

Abstract
Small ubiquitin-like modifier (SUMO1, 2, 3) is a group of proteins that conjugate to lysine residues of target proteins thereby modifying their activity, stability, and subcellular localization. A large number of SUMO target proteins are transcription factors and other nuclear proteins involved in gene expression. Furthermore, SUMO conjugation plays key roles in genome stability, quality control of newly synthesized proteins, proteasomal degradation of proteins and DNA damage repair. Any marked increase in levels of SUMO-conjugated proteins is therefore expected to have a major impact on the fate of cells. We show here that SUMO conjugation is activated in human astrocytic brain tumors. Levels of both SUMO1- and SUMO2/3-conjugated proteins were markedly increased in tumor samples. The effect was least pronounced in low-grade astrocytoma (WHO Grade II) and most pronounced in glioblastoma multiforme (WHO Grade IV). We also found a marked rise in levels of Ubc9, the only SUMO conjugation enzyme identified so far. Blocking SUMO1-3 conjugation in glioblastoma cells by silencing their expression blocked DNA synthesis, cell growth and clonogenic survival of cells. It also resulted in DNA-PK-dependent phosphorylation of H2AX, indicative of DNA double-strand damage, and G2/M cell cycle arrest. Collectively, these findings highlight the pivotal role of SUMO conjugation in DNA damage repair processes and imply that the SUMO conjugation pathway could be a new target of therapeutic intervention aimed at increasing the sensitivity of glioblastomas to radio- and chemotherapy.
Keywords: astrocytic brain tumors, DNA damage repair, DNA-PK, G2/M checkpoint, gene silencing, glioblastoma, H2AX, human, miRNA, SUMO conjugation, Ubc9
Introduction
Astrocytic tumors are the most common type of intrinsic brain tumors. They show a tendency for progression toward a more malignant phenotype. (1) Astrocytomas are classified according to the WHO malignancy scale, into low-grade astrocytoma (WHO Grade II, AII), anaplastic astrocytoma (WHO Grade III, AIII), and glioblastoma multiforme (WHO Grade IV, GBM). Treatment of these brain tumors presents the major challenge in neuro-oncology today. This is particularly evident in the case of GBM, the most common form of primary brain tumors, which carries a very poor prognosis even after surgical resection with subsequent radio- and chemotherapy.
The standard therapy for newly diagnosed glioblastoma is still surgical resection followed by radiotherapy and temozolomide treatment, but the median survival following therapy is only 12–14 months. Glioblastoma usually respond to radio- and chemotherapy, but recurrence is almost inevitable. This may be due to genetic alterations and the presence of glioblastoma stem cells. Several of the genetic alterations associated with glioblastoma play a role in repair of DNA damage and could therefore be responsible for the resistance to therapy. (2) The radioresistance of glioblastoma stem cells is caused by preferential activation of the DNA damage checkpoint response and an increase in the capacity for DNA damage repair. (3,4) DNA damage repair is a highly conserved process in which post-translational protein modifications with small ubiquitin-like modifier (SUMO1-3) play a fundamental role. (5) We therefore hypothesize that the SUMO conjugation pathway is highly activated in glioblastoma.
SUMO1-3 is a group of proteins that conjugate to lysine residues of target proteins thereby modulating their activity, stability and subcellular localization. A large number of SUMO conjugation target proteins are transcription factors or other nuclear proteins involved in gene expression and genome integrity. (6) A substantial change in levels of SUMO-conjugated proteins can, therefore, be expected to have a major impact on the fate of cells. The potential importance of the SUMO conjugation pathway as a therapeutic tumor target is already appreciated, (7) but contradictory findings have been published. (8–13)
To the best of our knowledge, no attempts have been made to establish whether levels of SUMO-conjugated proteins are elevated in tumors. Here, we show that levels of SUMO1- and SUMO2/3-conjugated proteins are markedly elevated in human astrocytic tumors. In addition, we provide evidence that blocking SUMO1-3 conjugation by silencing their expression suppresses DNA synthesis, cell growth, and clonogenic survival of glioblastoma cells, and triggers cell cycle arrest at G2/M and DNA damage. This suggests that the SUMO conjugation pathway has the potential for therapeutic intervention to block development of astrocytic brain tumors.
Materials and Methods
Ethics Statement
Informed patient consent was obtained according to the Helsinki declaration of ethical requirements, and consent was also given by the local ethics committee at the University of Cologne, Germany (Ethikkommission der Medizinischen Fakultaet der Universitaet zu Koeln; #03-170). The local ethics committee specifically approved this study. Informed patient consent was given in written form.
Tumor sampling
Surgical specimens were obtained from 58 patients with astrocytic tumors at the Department of General Neurosurgery, University of Cologne, Germany. Tumor samples were taken during neurosurgery, snap-frozen in liquid nitrogen immediately after excision, and stored at −80°C until analysis. All samples used for biochemical analyses were histologically evaluated using 10-μm cryostat sections stained with hematoxylin/eosin and compared with paraffin sections in order to ensure that representative tissues were taken. Only samples without evidence of necrosis were considered for the present study. Samples were classified according to the WHO classification of tumors of the nervous system, with 19 astrocytomas Grade II, 19 astrocytomas Grade III, and 20 glioblastoma multiforme. Peritumoral white matter samples, evaluated histologically to confirmed absence of tumor cells, served as controls.
Characterization of tumor samples
SUMO conjugation is a highly volatile process requiring various enzymes involved in conjugation (E1, E2, and E3) and de-conjugation (SENPs) processes. To guarantee that the SUMO conjugation profile is not changed during sample preparation and processing, tissue specimens dissected from brains during surgery must be frozen as quickly as possible, which means that samples from post-mortem brains cannot be used. Peritumoral white matter samples that were confirmed histologically to be free of tumor cells were used as controls. Brain tumors induce peritumoral vasogenic brain edema, (14) which may be associated with a decrease in ATP and an increase in lactate levels, (15) indicative of hypoxic conditions. Since prolonged hypoxia activates the SUMO conjugation pathway, (16) we evaluated VEGF mRNA levels in our control samples as a sensitive measure of hypoxic stress.
Quantitative PCR
Levels of vascular endothelial growth factor (VEGF) mRNA and β-actin mRNA were evaluated by quantitative PCR. Total RNA was extracted from approximately 20 mg of frozen samples using the Trizol reagent (Invitrogen). RNA was reversed transcribed to form cDNA using random hexamers and oligo(dT) as primers (Invitrogen). PCR reactions were run in a LightCycler 2.0 (Roche Diagnostics) using SYBR Green. The following sets of primers were used: VEGF, forward primer: TGAGGAGTCCAACATCACCA, reverse primer: TTTCTTGCGCTTTCGTTTTT; β-actin, forward primer: ATGCCATCCTGCGTCTGGACCT, reverse primer: CCGCCAGACAGCACTGTGTTG. For quantification, changes in VEGF mRNA levels were compared to β-actin mRNA levels.
Cell Culture and DNA Constructs
For our experiments, we used F98 rat glioblastoma cells (ATCC, CRL-2397) and MGR3 (17) human glioblastoma cells cultured in DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin (Invitrogen). A 4-hydroxytamoxifen (4-OHT)-inducible lentiviral system was used to express designed miRNAs. (18) A miRNA-based RNAi vector expressing three pre-miRNAs against SUMO1, SUMO2, and SUMO3 was first established in the vector pcDNA6.2-GW/EGFP-miR (Invitrogen) by inserting miR-SUMO1 (target sequence GGTCAGAGAATTGCTGATAAT) into the construct miR-SUMO2/3, (19) using BamHI and XhoI restriction sites. The fragment harboring GFP and miR-SUMO1/2/3 or miR-Neg, a pre-miRNA sequence not related to any mammalian gene, was then subcloned into the vector pF5xUAS to generate 5xUAS/miR-SUMO1/2/3 and 5xUAS/miR-Neg, respectively. In order to generate 4-OHT-inducible stable F98 and MGR3 cell lines, cells were infected with both 5xUAS/miRNA and GEV16 lentiviruses and selected with two antibiotics, hygromycin B (GEV16 selection; 10 μg/mL, Sigma-Aldrich) and puromycin (5XUAS/miRNA selection; 5 μg/mL, Sigma-Aldrich). Single cell clones were picked and screened for the knockdown efficiency of SUMO1/2/3 after induction with 100 nM 4-OHT by Western blot analysis. A single cell clone for 5XUAS/miR-Neg or 5XUAS/miR-SUMO1/2/3 (termed in the following miR-Neg and miR-SUMO, respectively) was expanded and used for further analysis.
Western Blot Analysis
Frozen tumor samples were weighed in the cold, and proteins were extracted with lysis buffer, as described before. (20) Cell cultures were processed in the same way to analyze levels of SUMO-conjugated proteins. Protein concentration in extracts was analyzed using the BCA protein assay (Thermo Scientific). Immunoblotting was performed using 4%–15% SDS-PAGE gels (Biorad) and the following primary antibodies: SUMO1 (by courtesy of Dr. John Hallenbeck, NIH), SUMO2/3 (Covance), Ubc9 (BD Biosciences), EGFR (Cell Signaling Technology), and β-actin as loading control (Sigma-Aldrich). For quantification of levels of SUMO1- and SUMO2/3-conjugated proteins, the high molecular area in each lane above the non-specific band at about 72 kDa was cropped, as indicated in 1 A and C. For image analysis, a very short exposure time was used so that the optical density of the smear of bands derived from control samples was just slightly above background optical density. Image analysis was performed using the ImageJ program (Wayne Rasband, NIH).
Effect of SUMO1-3 knockdown in MGR3 cells on levels of proteins related to DNA damage and cell cycle control were evaluated by Western blot analysis. MGR3 miR-Neg and miR-SUMO cells were exposed to 4-OHT (100 nM) for 5 days. Proteins were then extracted from cultures ± 4-OHT exposure. The following antibodies were used: p-H2AX (Ser139; Millipore), cyclin A, cyclin B1, cyclin E (Cell Signaling), and p-DNA-PK (Ser2056; by courtesy of Dr. Sandeep Burma, Texas). To evaluate the role of DNA-PK in H2AX phosphorylation, cells were first exposed to 4-OHT (100 nM) to activate expression of miR-Neg or miR-SUMO. Three days later 10 μM NU7026 (Sigma), a potent and highly specific inhibitor of DNA-PK, was added to cultures for additional 2 days.
Analysis of DNA Synthesis
F98 stable cells (miR-SUMO and miR-Neg) were plated at a density of 20,000 cells/cm2 grown at 37°C in 5% CO2 for 24 h prior to the addition of 4-OHT or vehicle. Forty-eight hours after adding 4-OHT, cells were detached with trypsin-EDTA, counted, re-suspended in culture medium, and re-plated in a 96-well plate at a density of 10,000 cells/well. 4-OHT (100 nM) or vehicle was added, and cells were maintained for an additional 4 hours in 5% CO2. 3H-thymidine (50 μCi, specific activity 46 Ci/mmol, ICN Biochemicals) was then added to the medium. Cells were harvested 24 hours later following detachment with trypsin-EDTA, and collected on filter paper. After drying overnight, cells were immersed in scintillation cocktail (ICN Biochemicals) and counted for 10 minutes in a liquid scintillation counter. Four test conditions were examined: 1) miR-Neg cells + 4-OHT; 2) miR-Neg cells + vehicle; 3) miR-SUMO cells + 4-OHT; and 4) miR-SUMO cells + vehicle. Uptake of 3H-thymidine for each test condition was recorded in units of cpm per 10,000 cells as means of 6 replicate observations.
Analysis of cell growth and clonogenic survival
The effect of SUMO1-3 knockdown on growth of MGR3 human glioblastoma cells was evaluated using the real time electronic system (RT-CES; ACEA Biosciences, San Diego). RT-CES allows continuous analysis of cell growth by impedance measurement. (21) MGR3 stable cells (miR-Neg and miR-SUMO) were seeded at a density of 2500 cells/well in 96-well E-plates in triplicate. Twenty–four hours after seeding, 100 nM 4-OHT was added to induce expression of miRNAs, and cell growth was measured at 15 minute intervals. Three independent experiments were performed for each cell line. In parallel experiments, we evaluated the effects of miR-SUMO expression on levels of SUMO1- and SUMO2/3-conjugated proteins. Cells were exposed to 100 nM 4-OHT to activate miRNA expression, and levels of SUMO1- and SUMO2/3-conjugated proteins were evaluated 1–4 days later by Western blot analysis. For clonogenic survival analysis, MGR3 miR-Neg or miR-SUMO cells were seeded at 2000 cells/well in 6-well plates in triplicate. Twenty-four hours after seeding, cells were incubated with or without 100 nM 4-OHT for 10 days. Cells were fixed with 4% PFA, and colonies were stained with 5% crystal violet solution. Three independent experiments were performed for each cell line.
Immunofluorescence staining
MGR3 miR-Neg and miR-SUMO cells were cultured in medium supplemented with/without 100 nM 4-OHT for 5 days and then fixed with paraformaldehyde. After blocking at room temperature for 1 hour, cells were incubated with anti-phospho-histone H2AX (p-Ser139; dilution 1:500, Millipore) at 4°C overnight. Cells were then incubated for 2 hours with Alexa Fluor 594-conjugated goat anti-mouse IgG antibody (dilution 1:400, Invitrogen) at room temperature. Images were captured on a Leica DM IRB microscope (Leica Microsystems). GFP fluorescence was also recorded as an indicator of miRNA expression.
Cell cycle analysis
MGR3 miR-Neg and miR-SUMO cells were cultured in medium supplemented with/without 100 nM 4-OHT for 5 days and then trypsinized and fixed with 70% ethanol for 2 hours on ice. For staining, cells were re-suspended in PBS supplemented with 100 μg/mL propidium iodide (PI, Sigma) and 200 μg/mL RNase (Sigma). Samples were analyzed by flow cytometry (BD FACSCanto II). Data were analyzed with FlowJo software (Tree Star).
Statistical analysis
All data are presented as means ± standard deviation. Statistical analysis was performed using ANOVA and Fisher’s PLSD test. A probability of 95% was taken to indicate significant difference between groups.
Results
Characterization of tumor samples
The epidermal growth factor receptor (EGFR) is almost selectively amplified in GBM and is associated with rapid tumor recurrence. (22,23) We found very strong expression of EGFR with various truncated forms in 47% of GBM samples and in one of the AIII samples (Figure S1), indicating that our samples are representative, and that our findings are comparable to those obtained by other groups. (23) Furthermore, the 170 kDa band of full-length EGFR appeared in almost all tumor samples, but at a much lower level. When we evaluated our control samples for signs of hypoxic stress by measuring VEGF mRNA levels, we did find that several of our controls had significantly higher levels of VEGF mRNA (P=0.0007; Figure S2). We therefore decided to compare levels of SUMO-conjugated proteins in astrocytomas to those found in control samples exhibiting low VEGF mRNA levels that had obviously not been exposed to hypoxia.
SUMO conjugation pathway is activated in astrocytic brain tumors
Several hundred SUMO-conjugated proteins have been identified so far in cell cultures exposed to stress conditions that are known to activate the SUMO conjugation pathway. (24) On Western blot analysis, these appear as a strong smear of bands at high molecular weight. To quantify levels of SUMO-conjugated proteins in tumor samples by Western blot analysis, we used a very short exposure time so that the smear of bands representing SUMO conjugated proteins in control samples was barely visible (Figure 1A, C). Levels of SUMO1-conjugated proteins were massively increased in astrocytic tumors. Compared to control samples, levels of SUMO1-conjugated proteins were elevated 12.3±7.9-fold, 17.4±9.2-fold, and 28.4±9.8-fold in AII, AIII, and GBM specimens, respectively (Figure 1A, B). Astrocytomas also exhibited high levels of SUMO2/3-conjugated proteins (Figure 1C). Compared to control specimens, levels of SUMO2/3-conjugated proteins were elevated 20.7±12.4-fold, 21.8±11.1-fold and 29.0±10.9-fold in AII, AIII, and GBM samples, respectively (Figure 1D).
Figure 1.

SUMO conjugation is activated in human astrocytoma malignancies. Western blot analysis depicts the pattern of SUMO1 and SUMO2/3 conjugation in astrocytoma samples in A (SUMO1) and C (SUMO2/3, same samples as A), which is summarized in B (SUMO1) and D (SUMO2/3). Tumor samples were taken during neurosurgery, and were processed and analyzed as described in detail in Materials and Methods. For quantification of levels of SUMO1- and SUMO2/3-conjugated proteins, the high molecular-weight area in each lane was cropped, as indicated in A and C. Data are presented as means ± SD. Statistical analysis was performed using ANOVA and Fisher’s PLSD test (* p<0.05; ** p<0.01; *** p<0.001). C – control (n=4); AII – WHO Grade II, low-grade astrocytoma (n=19); AIII – WHO Grade III, anaplastic astrocytoma (n=19); GBM – WHO Grade IV, glioblastoma multiforme (n=20).
Three enzymatic steps are involved in SUMO conjugation: an activating (E1), a conjugating (E2), and a ligating enzyme (E3). Ubc9 is the only E2 enzyme that has been identified so far. Its expression is markedly increased in malignant tissues of the colon, prostate, breast, head and neck, and lung, but decreased in metastatic breast, prostate, and lung cancer, (25) Ubc9 protein levels were significantly elevated in astrocytoma samples compared to control, with a 10±6.2-fold, 17±7.5-fold, and 20±7.1-fold increase in AII, AIII, and GBM, respectively (Figure 2A, B).
Figure 2.
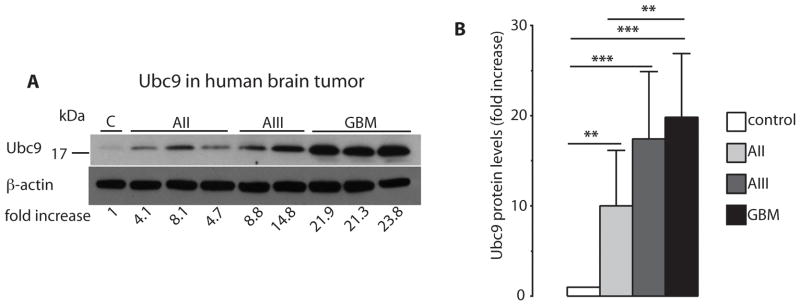
Levels of the SUMO conjugating enzyme Ubc9 are markedly increased in human astrocytoma malignancies. Samples were taken during neurosurgery, and processed and evaluated as described in Materials and Methods. Differences in Ubc9 protein levels in astrocytoma specimens are depicted in A (same samples as in Figure 1A and C) and summarized in B. Data are presented as means ± SD. Statistical analysis was performed using ANOVA and Fisher’s PLSD test (** p<0.01; *** p<0.001). C – control (n=4); AII – WHO Grade II, low-grade astrocytoma (n=19); AIII – WHO Grade III, anaplastic astrocytoma (n=19); GBM – WHO Grade IV, glioblastoma multiforme (n=20).
Silencing SUMO expression blocks DNA synthesis
To investigate whether DNA synthesis in glioblastoma cells is modified by SUMO conjugation, we evaluated the effects of suppressing SUMO conjugation by blocking its expression using an miRNA approach and incorporation of 3H-thymidine into DNA. To avoid the risk of adaptive processes occurring in cells with silenced SUMO expression during the long selection process for stable cell lines, we used an inducible miRNA expression system requiring two constructs. A schematic presentation of this inducible system is provided in Figure S3. F98 glioblastoma cells were stably transduced with two lentiviruses. The GEV16 transcription factor, constitutively expressed from the first construct driven by the ubiquitin promoter, is retained in the cytoplasm in the absence of the inducer 4-hydroxytamoxifen (4-OHT). In the presence of 4-OHT, GEV16 translocates to the nucleus and activates expression of miRNAs from 5xUAS/miRNA (Figure S3). We used this system to express GFP and SUMO1/2/3 miRNAs (miR-SUMO) from one construct. For controls, we used cells that, upon 4-OHT exposure, express miRNA not directed against any mammalian gene (miR-Neg). Two days after adding 4-OHT to cultures, SUMO1-3 expression was almost completely blocked in cells expressing miR-SUMO (Figure 3A, B). Exposure to 4-OHT did not produce major change in the incorporation of 3H-thymidine into DNA in cells expressing miR-Neg, but it blocked 3H-thymidine incorporation in miR-SUMO cells to about 13% of control (Figure 3C). This demonstrates the crucial role of SUMO conjugation in DNA synthesis in glioblastoma cells.
Figure 3.
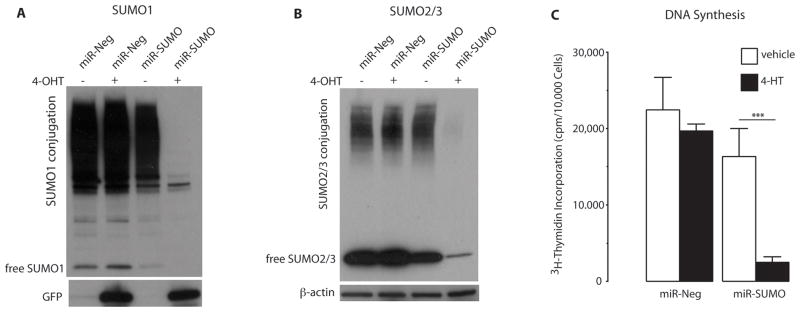
Blocking SUMO1-3 conjugation in F98 rat glioblastoma cells by silencing their expression suppressed DNA synthesis. Exposure to 4-hydroxytamoxifen (4-OHT) induced expression of miR-SUMO (3 miRNAs against SUMO1, SUMO2, and SUMO3) and miR-Neg (control miRNA) in stably transduced cells. Western blot analysis depicts SUMO1- (A) and SUMO2/3- (B) in cells without 4-OHT exposure (−) and with 4-OHT exposure (+), demonstrating depletion of SUMO1 and SUMO2/3 in miR-SUMO expressing cells. Uptake of 3H-thymidine as an indicator of DNA synthesis in transduced cells is shown in (C) and suggested that blocking SUMO1-3 conjugation by silencing their expression suppressed DNA synthesis. Data are presented as means±SD (n=6/group). Statistical analysis was performed using ANOVA and Fisher’s PLSD test (*** p < 0.001).
Silencing SUMO1-3 expression blocks growth and clonogenic survival of human glioblastoma cells
MGR3 miR-Neg or miR-SUMO cells were seeded as described above, and miRNA expression was induced 24 hours later by adding 100 nM 4-OHT. Western blot analysis indicated that 4-OHT-induced expression of miR-SUMO resulted in depletion of levels of high molecular weight SUMO1- and SUMO2/3-conjugated proteins and of free SUMO1 and SUMO2/3 (Figure 4A, B). Notably, levels of SUMO1-conjugated proteins were almost depleted already at day 1 after 4-OHT exposure (Figure 4A), while it took 2 days of exposure to markedly deplete levels of SUMO2/3-conjugated proteins (Figure 4B). Exposure to 4-OHT (Figure 4C, arrow) slightly reduced growth and clonogenic survival of cells expressing control miRNA (miR-Neg; Figure 4C, D), as expected. Tamoxifen exposure of glioblastoma cells has been shown to induce cell death, although at a higher concentration. (26) In cells with induced expression of miR-SUMO, in contrast, cell growth was completely blocked 2 days after addition of 4-OHT (Figure 4C) and clonogenic survival was markedly reduced (Figure 4D).
Figure 4.

Silencing SUMO1/2/3 expression blocks growth and clonogenic survival of glioblastoma cells. Western blot analysis indicates levels of SUMO1- and SUMO2/3-conjugated proteins in MGR3 human glioblastoma cells expressing control miRNA (miR-Neg) or SUMO1/2/3 miRNA (miR-SUMO) at indicated time points after adding 4-OHT (100 nM) (A, B). The strong bands at about 28 and 72 kDa are non-specific bands. Suppressed growth and clonogenic survival of cells expressing miR-SUMO is shown in C and D. Data are representative of three independent experiments. Arrow (C) indicates addition of 4-OHT.
Silencing SUMO1-3 expression activates H2AX phosphorylation and induces G2/M cell cycle arrest
To investigate the mechanisms underlying suppression of cell growth in miR-SUMO-expressing cells (Figure 4C), MGR3 cells expressing miR-Neg or miR-SUMO were analyzed 5 days after adding 4-OHT (100 nM) to the medium. To avoid overgrowth of control cells expressing miR-Neg, cultures were exposed to 4-OHT for 2 days, trypsinized and seeded at lower density for additional 3 days in medium supplemented with 4-OHT (100 nM).
First, we focused on phosphorylation of H2AX (p-H2AX), a hallmark of DNA double-strand breaks, (27) Silencing SUMO1-3 expression induced a massive rise in levels of p-H2AX (Figure 5A). H2AX phosphorylation is a key cellular response to DNA double-strand breaks, and p-H2AX binds to the sites of DNA breaks. (28) We therefore analyzed the subcellular localization of p-H2AX by immunofluorescence staining (Figure 5B). GFP fluorescence was used as indicator of miRNA expression. As shown in Figure 5B, knockdown of SUMO1-3 expression induced the formation of nuclear p-H2AX foci in miR-SUMO expressing cells.
Figure 5.
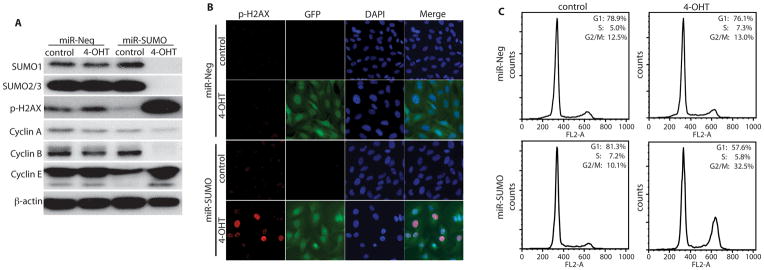
Silencing SUMO1/2/3 expression induced phosphorylation of H2AX and G2/M cell cycle arrest. Western blot analysis indicates levels of SUMO1 and SUMO2/3, phosphorylated H2AX (p-H2AX), and cyclin A, B1, E in MGR3 cells expressing control (miR-Neg) or SUMO1/2/3 miRNA (miR-SUMO) upon exposure to 4-OHT (100 nM) for 5 days (A). Immunofluorescence staining of phosphorylated H2AX revealed p-H2AX foci in the nuclei of cells in which SUMO1/2/3 expression was silenced, indicating DNA double-strand breaks (B). GFP and DAPI staining was used as indicator of miRNA expression and for identifying nuclei, respectively. FACS analysis demonstrates arrest of the cell cycle at G2/M in cells in which SUMO1/2/3 expression was silenced (C). Data are representative of three independent experiments.
To investigate the effects of silencing SUMO1/2/3 expression on cell division, we evaluated levels of cyclin A, B1, and E by Western blot analysis, and cell cycle by fluorescence-activated cell sorting (FACS). Cyclin B1 is an essential regulator for the G2 to M- phases transition. It has been shown that down-regulation of cyclin B1 induces G2/M cell cycle arrest. (29) Indeed, silencing SUMO1-3 expression induced a marked reduction in levels of cyclin B1 (Figure 5A) and an increase in the number of cells at the G2/M phase from 10.1% to 32.5% (Figure 5C).
Silencing SUMO1-3 expression activates DNA-PK
DNA damage-activated protein kinase (DNA-PK) is a kinase that phosphorylates H2AX under conditions of apoptotic DNA fragmentation, (30) and plays a role in the repair of DNA damage in glioblastoma cells exposed to radiation. (31) We therefore investigated whether DNA-PK is involved in phosphorylation of H2AX induced by silencing SUMO1-3 expression. Indeed, as shown in Figure 6A (arrow), a marked increase in levels of phosphorylated DNA-PK (p-DNA-PK) was observed in miR-SUMO cells at day 3 post exposure to 4-OHT. To further verify the role of DNA-PK in H2AX phosphorylation, cells were exposed to NU7026 (10 μM), a specific inhibitor of DNA-PK. (32) Exposing cells to NU7026 for 2 days almost completely blocked the increase of H2AX phosphorylation in miR-SUMO cells (Figure 6B). This suggested that DNA-PK plays a predominant role in the phosphorylation of H2AX in cells in which SUMO1-3 conjugation was blocked by silencing their expression.
Figure 6.
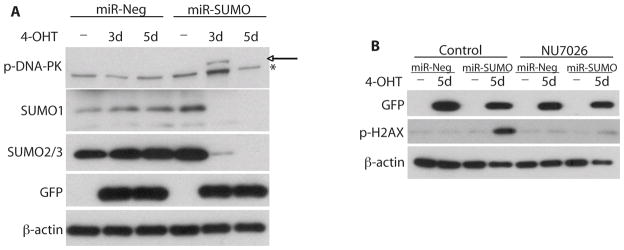
Blocking SUMO conjugation by silencing its expression resulted in DNA-PK-dependent phosphorylation of H2AX. Western blot analysis showed phosphorylation of DNA-PK in MGR3 miR-Neg and miR-SUMO cells at day 3 after adding 4-OHT to the medium (A). Arrow and asterisk indicate p-DNA-PK- and non-specific band, respectively. GFP was used as indicator of miRNA expression. Western blot analysis showed that the increase of phosphorylated H2AX (p-H2AX) is almost completely blocked in MGR3 miR-SUMO cells exposed to 4-OHT and the specific DNA-PK inhibitor NU7026 (B), suggesting that DNA-PK is the major kinase for the phosphorylation of H2AX induced by silencing SUMO expression. GFP was used as indicator of miRNA expression.
Discussion
Here we provide evidence that the SUMO conjugation pathway is activated in astrocytic tumors and that this effect is most pronounced in glioblastoma multiforme (GBM), the most aggressive form of all brain tumors, with poor prognosis even after radical surgical resection and subsequent aggressive radio- and chemotherapy. This is, to the best of our knowledge, the first study that demonstrates high levels of SUMO-conjugated proteins in any kind of cancer compared to normal tissue.
Hypoxia is a hallmark of astrocytic brain tumors. (33–35) Exposure of cells to hypoxia has been reported to activate SUMOylation and hypoxia-induced activation of SUMO conjugation is believed to be a protective stress response shielding cells from damage induced by hypoxia. (36) We therefore expect hypoxia to contribute to activation of SUMO conjugation in astrocytic brain tumors (Figure 1). On the other hand, we provided evidence here that the entire SUMO conjugation pathway was massively activated in astrocytic brain tumors, as illustrated by high levels of free and conjugated SUMO1 and SUMO2/3 and of the SUMO conjugating enzyme Ubc9 (Figures 1 and 2). This suggests that other mechanisms contribute to the activated SUMO conjugation pathway in human gliomas.
The observation that DNA synthesis was suppressed in glioblastoma cells by blocking SUMO1-3 conjugation through silencing their expression suggests that SUMO conjugation is essential for glioblastoma cell survival. Indeed, silencing SUMO1-3 expression blocked cell growth and clonogenic survival of glioblastoma cells. The rise in levels of phosphorylated H2AX and G2/M cell cycle arrest in miR-SUMO cells provide evidence that the inability of affected glioblastoma cells to repair DNA damage associated with cell division results in a block of the cell cycle at the G2/M checkpoint. Collectively, these data demonstrate the role of SUMO conjugation in glioblastoma cell division and suggest that the SUMO conjugation pathway has the potential as a target for therapeutic intervention to inhibit glioblastoma growth. After we have established that the SUMO conjugation pathway is massively activated in human glioblastomas and have provided evidence that an active SUMO conjugation pathway is required for glioblastoma cell survival, the next logical step is to identify proteins that are SUMO conjugated in glioblastomas and to elucidate the role of this post-translational protein modification for the growth of these tumors and resistance to therapy. This can best be done by quantitative proteomics analysis. (37)
Silencing SUMO1-3 expression in MRG3 human glioblastoma cells triggered DNA-PK activation, which in turn induced phosphorylation of H2AX, indicating DNA double-strand breaks, even in the absence of DNA damage-stimulating treatment (Figures 5 and 6). This suggests that DNA double-strand breaks occur spontaneously in dividing cells and require SUMO conjugation for repair. It has indeed been reported that DNA damage occurs during replication, as evidenced by H2AX phosphorylation (27,38) and that SUMO conjugation plays a pivotal role in DNA double-strand signaling and repair. (39) The SUMO ligases PIAS1 and PIAS4 bind to double-strand breaks and promote DNA double-strand break repair and confer resistance to radiation. (39) This implies that when the SUMO conjugation pathway is highly activated, cells have a high DNA damage repair capacity and a cell growth advantage over cells with a less active SUMO conjugation pathway. Nuclear extracts from radio-resistant glioblastoma cells have a much better DNA double-strand break repair proficiency than radio-sensitive glioblastoma cells, (40) suggesting a defining role of DNA repair capacity in glioblastoma growth. Therefore, the highly activated SUMO conjugation pathway in glioblastoma cells may contribute to the resistance of cells to radio- and chemotherapy.
Here, we have provided evidence that DNA-PK plays a role in silencing SUMO1-3 expression-induced H2AX phosphorylation, and that DNA double-strand breaks, spontaneously induced in replicating glioblastoma cells, cannot be repaired when SUMO1-3 expression is silenced (Figure 6). We did see phosphorylation and thus activation of DNA-PK 3 days after adding 4-OHT to cultures, but not after 5 days when H2AX phosphorylation was very pronounced (Figure 6A). One plausible explanation for this pattern is that H2AX phosphorylation is a process downstream of DNA-PK phosphorylation and that long-lasting phosphorylation of H2AX results when DNA double-strand repair is blocked in SUMO1-3 depleted cells. A long-lasting phosphorylation of H2AX has indeed been reported in cells when the DNA damage repair process is impaired. (41–43)
Here, we report that the SUMO conjugation pathway is markedly activated in glioblastomas and required for survival of glioblastoma cells. On the other hand, levels of the SUMO protease SENP3, which activates de-conjugation of SUMOylated proteins is markedly increased in primary human cancers of the colon, rectum, prostate, ovaries, and lungs. (10) Furthermore, SENP3-mediated de-SUMOylation correlates with accelerated cell proliferation under mild stress. (10) SENP3 expression is also responsible for HIF-1α transactivation under mild oxidative stress; and in nude mice xenografts overexpressing SENP3 produce larger tumors. (10) In addition, SENP1 is highly expressed in human prostate cancer specimens and correlate with HIF1α expression; indeed, SENP1 overexpression stabilizes HIF1α during hypoxia, (44) and it also facilitates high-grade prostatic neoplasia. (45) Collectively, these observations suggest a role of de-SUMOylation in cancer development. Since levels of the SUMO conjugating enzyme Ubc9 and levels of SUMO1- and SUMO2/3-conjugated proteins were markedly increased in human astrocytoma samples, we expect SUMO conjugation, and not de-conjugation, to be associated with cancer development in the astrocytic brain tumors under investigation.
In summary, this study shows that the SUMO conjugation pathway is activated in human astrocytic tumors and that this activation is most pronounced in glioblastoma multiforme. We also provide evidence that an active SUMO conjugation pathway is required for the functioning of glioblastoma cells because blocking SUMO conjugation suppressed cell growth and clonogenic survival, induced DNA double-strand breaks, and arrested the cell cycle at G2/M. This suggests that the SUMO conjugation pathway has the potential as target for therapeutic intervention aimed at blocking glioblastoma development by interfering with the DNA damage repair capacity of cells and thus increase the efficacy of radio- and chemotherapy.
Supplementary Material
Epidermal growth factor receptor (EGFR) amplification in human glioblastoma samples.
Vascular endothelial growth factor (VEGF) expression in peritumoral white matter.
Schematic presentation of the 4-hydroxytamoxifen (4-OHT)-inducible system used to express designed miRNAs.
Acknowledgments
The authors thank Dr. Werner Stenzel, Department of Neuropathology, University of Cologne, for the histophathological classification of tumor samples, Kathy Gage, Research Development Associate, for her excellent editorial contributions in the preparation of this manuscript, and Pei Miao and Andrew Barker for excellent technical assistance.
Grant Support
This study was supported by funds provided by the Department of Anesthesiology, Duke University Medical Center (to W.P.) and in part by R01 grant HL095552 from the National Institutes of Health (to W.P.). C.H. is supported by funds from the Bundesministerium fuer Bildung und Forschung (Center for Stroke Research Berlin).
Footnotes
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995;15:211–21. doi: 10.1002/glia.440150303. [DOI] [PubMed] [Google Scholar]
- 2.Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011;71:5945–9. doi: 10.1158/0008-5472.CAN-11-1245. [DOI] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. 2010;18:619–29. doi: 10.1016/j.ccr.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461–7. doi: 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- 6.Rosas-Acosta G, Russell WK, Deyrieux A, Russell DH, Wilson VG. A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol Cell Proteomics. 2005;4:56–72. doi: 10.1074/mcp.M400149-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mo YY, Yu Y, Theodosiou E, Ee PL, Beck WT. A role for Ubc9 in tumorigenesis. Oncogene. 2005;24:2677–83. doi: 10.1038/sj.onc.1208210. [DOI] [PubMed] [Google Scholar]
- 8.Kim KI, Baek SH. Small ubiquitin-like modifiers in cellular malignancy and metastasis. Int Rev Cell Mol Biol. 2009;273:265–311. doi: 10.1016/S1937-6448(08)01807-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhu S, Sachdeva M, Wu F, Lu Z, Mo YY. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene. 2010;29:1763–72. doi: 10.1038/onc.2009.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han Y, Huang C, Sun X, et al. SENP3-mediated de-conjugation of SUMO2/3 from promyelocytic leukemia is correlated with accelerated cell proliferation under mild oxidative stress. J Biol Chem. 2010;285:12906–15. doi: 10.1074/jbc.M109.071431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang C, Han Y, Wang Y, et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. Embo J. 2009;28:2748–62. doi: 10.1038/emboj.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Banerjee S. Differential PIAS3 expression in human malignancy. Oncol Rep. 2004;11:1319–24. [PubMed] [Google Scholar]
- 13.Brantley EC, Nabors LB, Gillespie GY, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 2008;14:4694–704. doi: 10.1158/1078-0432.CCR-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerstner ER, Duda DG, di Tomaso E, et al. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6:229–36. doi: 10.1038/nrclinonc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okada Y, Kloiber O, Hossmann KA. Regional metabolism in experimental brain tumors in cats: relationship with acid/base, water, and electrolyte homeostasis. J Neurosurg. 1992;77:917–26. doi: 10.3171/jns.1992.77.6.0917. [DOI] [PubMed] [Google Scholar]
- 16.Shao R, Zhang FP, Tian F, et al. Increase of SUMO-1 expression in response to hypoxia: direct interaction with HIF-1alpha in adult mouse brain and heart in vivo. FEBS Lett. 2004;569:293–300. doi: 10.1016/j.febslet.2004.05.079. [DOI] [PubMed] [Google Scholar]
- 17.Singh S, Okamura T, Ali-Osman F. Serine phosphorylation of glutathione S-transferase P1 (GSTP1) by PKCalpha enhances GSTP1-dependent cisplatin metabolism and resistance in human glioma cells. Biochem Pharmacol. 2010;80:1343–55. doi: 10.1016/j.bcp.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 18.Callus BA, Ekert PG, Heraud JE, et al. Cytoplasmic p53 is not required for PUMA-induced apoptosis. Cell Death Differ. 2008;15:213–5. doi: 10.1038/sj.cdd.4402245. author reply 5–6. [DOI] [PubMed] [Google Scholar]
- 19.Yang W, Paschen W. Gene expression and cell growth are modified by silencing SUMO2 and SUMO3 expression. Biochem Biophys Res Commun. 2009;382:215–8. doi: 10.1016/j.bbrc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Yang W, Sheng H, Warner DS, Paschen W. Transient global cerebral ischemia induces a massive increase in protein sumoylation. J Cereb Blood Flow Metab. 2008;28:269–79. doi: 10.1038/sj.jcbfm.9600523. [DOI] [PubMed] [Google Scholar]
- 21.Abassi YA, Jackson JA, Zhu J, O’Connell J, Wang X, Xu X. Label-free, real-time monitoring of IgE-mediated mast cell activation on microelectronic cell sensor arrays. J Immunol Methods. 2004;292:195–205. doi: 10.1016/j.jim.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 22.Malden LT, Novak U, Kaye AH, Burgess AW. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res. 1988;48:2711–4. [PubMed] [Google Scholar]
- 23.Schlegel J, Merdes A, Stumm G, et al. Amplification of the epidermal-growth-factor-receptor gene correlates with different growth behaviour in human glioblastoma. Int J Cancer. 1994;56:72–7. doi: 10.1002/ijc.2910560114. [DOI] [PubMed] [Google Scholar]
- 24.Bruderer R, Tatham MH, Plechanovova A, Matic I, Garg AK, Hay RT. Purification and identification of endogenous polySUMO conjugates. EMBO Rep. 2011;12:142–8. doi: 10.1038/embor.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moschos SJ, Jukic DM, Athanassiou C, et al. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum Pathol. 2010;41:1286–98. doi: 10.1016/j.humpath.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Hui AM, Zhang W, Chen W, et al. Agents with selective estrogen receptor (ER) modulator activity induce apoptosis in vitro and in vivo in ER-negative glioma cells. Cancer Res. 2004;64:9115–23. doi: 10.1158/0008-5472.CAN-04-2740. [DOI] [PubMed] [Google Scholar]
- 27.Furuta T, Takemura H, Liao ZY, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20303–12. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 28.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 29.Choi YH, Zhang L, Lee WH, Park KY. Genistein-induced G2/M arrest is associated with the inhibition of cyclin B1 and the induction of p21 in human breast carcinoma cells. Int J Oncol. 1998;13:391–6. doi: 10.3892/ijo.13.2.391. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee B, Kessinger C, Kobayashi J, et al. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair (Amst) 2006;5:575–90. doi: 10.1016/j.dnarep.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 31.Anderson JA, Harper JV, Cucinotta FA, O’Neill P. Participation of DNA-PKcs in DSB repair after exposure to high- and low-LET radiation. Radiat Res. 2010;174:195–205. doi: 10.1667/RR2071.1. [DOI] [PubMed] [Google Scholar]
- 32.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003;63:6008–15. [PubMed] [Google Scholar]
- 33.Ido K, Nakagawa T, Sakuma T, Takeuchi H, Sato K, Kubota T. Expression of vascular endothelial growth factor-A and mRNA stability factor HuR in human astrocytic tumors. Neuropathology. 2008;28:604–11. doi: 10.1111/j.1440-1789.2008.00926.x. [DOI] [PubMed] [Google Scholar]
- 34.Ziemer LS, Koch CJ, Maity A, Magarelli DP, Horan AM, Evans SM. Hypoxia and VEGF mRNA expression in human tumors. Neoplasia. 2001;3:500–8. doi: 10.1038/sj.neo.7900195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spence AM, Muzi M, Swanson KR, et al. Regional hypoxia in glioblastoma multiforme quantified with [18F]fluoromisonidazole positron emission tomography before radiotherapy: correlation with time to progression and survival. Clin Cancer Res. 2008;14:2623–30. doi: 10.1158/1078-0432.CCR-07-4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen HV, Chen JL, Zhong J, et al. SUMOylation attenuates sensitivity toward hypoxia- or desferroxamine-induced injury by modulating adaptive responses in salivary epithelial cells. Am J Pathol. 2006;168:1452–63. doi: 10.2353/ajpath.2006.050782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang W, Thompson JW, Wang Z, et al. Analysis of oxygen/glucose-deprivation-induced changes in SUMO3 conjugation using SILAC-based quantitative proteomics. Journal of proteome research. 2012;11:1108–17. doi: 10.1021/pr200834f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chowdhury D, Xu X, Zhong X, et al. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–9. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Britten RA, Liu D, Kuny S, Allalunis-Turner MJ. Differential level of DSB repair fidelity effected by nuclear protein extracts derived from radiosensitive and radioresistant human tumour cells. Br J Cancer. 1997;76:1440–7. doi: 10.1038/bjc.1997.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ouyang KJ, Woo LL, Zhu J, Huo D, Matunis MJ, Ellis NA. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 2009;7:e1000252. doi: 10.1371/journal.pbio.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li YJ, Stark JM, Chen DJ, Ann DK, Chen Y. Role of SUMO:SIM-mediated protein-protein interaction in non-homologous end joining. Oncogene. 2010;29:3509–18. doi: 10.1038/onc.2010.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012;26:1179–95. doi: 10.1101/gad.188284.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–95. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bawa-Khalfe T, Cheng J, Lin SH, Ittmann MM, Yeh ET. SENP1 induces prostatic intraepithelial neoplasia through multiple mechanisms. J Biol Chem. 2010;285:25859–66. doi: 10.1074/jbc.M110.134874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Epidermal growth factor receptor (EGFR) amplification in human glioblastoma samples.
Vascular endothelial growth factor (VEGF) expression in peritumoral white matter.
Schematic presentation of the 4-hydroxytamoxifen (4-OHT)-inducible system used to express designed miRNAs.