000; Vaudry et al., 2009). Moreover, in situ hybridization shows intense and apparently overlapping expression of short and hop receptor mRNAs in E10 telencephalon as well as E13 ventricular zone and cortical plate (Zhou et al., 2000). However, the relative expression levels of short and hop during early corticogenesis are undefined. Moreover, while evidence links PAC1 isoforms to anti-mitogenic effects from E13.5 onwards, functions of the PACAP system in early neurogenesis, when precursors proliferate to expand precursor pools, remain unresolved. Given that hop is pro-mitogenic, PACAP is a potential mitogen during this critical period. Here, assessing rat and mouse precursors, we tested the hypothesis that PACAP exhibits distinct mitogenic activities during corticogenesis, depending on PAC1 receptor isoforms. We found E10.5 precursors predominantly express hop, while the short mRNA is up-regulated and becomes dominant at E14.5. Blockade of hop exression using shRNA abolished PACAP mitogenic effects at E10.5. PACAP evokes calcium fluxes, increases phospho-PKC levels and stimulates proliferation at E10.5 but not E14.5, suggesting that control of mRNA isoform expression contributes to neurogenetic regulation.
Materials and Methods
Animals
Time-mated pregnant Sprague Dawley rats were obtained from Hilltop Lab Animals (Philadelphia, PA). Breeding pairs of PACAP KO mice on a C57BL/6 background were derived by Waschek as described (Colwell et al., 2004). Animals were managed by Robert Wood Johnson Animal Facility and maintenance, husbandry, transportation, housing and use were in compliance with Laboratory Animal Welfare Act (PL 89–544; PL-91–579) and NIH guidelines (NIH Manual Chapter 4206). Food and water were available ad libitum. The day of the plug was considered E0.5.
BrdU labeling of PACAP WT and KO mice
To maximize comparability, WT and KO littermates of either sex from heterozygous PACAP matings were analyzed. BrdU 100 μg/g was injected into E9.5 pregnant mice 1 h before sacrifice. Embryo brains were immersion fixed in 4% PFA for 30 min at 4 °C, processed for paraffin embedding, and sectioned coronally at 6 μm. Sections were immunostained for BrdU incorporation and counterstained with propidium iodide as previously reported (Mairet-Coello et al., 2009). Images of embryos identified by number alone (not genotype) were obtained from a Zeiss LSM 510-META confocal microscope. BrdU positive nuclei and total nuclei were counted blind on coronal sections in the middorsolateral cortex within a 100 μm wide sector based on the ventricular surface extending to the ventricular zone/intermediate zone boundary. BrdU labeling indices were calculated as the BrdU positive cells over total cells as described (Suh et al., 2001). Tissue from each embryo was used to determine genotype using touchdown PACAP PCR and primers described previously (Colwell et al., 2004).
Cell cultures
Embryos of either sex of E10.5 to E14.5 Sprague Dawley rats or E9.5 to E13.5 C57BL/6 mice were dissected under a dissecting microscope. E10.5 rat and E9.5 mouse telencephalic vesicles were incubated with trypsin (0.25 mg/ml, Sigma-Aldrich, St. Louis, MO) at 37 °C for 12 min followed by trypsin inhibitor (1 mg/ml) to aid in removing overlying epidermal ectoderm using dissecting forceps. The dorsolateral cortices were isolated and mechanically dissociated using a fire-polished glass pipette. Cells were plated on 2 μg/ml poly-D-lysine-coated 96-well plates (10,000 cells/well), 24-well plates (25,000 cells/well) or 35 mm dishes (80,000 cells/dish) in defined medium consisting of a 1:1 mixture of DMEM and F12, as described previously (Lu and DiCicco-Bloom, 1997; Mairet-Coello et al., 2009) containing insulin (10 μg/ml), bFGF (10 ng/ml) and BDNF (30 ng/ml, Peprotech, Rocky Hill, NJ) with PACAP38 (10 nM; American Peptide, Sunnyvale, CA) or its vehicle (0.01 N acetic acid). PACAP peptide was dissolved fresh for each experiment in sterile 0.01 N acetic acid to 100 μM and further diluted in medium to a final concentration of 10 nM. Preliminary PACAP dose-response studies (5 to 30 nM) indicated maximal mitogenic effects were obtained at 10 nM. To study the PLC pathway, cells were plated in medium containing PLC specific antagonist U-73122 (2 μM, Enzo, Farmingdale, NY) or inactive isomer U-73343 (2 μM, Enzo), and after 30 min, vehicle (0.01 N acetic acid) or PACAP (10 nM) were added. For retinoic acid (RA) experiments, cells were incubated with either all trans-RA (30 nM) (Sigma) or ethanol vehicle. RA was dissolved in 100% ethanol to 30 nM. For shRNA transfection using E10.5 rat precursors, 2 h after plating, 1 μg total DNA (mix of 0.5μg hop shRNA #1 and 0.5μg hop shRNA #2-see below) were transfected using Lipofectamine 2000 (Invitrogen) for 5 h as described earlier (Tury et al., 2011). BrdU was added at 22 h and cells were fixed at 24 h.
Expression vectors
To knockdown PAC1hop, the following oligonucleotides against different regions of rat PAC1 hop were synthesized, annealed and ligated into the RNAi-Ready-pSiren-DNR-DsRed-Express vector (Clontech). This vector places shRNA expression under control of the human U6 promoter and DsRed expression under control of the CMV promoter (Francone et al., 2010). Based on approaches reported previously (Francone et al., 2010), the following sequences were targeted:
PAC1hop shRNA #1, AATGCTACTGCAAGCCACA
PAC1hop shRNA #2, GCTGCGTGCAGAAATGCTA
Scramble control, AATGCACGCTCAGCACAAG
To test efficacy of shRNAs, we examined their ability to reduce PAC1 protein expression from rat PAC1 hop and short vectors when co-transfected in cell lines. pCI-neo-PAC1 short or hop constructs: full length rat PAC1 short or hop ((Spengler et al., 1993) gift from Dr. Journot) were inserted into mammalian expression vector pCI-neo (Promega) using EcoRI and NotI. This yields a pCI-neo PAC1 short or hop construct under control of the CMV promoter. All DNA constructs were verified by sequencing.
To test shRNA knock-down efficiency, pEAK-rapid human embryonic kidney 293 (HEK-293) cells were cultured in DMEM/F-12 containing 10% fetal calf serum, 25 mM HEPES and 1% penicillin/streptomycin for 2–3 days to 50–60% confluence. Cells were transfected with 1 μg of plasmid DNA per 4 cm2 of control vector, pCI-neo-PAC1 short or hop, in the absence or presence of 0.5μg PAC1 hop shRNA#1 plus 0.5 μg PAC hop shRNA #2 and 5μl of Lipofectamine 2000 in 1ml Optimem media (Invitrogen) for 5 hrs, after which, cells were washed and incubated with growth media for another 48 hrs. Cells were harvested for western blot analysis, and probed with a rabbit polyclonal anti-PAC1 IgG (H-55; Santa Cruz 30018) raised against the N-terminal extracellular domain that recognizes both rodent and human receptor proteins.
Immunocytochemistry
For triple immunofluorescence, cells were fixed in 4% paraformaldehyde (PFA) for 20 min at 2 h, 24 h or 48 h after plating, washed with PBS three times, incubated with anti-RC1 (1:1000, 40E-C, Developmental Studies Hybridoma Bank, San Francisco, CA) (RC1 recognizes an intermediate filament antigen in neuroepithelial cells and radial glial cells (Culican et al., 1990; Malatesta et al., 2003), currently considered a phosphorylated vimentin), anti-nestin (1:400, Chemicon, Billerica, MA), anti-beta-III tubulin (TuJ1, 1:1000, Convance, Princeton, NJ), anti-phospho-PKC (1:200, Cell Signaling) or anti-phospho-ERK (1:1000, Cell Signaling) in PBS containing 0.3% Triton X-100 and 2% NGS overnight followed by respective Alexa secondary antibodies (Invitrogen) as previously described (Lu and DiCicco-Bloom, 1997; Mairet-Coello et al., 2009). All culture dishes were provided a number so that the assessor was blind to experimental condition. The key was revealed only after data collection. Images were acquired with fluorescence microscope (Axiovert 200; Carl Zeiss MicroImaging) using 20× objective from 10 random fields per dish. Fluorescence image analysis was performed using AxioVisionLE software to determine mean fluorescence intensity. To define positive response, we set a fixed arbitrary threshold of fluorescence that was applied to all dishes within each experiment. TUNEL assay was performed using the Apoptosis Detection Kit (Chemicon, Billerica, MA).
DNA synthesis and cell division in vitro
Using 25,000 cells/well in 24-well plates, DNA synthesis was assessed using [3H] thymidine incorporation as a marker. 1 μCi/ml [3H] thymidine (Amersham/GE Healthcare, Pittsburgh, PA) was added to 24-well plates 4 h before harvesting at 24 h. Following aspiration of radiotracer-containing medium, cells were lifted with a trypsin-EDTA solution, and collected onto filter paper using a semi-automatic cell harvester (Skatron, Sterling, VA). After adding luminating solution Eco-Lite (MP Biomedicals), radioactivity was measured by scintillation spectrophotometry. Alternatively, BrdU (10 μM final) was added 2 h or 4 h before culture termination to define the BrdU labeling index at 24 h, the ratio of BrdU immunolabeled cells over total cells. Cells were counted blind under phase microscopy using 10× objective from 10 randomly selected fields in each of three dishes per group, or from 3–1cm strips of the dish, in fixed positions (stage micrometer) in the upper, middle and lower thirds of the dish, representing 3% of the dish surface.
To determine whether cells entering S phase in vitro subsequently underwent division between hours 24 and 36, a cohort of cells in each group was labeled using a 4 h BrdU pulse from hour 20 to 24. After washing cells twice with PBS, one set of cultures was fixed immediately to enumerate cells in S phase. A parallel set received culture media and were incubated another 12 h, after which they were fixed. Both groups at both time points were processed for BrdU immunostaining in parallel. The absolute number of BrdU-labeled cells per field was determined by counting blind labeled cells in 10 randomly selected fields per dish at 10× objective. To estimate total cell numbers at 24 h and 36 h, the sum of all cells counted in 10 random fields per dish was obtained at 10× objective.
Flow Cytometry
BrdU (10 μM) was added to E10.5 cells at 20 h. At 24 h, cells were fixed with 70% ethanol, then processed for BrdU immunostaining as well as propidium iodide DNA staining, using standard methods as reported previously (DiCicco-Bloom et al, 1990). Samples were analyzed on a Beckman Coulter Cell Counter, FC500 Cytomics. The figures were generated using CXP software.
Cell survival analysis
At 2 h and 24 h, 15 μg/ml fluorescein diacetate (FDA) (green fluorescence in living cells) (Vaudry et al., 2002c) and 15 μg/ml propidium iodide (red fluorescence in dead cells) were incubated with cells for 10 min as described previously (Mairet-Coello et al., 2009). At each time point, cells were counted blind under phase microscopy using 10× objective from 10 randomly selected fields in each of three dishes per group. Percentage cell survival was determined as the ratio of total green cells at 24 h over total green cells at 2 h.
Quantitative reverse transcription-PCR (Q-RT-PCR)
Total RNA was extracted from E10.5 and E14.5 rat cerebral cortices or from 24 h cultures of E10.5 cortical precursors (obtained from 50–75 embryos/isolation) using RNeasy Mini Kit (QIAGEN. Valencia, CA). DNA was digested by 15 min DNase I treatment. 0.5 μg RNA was reverse transcribed with M-MLV reverse transcriptase (Promega, Madison, MI). Quantitative reverse transcription PCR was performed using PCR Master Mix (Applied Biosystems, Foster City, CA) and reactions were performed in an ABI Prism 7000 Sequence Detection system (Applied Biosystems) as described (Mairet-Coello et al., 2009). The sequence of the primers:
PAC1 short forward 5'- AGTCGAGCATCTACTTACGGC-3'
PAC1 short reverse 5'-TTCCCTCTTGCTGACGTTCTC-3'
PAC1 hop forward 5'-ACTTCAGCTGCGTGCAGAAATGC-3'
PAC1 hop reverse 5'-GACGTTCTCTGGAGAGAAGGCAA-3'
Cyclin D1 forward, 5'-GGCCCAGCAGAACATCGAT-3'
Cyclin D1 reverse, 5'-GACCAGCTTCTTCCTCCACTTC-3'
Cyclin D3 forward, 5'-TCTCTGCCCAGTGACCATCA-3'
Cyclin D3 reverse, 5'-GGGCCCAAGACGTTTGG-3'
Cyclin E forward, 5'-AGCCCCCTGACCATTGTG-3'
Cyclin E reverse, 5'-TCGTTGACGTAGGCCACTTG-3'
Primers for rodent GAPDH were obtained from Applied Biosystem (proprietary sequences).
Primers worked with high specificity and non-specific PCR was ruled out by observing a clear peak in the melting curves. cDNAs encoding PAC1 short or PAC1 hop1 were provided by Dr. Journot, Montpellier, France and purified as reported previously (Nicot and DiCicco-Bloom, 2001). PACAP isoform values are presented as fold difference compared to E10.5 hop mRNA levels, which were arbitrarily set at 100%.
Western blot analysis
Proteins were extracted from 24 h E10.5 rat cortical precursors (50–75 embryos for each experiment). Equal amounts of proteins (10–30 μg) were loaded on 12% SDS-polyacrylamide gels and were electrotransfered onto polyvinylidene difluoride membranes. Following incubation with blocking buffer, filters were incubated overnight at 4 ° with primary antibodies (Santa Cruz, CA): anti-PAC1 (1:300; H-55), anti-cyclin D1 (1:500), anti-cyclin D2 (1:1000), and anti-actin (1:1000), as previously described (Mairet-Coello et al., 2009). Incubations with horseradish peroxidase conjugated secondary antibodies (1:1000) were performed for 1 h at room temperature, and visualization was performed using chemiluminescence (ECL, Amersham Biosciences). Autoradiographic films were analyzed using the Bio-Rad Gel Doc 2000 with Quantity One version 4.2.1 software (Bio-Rad). To control for loading, blots were stripped and reanalyzed for β-actin.
Measurement of intracellular calcium
Culture medium was removed from 96-well plates and cortical precursor cells were washed with Balanced Salt Solution (pH=7.2) (Pan et al., 2000). Cells were incubated with Ca2+ indicator Fluo-4 AM (3 μM, Molecular Probes, Eugene, OR) for 30 min at 37 °C. Following vehicle, PACAP38 (10 nM) or KCl (40 mM) addition, fluorescence intensity was measured at a 37 °C/5% CO2 atmosphere using excitation wavelength at 488±5 nm and emission wavelength at 515±5 nm using BD Pathway 855 BioImager with 20× objective (Olympus Plan fluo 0.75 NA). Images were acquired at 1 s intervals and recording lasted for 110 s after adding reagents. The mean time-dependent fluorescence intensity of individual cells was measured using Image J software after background subtraction.
Statistical analysis
Data are expressed as mean±SEM. Statistical comparisons were made by unpaired Student's t test or one-way ANOVA using Excel (Microsoft) and GraphPad Prism (GraphPad Software, La Jolla, CA).
Results
Deletion of the PACAP gene (ADCYAP1) results in decreased proliferation in the early embryonic ventricular zone (VZ)
Previous studies indicate that PACAP inhibits cortical precursor proliferation at E13.5–17.5 (Lu and DiCicco-Bloom, 1997; Suh et al., 2001; Nicot et al., 2002; Tury et al., 2011). To begin defining effects on early neurogenesis, we compared the proportion of cells in S phase in the VZ of WT and PACAP−/− mice at E9.5 using BrdU immunhistochemistry [Figure 1]. At E9.5, the rostral neural tube has closed to form the three primitive brain vesicles. The prosencephalic vesicle consists of only the VZ and exhibits a 4–5 cell thick layer. The BrdU labeling index (LI) in the VZ of the PACAP−/− prosencephalon was reduced to 47% compared to the WT control LI of 60%, suggesting that PACAP has a proliferative role at this age. Since the reduction of BrdU LI in the PACAP−/− mice may result from decreased cell survival, we performed TUNEL staining in the same region [Figure 1 d–f]. There was no statistically significant difference in TUNEL positive cells between WT and PACAP−/−, suggesting cell survival was not affected. However, to further analyze PACAP function at this age, we established a novel culture model of young precursors to more directly define PACAP effects during early neurogenesis.
Figure 1.

The telencephalic vesicles of E9.5 PACAP −/− mice exhibit reduced S phase labeling but no change in cell survival. BrdU immunohistochemical staining of E9.5 wild type (WT) (a) and PACAP−/− (b) ventricle zone (VZ) analyzed on coronal sections from the mid-dorsolateral cortex. BrdU-positive cells (green) and total cells (propidium iodide; red) were counted blind in the VZ on these confocal images. (c) Quantification of BrdU labeling index (LI). PACAP−/− exhibited a 47% LI compared to 60% in the WT. TUNEL staining of E9.5 WT (d) and PACAP−/− (e) VZ. TUNEL-postive cells (green) and total cells (DAPI; blue) were counted blind. (f) Quantification of TUNEL labeled cells per section. n=3 for each genotype. Results are expressed as mean ± SEM, **P<0.01. Scale bar, 10 μm.
Characterization of precursor cell culture
At E10.5 the rat cerebral cortex (similar to E9.5 mouse; Figure 1) consists primarily of VZ precursors, with the majority proliferating [Figure 2a]. To maintain precursor survival, we used defined media containing multiple trophic factors including insulin, bFGF and BDNF. In the absence of trophic factors, early precursors were unable to survive for 20 h even when incubated in Neurobasal plus B27 media (not shown). In this model, 96.2±3.2% of the E10.5 precursors expressed RC1 and 98.1±5.4% expressed nestin at 24 h, both markers of neural precursors [Figure 2b, c]; 15.6±2.2% of the cells expressed β III tubulin (TuJ1), an early marker of postmitotic neurons [Figure 2d, e] and this percentage increased to 19.8±2.9% at 48h, suggesting the differentiation potential of these precursors. In contrast, neither 24 h nor 48 h cultures exhibited glial antigens, including markers of astrocytes, glial fibrillary acidic protein (GFAP), nor oligodendrocyte progenitors, NG2 (not shown). Under identical conditions, cultures of E11.5 and E12.5 precursors at 24 h exhibited diminished precursor markers compared to the younger age (92.1% and 88.4% expressing RC1; 87.9% and 86.7% expressing nestin, respectively) and an increase in neuronal protein (18.1% and 43.9% β III tubulin, respectively) [Figure 2f], suggesting age dependent differences in developmental capacity at the time of plating, as reported previously (Romito-DiGiacomo et al., 2007).
Figure 2.

Characterization of a new precursor culture model. (a) Photomicrograph of a rat embryo at E10.5. White box indicates the telencephalic region dissected for culture. Scale bar, 1000 μm. (b–f) Cortical cultures consist of neural precursors and differentiating neurons at 24 h. Immunocytochemical analysis of cell type specific markers: (b) RC1 (c) nestin (d) β III tubulin (e) merged. Arrowheads indicate RC1 and nestin double positive cells, double arrowheads indicate β III tubulin positive cells, and arrows indicate triple positive cells. (e) Scale bar, 10 μm. (f) Examples of typical triple immunostaining of E10.5–E12.5 precursors are shown. Cells identified by arrowheads are magnified in the insets, with dashed lines emphasizing their general morphology. Below, the percentage of labeled cells at 24 h is quantified. Note that a greater proportion of cells exhibit β III tubulin at E12.5, when we observe many cells with extended processes. Data were obtained from three separate experiments, 100–150 cells analyzed per staining. Scale bar, 10 μm.
PACAP elicits age-dependent effects on DNA synthesis in both rat and mouse
To define PACAP proliferative activity, we examined effects of exogenous PACAP using rodent cortical precursors isolated at different embryonic ages under identical culture conditions. Initial studies employed rat, rather than mouse, precursors based on the more extensive previous work in the former model (Lu and DiCicco-Bloom, 1997; Vaccarino et al., 1999; Nicot and DiCicco-Bloom, 2001; Suh et al., 2001; Carey et al., 2002; Noctor et al., 2004; Schaar et al., 2004; Malagelada et al., 2011; Tury et al., 2011). In E10.5 rat precursors incubated for 24 h, PACAP exposure elicited a 33% increase in [3H] thymidine incorporation [Figure 3a rat], indicating the peptide increased the amount of DNA synthesis. However, in cultures isolated from embryos just one day older (E11.5), PACAP elicited no effects on [3H] thymidine incorporation. Subsequently, at E12.5 and E14.5, PACAP inhibited mitosis by 23% and 39% respectively, suggesting mitogenic effects are developmental stage dependent [Figure 3a]. The anti-mitogenic effects elicited at older ages are consistent with previous studies that used media lacking the panel of trophic factors (Lu and DiCicco-Bloom, 1997; Nicot and DiCicco-Bloom, 2001; Suh et al., 2001; Tury et al., 2011).
Figure 3.

PACAP exhibits age-dependent effects on DNA synthesis and promotes G1 cyclins and G1/S phase progression in vitro. (a) Effects of PACAP exposure on DNA synthesis at 24 h in rat and mouse precursors from different ages. The timeline above the graphs details the experimental paradigm. DNA synthesis was assessed using [3H] thymidine incorporation. Data were obtained from five experiments for each species, using three wells (25,000 cells/well) per group for each experiment. Data are expressed as percent control: control values ranged from 4000–12000cpm, *P<0.05, **P<0.01. (b) PACAP treatment increases the proportion of E10.5 rat precursor cells engaged in S phase. BrdU positive cells (green) and total cells (phase) were counted in 10 randomly selected fields. Scale bar, 10 μm. PACAP exposure increased the BrdU LI from 36% in control to 45% in the PACAP group at 24 h. (c) Flow cytometric analysis of BrdU immunolabeling of control and PACAP treated E10.5 rat precursors. PACAP treatment increased S phase labeling from 43% to 50% at 24 h. (d) Flow cytometric analysis of DNA content using propidium iodide indicates more cells were engaged in S phase in response to PACAP exposure at 24 h. (e) Real time PCR analysis indicates that PACAP treatment increased cyclin D1 mRNA by 221%, cyclin mRNA D2 by 116% and cyclin mRNA D3 by 118%, but not cyclin E (not shown), at 24 h. Data were obtained using total RNA (50–75 embryos for each isolation) obtained from 3 separate experiments. (f) PACAP treatment increased protein levels of cyclin D1 by 58% and D2 by 74% at 24 h, as defined by western blot analysis. Quantifications were performed on 3–4 separate blots for each protein, cyclins D1, D2, D3 and E.
To allow comparison to genetic deletion mutants, we extended study to the mouse, examining comparable developmental stages, E9.5 to E13.5. In E9.5 mouse precursors, PACAP exposure increased [3H] thymidine incorporation by 23% compared to vehicle, stimulation that was comparable to that in E10.5 rat. Similarly, PACAP elicited no effect at E10.5 (E11.5 in rat) and induced a 30% decrease on E13.5 (E14.5 in rat), suggesting mitogenic effects are developmental stage dependent in both species [Figure 3a mouse]. However, since PACAP elicited similar effects in both species, we continued our studies using rat cortical precursors, because they are more convenient to isolate and maintain, and allow comparison with extensive previous studies.
As PACAP might potentially activate three related receptors, including PAC1, VPAC1 and VPAC2, to elicit mitogenic stimulation, we examined the effects of related peptide, vasoactive intestinal peptide (VIP), because at physiological concentrations, it activates only VPAC1 and VPAC2 receptors, having only low affinity for PAC1(Arimura, 1992). VIP (10 nM) treatment did not elicit any change in DNA synthesis of E10.5 rat precursors at 24 h (Con=100±8.2%,VIP=107±6.4%; mean [3H] thymidine incorporation, expressed as % Control±SEM), suggesting that if they are present, neither VPAC receptor links to mitogenic regulation at this stage. This also implies that PACAP acts via the PAC1 receptor at E10.5 to stimulate [3H] thymidine incorporation.
PACAP promotes G1/S progression and cell division without altering cell survival at E10.5
While PACAP increased DNA synthesis by 33% in E10.5 cultures [Figure 3a], this effect may be attributable to either promoting G1/S progression, or alternatively, preventing precursor cell death, since PACAP has well documented neurotrophic activity (Waschek, 2002; Ohta et al., 2006; Vaudry et al., 2009). To begin addressing this question, E10.5 precursors engaged in S phase were labeled with a 4 h BrdU pulse to define the labeling index at 24 h. After PACAP exposure, the BrdU LI was increased from 36% in control to 45% in the PACAP group, as assessed by immunocytochemistry of precursor cultures [Figure 3b], consistent with PACAP increasing G1/S progression. To verfiy these changes, we also employed alternative measures, specifically, flow cytometric analysis of cultured cells after 24 h incubation. Consistent with BrdU immunocytochemistry, BrdU flow cytometry indicated that PACAP treatment also increased the S-phase percentage from 43% in control to 50% after peptide exposure. [Figure 3c]. Moreover, by analyzing single cell levels of propidium iodide that quantitatively binds DNA, we also observed that the fraction of cells engaged in S phase increased from 38% in control to 45% in the PACAP treated cultures [Figure 3d]. This 7% increase represents a change of comparable magnitude to that observed with both BrdU based methods [Figure 3b– a 9% increase; Fig 3c– a 7% increase] and is consistent with the peptide increasing G1/S phase progression in vitro.
If PACAP enhanced G1 to S phase progression, the peptide might act via cell cycle machinery, especially G1 cyclins. Although limited tissue availability hampered analyses, we began examining this issue by performing real-time PCR and found that cyclins D1, D2 and D3 (but not cyclin E, not shown) were all increased at 24 h by PACAP exposure of E10.5 precursors [Figure 3e]. These changes in D cyclins at the transcriptional level encouraged us to examine PACAP effects at the translational level as well, by performing western blot analyses using the very limited tissues from E10.5 cortices. Consistent with the real-time PCR results, PACAP also increased cyclin D1 and D2 protein expression levels by 58% and 74% respectively [Figure 3f], suggesting that PACAP upregulates G1 D cyclins to promote G1/S phase progression.
To determine whether cells engaged in S phase were able to complete mitosis and divide, we labeled a cohort of precursors by BrdU exposure from 20 h to 24 h, and following PBS washes, cells were either fixed immediately, to enumerate cells in S phase, or alternatively, were incubated another 12 h in either control or PACAP containing media. If cells that were engaged in S phase (incorporated BrdU) at 24 h subsequently underwent division, the absolute number of BrdU labeled cells would increase from 24 h to 36 h. Indeed, at 36 h, both control and PACAP groups demonstrated an increase in BrdU+ cells, indicating that cells successfully underwent division, with the PACAP group producing more new cells than control [Figure 4a]. To examine the potential role of cell survival, we assessed the total numbers of cells by phase microscopy as well as by using the FDA/PI assay. At 24 h, when the BrdU LI was increased by PACAP [Figure 3b], total cell numbers were not different between control and PACAP exposed cultures [Figure 4b, 24 h], consistent with PACAP promoting S phase entry. Further, there was no difference at 24 h in cell survival among groups compared to the number of cells 2 h after plating [Figure 4c], suggesting that PACAP did not exhibit trophic activity during this period. However, while there were no differences in control and PACAP group cell numbers or cell survival at 24 h, differences did emerge at 36 h. The number of cells in control media at 36 h was similar to its 24 h value [Figure 4b, compare Con 24 h to 36 h]. In contrast, the PACAP treated group displayed a 61% increase in total cells compared to its 24 h value [Figure 4b, PACAP 36 h], suggesting that enhanced S phase entry induced by PACAP at 24 h resulted in more neurogenesis 12 h later. It should be noted that while both control and PACAP treated groups exhibited S phase entry and division of these labeled cells, i.e., cell division, [Figure 3a], the total cell population did not necessarily increase in both groups [Figure 3b]: We know that there is ongoing cell death in both groups, with only ~80% of plated cells surviving at 24 h [Figure 3c]. We suspect that the enhanced proliferation in the PACAP group exceeded the losses that are ongoing in the model. In aggregate, these data suggest that PACAP promotes precursor proliferation by increasing the proportion of cells that enter S phase and complete cell division.
Figure 4.

Effects of PACAP on cell division, cell number and cell survival. (a) Quantification of total BrdU positive cells: vehicle (control) or PACAP was added at cell plating. Precursors in S phase were labeled with BrdU from hour 20 to 24, and one group of sister control and PACAP cultures were fixed at 24 h. A parallel set of control and PACAP cultures were rinsed twice with PBS, and incubated in fresh control or PACAP medium for another 12 h and then fixed at 36 h. At 24 h, 59% more cells were in S phase in the PACAP exposed group compared to control, consistent with increased BrdU LI in Figure 3b. At 36 h, the number of BrdU+ cells increased in both groups, indicating that cells that entered S phase subsequently went on to divide. (b) Quantification of total cell number in control and PACAP-treated cultures at 24 h and 36 h. The y-axis corresponds to the total number of cells counted in 10 random fields. Whereas no difference in cell number among groups was observed at 24 h, a 61% increase was detected in the PACAP group at 36 h compared to its 24 h value, suggeting cell proliferation. Note there was no change in control cell numbers between 24 h and 36 h. (c) Effects of PACAP on cell survival and cell death. PI and FDA were used to distinguish dead cells (arrowheads) and living cells (arrows) respectively. Scale bar, 10μm. Percentage cell survival was determined as the ratio of
FDA-stained cells at 24 h over 2 h. Results are expressed as mean ± SEM, *P<0.05, **P<0.01. Data were derived from three different experiments, two dishes per group per experiment.
Developmental transition of PAC1 receptor isoform expression during neurogenesis
Since PACAP elicited opposing effects on proliferation of precursors at different ages, we speculated the effects may reflect expression of distinct PAC1 isoforms. Thus, we examined expression of short and hop isoforms in E10.5 and E14.5 rat cortices using traditional RT-PCR (not shown) and found that both variants were present, consistent with previous studies (Waschek et al., 1998; Zhou et al., 2000; Suh et al., 2001).To quantify stage-dependent expression of the isoforms, we performed quantitative real-time PCR. Using standard curves for both short and hop cDNAs, we assessed short and hop mRNA expression levels at E10.5 and E14.5. There was 24-fold greater expression of the hop than the short isoform at E10.5 [Figure 5a left panel]. In contrast, by E14.5 the short isoform mRNA was up-regulated markedly, while hop isoform changed little, reversing the ratio, so that short was 15-fold greater than hop [Figure 5a right panel]. The developmental transition in the ratio of PAC1 isoform mRNAs suggested that change in their expression may underlie the stage specific mitogenic effects of PACAP.
Figure 5.

Developmental transition of PAC1 receptor mRNA isoform expression during corticogenesis and regulation by retinoic acid. (a) At E10.5, the level of the hop isoform is 24-fold greater than short; at E14.5, short is 15-fold greater than hop. Values are presented as fold difference when compared with the E10.5 hop mRNA levels arbitrarily set at 100. Values are representative of three experiments, three to eight animals per group for each experiment. ***P<0.001. (b) RA exposure increased differentially the short and hop isoforms at 24 h, detected using real time PCR. Data represent three experiments, two dishes per group, N=6 (2 × 106 cells/dish) for RNA quantification. (c) E10.5 precursors were incubated with either vehicle (ethanol) or RA (30 nM) at plating. At 24 h, cultures received either PACAP vehicle (Con) or PACAP (10 nM) and DNA synthesis was assessed at 48 h. In the presence of RA, PACAP increased precursor DNA synthesis, while the peptide had no effect in the RA vehicle group. Note that RA alone had no effect on DNA synthesis.; Data represent 3 wells per group (25,000 cells/well) in each of 3 experiments for [3H] thymidine incorporation. *P<0.05, ***P<0.001.
Retinoic acid treatment maintains hop isoform expression and PACAP mitogenic activity
The foregoing data suggest that expression of the hop isoform, known to activate PLC pathways, contributes to PACAP mitogenic stimulation in early precursors. Indeed, in E14.5 precursors, this stimulatory mechanism was established by overexpressing the hop isoform using transfection (Nicot and DiCicco-Bloom, 2001). To examine E10.5 precursors, we first asked whether a stimulus that promoted hop expression would also sustain PACAP mitogenic stimulation during development. We explored several hormone pathways known to affect early neurogenesis including steroids and retinoic acid (RA) (Studer et al., 1994; Haskell and LaMantia, 2005), as the latter is known to modulate PAC1 expression levels (Waschek et al., 1997). To examine RA effects, E10.5 rat precursors were incubated with vehicle (ethanol) or RA (30 nM) for 24 h, and real time PCR was performed. RA treatment increased the levels of the hop isoform 3-fold, whereas the short isoform was only enhanced by 50% [Figure 5b]. To define the effects of PACAP after altering the receptor isoform ratio, we then added PACAP or its vehicle for another 24 h, and measured DNA synthesis. It should be noted that after 24 h in culture, E10.5 precursors may undergo the same transition in mitogenic response as precursors in vivo: E10.5 precursors first incubated for 24 h exhibited no response to addition of PACAP [Figure 5c vehicle]. These data may suggest that incubation of E10.5 precursors allows them to become developmentally equivalent to E11.5 in PACAP response [Figure 3a]. In contrast, in the RA treated group in which the hop isoform exceeded the short isoform, PACAP exposure increased DNA synthesis by 21% [Figure 5c RA], indicating that the increased hop to short ratio correlates with PACAP mitogenic stimulation. On the other hand, RA treatment alone did not stimulate DNA synthesis [Figure 5c]. While RA is normally a potent inducer of neuronal differentiation and not of proliferation during development, it surprisingly appears to serve a different function in our culture system, conferring a sensitivity to a pro-mitotic action of PACAP. Regardless, to more directly assess the role of the hop receptor isoform to PACAP stimulation, we turned to molecular approaches.
Knockdown of hop expression blocks PACAP's pro-mitogenic activity
To examine whether high levels of hop expression are required for PACAP mitogenic stimulation in early precursors, we designed 2 hop short hairpin RNAs (shRNA) to rat PAC1hop. To first define the efficacy of the hop shRNAs, we examined their effects on rat PAC1 hop and short expression vectors. These studies were performed in human HEK-293 cells which only express human PAC1, likely the short isoform, which is not targeted by rat hop shRNAs. When the empty control vector was expressed in HEK cells, the endogenous human PAC1 protein was observed [Figure 6a, left] since the available PAC1 antibodies identify both rodent and human receptor proteins. Transfection of HEK cells with the rat hop expression vector alone increased levels of total PAC1 protein 2-fold [Figure 6a, left]. When hop protein was coexpressed with either shRNA#1 or shRNA#2, the increased levels were modestly decreased, whereas transfections of both shRNAs reduced overexpressed proteins levels by more than 75%, approaching those of endogenous human PAC1 [Figure 6a, left]. In marked contrast, the hop shRNAs did not block rat PAC1 short isoform overexpession [Figure 6a, right], suggesting the shRNAs specifically targeted hop specific sequences. Therefore, we used the combination of both #1 and #2 shRNAs in neuronal cultures. We silenced hop expression in E10.5 precursors, in the absence or presence of PACAP for 24h, and compared BrdU LI to control shRNA. Control shRNA does not correspond to known rodent transcripts. The shRNA vector transfected cells were identified by red fluorescence from the pSiren-DNR-DsRed-Express vectors [Fig. 6b]. In the control shRNA group, PACAP elicited a significant increase in BrdU LI compared to vehicle (Vehicle=30.8%, PACAP=39.3%). In contrast, in the presence of hop shRNAs, PACAP no longer stimulated S phase entry in early cortical precursors [Fig. 6b quantification]. The hop shRNA alone (Vehicle) also had no effect compared to control shRNA vector, suggesting that ongoing S phase entry does not depend on hop signaling. These observations suggest that PACAP mitogenic stimulation requires hop isoform expression. The blockade of PACAP stimulation in the presence of hop isoform knock down is consistent with the converse experiment, in which enhanced hop expression elicited by RA treatment was associated with sustained PACAP stimulation of DNA synthesis [Figure 5b, c]. Since PACAP mitogenic activity through the hop isoform is known to depend on PLC pathways in other models, we next defined signaling transduction pathways activated by PACAP in young neural precursors.
Figure 6.

shRNA knockdown of PAC1 hop expression. (a) pEAK rapid HEK-293 cells were transfected with control vector, or full length rat hop or short expression vectors without and with hop shRNAs. PAC1 antibody detects both hop and short proteins including both human as well as rat. Data were derived from three experiments. Transfection efficiency was 50–60% per group. (b) E10.5 precursors were transfected with either control shRNA or both hop shRNAs 2 h after plating, in the absence of PACAP (Vehicle) or the presence of PACAP (10 nM). BrdU was added at 22 h, and cells were fixed at 24 h. Cells that were transfected with control shRNA or hop shRNAs can be visualized by red fluorescence. The four groups were analyzed by one-way ANOVA, followed by Tukey's multiple comparison test. Six transfections from three experiments were performed, and transfection efficiency was 10–15% in E10.5 precursors. The number of cells analyzed from each experiment was 20–35 per dish. Scale bar, 10 μm. *p<0.05, **P<0.01.
The role of PLC activation in PACAP's pro-mitogenic effects
In previous studies, PACAP mitogenic stimulation has been associated with activation of PLC that triggers the PKC pathway (Spengler et al., 1993; Lu et al., 1998; Nicot and DiCicco-Bloom, 2001). Since hop is the dominant receptor isoform at E10.5, we hypothesized that PLC activation may underlie PACAP stimulation. To investigate the role of PLC, we employed a membrane-permeable PLC specific inhibitor, U-73122 and the inactive form U-73343 as a negative control. We incubated E10.5 cultures for 16 h only to avoid overall cell toxicity. In the presence of inactive drug, PACAP elicited a 22% increase in [3H] incorporation (an increase at 16 h that is proportional to 33% at 24 h) whereas PACAP elicited no change when the PLC inhibitor was present, suggesting that pro-mitogenic effects were mediated via the PLC pathway [Figure 7]. Nonetheless, because of possible cell toxicity, we turned to more direct methods to detect cellular responses.
Figure 7.
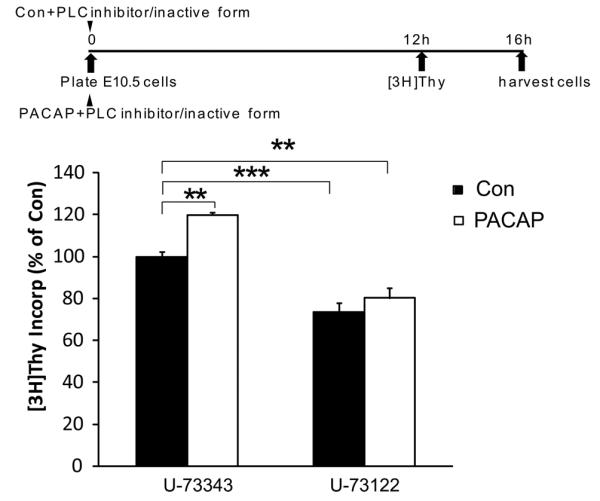
The role of PLC activation in PACAP's pro-mitogenic effects at E10.5. In the presence of the inactive analogue U-73343 (2 μM) for 16 h, PACAP increased DNA synthesis by 22%, while the peptide-induced increase in DNA synthesis was blocked by PLC antagonist U-73122 (2 μM). All data bars are normalized to the control treatment (vehicle) in the presence of inactive analogue U-73343, which was arbitrarily set at 100%. Control values ranged from 2800–7500 cpm. The four groups were analyzed by one-way ANOVA, followed by Tukey's multiple comparison test. Data were derived from four experiments, three wells per group for each experiment, **P<0.01, ***P<0.001.
PACAP induces intracellular calcium oscillations in E10.5 but not E14.5 cortical precursors
Activated PLC can hydrolyze phosphatidylinositol 4,5-bisphosphate to generate two second messengers, diacyl glycerol (DAG) and inositol 1,4,5 tris-phosphate (IP3). The latter signal engages the IP3 receptor and elicits release of Ca2+ from the endoplasmic reticulum. Ca2+ is a well-known stimulator of proliferation in multiple systems (Berridge et al., 2000; Lewis, 2003; Weissman et al., 2004). As the foregoing evidence raised the possibility of PLC signaling in PACAP mitogenic activity, we examined this downstream mediator, intracellular Ca2+. We monitored intracellular Ca2+ changes upon PACAP activation in E10.5 and E14.5 cortical precursors in parallel using Ca2+ fluorescence indicator Fluo-4 AM. PACAP triggered an increase of cytosolic Ca2+ in most E10.5 cortical precursors, suggesting the peptide activated the inositol phospholipid signaling pathway (Spengler et al., 1993; DiCicco-Bloom et al., 2000; Nicot and DiCicco-Bloom, 2001; Hegg et al., 2003) [Figure 8a, d]. Interestingly, the majority of E10.5 cells exhibited Ca2+ oscillations [Table 1]. In contrast, PACAP did not elicit Ca2+ changes in E14.5 cortical precursors [Figure 8b, e]. However, both E10.5 and E14.5 precursors were able to respond to 40 mM KCl [Figure 8c, f, g], suggesting that a depolarization induced Ca2+ response, specifically influx, was maintained by cells at both stages. We did note that PACAP induced complex Ca2+ oscillation curves, especially compared to those elicited by KCl [Figure 8d, f, g], but did not pursue this further as others have defined mechanisms of IP3 induced Ca2+ oscillations (Berridge et al., 2003; Hogan et al., 2010).
Figure 8.

PACAP induces intracellular Ca2+ fluxes in E10.5 but not E14.5 cortical precursors. (a–c) Representative color-coded images of intracellular Ca2+ fluxes. Cells were pre-loaded with Ca2+ fluorescent indicator Fluo-4 AM and recorded for 110 s after reagent addition. (d–g) Representative traces of intracellular [Ca2+] changes, which are reflected by △F/F0, △F=F-F0; F0 is fluorescence intensity before addition of reagents. PACAP (10 nM) exposure increased intracellular calcium levels in E10.5 (a, d) but not E14.5 precursors (b, e) at 2 h culture. KCl (40 mM) elicited increased intracellular Ca2+ in both E10.5 (f) and E14.5 (c, g). Cells were monitored by BD Pathway 855 BioImager, at 37°C/5% CO2. Data were derived from three independent experiments using parallel cultures of E10.5 and E14.5 precursors in 96 well plates. 100–150 cells per group were randomly chosen in each experiment and fluorescence intensity values were quantified.
Table 1.
Calcium fluxes and oscillations in E10.5 and E14.5 cortical precursors.
| Calcium Fluxes | E10.5 | E14.5 |
|---|---|---|
| PACAP responsive cells | 201/220 (91%) | 7/306 (2%) |
| PACAP induced oscillations | 172/201 (86%) | 0/306 (0%) |
| KCl responsive cells | 137/168 (82%) | 398/412 (97%) |
Precursors were pre-loaded with Ca2+ fluorescent indicator Fluo-4 AM and recorded on a BD Pathway 855 BioImager for 110 s after addition of PACAP (10 nM) or KCl (40 mM).
PACAP activates PKC but not MAPK during early neurogenesis
Another signal downstream of PLC production of DAG and Ca2+ elevation is PKC (Nishizuka, 1984). Initial activation of PKC can be mediated by its phosphorylation, which may serve as a molecular marker (Pearce et al., 2010). PACAP treatment (30 min) produced a two-fold increase in phospho-PKC immunoreactivity in E10.5 cortical precursors, a response nearly identical to that elicited by the potent PKC agonist 12-O-tetradecanoylphorbol-13-acetate (TPA) [Figure 9]. In contrast, PACAP did not elicit changes in phospho-PKC levels of E14.5 precursors, although a robust 3-fold response was elicited by TPA [Figure 9 E14.5]. In addition, since PACAP has been shown to activate MAPK pathways in several culture systems (Barrie et al., 1997; Vaudry et al., 2002a), we performed similar studies and assessed phosphorylated ERK. However, PACAP did not elicit increased phospho-ERK in either E10.5 or E14.5 cultures, while bFGF, a relevant cortical mitogen (Vaccarino et al., 1999; Li and DiCicco-Bloom, 2004), elicited robust ERK activation in parallel dishes at both ages [Figure 10], suggesting PACAP mitogenic signaling is independent of ERK pathways in early precursors, consistent with previous work (Gerdin and Eiden, 2007).
Figure 9.

PACAP activates PKC in early E10.5 cortical precursors but not in E14.5 cells. (a) Patterns of phospho-PKC staining in E10.5 and E14.5 cortical precursors 30 min after treatment with vehicle (Con), PACAP (10 nM) and PKC agonist TPA (200 nM). PACAP exposure increased phospho-PKC+ cells by 2-fold compared to vehicle in 2 h cultures, comparable to the PKC agonist at E10.5. In contrast, PACAP did not elicit changes in phospho-PKC+ cells at E14.5, while both ages responded to agonist TPA. Positive cells are indicated by arrows and negative cells by arrowheads. (b) Quantification of phospho-PKC immunostaining. Cells were plated in 35 mm dishes in defined media without growth factors, and reagents were added at 2 h for 30 min. Data are representative of three experiments, three dishes per group per experiment. ***P<0.001
Figure 10.

PACAP did not activate phospho-ERK while bFGF exposure led to ERK activation in both E10.5 and E14.5 precursors. (a) Examples of phospho-ERK staining in E10.5 and E14.5 10 min after treatment with vehicle (Con), PACAP (10 nM) and bFGF (10 ng/ml). (b) Qunatification of phospho-ERK immunostaining. Cells were plated in 35 mm dishes in defined media without growth factors, and reagents were added at 2 h for 10 min. Data are representative of three experiments, three dishes per group.**P<0.01, ***P<0.001. Scale bar, 10 μm.
Discussion
Our observations provide compelling evidence that PACAP contributes to neuronal precursor proliferation through age-dependent bi-directional activity during cortical development. PACAP exhibits pro-mitogenic effects during early neurogenesis, while the peptide may restrain proliferation from E12.5 onward. The temporally specific effects on proliferation are mediated by developmental expression of PAC1 receptor isoforms, with hop predominating early followed by short expression later. PACAP signaling provides a novel example in the developing nervous system that alternative splicing of receptor mRNA isoforms differentially controls cortical proliferation.
During early neurogenesis, when stem cells are proliferating to enlarge precursor pools, PACAP enhances proliferation through the hop isoform: it increases DNA synthesis, and promotes G1/S progression and cell division without affecting survival. The stimulation elicited by PACAP in vitro is consistent with reduced mitotic labeling in the KO in vivo, where the 13% reduction is comparable to the 14% decrease observed in the bFGF/FGF2 null (Vaccarino et al., 1999). However, several extracellular factors (FGF2, Wnts, Shh, BMPs) that regulate proliferation and neurogenesis co-exist in developing cortex (Stevens et al., 2010), and may partially compensate one another. While PACAP may elicit modest effects due to a selectively responsive cell subset, PAC1 expression appears to be nearly universal (Basille et al., 2000; Suh et al., 2001; Nishimoto et al., 2007), though PAC1 isoform selective reagents have not yet been developed.
During early neurogenesis when hop predominates, PACAP activates PLC and calcium fluxes, enhancing PKC phosphorylation and G1 stage D cyclins. Significantly, the important role for the PAC1 hop isoform in early precursors is indicated by the blockade of PACAP mtiogenic stimulation in presence of hop shRNAs. To our knowledge, this is the first use of knock down techniques to define activities of hop isoform, and for defining roles of third intracellular loops of GPCRs. Conversely, maintaining elevated hop/short ratios through RA treatment, sustained pro-mitogenic activity beyond this early developmental window. However, with increasing age and short expression, PACAP promotes cell cycle exit. PACAP induces cell cycle exit by selective increases in cyclin dependent kinase inhibitor p57kip2 (not p27kip1) activity, and association with CDK2/cyclin E complex (Carey et al., 2002; Tury et al., 2011).
One caveat of these studies is use of wild type cells, which release PACAP as an autocrine regulator of proliferation (Lu and DiCicco-Bloom, 1997; Suh et al., 2001). We attempted to compare roles of exogenous and endogenous PACAP by dissecting littermates from heterozygote crosses, but too few E9.5 cells were available for single cortex analysis. However, we assessed E14.5 WT and PACAP KO precursors and found that PACAP elicited ~35% reductions in DNA synthesis (WT: Con=100±9%, PACAP=62±6%; KO: Con=100±7%, PACAP=67±8%; N=6 wells, two experiments), consistent with previous anti-mitogenic effects (Lu and DiCicco-Bloom, 1997; Lu et al., 1998; Suh et al., 2001; Carey et al., 2002). Significantly, there was no difference in survival of PACAP KO precursors compared to WT littermates, suggesting trophic mechanisms were not involved at E14.5, as we observed at E10.5 [Figures 1, 4]. Thus, in the absence of peptide, PACAP response systems appear to function normally, as suggested by previous expression analyses (Girard et al., 2006).
Current neural precursor/stem cell cultures include adherent cells, neurospheres, mixed cell co-cultures, and serum-free suspension cultures (Qian et al., 1998; Shen et al., 2004; Watanabe et al., 2005; Shen et al., 2006). These systems may be technically demanding, requiring cell shaking and centrifugation (Qian et al., 1998), preplating with endothelial cells (Shen et al., 2004), or incubation with soluble factors (Watanabe et al., 2005). Procedures for our culture system are relatively less complicated. Further the precursors are ready for immediate use, an attribute that may preserve comparability to their in vivo state, and may mitigate against emergence of traits reflecting adaptation to culture. The defined culture medium contains a restricted panel of factors (insulin, bFGF and BDNF). While this simplified medium may not support certain developmental events, we were able to test effects of steroids and RA, as well as employ shRNA knockdown techniques. One possible limitation is use of trypsin to separate overlying epidermis from brain, since this may alter surface receptors or ion channels. There may be variability in dissecting cortex away from the medial (future hippocampus) and ventral (future ganglionic eminences) telencephalic vesicles, since specific boundary markers are unavailable. Nonetheless, PACAP's consistent stimulatory effect at E10.5 and its opposite activity at E14.5 suggests the temporal transition in PAC1 isoforms may control forebrain development. Future studies should define the precise localization of PAC1 isoform expression in combination with lineage specific markers for cortex, hippocampus and basal ganglia.
Our observations suggest that precursors in vitro undergo the developmental transition in response to PACAP observed in vivo. While E10.5 cells exhibited mitotic stimulation in response to PACAP, if first cultured for 24 h, PACAP then elicited no increase in DNA synthesis, a response that is identical to that of cells obtained directly from E11.5 embryos, though additional studies may be required. The fact that RA induced PAC1 hop isoform expression and maintained PACAP mitogenic activity indicates that these cells are plastic and responsive to environmental cues. We chose RA because previous studies indicate it regulates PACAP receptors (Waschek et al., 1997). RA can promote the transition from radial glial symmetric division to asymmetric production of intermediate cortical precursors and neurons (Siegenthaler et al., 2009). Thus it is possible RA induces changes in the proportions of cortical precursors that may influence PACAP effects, an interesting direction for further studies. Regardless, our culture system provides a different platform in which to elucidate intrinsic and extrinsic regulation of neural stem cells and may provide insights into stem cell division patterns, potency and windows of plasticity.
Understanding regulation of PACAP pro-mitogenic activity could provide a strategy for stem cell based regenerative therapy since PACAP stimulates neural stem cell proliferation and promotes neurogenesis in vitro and in vivo (Mercer et al., 2004; Ohta et al., 2006; Scharf et al., 2008; Fang et al., 2010), though it can also modulate gliogenesis (including oligodendrocytes and astrocytes) under different conditions (Lee et al., 2001; Nishimoto et al., 2007). It remains uncertain whether short (Ohta et al., 2006; Scharf et al., 2008) or hop (Mercer et al., 2004) isoforms mediated proliferative stimulation since both forms were detected and both cAMP and PLC/PKC pathways were activated in adult subventricular zone and dentate gyrus, suggesting both regional and temporal specificity is involved. Our study demonstrates that PACAP pro-mitogenic activity is mediated through the hop isoform and that developmental upregulation of the short isoform may restrain precursor proliferation in developing cortex. Furthermore, while cAMP and ERK activation are both downstream of PACAP in some systems (Gerdin and Eiden, 2007), our studies suggest hop isoform stimulates early cortical precursor proliferation through PLC/PKC pathways and G1 cyclins, though we cannot exclude involvement of other signaling mechanisms. In addition, while continued expression of hop mRNA (and presumably protein) in E14.5 precursors would predict a phospho-PKC response to PACAP treatment at this age, its absence raises several questions regarding the coupling of PAC1 hop to G proteins, age dependent expression of G protein family members, downstream signaling, or inhibitory cross talk between short and hop isoforms that warrant future study.
The model of a developmental transition in PAC1 splice isoform expression provides an important target for future exploration of mRNA splicing mechanisms. Developmental control of neurogenesis through RNA splicing of receptors that regulate proliferation is not commonly reported in the nervous system. However, alternative isoforms of cytosolic adaptor protein NUMB regulate proliferation and differentiation in P19 carcinoma cells (Verdi et al., 1999) and cortical precursors (Bani-Yaghoub et al., 2007). Similarly in cancer cells, the predominant fibroblast growth factor receptor 3 (FGFR3) elicits stimulation, whereas a normal FGFR3 splice variant (△8–10) inhibits proliferation. The regulation of proliferation depended on the ratio of different splice isoforms, with loss of the inhibitory variant leading to uncontrolled proliferation (Tomlinson et al., 2005). In adult cerebral cortex, antagonistic effects of dopamine in the synapse also depend on the ratio of dopamine D2 receptor isoforms, D2L and D2R (Picetti et al., 1997), which are generated by alternative splicing of a 29 amino acid insert in the third intracellular loop (Usiello et al., 2000), similar to PAC1. Control of pre-mRNA splicing is a complex process that involves many layers of regulation (Li et al., 2007; Greenberg et al., 2009). Recent research indicates that alternative splicing is frequent during embryonic development (E8.5–E11.5 in mouse) including the PACAP system, and alternative splicing is disproportionately involved in neurogenesis (Revil et al., 2010). Thus defining splicing regulation may provide insights into neuropsychiatric disorders, such as schizophrenia, PTSD and autism (Hashimoto et al., 2007; Nijmeijer et al., 2010; Ressler et al., 2011)
In conclusion, PACAP signaling influences precursor mitosis during brain development. The developmental switch in PAC1 hop and short isoforms converts PACAP mitogenic stimulation to inhibition. It is likely that the PAC1 receptor is but one of many signals governing corticogenesis that is subject to regulation by mechanisms controlling mRNA splicing.
Acknowledgments
This work was supported by NIH grant NS32401 (E.D-B), HL069000, AG028614 (J.M.), HD34475, HD06576 and HD04612 (J.W.) and UMDNJ Foundation (#62-09) (Z.P.). We thank Karl Herrup for valuable critical review, Richard Mains for inspiring guidance and discussion, Betty Eipper and Yanping Wang for technical support.
Footnotes
No conflict of interest
References
- Arimura A. Pituitary adenylate cyclase activating polypeptide (PACAP): discovery and current status of research. Regul Pept. 1992;37:287–303. [PubMed] [Google Scholar]
- Bani-Yaghoub M, Kubu CJ, Cowling R, Rochira J, Nikopoulos GN, Bellum S, Verdi JM. A switch in numb isoforms is a critical step in cortical development. Dev Dyn. 2007;236:696–705. doi: 10.1002/dvdy.21072. [DOI] [PubMed] [Google Scholar]
- Barrie AP, Clohessy AM, Buensuceso CS, Rogers MV, Allen JM. Pituitary adenylyl cyclase-activating peptide stimulates extracellular signal-regulated kinase 1 or 2 (ERK1/2) activity in a Ras-independent, mitogen-activated protein Kinase/ERK kinase 1 or 2-dependent manner in PC12 cells. J Biol Chem. 1997;272:19666–19671. doi: 10.1074/jbc.272.32.19666. [DOI] [PubMed] [Google Scholar]
- Basille M, Vaudry D, Coulouarn Y, Jegou S, Lihrmann I, Fournier A, Vaudry H, Gonzalez B. Comparative distribution of pituitary adenylate cyclase-activating polypeptide (PACAP) binding sites and PACAP receptor mRNAs in the rat brain during development. J Comp Neurol. 2000;425:495–509. [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Carey RG, Li B, DiCicco-Bloom E. Pituitary adenylate cyclase activating polypeptide anti-mitogenic signaling in cerebral cortical progenitors is regulated by p57Kip2-dependent CDK2 activity. J Neurosci. 2002;22:1583–1591. doi: 10.1523/JNEUROSCI.22-05-01583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Waschek JA. Selective deficits in the circadian light response in mice lacking PACAP. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1194–1201. doi: 10.1152/ajpregu.00268.2004. [DOI] [PubMed] [Google Scholar]
- Culican SM, Baumrind NL, Yamamoto M, Pearlman AL. Cortical radial glia: identification in tissue culture and evidence for their transformation to astrocytes. J Neurosci. 1990;10:684–692. doi: 10.1523/JNEUROSCI.10-02-00684.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCicco-Bloom E, Deutsch PJ, Maltzman J, Zhang J, Pintar JE, Zheng J, Friedman WF, Zhou X, Zaremba T. Autocrine expression and ontogenetic functions of the PACAP ligand/receptor system during sympathetic development. Dev Biol. 2000;219:197–213. doi: 10.1006/dbio.2000.9604. [DOI] [PubMed] [Google Scholar]
- Fang KM, Chen JK, Hung SC, Chen MC, Wu YT, Wu TJ, Lin HI, Chen CH, Cheng H, Yang CS, Tzeng SF. Effects of combinatorial treatment with pituitary adenylate cyclase activating peptide and human mesenchymal stem cells on spinal cord tissue repair. PLoS One. 2010;5:e15299. doi: 10.1371/journal.pone.0015299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francone VP, Ifrim MF, Rajagopal C, Leddy CJ, Wang Y, Carson JH, Mains RE, Eipper BA. Signaling from the secretory granule to the nucleus: Uhmk1 and PAM. Mol Endocrinol. 2010;24:1543–1558. doi: 10.1210/me.2009-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdin MJ, Eiden LE. Regulation of PC12 cell differentiation by cAMP signaling to ERK independent of PKA: do all the connections add up? Sci STKE. 2007;2007:pe15. doi: 10.1126/stke.3822007pe15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard BA, Lelievre V, Braas KM, Razinia T, Vizzard MA, Ioffe Y, El Meskini R, Ronnett GV, Waschek JA, May V. Noncompensation in peptide/receptor gene expression and distinct behavioral phenotypes in VIP- and PACAP-deficient mice. J Neurochem. 2006;99:499–513. doi: 10.1111/j.1471-4159.2006.04112.x. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Xu B, Lu B, Hempstead BL. New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci. 2009;29:12764–12767. doi: 10.1523/JNEUROSCI.3566-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Arimura A, Gozes I, Journot L, Laburthe M, Pisegna JR, Rawlings SR, Robberecht P, Said SI, Sreedharan SP, Wank SA, Waschek JA. International Union of Pharmacology. XVIII. Nomenclature of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide. Pharmacol Rev. 1998;50:265–270. [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, et al. Pituitary adenylate cyclase-activating polypeptide is associated with schizophrenia. Mol Psychiatry. 2007;12:1026–1032. doi: 10.1038/sj.mp.4001982. [DOI] [PubMed] [Google Scholar]
- Haskell GT, LaMantia AS. Retinoic acid signaling identifies a distinct precursor population in the developing and adult forebrain. J Neurosci. 2005;25:7636–7647. doi: 10.1523/JNEUROSCI.0485-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegg CC, Au E, Roskams AJ, Lucero MT. PACAP is present in the olfactory system and evokes calcium transients in olfactory receptor neurons. J Neurophysiol. 2003;90:2711–2719. doi: 10.1152/jn.00288.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Lelievre V, Zhao P, Torres M, Rodriguez W, Byun JY, Doshi S, Ioffe Y, Gupta G, de los Monteros AE, de Vellis J, Waschek J. Pituitary adenylyl cyclase-activating polypeptide stimulates DNA synthesis but delays maturation of oligodendrocyte progenitors. J Neurosci. 2001;21:3849–3859. doi: 10.1523/JNEUROSCI.21-11-03849.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS. Calcium oscillations in T-cells: mechanisms and consequences for gene expression. Biochem Soc Trans. 2003;31:925–929. doi: 10.1042/bst0310925. [DOI] [PubMed] [Google Scholar]
- Li B, DiCicco-Bloom E. Basic fibroblast growth factor exhibits dual and rapid regulation of cyclin D1 and p27 to stimulate proliferation of rat cerebral cortical precursors. Dev Neurosci. 2004;26:197–207. doi: 10.1159/000082137. [DOI] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Lu N, DiCicco-Bloom E. Pituitary adenylate cyclase-activating polypeptide is an autocrine inhibitor of mitosis in cultured cortical precursor cells. Proc Natl Acad Sci U S A. 1997;94:3357–3362. doi: 10.1073/pnas.94.7.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu N, Zhou R, DiCicco-Bloom E. Opposing mitogenic regulation by PACAP in sympathetic and cerebral cortical precursors correlates with differential expression of PACAP receptor (PAC1-R) isoforms. J Neurosci Res. 1998;53:651–662. doi: 10.1002/(SICI)1097-4547(19980915)53:6<651::AID-JNR3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Mairet-Coello G, Tury A, DiCicco-Bloom E. Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J Neurosci. 2009;29:775–788. doi: 10.1523/JNEUROSCI.1700-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Lopez-Toledano MA, Willett RT, Jin ZH, Shelanski ML, Greene LA. RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J Neurosci. 2011;31:3186–3196. doi: 10.1523/JNEUROSCI.4011-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Gotz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Mercer A, Ronnholm H, Holmberg J, Lundh H, Heidrich J, Zachrisson O, Ossoinak A, Frisen J, Patrone C. PACAP promotes neural stem cell proliferation in adult mouse brain. J Neurosci Res. 2004;76:205–215. doi: 10.1002/jnr.20038. [DOI] [PubMed] [Google Scholar]
- Nicot A, DiCicco-Bloom E. Regulation of neuroblast mitosis is determined by PACAP receptor isoform expression. Proc Natl Acad Sci U S A. 2001;98:4758–4763. doi: 10.1073/pnas.071465398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot A, Lelievre V, Tam J, Waschek JA, DiCicco-Bloom E. Pituitary adenylate cyclase-activating polypeptide and sonic hedgehog interact to control cerebellar granule precursor cell proliferation. J Neurosci. 2002;22:9244–9254. doi: 10.1523/JNEUROSCI.22-21-09244.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijmeijer JS, et al. Identifying loci for the overlap between attention-deficit/hyperactivity disorder and autism spectrum disorder using a genome-wide QTL linkage approach. J Am Acad Child Adolesc Psychiatry. 2010;49:675–685. doi: 10.1016/j.jaac.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto M, Furuta A, Aoki S, Kudo Y, Miyakawa H, Wada K. PACAP/PAC1 autocrine system promotes proliferation and astrogenesis in neural progenitor cells. Glia. 2007;55:317–327. doi: 10.1002/glia.20461. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308:693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Ohta S, Gregg C, Weiss S. Pituitary adenylate cyclase-activating polypeptide regulates forebrain neural stem cells and neurogenesis in vitro and in vivo. J Neurosci Res. 2006;84:1177–1186. doi: 10.1002/jnr.21026. [DOI] [PubMed] [Google Scholar]
- Pan Z, Damron D, Nieminen AL, Bhat MB, Ma J. Depletion of intracellular Ca2+ by caffeine and ryanodine induces apoptosis of chinese hamster ovary cells transfected with ryanodine receptor. J Biol Chem. 2000;275:19978–19984. doi: 10.1074/jbc.M908329199. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Picetti R, Saiardi A, Abdel Samad T, Bozzi Y, Baik JH, Borrelli E. Dopamine D2 receptors in signal transduction and behavior. Crit Rev Neurobiol. 1997;11:121–142. doi: 10.1615/critrevneurobiol.v11.i2-3.20. [DOI] [PubMed] [Google Scholar]
- Qian X, Goderie SK, Shen Q, Stern JH, Temple S. Intrinsic programs of patterned cell lineages in isolated vertebrate CNS ventricular zone cells. Development. 1998;125:3143–3152. doi: 10.1242/dev.125.16.3143. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, Norrholm SD, Kilaru V, Smith AK, Myers AJ, Ramirez M, Engel A, Hammack SE, Toufexis D, Braas KM, Binder EB, May V. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revil T, Gaffney D, Dias C, Majewski J, Jerome-Majewska LA. Alternative splicing is frequent during early embryonic development in mouse. BMC Genomics. 2010;11:399. doi: 10.1186/1471-2164-11-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romito-DiGiacomo RR, Menegay H, Cicero SA, Herrup K. Effects of Alzheimer's disease on different cortical layers: the role of intrinsic differences in Abeta susceptibility. J Neurosci. 2007;27:8496–8504. doi: 10.1523/JNEUROSCI.1008-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar BT, Kinoshita K, McConnell SK. Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron. 2004;41:203–213. doi: 10.1016/s0896-6273(03)00843-2. [DOI] [PubMed] [Google Scholar]
- Scharf E, May V, Braas KM, Shutz KC, Mao-Draayer Y. Pituitary adenylate cyclase-activating polypeptide (PACAP) and vasoactive intestinal peptide (VIP) regulate murine neural progenitor cell survival, proliferation, and differentiation. J Mol Neurosci. 2008;36:79–88. doi: 10.1007/s12031-008-9097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, Temple S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci. 2006;9:743–751. doi: 10.1038/nn1694. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, Napoli JL, Peterson AS, Pleasure SJ. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. doi: 10.1016/j.cell.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, Journot L. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993;365:170–175. doi: 10.1038/365170a0. [DOI] [PubMed] [Google Scholar]
- Stevens HE, Smith KM, Rash BG, Vaccarino FM. Neural stem cell regulation, fibroblast growth factors, and the developmental origins of neuropsychiatric disorders. Front Neurosci. 2010;4 doi: 10.3389/fnins.2010.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer M, Popperl H, Marshall H, Kuroiwa A, Krumlauf R. Role of a conserved retinoic acid response element in rhombomere restriction of Hoxb-1. Science. 1994;265:1728–1732. doi: 10.1126/science.7916164. [DOI] [PubMed] [Google Scholar]
- Suh J, Lu N, Nicot A, Tatsuno I, DiCicco-Bloom E. PACAP is an anti-mitogenic signal in developing cerebral cortex. Nat Neurosci. 2001;4:123–124. doi: 10.1038/83936. [DOI] [PubMed] [Google Scholar]
- Tomlinson DC, L'Hote CG, Kennedy W, Pitt E, Knowles MA. Alternative splicing of fibroblast growth factor receptor 3 produces a secreted isoform that inhibits fibroblast growth factor-induced proliferation and is repressed in urothelial carcinoma cell lines. Cancer Res. 2005;65:10441–10449. doi: 10.1158/0008-5472.CAN-05-1718. [DOI] [PubMed] [Google Scholar]
- Tury A, Mairet-Coello G, Dicicco-Bloom E. The Cyclin-Dependent Kinase Inhibitor p57Kip2 Regulates Cell Cycle Exit, Differentiation, and Migration of Embryonic Cerebral Cortical Precursors. Cereb Cortex. 2011;21:1840–1856. doi: 10.1093/cercor/bhq254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002a;296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Chen Y, Ravni A, Hamelink C, Elkahloun AG, Eiden LE. Analysis of the PC12 cell transcriptome after differentiation with pituitary adenylate cyclase-activating polypeptide (PACAP) J Neurochem. 2002b;83:1272–1284. doi: 10.1046/j.1471-4159.2002.01242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Rousselle C, Basille M, Falluel-Morel A, Pamantung TF, Fontaine M, Fournier A, Vaudry H, Gonzalez BJ. Pituitary adenylate cyclase-activating polypeptide protects rat cerebellar granule neurons against ethanol-induced apoptotic cell death. Proc Natl Acad Sci U S A. 2002c;99:6398–6403. doi: 10.1073/pnas.082112699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, Vaudry H. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol Rev. 2009;61:283–357. doi: 10.1124/pr.109.001370. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Bashirullah A, Goldhawk DE, Kubu CJ, Jamali M, Meakin SO, Lipshitz HD. Distinct human NUMB isoforms regulate differentiation vs. proliferation in the neuronal lineage. Proc Natl Acad Sci U S A. 1999;96:10472–10476. doi: 10.1073/pnas.96.18.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschek JA. Multiple actions of pituitary adenylyl cyclase activating peptide in nervous system development and regeneration. Dev Neurosci. 2002;24:14–23. doi: 10.1159/000064942. [DOI] [PubMed] [Google Scholar]
- Waschek JA, Lelievre V, Bravo DT, Nguyen T, Muller JM. Retinoic acid regulation of the VIP and PACAP autocrine ligand and receptor system in human neuroblastoma cell lines. Peptides. 1997;18:835–841. doi: 10.1016/s0196-9781(97)00015-6. [DOI] [PubMed] [Google Scholar]
- Waschek JA, Casillas RA, Nguyen TB, DiCicco-Bloom EM, Carpenter EM, Rodriguez WI. Neural tube expression of pituitary adenylate cyclase-activating peptide (PACAP) and receptor: potential role in patterning and neurogenesis. Proc Natl Acad Sci U S A. 1998;95:9602–9607. doi: 10.1073/pnas.95.16.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- Weissman TA, Riquelme PA, Ivic L, Flint AC, Kriegstein AR. Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron. 2004;43:647–661. doi: 10.1016/j.neuron.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Zhou C, Kikuyama S, Nakajo S, Hirabayashi T, Mizushima H, Shioda S. Splice variants of PAC(1) receptor during early neural development of rats. Peptides. 2000;21:1177–1183. doi: 10.1016/s0196-9781(00)00257-6. [DOI] [PubMed] [Google Scholar]