

Abstract
Blood pressure (BP) is regulated by multiple neuronal, hormonal, renal and vascular control mechanisms. Changes in signaling mechanisms in the endothelium, vascular smooth muscle (VSM) and extracellular matrix cause alterations in vascular tone and blood vessel remodeling and may lead to persistent increases in vascular resistance and hypertension (HTN). In VSM, activation of surface receptors by vasoconstrictor stimuli causes an increase in intracellular free Ca2+ concentration ([Ca2+]i), which forms a complex with calmodulin, activates myosin light chain (MLC) kinase and leads to MLC phosphorylation, actin-myosin interaction and VSM contraction. Vasoconstrictor agonists could also increase the production of diacylglycerol which activates protein kinase C (PKC). PKC is a family of Ca2+-dependent and Ca2+-independent isozymes that have different distributions in various blood vessels, and undergo translocation from the cytosol to the plasma membrane, cytoskeleton or the nucleus during cell activation. In VSM, PKC translocation to the cell surface may trigger a cascade of biochemical events leading to activation of mitogen-activated protein kinase (MAPK) and MAPK kinase (MEK), a pathway that ultimately increases the myofilament force sensitivity to [Ca2+]i, and enhances actin-myosin interaction and VSM contraction. PKC translocation to the nucleus may induce transactivation of various genes and promote VSM growth and proliferation. PKC could also affect endothelium-derived relaxing and contracting factors as well as matrix metalloproteinase (MMPs) in the extracellular matrix further affecting vascular reactivity and remodeling. In addition to vasoactive factors, reactive oxygen species, inflammatory cytokines and other metabolic factors could affect PKC activity. Increased PKC expression and activity have been observed in vascular disease and in certain forms of experimental and human HTN. Targeting of vascular PKC using PKC inhibitors may function in concert with antioxidants, MMP inhibitors and cytokine antagonists to reduce VSM hyperactivity in certain forms of HTN that do not respond to Ca2+ channel blockers.
Keywords: calcium, endothelium, vascular smooth muscle, hypertension
INTRODUCTION
Hypertension (HTN) is major cardiovascular and renal disease affecting a large proportion of the population in the Western World. Several factors contribute to increased blood pressure (BP) including neuronal, hormonal, renal and vascular mechanisms. Understanding the physiological mechanisms that control BP would help define the pathological changes in HTN, and help design specific approaches to manage the increases in BP and HTN.
Blood vessels play a major role in the control of vascular tone. The blood vessel wall has three layers; the tunica intima made of a single layer of endothelial cells (ECs), the tunica media made of several layers of vascular smooth muscle cells (VSMCs), and the adventitia made of fibroblasts, connective tissue and extracellular matrix (ECM). ECs release vasodilator and vasoconstrictor mediators that control the vessel diameter. The ability of VSMCs to contract and relax plays an important role in the regulation of the vessel diameter and blood flow to various tissues and organs. The ECM proteins provide structural integrity to the vessel wall and are regulated by proteolytic enzymes such as matrix metalloproteinases (MMPs).
This review will focus on how vasoconstrictor agonists affect the mechanisms of VSM contraction particularly protein kinase C (PKC), and the changes in these mechanisms in vascular disease such as HTN. The role of PKC in the regulation of EC function and ECM will be briefly discussed. In addition to changes in vasoactive factors, changes in reactive oxygen species (ROS) [1–3], MMPs and inflammatory cytokines [4–6] in the plasma and vascular tissues [7–9] have been observed in HTN and coronary artery disease. The effects of ROS [2, 3], MMPs [10–12] and cytokines [13–15] in HTN may be partly related to their effects on PKC and consequent changes in vascular reactivity, growth and remodeling. Understanding the role of PKC as a major regulator of VSM function, the PKC isoforms, their protein substrates and subcellular distribution, and their interaction with other factors such as ROS, MMPs and cytokines would provide important information regarding the benefits of determining PKC activity in the diagnosis of VSM hyperactivity disorders and the potential usefulness of PKC inhibitors in the management of vascular disease such as HTN.
Mechanisms of VSM Contraction
Ca2+ is a major determinant of VSM contraction. VSM activation by physiological or pharmacological agonist triggers an increase in intracellular free Ca2+ concentration ([Ca2+]i) due to Ca2+ release from the sarcoplasmic reticulum and Ca2+ influx from the extracellular space through plasma membrane Ca2+ channels. Four Ca2+ ions bind to calmodulin (CAM) to form a Ca2+-CAM complex, which activates myosin light chain (MLC) kinase, and in turn causes the phosphorylation of the 20 kDa MLC, stimulates actin-myosin interaction and promotes VSM contraction (Fig. 1). During VSM relaxation, removal of the vasoconstrictive agonist causes a decrease in [Ca2+]i due to Ca2+ extrusion via the plasmalemmal Ca2+ pump (PMCA) and the Na+-Ca2+ exchanger, as well as Ca2+ reuptake via the sarcoplasmic reticulum Ca2+ pump (SERCA). The decrease in [Ca2+]i allows the dissociation of the Ca2+-CAM complex, and the remaining phosphorylated MLC is dephosphorylated by MLC phosphatase, leading to detachment of actin-myosin crossbridges and VSM relaxation [16–19].
Fig. 1.

Mechanisms of VSM contraction. The interaction of an agonist (A) such as phenylephrine with its specific α-adrenergic receptor (R) activates phospholipase C (PLCβ) and stimulates the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 stimulates Ca2+ release from the sarcoplasmic reticulum (SR). Agonists also stimulate Ca2+ influx through Ca2+ channels. Ca2+ binds calmodulin (CAM), activates MLC kinase (MLCK), causes MLC phosphorylation, and initiates VSM contraction. DAG activates PKC. PKC-induced phosphorylation of CPI-17, inhibits MLC phosphatase and increases MLC phosphorylation and VSM contraction. PKC-induced phosphorylation of the actin-binding protein calponin (CaP) allows more actin to bind myosin and enhances contraction. PKC may also activate a protein kinase cascade involving Raf, MAPK kinase (MEK) and MAPK, leading to phosphorylation of the actin-binding protein caldesmon (CaD) and enhanced contraction. Activation of RhoA/Rho-kinase inhibits MLC phosphatase and further enhances the Ca2+ sensitivity of contractile proteins. AA, arachidonic acid; G, heterotrimeric GTP-binding protein; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PLD, phopholipase D; PS, phosphatidylserine. Dashed line indicates inhibition.
Ca2+-dependent VSM contraction is usually observed during VSM depolarization by mechanical stretch, nerve stimuli, electrical stimulation, or in the presence of high KCl solution. VSM depolarization activates voltage-gated Ca2+ channels (VGCCs) and because of the large concentration gradient between extracellular Ca2+ (millimolar) and intracellular [Ca2+]i (nanomolar), the opening of VGCCs facilitates Ca2+ influx, and leads to MLC phosphorylation and VSM contraction. In contrast with membrane depolarization, physiological agonists such as norepinephrine, prostaglandin F2α and thromboxane A2 activate other intracellular signaling pathways in addition to Ca2+ channels. In VSM, the interaction of an agonist with its specific receptor causes activation of phospholipase C (PLC), and promotes the hydrolysis of phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) [20, 21]. Because IP3 is water soluble it diffuses in the cytosol, stimulates IP3 receptors in the sarcoplasmic reticulum and causes Ca2+ release from the intracellular stores, transient increase in [Ca2+]i and VSM contraction. Agonists also activate receptor-operated (ROCs) and store-operated Ca2+ channels (SOCs), causing maintained Ca2+ influx, increased [Ca2+]i, MLC phosphorylation and VSM contraction (Fig. 1). However, Ca2+-dependent MLC phosphorylation may not be the only mechanism involved in VSM contraction. For instance, Ca2+ channel blockers such as nifedipine, verapamil or diltiazem do not completely inhibit agonist-induced VSM contraction. Also, agonist-induced maintained contraction has been observed in certain blood vessels incubated in Ca2+-free solution and in the absence of detectable increases in [Ca2+]i [17, 22–24]. Agonist-induced dissociations between [Ca2+]i and force, between [Ca2+]i and MLC phosphorylation, and between MLC phosphorylation and force have also been observed in several vascular preparations, suggesting activation of additional signaling pathways that cause sensitization of the contractile myofilaments to [Ca2+]i including Rho-kinase and protein kinase C (PKC) [16, 18].
PKC Isoforms
PKC was first described as a Ca2+-activated phospholipid-dependent protein kinase [25], but subsequent biochemical analysis and molecular cloning revealed a family of different PKC isozymes of closely related structure. The PKC molecule is a single polypeptide, comprised of N-terminal regulatory domain and C-terminal catalytic domain (Fig. 2) separated by a hinge region that becomes proteolytically labile when the enzyme is membrane-bound [26]. Classic PKCs have four conserved regions (C1–C4) and five variable regions (V1–V5). The C1 region contains a tandem repeat of the characteristic cysteine-rich zinc-finger-like sequence. The sequence Cys-X2-Cys-X13(14)-Cys-X7-Cys-X7-Cys, where X represents any amino acid, is conserved among different PKCs, and each 30-residue sequence of this type is independently folded and binds a zinc ion [27]. The Cys-rich motif is duplicated in most PKCs and may form the DAG/phorbol ester-binding site. The Cys-rich motif is also immediately preceded by an autoinhibitory pseudosubstrate sequence. The C1 region also contains the phosphatidylserine recognition site [26]. In Ca2+-dependent PKCs, the C2 region is rich in acidic residues and has a Ca2+-binding site. The C3 and C4 regions contain the adenosine triphosphate (ATP) and substrate binding sites. Similar to other protein kinases, all PKCs have an ATP-binding sequence, Gly-X-Gly-X-X-Gly-----Lys [21, 26] (Fig. 2).
Fig. 2.
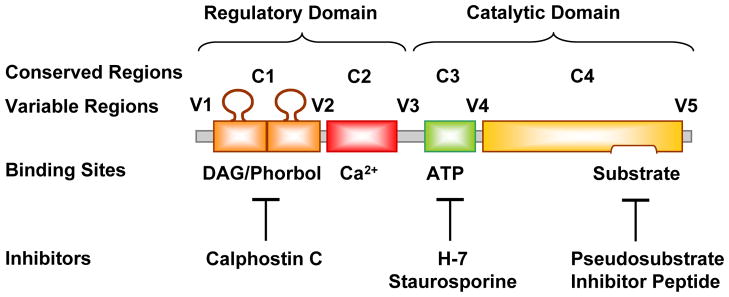
Biochemical structure of a classic PKC, and sites of action of activators and inhibitors. A classic PKC molecule has four conserved (C1–C4) and five variable (V1–V5) regions. C1 region contains binding sites for DAG, phorbol ester, and phosphatidylserine, and for the PKC inhibitor calphostin C. C2 region contains Ca2+-binding site. C3 regions contain binding sites for ATP and the PKC inhibitors H-7 and staurosporine. C4 region contains binding sites for physiological PKC substrate. Endogenous pseudosubstrate and exogenous pseudosubstrate inhibitor peptide bind to the catalytic domain and prevents PKC from phosphorylating the true substrate. Upon activation, the PKC molecule unfolds to remove endogenous pseudosubstrate, and bring ATP into proximity with the substrate.
PKC isoforms are generally classified into three subgroups, conventional or classic cPKs, novel nPKCs, and atypical aPKCs (Table 1). cPKCs include α, βI, βII, and γ PKC. They have the traditional 4 conserved regions (C1–C4) and the 5 variable regions (V1–V5). The cDNA clones for α, βI, βII and γ PKC were isolated from bovine [28, 29], rat [30], rabbit [31] and human brain libraries [28]. βI and βII cDNAs are derived from a single mRNA transcript by alternative splicing, and differ only in ~50 amino acid residues in their carboxyl-terminal end in the variable region V5 [30, 31]. Because α, βI, βII and γ-PKC have a phorbol ester biding site, they are down-regulated by prolonged exposure to phorbol esters. nPKCs include δ, ε, η(L) and θPKC. They lack the C2 region and are Ca2+-independent [30].ε-PKC differs from α-, βI-, βII-, and γ-PKC in that the V1 region is extended while the C2 region is deleted [32]. η-PKC shows the highest sequence similarity to ε-PKC with 59.4% identity [33]. PKC L, is the human homologue of mouse η-PKC [34]. θ-PKC consists of 707 amino acids and shows the highest sequence similarity to δ-PKC (67% identity) [33]. aPKCs include ζ and λ/ι PKC, and characteristically have only one Cys-rich zinc finger-like motif. aPKCs are dependent on phosphatidylserine, but are not affected by DAG, phorbol esters or Ca2+, and therefore do not translocate or downregulate in response to phorbol esters or DAG [30] (Table 1).
Table 1.
Representative PKC Distribution and Subcellular Distribution in Blood Vessels
| PKC | MW (kDa) | Blood Vessel | Resting Cell | Activated Cell | Ref |
|---|---|---|---|---|---|
| Classic | |||||
| α | 74–82 | Rat aorta Rat Carotid artery Rat mesenteric artery Porcine coronary artery Bovine aorta Ferret portal vein |
Cytosolic Cytosolic Cytosolic/membrane Cytosolic Cytosolic Cytosolic |
Nuclear Membrane Cytosolic/Membrane Membrane Membrane Surface membrane |
[178] [179] [180] [78] [181] [99] |
| β | 80–82 | Rat aorta Rat Carotid artery |
Cytosolic Cytosolic |
Nuclear Membrane |
[178] [179] |
| γ | 70–82 | Rat mesenteric artery | Cytosolic | Cytosolic | [180] |
| Novel | |||||
| δ | 76–82 | Rat aorta Rat mesenteric artery |
Cytoskeleton/organelle Membrane |
Cytoskeleton/organelle Membrane |
[83] [180] |
| ε | 90–97 | Rat mesenteric artery Ferret aorta Porcine coronary artery |
Cytosolic/membrane Cytosol Cytosolic |
Cytosolic/membrane Surface membrane Membrane |
[180] [56] [78] |
| η | NIH 3T3 fibroblasts | Cytosolic/membrane | Membrane | [182] | |
| Atypical | |||||
| ζ | 64–82 | Rat aorta Rat mesenteric artery Ferret aorta, portal vein |
Perinuclear Cytosolic Perinuclear |
Intranuclear Cytosolic Intranuclear |
[83] [180] [56] |
| λ/ι | 70 | Rabbit femoral artery Rabbit portal vein |
Cytosolic | Cytosolic | [183] |
PKC Substrates
The inactive PKC molecule is folded such that the basic autoinhibitory pseudosubstrate is tightly bound to the acidic patch in the substrate-binding site, a conformation that protects it from proteolysis. When PKC is activated by phosphatidylserine, DAG and Ca2+ the pseudosubstrate becomes unmasked and liable to proteolysis. This is supported by the observation pseudosubstrate antibodies activate PKC likely by removing the pseudosubstrate from the active substrate binding site [35]. Activated PKC phosphorylates arginine-rich protein substrates, and these peptides neutralize the acidic patch that maintains the pseudosubstrate in the active site, thus displacing the basic pseudosubstrate from the substrate-binding site in the catalytic domain [26, 36, 37]. The amino acid sequence in the vicinity of the substrate phosphorylation site may provide a substrate recognition guide for PKC and structure-function studies of synthetic peptide substrates suggest that PKC requires basic residue determinants in common with other serine/threonine protein kinases [37].
Common PKC substrates include lysine-rich histone and myelin basic protein [25]. α-, β-, γ-, and ζ-PKC are potent histone IIIS kinases. δ-, ε-, and η-PKC do not adequately phosphorylate histone IIIS, but readily phosphorylate myelin basic protein [32, 38, 39]. However, removal of the regulatory domain of ε-PKC by limited proteolysis generates a catalytic fragment that can phosphorylate histone IIIS [32].
Myristoylated, alanine-rich C kinase substrate (MARCKS) is an 87-kDa protein and a major PKC substrate that binds F-actin and bridges cytoskeletal actin to the plasma membrane [40, 41]. Also, PKC-induced phosphorylation of the inhibitory GTP-binding protein Gi facilitates the dissociation of its αi subunit from adenylyl cyclase and leads to increased adenylyl cyclase activity [42]. Other PKC substrates include plasma membrane ion channels and pumps. PKC inhibits Ca2+-dependent large conductance K+ channel (BKCa) in pulmonary VSM [43]. Also, thromboxane A2 may inhibit voltage-gated K+ channels and pulmonary vasoconstriction via a mechanism involving ζ-PKC [44]. PKC-induced phosphorylation of SERCA promotes Ca2+ uptake, and activation of PMCA promotes Ca2+ extrusion, leading to reduction in agonist-induced increase in VSM [Ca2+]i [45]. PKC may phosphorylate the α1 subunit of Na+/K+-ATPase, and activate the Na+/H+ exchanger and thereby increase cytoplasmic pH and cause cell alkalinization [46, 47].
PKC substrates also include cytoskeletal and regulatory proteins in VSM. PKC-induced phosphorylation of vinculin, a cytoskeletal protein localized at adhesion plaques, could affect cell shape and adhesion properties [48]. PKC also causes phosphorylation of the CPI-17 regulatory protein leading to inhibition of MLC phosphatase, increased MLC phosphorylation and enhanced VSM contraction [49]. Also, α-PKC phosphorylates the actin-binding protein calponin, allowing more actin to interact with myosin and further VSM contraction [29]. However, PKC could also phosphorylate the 20-kDa MLC and MLC kinase, leading to inhibition of Ca2+-dependent actin-myosin interaction and VSM contraction [50].
PKC Distribution
PKC isoforms are expressed in various vascular beds (Table 1). α-PKC is a universally expressed in almost all blood vessels examined. γ-PKC is expressed mainly in neurons and nerve endings of blood vessels. δ-PKC is mainly associated with the cytoskeleton. ζ-PKC is universally expressed in many vascular tissues. η/L-PKC is expressed in the lung, skin, heart and brain, θ-PKC in skeletal muscle and ι/λ-PKC in the testis and ovary [39].
In resting cells, α, β and γ-PKC are localized mainly in the cytosolic fraction, and activated PKC undergoes translocation from the cytosolic to the particulate and membrane fraction [51, 52]. Activated α-,β- and γ-PKC usually undergo translocation from the cytosol to the cell membrane [53] (Table 1). However, in normal fibroblasts, α-PKC is tightly associated with the cytoskeleton and organized into plasmalemmal focal contacts which are composed of structural proteins such as vinculin, talin, integrin and α-actinin that allow the attachment of cytoskeletal microfilaments to the plasma membrane [54]. In neural cells, βI-PKC is associated with the plasma membrane, while βII-PKC is localized in the Golgi complex [21]. In the cerebellum, γ-PKC is present in the cell bodies, dendrites and axons of Purkinje’s cells. Immuno-electron microscopy revealed that γ-PKC is associated with most cell membranous structures, except the nucleus [55].
Because δ-PKC is localized in the vicinity of the cytoskeleton, it is often identified in the particulate fraction of both resting and activated cells. In contrast, ε-PKC undergoes translocation from the cytosol to the surface membrane during VSM activation. ζ-PKC is localized in the vicinity of the nucleus in both resting and activated mature VSMCs [56]. However, in the developing embryo, ζ-PKC may have different distribution and function and may play a role in perinatal pulmonary vasoconstriction [57].
Different physico-chemical forces may drive PKC translocation including simple diffusion or specific targeting mechanisms that allow tight binding of PKC to its target substrate. Some of the targeting mechanisms include conformation changes and altered hydrophobicity, lipid modification, phosphorylation and targeting sequences. For instance, binding of Ca2+ or DAG to PKC may cause conformational changes that unfolds the PKC molecule and result in exposure of the substrate region and increased PKC hydrophobicity and binding to membrane lipids [26]. Also, modification in the lipid component of a protein could influence its subcellular distribution. The VSM plasma membrane is composed of several domains of focal adhesions alternating with zones rich in caveolae, and both harbor a subset of membrane-associated proteins. Also, the plasma membrane lipids are segregated into cholesterol-rich lipid rafts and glycerophospholipid-rich non-raft regions, an arrangement that is critical for preserving the membrane protein architecture and for the translocation of proteins to the plasma membrane. In VSMC membrane, lipid segregation is supported by annexins that target membrane sites of distinct lipid composition, and each annexin requires different [Ca2+] for its translocation to the plasma membrane, thus allowing a spatially confined graded response to external stimuli and plasmalemmal localization of PKC [58]. Protein phosphorylation could also change their conformation or electric charge and consequently affect their lipid affinity and binding to the plasma membrane. While myristoylation of MARCKS is essential for its binding to actin and the plasma membrane, its phosphorylation by PKC may have an electrostatic effect that affects the protein affinity to the plasma membrane and consequently interferes with its actin cross-linking and causes its displacement from the plasma membrane. This is supported by the observation that dephosphorylation of MARCKS causes its re-association with the plasma membrane via its stably attached myristic acid membrane-targeting moiety [59]. Phosphorylation of PKC itself via autophosphorylation or by a putative PKC kinase may also determine its localization and full activation, and PKC phosphorylation sites have been identified in the catalytic domain of α-, β- and δ-PKC [60]. Also, binding sites for arginine-rich polypeptides have been identified in the PKC molecule distal to its catalytic site allowing targeting of PKC to target substrates at specific subcellular locations [61]. Receptors for activated C-kinase (RACKs) may target PKC to cytoskeletal elements, while a peptide inhibitor derived from the PKC binding proteins annexin I and RACKI may interfere with translocation of β-PKC [62].
PKC Function
PKC is involved in many physiological functions including secretion and exocytosis, modulation of ion channel, gene expression and cell growth and proliferation [21, 39]. For example, transfection of a vector containing the full-length cDNA encoding βI-PKC in rat fibroblasts led to overexpression of the isozyme and caused cell growth abnormalities that mimicked the effects of the tumor promoter phorbol esters. However, these cell lines did not exhibit the typical characteristics of malignantly transformed fibroblasts. Hence, the overproduction of PKC per se may not be sufficient to cause cancer, although it may facilitate the cell conversion to malignancy by genotoxic agents [63].
PKC may exert negative-feedback control over cell signaling by downregulation of surface receptors and/or inhibition of agonist-induced activation of PLC and phosphoinositide hydrolysis [21]. Also, PKC may play a role in VSM contraction [18, 21, 39, 64]. PKC activators such as DAG analogs and phorbol esters cause contraction in isolated blood vessels ex vivo [17, 18, 39]. Phorbol ester-induced vascular contraction is not associated with detectable increases in [Ca2+]I, and a role of Ca2+-independent ε-PKC has been suggested [24, 56]. Also, PKC inhibitors inhibit agonist-induced contraction of coronary VSM [17, 64]. However, PKC may induce phosphorylation of MLC kinase leading to inhibition of VSM contraction [50].
PKC-induced phosphorylation of certain substrates may activate a cascade of protein kinases that enhance VSM contraction [65]. PKC-induced phosphorylation of CPI-17 promotes the inhibition of MLC phosphatase and leads to further increases in MLC phosphorylation and VSM contraction (Fig. 1) [49]. α-PKC-induced phosphorylation of the actin binding protein calponin could reverse the calponin-mediated inhibition of actin-activated myosin ATPase, thus allowing more actin to interact with myosin and enhance VSM contraction (Fig. 1) [18, 29].
Mitogen-activated protein kinase (MAPK) is a Ser/Thr protein kinase that requires dual phosphorylation at both the Thr and Tyr residues for its activation. In quiescent undifferentiated VSMCs, MAPK is mainly in the cytosol, but upon cell activation by a growth factor or a mitogen, MAPK undergoes translocation from the cytosol to the nucleus where it promotes gene expression and cell growth [66]. Importantly, tyrosine kinase and MAPK activities have been identified in differentiated contractile VSM, suggesting a role in VSM contraction [65]. Activation of differentiated VSMCs with the α-adrenergic agonist phenylephrine is associated with an initial translocation of MAPK from the cytosol to the surface membrane. However, during maintained VSM activation MAPK undergoes redistribution from the surface membrane to the cytoskeleton [65]. It is likely that agonist-induced activation and generation of DAG at the surface membrane promotes translocation of the Ca2+-independent ε-PKC from the cytosol to the surface membrane, where it becomes fully activated. Activated ε-PKC in turn promotes translocation of both MAPK kinase (MEK) and MAPK from the cytosol to the surface membrane to form a protein kinase complex. PKC then induces phosphorylation and activation of MEK, which in turn causes phosphorylation of MAPK at both Thr and Tyr residues [67]. Tyr-phosphorylated MAPK is then targeted to the cytoskeleton, where it induces phosphorylation of the actin-binding protein caldesmon [68, 69]. The phosphorylation of caldesmon reverses its inhibition of actin-mediated MgATPase activity leading to further increases in actin-myosin crossbridge cycling and VSM contraction (Fig. 1) [18, 65].
PKC Activators
PKC isoforms have different sensitivity to Ca2+, phosphatidylserine, DAG and other phospholipid products. Ca2+-dependent PKCs bind Ca2+ in a phospholipid-dependent manner such that Ca2+ may form a “bridge” holding the PKC-phospholipid complex at the plasma membrane [70]. Phosphatidylserine is required for activation of most PKCs. Phosphatidylinositol and phosphatidic acid may activate PKC, but may require high Ca2+ concentrations. DAG activates Ca2+-independent PKCs and reduces the Ca2+ requirement for activation and membrane associationof Ca2+-dependent PKCs [21].
Lipids derived from sources other than glycerolipid hydrolysis such as cis-unsaturated free fatty acids and lysophosphatidylcholine, ceramide (a sphingomyelinase product), phosphatidylinositol 3,4,5-trisphosphate and cholesterol sulfate may also activate PKC [71]. Other PKC activators include phorbol esters such as 12-o-tetradecanoylphorbol-13-acetate (TPA), phorbol myristate acetate (PMA) and phorbol 12,13-dibutyrate (PDBu). Phorbol esters reduce the apparent Km of PKC for Ca2+ and stabilize it in the membrane-bound form [39].
Bryostatin, a marine natural product, binds to and activates PKC and is more potent than PMA in translocating δ- and ε-PKC but is not a carcinogen or a complete tumor promoter [72]. Oxidized low density lipoprotein (LDL) increases the activity of α- and ε-PKC in coronary VSM, and promotes coronary artery vasoconstriction and atherogenesis [73]. γ-Radiation may activate α- and ε-PKC, and in turn promote smooth muscle cell apoptosis [74].
PKC activity and affinity for its substrate could be modified by its phosphorylation by other protein kinases or even by its own autophosphorylation [75–77]. α-, βI- and βII-PKC are expressed as inactive precursors that require phosphorylation by a “PKC kinase” for permissive activation. Phosphorylation of α-PKC may prevent its down-regulation during prolonged exposure to phorbol ester [76]. Also, phosphorylation of βII-PKC at the C-terminus allows it to bind ATP and substrate with higher affinity. Phosphorylation of structure determinants in the regulatory domain of PKC may increase its affinity to Ca2+ [77]. Autophosphorylation of the Ca2+-independent δ-PKC at Ser-643 may occur in vivo: and consequently control the activity and biological function of δ-PKC [75].
PKC Inhibitors
Several PKC inhibitors with different affinity, efficacy and specificity have been developed (Table 2). PKC inhibitors acting on the catalytic domain by competing with ATP are not specific and inhibit other protein kinases. PKC inhibitors acting on the regulatory domain by competing at the DAG/phorbol ester or the phosphatidylserine binding site may be more specific. While prolonged exposure to phorbol esters can downregulate α-, β-, γ-, and ε-PKC [78], the tumor promoting actions of phorbol esters limit their use.
Table 2.
Representative PKC Inhibitors
| Chemical Group | Example | Site of Action | Specificity |
|---|---|---|---|
| 1-(5-isoquinolinesulfonyl)-2-methylpiperazines | H-7 | Catalytic domain Compete with ATP at the ATP binding site |
Also, inhibits cyclic AMP and cyclic GMP-dependent protein kinases |
| Microbial Alkaloids, Products of Streptomyces | Staurosporine SCH47112 | Catalytic domain, ATP binding site | Also, inhibits MLC kinase and tyrosine kinase |
| Benzophenanthridine Alkaloids | Chelerythrine | Catalytic domain | Competitive inhibitor with histone IIIS |
| Indocarbazoles | Gö6976 | Catalytic domain | Ca2+-dependent α-and βI-PKC |
| Bisindolylmaleimide Staurosporine Analogs |
GF109203X Ro-318220 Midostaurin (PKC412, CGP41251) Ruboxistaurin (LY333531) |
Catalytic domain | PKC isozymes α, βI, βII, γ, δ and ε. Ruboxistaurin mesylate is a selective antagonist of PKC βI and PKC βII. |
| Perylenequinone Metabolites from Cladosporium cladosporioides | Calphostin C (UCN-1028A) | Regulatory domain | Binds to the regulatory domain at DAG/phorbol ester binding site |
| Membrane lipids | Sphingosine | Regulatory domain | Competitive inhibitor with phosphatidylserine |
| Other: | Adriamycin Aminoacridine Apigenin Cercosporin Chlorpromazine Dexniguldipine Polymixin B Sangivamycin Tamoxifen Trifluoperazine UCN-01, UCN-02 |
The pseudosubstrate region in the regulatory domain of PKC contains an amino acid sequence between the 19 and 36 residues that resembles the substrate phosphorylation site. Synthetic pseudosubstrate inhibitor peptides (19 to 36) inhibit specific PKCs by exploiting their substrate specificity without interfering with ATP binding. These synthetic peptides inhibits both PKC substrate phosphorylation and PKC autophosphorylation [37]. Also, myr-ψPKC, a myristoylated peptide based on the substrate motif of α- and β-PKC, inhibits TPA-induced PKC activation and phosphorylation of MARCKS [79].
In smooth muscle, α-Tocopherol inhibits the expression, activity and phosphorylation of α-PKC, while β-tocopherol protects PKC from the inhibitory effects of α-tocopherol [80].
Short interference RNA (siRNA) can prevent the expression of a specific PKC isoform and thereby determine its role in a specific cellular function. Antisense techniques, knockout mice and transgenic animals have also been used to study the effects of downregulation of a specific PKC isoform in vivo.
PKC and Hypertension
HTN is a multifactorial disorder that involves changes in the neural, hormonal, renal and vascular control mechanisms of BP [81]. Increases in the amount and activity of PKC could cause disturbance in one or more of these physiological control mechanisms, leading to persistent increases in BP and HTN. PKC could promote VSM growth, proliferation and contraction pathways. The relation between PKC and HTN could also involve changes in the vascular endothelium, ECM and MMPs-mediated vascular remodeling, oxidative stress and free radicals, renal hemodynamics and renin-angioensin system, neuronal changes and sympathetic hyperactivity, vascular inflammation and potential interactions with inflammatory cytokines, and other metabolic factors (Fig. 3).
Fig. 3.

Role of PKC in hypertension. Genetic, dietary and environmental risk factors, lead to vascular, neural and renal dysfunction, and increased release of various mediators from endothelial cells (ROS, ET-1, ANG II), sympathetic neurons (norepinephrine) and the kidney (ANG II). These mediators could stimulate VSM and activate PKC, as well as Ca2+, Rho kinase, and MAPK thereby induce vasoconstriction and VSM growth and proliferation. The interaction of PKC with matrix metalloproteinases (MMPs) in the extracellular matrix (ECM) could contribute to vascular remodeling. Activation of the renin-angiotensin system (RAS) and increased ANG II production induce salt and water retention and increase plasma volume. Persistent increases in peripheral vascular resistance and plasma volume lead to hypertension.
PKC and VSM Growth and Reactivity in Hypertension
Increased expression/activity of PKC could promote trophic changes in VSM and lead to increases in the vessel wall thickness and hypertrophic remodeling (Fig. 3). Overexpression of α-PKC in A7r5 VSMC line stimulates cell proliferation [82]. Also, the localization of ζ-PKC in the vicinity of the nucleus suggests a role in VSM growth and the hypertrophic remodeling associated with HTN [56, 83]. Combined increases in PKC activity and [Ca2+]i could exert trophic effects in both the vasculature and the heart, leading to narrowing of the arterial lumen and cardiac hypertrophy in long-standing HTN [47]. Increased PKC expression and activity could also enhance vasoconstriction and increase vascular resistance and BP (Fig. 3). α-PKC activation has been shown to enhance VSM contraction, and its overexpression in VSM may be involved in HTN [56, 83]. Also, the Ca2+-independent ε-PKC may enhance the myofilament force sensitivity to [Ca2+]i in VSM and promote the vasoconstriction associated with HTN [18, 56]. The localization of δ-PKC in the cytoskeleton suggests that it may play a role in the vascular remodeling observed in HTN [39].
PKC in Genetic Hypertension
Genetic linkage studies in certain families have supported the genetic origin of HTN. For instance, mutations in BMPR2 gene, which encodes a bone morphogenetic protein receptor II, a TGF-β superfamily member, have been linked to 55% of familial pulmonary arterial HTN [84–86]. Mice carrying BMPR2 heterozygous alleles (BMPR2+/−) are genetically equivalent to mutant human gene and develop pulmonary arterial HTN under stress conditions [87]. Proteomics studies on mouse tissues have identified β-PKC as one of the signaling pathways associated with BMPR2 [88], suggesting a role of PKC in genetic HTN.
PKC may also play a role in spontaneously hypertensive rats (SHR). Norepinephrine-induced contraction is more readily inhibited by the PKC inhibitor 1-(5-isoquinolinesulfonyl)-2-methylpiperazine (H-7) in the aorta of SHR than Wistar-Kyoto rats (WKY). Also, treatment of the aortic segments with H-7 causes a shift to the right in the concentration-contraction curve of the PKC activator TPA in the aorta of SHR, but not WKY [89]. The PKC activator PDBu also produces contraction and greater reduction in cytosolic PKC in the aorta of SHR than WKY [90]. In SHR, γ-interferon restores PKC level to that in normal control rat, suggesting an interaction between PKC and cytokines in genetic HTN [91].
The role of PKC in genetic HTN has been further studied by measuring vascular contraction and PKC activity during the development of HTN in young (5–6 weeks) SHR. High KCl-induced contraction in intact mesenteric arteries and the Ca2+-force relationship in vessels permeabilized with α-toxin were not different in SHR and WKY rats. Treatment with the PKC activator PDBu caused greater enhancement of high KCl-induced contraction in intact vessels and the Ca2+-force relationship in permeabilized vessels of SHR than those of WKY. The PKC inhibitors H-7 and calphostin C caused greater inhibition of contraction in blood vessels of SHR than WKY. These data support that PKC enhances the Ca2+ sensitivity of the contractile proteins in VSM to a greater extent in blood vessels of young prehypertensive SHR than WKY. The data also suggest that PKC activation in VSM occurs before overt HTN, and support a causative role of PKC in the development of genetic HTN [92].
To further examine potential inborn differences in vascular PKC before the onset of HTN, studies have compared VSM proliferation in cells from young (1–2 week) SHR and WKY rats. In cultured aortic VSM from SHR and WKY rats, both ANG II and endothelin-1 (ET-1) enhanced thymidine incorporation into DNA, an indicator of DNA synthesis. Treatment of VSMCs with the PKC inhibitor chelerythrine caused greater suppression of ANG II and ET-1 induced DNA synthesis and VSM growth in cells of SHR than WKY, suggesting an inborn increase in PKC activity in VSMCs of SHR [93].
In a study assessing the role of PKC in the changes in vascular tone associated with genetic HTN in vivo, it was found that perfusing the PKC activator PDBu in the hindlimb of anesthetized SHR and WKY rats caused prolonged vasoconstriction and increased perfusion pressure. The PDBu-induced vasoconstriction and increased perfusion pressure were inhibited by the PKC inhibitor staurosporine to a greater extent in SHR than WKY rats, supporting a role of PKC in the regulation of vascular function and BP in vivo, and increased PKC expression and activity in VSM of rat models of genetic HTN [94].
Sex differences in the expression and activity of PKC have been observed in VSM of WKY and SHR. VSM contraction and the expression and activity of α-, δ- and ζ-PKC in response to the phorbol ester PDBu are less in intact female than intact male WKY, and these sex differences are greater in VSM from SHR than WKY rats [95]. PDBu-induced contraction and PKC activity were similar in castrated and intact male rats, but greater in ovariectomized (OVX) than in intact female rats. Treatment of OVX females with 17β-estradiol subcutaneous implants caused reduction in PDBu contraction and PKC activity, that were greater in SHR than WKY rats. These data suggested sex-related reduction in VSM contraction and the expression and activity of α-, δ- and ζ-PKC in female compared with male rats, and that these differences are likely mediated by estrogen and are enhanced in genetic HTN [95].
PKC and Human Essential Hypertension
Studies have shown an increase in oxidative stress and growth response in VSMCs from resistance arteries of patients with essential HTN as compared to cells from normotensive controls. ANG II increases ROS to a greater extent in VSM from hypertensive than normotensive subjects. Also, ANG II increases phospholipase D (PLD) activity and DNA and protein synthesis to a greater extent in VSMCs from hypertensive than normotensive subjects, and the ANG II effects are partially inhibited by treating the cells with the PKC inhibitors chelerythrine and calphostin C. These data suggest that the increased oxidative stress and growth-promoting effects of ANG II in VSMCs from hypertensive patients may involve increased activity of PLD- and PKC-dependent pathways and further support a role of these pathways in the vascular remodeling associated with HTN [96].
One of the properties of PKC is that it undergoes translocation from the cytosol to cell membrane during VSM activation, a property that can be used in the diagnosis and prognosis of VSM hyperactivity in HTN. However, the subcellular distribution of PKC may vary depending on the type and abundance of membrane lipids. Studies have shown increased cholesterol/phospholipid ratio, higher levels of monounsaturated fatty acids, and lower levels of polyunsaturated fatty acids in erythrocyte membranes from elderly hypertensive subjects as compared to normotensive controls. On the other hand, the levels of activated membrane-associated PKC are not increased, but rather decreased in erythrocytes of elderly hypertensive subjects, which may not be related to the etiopathology of HTN, but represent an adaptive compensatory mechanism to HTN [97].
PKC and Aortic Constriction-Induced Hypertension
PKC activation and translocation are increased in a rat model of pressure overload and left ventricular hypertrophy produced by banding or clipping of the aorta [83]. The increased PKC activity is associated with increased tritiated phorbol ester ([3H]PDBu) binding and PKC concentration in both the cytosolic and membrane fractions [98]. Immunoblot analysis has revealed that the increased PKC activity is mainly due to increases in the amount of βI-, βII- and ε-PKC in the surface membrane and nuclear-cytoskeletal fractions [98]. Imaging of the subcellular distribution of PKC revealed that in VSMCs of normotensive rats α-PKC is mainly in the cytosol, while ζ-PKC is in the perinuclear area [56, 99]. In VSMCs of hypertensive rats, α-PKC is activated and localized at the surface membrane, while ζ-PKC is localized in the nucleus [83].
PKC: Endothelial Dysfunction and Hypertension
Changes in PKC activity in the endothelium could contribute to the regulation of vascular function and BP. Studies have suggested a role of PKC in the endothelial cell dysfunction observed in blood vessels of SHR and deoxycorticosterone acetate (DOCA)-salt hypertensive rats [100, 101]. NO is one of the major vasodilators produced by the endothelium. Activated endothelial NO synthase (eNOS) catalyzes the transformation of L-arginine to L-citrulline and the production of NO. Mice deficient in eNOS are hypertensive and lack NO-mediated vasodilation [102]. PKC activation may affect NOS activity and NO production or bioactivity. PKC may cause phosphorylation of Thr-495 and dephosphorylation of Ser-1175 in eNOS and in turn inhibit NO production [103, 104]. Specifically, α- and δ-PKC phosphorylate eNOS at Ser-1175 and increase NO production [105, 106]. PKC may also play a role in eNOS “uncoupling”, a process in which eNOS is over-expressed or hyperactivated in an attempt to produce more NO to reduce vascular tone, but instead produces superoxide (O2−•) [107, 108]. In SHR, oral administration of the PKC inhibitor midostaurin, a staurosporine analog, reverses aortic eNOS “uncoupling”, and causes up-regulation of eNOS expression and diminished production of ROS. Also, aortic levels of (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4), a NOS cofactor, are reduced in SHR compared with WKY. In addition, midostaurin lowered BP in SHR and, to a lesser extent in WKY [109], supporting potential benefits of PKC inhibitors in genetic HTN.
PKC: Oxidative Stress and Hypertension
Oxidative stress has been demonstrated in most forms of HTN including essential and renovascular HTN. Increased O2−• production decreases NO bioactivity, and in turn increases vasoconstriction and vascular resistance in HTN [1–3]. The HTN-associated increase in O2−• production in HTN may partly involve PKC. In isolated arteries, high pressure induces O2−• production via PKC-dependent activation of NADPH oxidase [3]. Also, O2−• production is increased in sympathetic neurons of DOCA-salt hypertensive rats via activation of NADPH oxidase [110]. Studies have also shown that the impaired vasodilation and increased vascular O2−• production in the 2 kidney-1 clip (2K-1C) rat model of renovascular HTN are likely related to PKC-mediated activation of membrane-associated NADPH-dependent oxidase [2, 3, 111].
PKC: MMPs and Vascular Remodeling in Hypertension
PKC may play a signaling role in the expression and activity of MMPs and consequently affects ECM composition and vascular remodeling. MMPs are a family of zinc-containing proteases that play a role in the degradation of ECM proteins [112–114], and may have additional effects on the endothelium and VSM [115, 116]. MMPs activity is regulated at the transcription level as well as by activation of their pro-form, interaction with specific ECM components, and inhibition by endogenous tissue inhibitors of MMPs (TIMPs). Changes in hemodynamics, vessel injury, inflammatory cytokines and ROS could upregulate MMPs and promote vascular remodeling and HTN. Some studies have shown that the plasma levels and activity of MMP-2, MMP-9 and TIMP-1 are elevated in hypertensive patients [117]. Other studies have shown that the plasma levels of active MMP-2 and -9 are decreased in patients with essential HTN, and treatment with amlodipine normalized MMP-9 plasma levels [118]. These findings suggested a relationship between abnormal ECM metabolism and HTN, and that antihypertensive treatment may modulate collagen metabolism. In a study examining the serum levels of carboxy-terminal telopeptide of collagen type I (CITP) as a marker of extracellular collagen type I degradation, MMP-1 (collagenase), TIMP-1, and MMP-1–TIMP-1 complex, baseline free MMP-1 was decreased and baseline free TIMP-1 was increased in hypertensive compared with normotensive subjects. Hypertensive patients treated with the angiotensin-converting enzyme (ACE) inhibitor lisinopril for 1 year showed an increase in free MMP-1, a decrease in free TIMP-1, and an increase in serum CITP. These findings suggest that systemic extracellular degradation of collagen type I is depressed in patients with essential HTN and may facilitate organ fibrosis, and this can be normalized by treatment with lisinopril [7]. Also, gelatin zymographic analysis of in internal mammary artery from normotensive and hypertensive patients undergoing coronary artery bypass surgery, indicated a decrease in activity of MMP-2 and -9 in HTN. MMP-1 activity was also decreased by 4-fold without a change in protein levels. Immunoblot analysis revealed a decrease in the tissue levels of extracellular matrix metalloproteinase inducer (EMMPRIN), MMP activator protein (MT1-MMP) and MMP-9 in HTN. Also, measurement of plasma markers of collagen synthesis (procollagen type I amino-terminal propeptide [PINP]) and collagen degradation (carboxy-terminal telopeptide of collagen type I [ICTP]) has shown no changes in PINP levels but decreased degradation of collagen in HTN. These data demonstrate that MMP-1 and -9, MMP inducer and activator proteins are downregulated in HTN, and may result in increased collagen deposition in HTN [8].
Studies have shown that the total wall thickness and the medial area are increased in the aorta but not vena cava of DOCA-salt versus sham rats. In HTN, MMP-2 expression and activity were increased in the aorta but not vena cava, while MMP-9 was weakly expressed in both vessels. TIMP-2 expression was increased in the aorta of DOCA-salt rats compared to sham, but barely detectable in vena cava of DOCA-salt and sham or rats. These data suggest a link between MMPs and vascular remodeling in the aorta of DOCA-salt hypertensive rats. The increase in TIMP-2 expression in the aorta of DOCA-salt rats may be an adaptive mechanism to the high levels of MMP-2 [9]. Other studies have shown that in wild-type mice treated with ANG II and a 5% NaCl diet for 10 days, the onset of HTN is accompanied by increased MMP-9 activity in conductance vessels. In contrast, in MMP-9(−/−) mice, the absence of MMP-9 activity is associated with vessel stiffness and increased pulse pressure, suggesting that in early stages of HTN, MMP-9 activation may preserve vessel compliance and alleviate BP increase [119].
Growth factors and cytokines such as nuclear factor κB and IL-1α stimulate VSMCs to secrete MMP-1, -3, -9, and these effects may be dependent on activation of ζ-PKC, and may contribute to inhibition of VSMC proliferation and vascular remodeling [10]. PKC also increases MMP-2 secretion in endothelial cells [120], and PKC-α plays a critical role in MMP-9 secretion in bovine capillary endothelial cells through ERK1/2 signaling [11]. PKC-β plays a signaling role in the expression and activity of MMP-1 and -3 in human coronary artery endothelial cells [121]. In cardiac microvascular endothelial cells, IL-1β activates α-PKC and βI-PKC and increases the expression and activity of MMP-2, and inhibition of α-PKC and βI-PKC abrogates the IL-1β stimulated increase in MMP-2 [12].
PKC in Salt-Sensitive Hypertension
Increased dietary sodium intake causes HTN in salt-sensitive individuals [122, 123]. Studies have shown an increase in BP and the heart to body weight ratio in DOCA salt-sensitive hypertensive rats compared to control rats. Also, α-, γ- and ε-PKC are upregulated while δ-PKC is not altered in cardiac extracts of DOCA-salt rats compared to controls. On the other hand, δ-PKC is increased in cardiac fibroblasts from DOCA-salt rats compared to controls. These data suggest cell-specific increase in the expression of α, γ, δ or ε-PKC in the hearts of DOCA-salt hypertensive rats [124]. Also, the PKC inhibitor GF109203X (2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide) decreases both basal tone and MAPK (ERK1/2) activity in DOCA-salt rats, suggesting that the increased basal vascular tone and MAPK activity in DOCA-salt hypertensive rats may involve PKC [125].
Changes in cardiac PKC have also been observed in Dahl salt-sensitive hypertensive rats. Marinobufagenin, an endogenous ligand of the α1 subunit of the cardiac Na/K-ATPase, is increased in sodium-loaded Dahl-salt-sensitive rats, and PKC-induced phosphorylation of the α1 Na/K-ATPase may increase its sensitivity to marinobufagenin, and further contribute to the increased BP in this rat model [111].
PKC may also affect the renin-angiotensin-aldosterone system and the renal control mechanism of BP. Infusion of ANG II in rats causes HTN, vascular endothelial dysfunction and increased vascular O2−• production. Some of the vascular effects of ANG II may be mediated by increased endothelial cell release of ET-1, which in turn activates PKC [126–128]. Interestingly, ANG II-induced ET-1 production and PKC activity are greater in blood vessels of SHR than normotensive control rats [129]. Other studies have shown that cytosolic PKC activity is higher in aortic VSM from SHR than those from WKY or SHR treated with the angiotensin-converting enzyme (ACE) inhibitor enalapril, and the changes in vascular PKC activity were paralleled by changes in BP. Membrane-bound PKC activity was detected in aortic VSM of SHR, but not in that of the WKY or enalapril-treated SHR. Also, α-PKC mRNA expression and protein amount were greater in aortic VSM from SHR than those from WKY or enalapril-treated SHR, suggesting that the beneficial effects of ACE inhibitors in HTN may in part involve changes in expression and activity of α-PKC in VSM [130]. Other studies have shown that PKC could affect the Na+/Ca2+ exchange mechanism in the renal arterioles leading to defective renal vasodilation and salt-sensitive HTN [131].
PKC may also affect the renal tubular cells and the kidney function. In renal tubular epithelial cells, δand ζ-PKC are localized in the plasma membrane whereas α- and ε-PKC are cytosolic. Dopamine, an intrarenal modulator of sodium metabolism and BP, causes translocation of α- and ε-PKC to the plasma membrane [132, 133], supporting a role of PKC in the control of renal sodium and water reabsorption and BP [134].
PKC: Neuronal Dysfunction and Hypertension
PKC may play a role in the neural control mechanisms of BP. The expression and redistribution of PKC isozymes are increased in brain tissue of SHR [135]. Also, sympathetic nerves are known to control VSM contraction by releasing chemical transmitters such as norepinephrine, which in turn trigger the increase in [Ca2+]i and PKC activity. Polymorphisms in human tyrosine hydroxylase gene have been associated with increased sympathetic activity, norepinephrine release and HTN [136], and the role of PKC in these hypertensive subjects remains to be investigated.
PKC: Metabolic Dysfunction and Hypertension
Metabolic disorders are often associated with hyperglycemia and glucose intolerance, insulin resistance, central and overall obesity, dyslipidemia (increased triglyceride and decreased high-density lipoprotein (HDL) cholesterol levels), and different vascular manifestations and complications including HTN. Evidence suggests a role of PKC in these metabolic disorders. For example, glucose-induced increase in endothelial cell permeability is associated with activation of α-PKC [137]. Also, glucose, via activation of PKC, may affect the Na+/H+ exchanger mRNA expression and activity in VSMCs [138]. Importantly, an antisense complementary to the mRNA initiation codon regions for α- and β-PKC causes downregulation of these PKC isoforms and inhibits insulin-induced glucose uptake in rat adipocytes [139]. Also, inhibitors of β-PKC ameliorate the vascular dysfunction in rat models of diabetes and attenuate the progression of experimental diabetic nephropathy and HTN [140].
PKC: Vascular Inflammation and Hypertension
Vascular inflammation may play a role in cardiovascular disease [4, 141]. Plasma levels of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6 are increased in patients with HTN and coronary artery disease [5, 6, 142–145]. Also, infusion of ANG II does not induce HTN in IL-6 knockout mice, supporting a role of IL-6 in HTN [146]. In isolated pulmonary artery, hypoxia causes upregulation of TNF-α and IL-1β, a process that is dependent on PKC activation and promotes pulmonary vasoconstriction [13]. Also, TNF-α activates PKC and mitogenic signaling in cultured VSMCs [14], and inhibition of PKC-δ blocks high glucose-induced secretion of TNF-α in cultured rat and human aortic VSMCs [15].
PKC and Pulmonary Hypertension
PKC exert specific effects on the pulmonary vessels and may play a role in pulmonary HTN. Both insulin-like growth factor I and PKC activation stimulate proliferation of pulmonary artery VSMCs. PKC is also one of the signaling pathways involved in hypoxia-induced pulmonary artery VSMC proliferation, and chronic hypoxia may increase PKC activity and promote growth in pulmonary artery adventitial fibroblasts [147]. Mice deficient in ε-PKC show decreased hypoxic pulmonary vasoconstriction [148]. Also, ET-1 is a potent pulmonary vasoconstrictor, and endothelin receptor antagonists have shown benefits in patients with pulmonary HTN [149, 150]. ET-1 induced pulmonary vasoconstriction is partly mediated by PKC, and PKC inhibitors decrease ET-1 induced pulmonary artery contraction [151].
PKC and Hypertension-in-Pregnancy and Preeclampsia
Normal pregnancy is often associated with decreased BP, increased uterine blood flow and decreased vascular responses to vasoconstrictors [152, 153]. Uterine artery from pregnant sheep and aorta of late pregnant rats show decreased vascular contraction and PKC activity [154, 155]. Also, the expression, activation and translocation of the Ca2+-dependent α-PKC and the Ca2+-independent δ- and ζ-PKC are reduced in the aorta of late pregnant compared with nonpregnant rats [155, 156].
In 5% to 7% of pregnancies, women develop a condition called preeclampsia characterized by proteinuria and severe increases in BP [153]. Studies in animal models of HTN in pregnancy have provided useful information regarding the potential causes of preeclampsia. BP is greater in late pregnant rats treated with the NO synthase inhibitor L-NAME, compared with normal pregnant or virgin rats nontreated or treated with L-NAME [157]. Also, phenylephrine-induced contraction is greater in aortas from L-NAME-treated pregnant rats compared with normal pregnant or virgin rats [157, 158]. Additionally, expression and activity of vascular α- and δ-PKC are enhanced in L-NAME-treated compared with non-treated pregnant rats [155, 156], suggesting a role of α- and δ-PKC in the increased vasoconstriction and vascular resistance during HTN in pregnancy [155, 156].
PKC may also play a role in the changes in ANG II receptor-mediated signaling during preeclampsia. In cultured neonatal rat cardiomyocytes, immunoglobulin from preeclamptic women enhances angiotensin type 1 (AT1) receptor-mediated chronotropic response, while immunoglobulin from control subjects has no effect, and the chronotropic effects of imunoglobulin are prevented by the PKC inhibitor calphostin C. Also, confocal microscopy of VSMCs has shown colocalization of purified IgG from preeclamptic women and AT1 receptor antibody. These findings have suggested that preeclamptic women develop auto-antibodies that stimulate AT1 receptor, a process that may to be mediated by PKC [159].
Experimental studies have suggested that reduction in uteroplacental perfusion pressure and the ensuing placental ischemia or hypoxia during late pregnancy may increase the release of cytokines into the maternal circulation, which in turn cause generalized vascular changes and HTN [153, 160–164]. Plasma levels of TNF-α are elevated in women with preeclampsia [162, 163]. Sources other than the placenta may also contribute to the elevated serum levels of TNF-α in preeclamptic women [165]. Interestingly, infusion of TNF-α or IL-6 in pregnant rats to reach plasma levels similar to those observed in preeclampsia, are associated with inceassed BP and systemic vasoconstriction [166, 167]. Also, treatment of aortic segments from pregnant rats with TNF-α or IL-6 enhances reactivity to vasoconstrictor stimuli [168, 169]. Cytokines may increase the expression and activity of vascular PKC leading to increased myofilament force sensitivity to [Ca2+]i and enhanced VSM contraction. Other vasoactive factors such as soluble fms-like tyrosine kinase-1 (sFlt-1) and soluble endoglin (sEng) may be released during reduction of uteroplacental perfusion pressure [170, 171] and their effects on PKC need to be examined.
PKC Inhibitors as Modulators of Vascular Function in Hypertension
The effects of PKC inhibitors on VSM contraction has been examined in isolated blood vessels, but the in vivo effects of PKC inhibitors have not been fully examined. Dahl-salt-sensitive rats on high NaCl (8%) diet exhibit an increase in BP, excretion of the endogenous inhibitor of α1 Na/K-ATPase marinobufagenin, left ventricular weight, and myocardial Na/K-ATPase and βII-PKC and δ-PKC. Treatment of Dahl-salt rats with cicletanine causes reduction in BP and left ventricular weight, decreased sensitivity of Na/K-ATPase to marinobufagenin, no increase in βII-PKC, and reduced phorbol diacetate-induced Na/K-ATPase phosphorylation. The cicletanine-induced decrease in BP may be due to targeting of PKC-induced phosphorylation of cardiac α1 Na/K-ATPase [111]. In isolated human mesenteric artery, marinobufagenin induces sustained vasoconstriction, possibly due to inhibition of the plasmalemmal Na/K-ATPase activity. Treatment of the vessel with cicletanine inhibited marinobufagenin-induced contraction and attenuated marinobufagenin-induced Na/K-ATPase inhibition, and the effects of cicletanine were prevented by the PKC activator phorbol diacetate. Similarly, in rat brain cicletanine inhibits PKC activity, and these inhibitory effects on PKC are prevented in the presence of phorbol diacetate. These data suggest that PKC is involved in the regulation of Na/K-ATPase and vascular tone, and may represent a potential target for therapeutic intervention in HTN [172].
It is important to note that HTN is a multifactorial disease, and PKC inhibitors alone may not be sufficient to manage HTN. However, PKC inhibitors may decrease the VSM growth and hyperactivity associated with HTN particularly when used with other therapeutic modalities. PKC inhibitors could potentiate the inhibitory effects of Ca2+ channel blockers on vasoconstriction. Targeting Ca2+-independent PKCs could be beneficial in Ca2+ antagonist-resistant forms of HTN. The effects of PKC inhibitors in reducing vasoconstriction and BP could also be potentiated by Rho-kinase and MAPK inhibitors. RhoA/Rho-kinase causes inhibition of MLC phosphatase and thereby enhances Ca2+-MLC kinase dependent VSM contraction, and may play a role in the development and progression of HTN [146, 173]. The interaction between PKC and other pathways such as ROS, MMPs and inflammatory cytokines could also be associated with vascular disease. The combined use of isoform-specific PKC inhibitors with antioxidants, MMPs inhibitors and cytokine antagonists may provide a multi-prong approach for treatment of Ca2+ antagonist-insensitive forms of HTN.
Upregulation of PKC could play a role not only in vascular disease such as HTN and atherogenesis, but also in metabolic disorders, insulin resistance and cancer in what has been termed as the “PKC syndrome’” [174]. Therefore, it is important to further test the effects of PKC inhibitors in vivo and in animal models of HTN with other co-morbidities such as hypercholesterolemia and diabetes. Although the first generation of PKC inhibitors may not be very selective, newly-developed PKC inhibitors are more specific, and further experimental studies and clinical trials are needed before these compounds can be used safely in human. Certain PKC inhibitors such as ruboxistaurin (LY333531), a selective β-PKC inhibitor, have shown promise in clinical trials for diabetic retinopathy, macular edema and microvascular complications [175–177]. Using similar strategies to develop specific inhibitors of α-, δ- or ε-PKC isoform with improved enzyme selectivity and pharmakokinetics may lead to new therapies for HTN.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Blood Institute (HL-65998, HL-98724, HL-111775) and The Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD-60702).
List of abbreviations
- ANG II
angiotensin II
- ATP
adenosine triphosphate
- BP
blood pressure
- CPI-17
PKC-potentiated phosphatase inhibitor protein-17 kDa
- CAM
calmodulin
- DAG
diacylglycerol
- EC
endothelial cell
- ET-1
endothelin-1
- HTN
hypertension
- IP3
inositol 1,4,5-trisphosphate
- MAPK
mitogen-activated protein kinase
- MARCKs
myristoylated alanine-rich C-kinase substrate
- MMP
matrix metalloproteinase
- MEK
MAPK kinase
- MLC
myosin light chain
- NADPH
nicotinamide adenine dinucleotide phosphate
- O2−•
superoxide
- PDBu
phorbol 12,13-dibutyrate
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC
phospholipase C
- PKC
protein kinase C
- PMA
phorbol myristate acetate
- RACKs
receptors for activated C-kinase
- RAS
renin-angiotensin system
- Rho-kinase
Rho-associated kinase
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rat
- TPA
12-o-tetradecanoylphorbol-13-acetate
- VSMC
vascular smooth muscle cell
- WKY
Wistar-Kyoto
References
- 1.Cardillo C, Kilcoyne CM, Quyyumi AA, Cannon RO, 3rd, Panza JA. Selective defect in nitric oxide synthesis may explain the impaired endothelium-dependent vasodilation in patients with essential hypertension. Circulation. 1998;97:851–856. doi: 10.1161/01.cir.97.9.851. [DOI] [PubMed] [Google Scholar]
- 2.Heitzer T, Wenzel U, Hink U, Krollner D, Skatchkov M, Stahl RA, MacHarzina R, Brasen JH, Meinertz T, Munzel T. Increased NAD(P)H oxidase-mediated superoxide production in renovascular hypertension: evidence for an involvement of protein kinase C. Kidney Int. 1999;55:252–260. doi: 10.1046/j.1523-1755.1999.00229.x. [DOI] [PubMed] [Google Scholar]
- 3.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108:1253–1258. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 4.Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 5.Nijm J, Wikby A, Tompa A, Olsson AG, Jonasson L. Circulating levels of proinflammatory cytokines and neutrophil-platelet aggregates in patients with coronary artery disease. Am J Cardiol. 2005;95:452–456. doi: 10.1016/j.amjcard.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 6.McLachlan CS, Chua WC, Wong PT, Kah TL, Chen C, El Oakley RM. Homocysteine is positively associated with cytokine IL-18 plasma levels in coronary artery bypass surgery patients. Biofactors. 2005;23:69–73. doi: 10.1002/biof.5520230202. [DOI] [PubMed] [Google Scholar]
- 7.Laviades C, Varo N, Fernandez J, Mayor G, Gil MJ, Monreal I, Diez J. Abnormalities of the extracellular degradation of collagen type I in essential hypertension. Circulation. 1998;98:535–540. doi: 10.1161/01.cir.98.6.535. [DOI] [PubMed] [Google Scholar]
- 8.Ergul A, Portik-Dobos V, Hutchinson J, Franco J, Anstadt MP. Downregulation of vascular matrix metalloproteinase inducer and activator proteins in hypertensive patients. Am J Hypertens. 2004;17:775–782. doi: 10.1016/j.amjhyper.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 9.Watts SW, Rondelli C, Thakali K, Li X, Uhal B, Pervaiz MH, Watson RE, Fink GD. Morphological and biochemical characterization of remodeling in aorta and vena cava of DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H2438–2448. doi: 10.1152/ajpheart.00900.2006. [DOI] [PubMed] [Google Scholar]
- 10.Hussain S, Assender JW, Bond M, Wong LF, Murphy D, Newby AC. Activation of protein kinase Czeta is essential for cytokine-induced metalloproteinase-1, -3, and -9 secretion from rabbit smooth muscle cells and inhibits proliferation. J Biol Chem. 2002;277:27345–27352. doi: 10.1074/jbc.M111890200. [DOI] [PubMed] [Google Scholar]
- 11.Park MJ, Park IC, Lee HC, Woo SH, Lee JY, Hong YJ, Rhee CH, Lee YS, Lee SH, Shim BS, Kuroki T, Hong SI. Protein kinase C-alpha activation by phorbol ester induces secretion of gelatinase B/MMP-9 through ERK 1/2 pathway in capillary endothelial cells. Int J Oncol. 2003;22:137–143. [PubMed] [Google Scholar]
- 12.Mountain DJ, Singh M, Menon B, Singh K. Interleukin-1beta increases expression and activity of matrix metalloproteinase-2 in cardiac microvascular endothelial cells: role of PKCalpha/beta1 and MAPKs. Am J Physiol Cell Physiol. 2007;292:C867–875. doi: 10.1152/ajpcell.00161.2006. [DOI] [PubMed] [Google Scholar]
- 13.Tsai BM, Wang M, Pitcher JM, Meldrum KK, Meldrum DR. Hypoxic pulmonary vasoconstriction and pulmonary artery tissue cytokine expression are mediated by protein kinase C. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1215–1219. doi: 10.1152/ajplung.00179.2004. [DOI] [PubMed] [Google Scholar]
- 14.Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Srivastava SK. Aldose reductase mediates the mitogenic signals of cytokines. Chem Biol Interact. 2003;143–144:587–596. doi: 10.1016/s0009-2797(02)00194-1. [DOI] [PubMed] [Google Scholar]
- 15.Ramana KV, Tammali R, Reddy AB, Bhatnagar A, Srivastava SK. Aldose reductase-regulated tumor necrosis factor-alpha production is essential for high glucose-induced vascular smooth muscle cell growth. Endocrinology. 2007;148:4371–4384. doi: 10.1210/en.2007-0512. [DOI] [PubMed] [Google Scholar]
- 16.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 17.Khalil RA, van Breemen C. Sustained contraction of vascular smooth muscle: calcium influx or C-kinase activation? J Pharmacol Exp Ther. 1988;244:537–542. [PubMed] [Google Scholar]
- 18.Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- 19.Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol. 2005;70:1537–1547. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–321. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 21.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 22.Morgan KG, Khalil RA, Suematsu E, Katsuyama H. Calcium-dependent and calcium-independent pathways of signal transduction in smooth muscle. Jpn J Pharmacol. 1992;58(Suppl 2):47P-53P. [PubMed] [Google Scholar]
- 23.Nishimura J, Khalil RA, van Breemen C. Agonist-induced vascular tone. Hypertension. 1989;13:835–844. doi: 10.1161/01.hyp.13.6.835. [DOI] [PubMed] [Google Scholar]
- 24.Jiang MJ, Morgan KG. Intracellular calcium levels in phorbol ester-induced contractions of vascular muscle. Am J Physiol. 1987;253:H1365–1371. doi: 10.1152/ajpheart.1987.253.6.H1365. [DOI] [PubMed] [Google Scholar]
- 25.Takai Y, Kishimoto A, Iwasa Y, Kawahara Y, Mori T, Nishizuka Y. Calcium-dependent activation of a multifunctional protein kinase by membrane phospholipids. J Biol Chem. 1979;254:3692–3695. [PubMed] [Google Scholar]
- 26.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 27.Klevit RE, Herriott JR, Horvath SJ. Solution structure of a zinc finger domain of yeast ADR1. Proteins. 1990;7:215–226. doi: 10.1002/prot.340070303. [DOI] [PubMed] [Google Scholar]
- 28.Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science. 1986;233:859–866. doi: 10.1126/science.3755548. [DOI] [PubMed] [Google Scholar]
- 29.Parker CA, Takahashi K, Tao T, Morgan KG. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. Am J Physiol. 1994;267:C1262–1270. doi: 10.1152/ajpcell.1994.267.5.C1262. [DOI] [PubMed] [Google Scholar]
- 30.Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. Protein kinase C zeta subspecies from rat brain: its structure, expression, and properties. Proc Natl Acad Sci U S A. 1989;86:3099–3103. doi: 10.1073/pnas.86.9.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohno S, Konno Y, Akita Y, Yano A, Suzuki K. A point mutation at the putative ATP-binding site of protein kinase C alpha abolishes the kinase activity and renders it down-regulation-insensitive. A molecular link between autophosphorylation and down-regulation. J Biol Chem. 1990;265:6296–6300. [PubMed] [Google Scholar]
- 32.Schaap D, Parker PJ, Bristol A, Kriz R, Knopf J. Unique substrate specificity and regulatory properties of PKC-epsilon: a rationale for diversity. FEBS Lett. 1989;243:351–357. doi: 10.1016/0014-5793(89)80160-7. [DOI] [PubMed] [Google Scholar]
- 33.Osada S, Mizuno K, Saido TC, Suzuki K, Kuroki T, Ohno S. A new member of the protein kinase C family, nPKC theta, predominantly expressed in skeletal muscle. Mol Cell Biol. 1992;12:3930–3938. doi: 10.1128/mcb.12.9.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bacher N, Zisman Y, Berent E, Livneh E. Isolation and characterization of PKC-L, a new member of the protein kinase C-related gene family specifically expressed in lung, skin, and heart. Mol Cell Biol. 1991;11:126–133. doi: 10.1128/mcb.11.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makowske M, Rosen OM. Complete activation of protein kinase C by an antipeptide antibody directed against the pseudosubstrate prototope. J Biol Chem. 1989;264:16155–16159. [PubMed] [Google Scholar]
- 36.Orr JW, Keranen LM, Newton AC. Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. J Biol Chem. 1992;267:15263–15266. [PubMed] [Google Scholar]
- 37.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 38.Dekker LV, McIntyre P, Parker PJ. Mutagenesis of the regulatory domain of rat protein kinase C-eta. A molecular basis for restricted histone kinase activity. J Biol Chem. 1993;268:19498–19504. [PubMed] [Google Scholar]
- 39.Kanashiro CA, Khalil RA. Signal transduction by protein kinase C in mammalian cells. Clin Exp Pharmacol Physiol. 1998;25:974–985. doi: 10.1111/j.1440-1681.1998.tb02170.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang JK, Walaas SI, Sihra TS, Aderem A, Greengard P. Phosphorylation and associated translocation of the 87-kDa protein, a major protein kinase C substrate, in isolated nerve terminals. Proc Natl Acad Sci U S A. 1989;86:2253–2256. doi: 10.1073/pnas.86.7.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–622. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 42.Katada T, Gilman AG, Watanabe Y, Bauer S, Jakobs KH. Protein kinase C phosphorylates the inhibitory guanine-nucleotide-binding regulatory component and apparently suppresses its function in hormonal inhibition of adenylate cyclase. Eur J Biochem. 1985;151:431–437. doi: 10.1111/j.1432-1033.1985.tb09120.x. [DOI] [PubMed] [Google Scholar]
- 43.Barman SA, Zhu S, White RE. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004;286:L149–155. doi: 10.1152/ajplung.00207.2003. [DOI] [PubMed] [Google Scholar]
- 44.Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93:656–663. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- 45.Limas CJ. Phosphorylation of cardiac sarcoplasmic reticulum by a calcium-activated, phospholipid-dependent protein kinase. Biochem Biophys Res Commun. 1980;96:1378–1383. doi: 10.1016/0006-291x(80)90103-5. [DOI] [PubMed] [Google Scholar]
- 46.Rosoff PM, Stein LF, Cantley LC. Phorbol esters induce differentiation in a pre-B-lymphocyte cell line by enhancing Na+/H+ exchange. J Biol Chem. 1984;259:7056–7060. [PubMed] [Google Scholar]
- 47.Aviv A. Cytosolic Ca2+, Na+/H+ antiport, protein kinase C trio in essential hypertension. Am J Hypertens. 1994;7:205–212. doi: 10.1093/ajh/7.2.205. [DOI] [PubMed] [Google Scholar]
- 48.Schwienbacher C, Jockusch BM, Rudiger M. Intramolecular interactions regulate serine/threonine phosphorylation of vinculin. FEBS Lett. 1996;384:71–74. doi: 10.1016/0014-5793(96)00286-4. [DOI] [PubMed] [Google Scholar]
- 49.Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J Physiol. 2001;535:553–564. doi: 10.1111/j.1469-7793.2001.t01-1-00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inagaki M, Yokokura H, Itoh T, Kanmura Y, Kuriyama H, Hidaka H. Purified rabbit brain protein kinase C relaxes skinned vascular smooth muscle and phosphorylates myosin light chain. Arch Biochem Biophys. 1987;254:136–141. doi: 10.1016/0003-9861(87)90089-0. [DOI] [PubMed] [Google Scholar]
- 51.Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 52.Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J. 1998;12:35–42. [PubMed] [Google Scholar]
- 53.Kraft AS, Anderson WB. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 1983;301:621–623. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]
- 54.Hyatt SL, Klauck T, Jaken S. Protein kinase C is localized in focal contacts of normal but not transformed fibroblasts. Mol Carcinog. 1990;3:45–53. doi: 10.1002/mc.2940030202. [DOI] [PubMed] [Google Scholar]
- 55.Kose A, Saito N, Ito H, Kikkawa U, Nishizuka Y, Tanaka C. Electron microscopic localization of type I protein kinase C in rat Purkinje cells. J Neurosci. 1988;8:4262–4268. doi: 10.1523/JNEUROSCI.08-11-04262.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khalil RA, Lajoie C, Resnick MS, Morgan KG. Ca(2+)-independent isoforms of protein kinase C differentially translocate in smooth muscle. Am J Physiol. 1992;263:C714–719. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- 57.Cogolludo A, Moreno L, Lodi F, Tamargo J, Perez-Vizcaino F. Postnatal maturational shift from PKCzeta and voltage-gated K+ channels to RhoA/Rho kinase in pulmonary vasoconstriction. Cardiovasc Res. 2005;66:84–93. doi: 10.1016/j.cardiores.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 58.Draeger A, Wray S, Babiychuk EB. Domain architecture of the smooth-muscle plasma membrane: regulation by annexins. Biochem J. 2005;387:309–314. doi: 10.1042/BJ20041363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thelen M, Rosen A, Nairn AC, Aderem A. Regulation by phosphorylation of reversible association of a myristoylated protein kinase C substrate with the plasma membrane. Nature. 1991;351:320–322. doi: 10.1038/351320a0. [DOI] [PubMed] [Google Scholar]
- 60.Cazaubon SM, Parker PJ. Identification of the phosphorylated region responsible for the permissive activation of protein kinase C. J Biol Chem. 1993;268:17559–17563. [PubMed] [Google Scholar]
- 61.Leventhal PS, Bertics PJ. Activation of protein kinase C by selective binding of arginine-rich polypeptides. J Biol Chem. 1993;268:13906–13913. [PubMed] [Google Scholar]
- 62.Ron D, Mochly-Rosen D. Agonists and antagonists of protein kinase C function, derived from its binding proteins. J Biol Chem. 1994;269:21395–21398. [PubMed] [Google Scholar]
- 63.Housey GM, Johnson MD, Hsiao WL, O’Brian CA, Murphy JP, Kirschmeier P, Weinstein IB. Overproduction of protein kinase C causes disordered growth control in rat fibroblasts. Cell. 1988;52:343–354. doi: 10.1016/s0092-8674(88)80027-8. [DOI] [PubMed] [Google Scholar]
- 64.Dallas A, Khalil RA. Ca2+ antagonist-insensitive coronary smooth muscle contraction involves activation of epsilon-protein kinase C-dependent pathway. Am J Physiol Cell Physiol. 2003;285:C1454–1463. doi: 10.1152/ajpcell.00066.2003. [DOI] [PubMed] [Google Scholar]
- 65.Khalil RA, Menice CB, Wang CL, Morgan KG. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ Res. 1995;76:1101–1108. doi: 10.1161/01.res.76.6.1101. [DOI] [PubMed] [Google Scholar]
- 66.Mii S, Khalil RA, Morgan KG, Ware JA, Kent KC. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. Am J Physiol. 1996;270:H142–150. doi: 10.1152/ajpheart.1996.270.1.H142. [DOI] [PubMed] [Google Scholar]
- 67.Adam LP, Gapinski CJ, Hathaway DR. Phosphorylation sequences in h-caldesmon from phorbol ester-stimulated canine aortas. FEBS Lett. 1992;302:223–226. doi: 10.1016/0014-5793(92)80446-n. [DOI] [PubMed] [Google Scholar]
- 68.D’Angelo G, Graceffa P, Wang CA, Wrangle J, Adam LP. Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Quantitation using novel anti-phosphopeptide antibodies. J Biol Chem. 1999;274:30115–30121. doi: 10.1074/jbc.274.42.30115. [DOI] [PubMed] [Google Scholar]
- 69.Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C718–726. doi: 10.1152/ajpcell.2000.278.4.C718. [DOI] [PubMed] [Google Scholar]
- 70.Bazzi MD, Nelsestuen GL. Protein kinase C interaction with calcium: a phospholipid-dependent process. Biochemistry. 1990;29:7624–7630. doi: 10.1021/bi00485a012. [DOI] [PubMed] [Google Scholar]
- 71.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 72.Szallasi Z, Smith CB, Pettit GR, Blumberg PM. Differential regulation of protein kinase C isozymes by bryostatin 1 and phorbol 12-myristate 13-acetate in NIH 3T3 fibroblasts. J Biol Chem. 1994;269:2118–2124. [PubMed] [Google Scholar]
- 73.Giardina JB, Tanner DJ, Khalil RA. Oxidized-LDL enhances coronary vasoconstriction by increasing the activity of protein kinase C isoforms alpha and epsilon. Hypertension. 2001;37:561–568. doi: 10.1161/01.hyp.37.2.561. [DOI] [PubMed] [Google Scholar]
- 74.Claro S, Kanashiro CA, Oshiro ME, Ferreira AT, Khalil RA. alpha- and epsilon-protein kinase C activity during smooth muscle cell apoptosis in response to gamma-radiation. J Pharmacol Exp Ther. 2007;322:964–972. doi: 10.1124/jpet.107.125930. [DOI] [PubMed] [Google Scholar]
- 75.Li W, Zhang J, Bottaro DP, Pierce JH. Identification of serine 643 of protein kinase C-delta as an important autophosphorylation site for its enzymatic activity. J Biol Chem. 1997;272:24550–24555. doi: 10.1074/jbc.272.39.24550. [DOI] [PubMed] [Google Scholar]
- 76.Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- 77.Edwards AS, Newton AC. Phosphorylation at conserved carboxyl-terminal hydrophobic motif regulates the catalytic and regulatory domains of protein kinase C. J Biol Chem. 1997;272:18382–18390. doi: 10.1074/jbc.272.29.18382. [DOI] [PubMed] [Google Scholar]
- 78.Kanashiro CA, Altirkawi KA, Khalil RA. Preconditioning of coronary artery against vasoconstriction by endothelin-1 and prostaglandin F2alpha during repeated downregulation of epsilon-protein kinase C. J Cardiovasc Pharmacol. 2000;35:491–501. doi: 10.1097/00005344-200003000-00021. [DOI] [PubMed] [Google Scholar]
- 79.Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J Biol Chem. 1993;268:1982–1986. [PubMed] [Google Scholar]
- 80.Clement S, Tasinato A, Boscoboinik D, Azzi A. The effect of alpha-tocopherol on the synthesis, phosphorylation and activity of protein kinase C in smooth muscle cells after phorbol 12-myristate 13-acetate down-regulation. Eur J Biochem. 1997;246:745–749. doi: 10.1111/j.1432-1033.1997.t01-2-00745.x. [DOI] [PubMed] [Google Scholar]
- 81.Cain AE, Khalil RA. Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin Nephrol. 2002;22:3–16. [PubMed] [Google Scholar]
- 82.Wang S, Desai D, Wright G, Niles RM, Wright GL. Effects of protein kinase C alpha overexpression on A7r5 smooth muscle cell proliferation and differentiation. Exp Cell Res. 1997;236:117–126. doi: 10.1006/excr.1997.3714. [DOI] [PubMed] [Google Scholar]
- 83.Liou YM, Morgan KG. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. Am J Physiol. 1994;267:C980–989. doi: 10.1152/ajpcell.1994.267.4.C980. [DOI] [PubMed] [Google Scholar]
- 84.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, Galie N, Manes A, Corris P, Simonneau G, Humbert M, Morrell NW, Trembath RC. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat. 2006;27:212–213. doi: 10.1002/humu.9398. [DOI] [PubMed] [Google Scholar]
- 87.Song Y, Jones JE, Beppu H, Keaney JF, Jr, Loscalzo J, Zhang YY. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation. 2005;112:553–562. doi: 10.1161/CIRCULATIONAHA.104.492488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hassel S, Eichner A, Yakymovych M, Hellman U, Knaus P, Souchelnytskyi S. Proteins associated with type II bone morphogenetic protein receptor (BMPR-II) and identified by two-dimensional gel electrophoresis and mass spectrometry. Proteomics. 2004;4:1346–1358. doi: 10.1002/pmic.200300770. [DOI] [PubMed] [Google Scholar]
- 89.Shibata R, Morita S, Nagai K, Miyata S, Iwasaki T. Effects of H-7 (protein kinase inhibitor) and phorbol ester on aortic strips from spontaneously hypertensive rats. Eur J Pharmacol. 1990;175:261–271. doi: 10.1016/0014-2999(90)90563-l. [DOI] [PubMed] [Google Scholar]
- 90.Bazan E, Campbell AK, Rapoport RM. Protein kinase C activity in blood vessels from normotensive and spontaneously hypertensive rats. Eur J Pharmacol. 1992;227:343–348. doi: 10.1016/0922-4106(92)90014-m. [DOI] [PubMed] [Google Scholar]
- 91.Sauro MD, Hadden JW. Gamma-interferon corrects aberrant protein kinase C levels and immunosuppression in the spontaneously hypertensive rat. Int J Immunopharmacol. 1992;14:1421–1427. doi: 10.1016/0192-0561(92)90014-c. [DOI] [PubMed] [Google Scholar]
- 92.Sasajima H, Shima H, Toyoda Y, Kimura K, Yoshikawa A, Hano T, Nishio I. Increased Ca2+ sensitivity of contractile elements via protein kinase C in alpha-toxin permeabilized SMA from young spontaneously hypertensive rats. Cardiovasc Res. 1997;36:86–91. doi: 10.1016/s0008-6363(97)00131-4. [DOI] [PubMed] [Google Scholar]
- 93.Rosen B, Barg J, Zimlichman R. The effects of angiotensin II, endothelin-1, and protein kinase C inhibitor on DNA synthesis and intracellular calcium mobilization in vascular smooth muscle cells from young normotensive and spontaneously hypertensive rats. Am J Hypertens. 1999;12:1243–1251. doi: 10.1016/s0895-7061(99)00158-2. [DOI] [PubMed] [Google Scholar]
- 94.Bilder GE, Kasiewski CJ, Perrone MH. Phorbol-12,13-dibutyrate-induced vasoconstriction in vivo: characterization of response in genetic hypertension. J Pharmacol Exp Ther. 1990;252:526–530. [PubMed] [Google Scholar]
- 95.Kanashiro CA, Khalil RA. Gender-related distinctions in protein kinase C activity in rat vascular smooth muscle. Am J Physiol Cell Physiol. 2001;280:C34–45. doi: 10.1152/ajpcell.2001.280.1.C34. [DOI] [PubMed] [Google Scholar]
- 96.Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. 2001;19:1245–1254. doi: 10.1097/00004872-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 97.Escriba PV, Sanchez-Dominguez JM, Alemany R, Perona JS, Ruiz-Gutierrez V. Alteration of lipids, G proteins, and PKC in cell membranes of elderly hypertensives. Hypertension. 2003;41:176–182. doi: 10.1161/01.hyp.0000047647.72162.a8. [DOI] [PubMed] [Google Scholar]
- 98.Gu X, Bishop SP. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circ Res. 1994;75:926–931. doi: 10.1161/01.res.75.5.926. [DOI] [PubMed] [Google Scholar]
- 99.Khalil RA, Lajoie C, Morgan KG. In situ determination of [Ca2+]i threshold for translocation of the alpha-protein kinase C isoform. Am J Physiol. 1994;266:C1544–1551. doi: 10.1152/ajpcell.1994.266.6.C1544. [DOI] [PubMed] [Google Scholar]
- 100.Fatehi-Hassanabad Z, Fatehi M, Shahidi MI. Endothelial dysfunction in aortic rings and mesenteric beds isolated from deoxycorticosterone acetate hypertensive rats: possible involvement of protein kinase C. Eur J Pharmacol. 2004;494:199–204. doi: 10.1016/j.ejphar.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 101.Soloviev AI, Parshikov AV, Stefanov AV. Evidence for the involvement of protein kinase C in depression of endothelium-dependent vascular responses in spontaneously hypertensive rats. J Vasc Res. 1998;35:325–331. doi: 10.1159/000025602. [DOI] [PubMed] [Google Scholar]
- 102.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 103.Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- 104.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 105.Motley ED, Eguchi K, Patterson MM, Palmer PD, Suzuki H, Eguchi S. Mechanism of endothelial nitric oxide synthase phosphorylation and activation by thrombin. Hypertension. 2007;49:577–583. doi: 10.1161/01.HYP.0000255954.80025.34. [DOI] [PubMed] [Google Scholar]
- 106.Partovian C, Zhuang Z, Moodie K, Lin M, Ouchi N, Sessa WC, Walsh K, Simons M. PKCalpha activates eNOS and increases arterial blood flow in vivo. Circ Res. 2005;97:482–487. doi: 10.1161/01.RES.0000179775.04114.45. [DOI] [PubMed] [Google Scholar]
- 107.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 109.Li H, Witte K, August M, Brausch I, Godtel-Armbrust U, Habermeier A, Closs EI, Oelze M, Munzel T, Forstermann U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol. 2006;47:2536–2544. doi: 10.1016/j.jacc.2006.01.071. [DOI] [PubMed] [Google Scholar]
- 110.Dai X, Cao X, Kreulen DL. Superoxide anion is elevated in sympathetic neurons in DOCA-salt hypertension via activation of NADPH oxidase. Am J Physiol Heart Circ Physiol. 2006;290:H1019–1026. doi: 10.1152/ajpheart.00052.2005. [DOI] [PubMed] [Google Scholar]
- 111.Fedorova OV, Talan MI, Agalakova NI, Droy-Lefaix MT, Lakatta EG, Bagrov AY. Myocardial PKC beta2 and the sensitivity of Na/K-ATPase to marinobufagenin are reduced by cicletanine in Dahl hypertension. Hypertension. 2003;41:505–511. doi: 10.1161/01.HYP.0000053446.43894.9F. [DOI] [PubMed] [Google Scholar]
- 112.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 113.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 114.Benjamin MM, Khalil RA. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. EXS. 2012;103:209–279. doi: 10.1007/978-3-0348-0364-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chew DK, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–1010. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 116.Raffetto JD, Ross RL, Khalil RA. Matrix metalloproteinase 2-induced venous dilation via hyperpolarization and activation of K+ channels: relevance to varicose vein formation. J Vasc Surg. 2007;45:373–380. doi: 10.1016/j.jvs.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Derosa G, D’Angelo A, Ciccarelli L, Piccinni MN, Pricolo F, Salvadeo S, Montagna L, Gravina A, Ferrari I, Galli S, Paniga S, Tinelli C, Cicero AF. Matrix metalloproteinase-2, -9, and tissue inhibitor of metalloproteinase-1 in patients with hypertension. Endothelium. 2006;13:227–231. doi: 10.1080/10623320600780942. [DOI] [PubMed] [Google Scholar]
- 118.Zervoudaki A, Economou E, Stefanadis C, Pitsavos C, Tsioufis K, Aggeli C, Vasiliadou K, Toutouza M, Toutouzas P. Plasma levels of active extracellular matrix metalloproteinases 2 and 9 in patients with essential hypertension before and after antihypertensive treatment. J Hum Hypertens. 2003;17:119–124. doi: 10.1038/sj.jhh.1001518. [DOI] [PubMed] [Google Scholar]
- 119.Flamant M, Placier S, Dubroca C, Esposito B, Lopes I, Chatziantoniou C, Tedgui A, Dussaule JC, Lehoux S. Role of matrix metalloproteinases in early hypertensive vascular remodeling. Hypertension. 2007;50:212–218. doi: 10.1161/HYPERTENSIONAHA.107.089631. [DOI] [PubMed] [Google Scholar]
- 120.Papadimitriou E, Waters CR, Manolopoulos VG, Unsworth BR, Maragoudakis ME, Lelkes PI. Regulation of extracellular matrix remodeling and MMP-2 activation in cultured rat adrenal medullary endothelial cells. Endothelium. 2001;8:243–253. doi: 10.3109/10623320109090801. [DOI] [PubMed] [Google Scholar]
- 121.Li D, Liu L, Chen H, Sawamura T, Ranganathan S, Mehta JL. LOX-1 mediates oxidized low-density lipoprotein-induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation. 2003;107:612–617. doi: 10.1161/01.cir.0000047276.52039.fb. [DOI] [PubMed] [Google Scholar]
- 122.Smith L, Payne JA, Sedeek MH, Granger JP, Khalil RA. Endothelin-induced increases in Ca2+ entry mechanisms of vascular contraction are enhanced during high-salt diet. Hypertension. 2003;41:787–793. doi: 10.1161/01.HYP.0000051643.05700.56. [DOI] [PubMed] [Google Scholar]
- 123.Khalil RA. Dietary salt and hypertension: new molecular targets add more spice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R509–513. doi: 10.1152/ajpregu.00600.2005. [DOI] [PubMed] [Google Scholar]
- 124.Fareh J, Touyz RM, Schiffrin EL, Thibault G. Altered cardiac endothelin receptors and protein kinase C in deoxycorticosterone-salt hypertensive rats. J Mol Cell Cardiol. 2000;32:665–676. doi: 10.1006/jmcc.2000.1110. [DOI] [PubMed] [Google Scholar]
- 125.Kim J, Lee YR, Lee CH, Choi WH, Lee CK, Bae YM, Cho S, Kim B. Mitogen-activated protein kinase contributes to elevated basal tone in aortic smooth muscle from hypertensive rats. Eur J Pharmacol. 2005;514:209–215. doi: 10.1016/j.ejphar.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 126.Sirous ZN, Fleming JB, Khalil RA. Endothelin-1 enhances eicosanoids-induced coronary smooth muscle contraction by activating specific protein kinase C isoforms. Hypertension. 2001;37:497–504. doi: 10.1161/01.hyp.37.2.497. [DOI] [PubMed] [Google Scholar]
- 127.Cain AE, Tanner DM, Khalil RA. Endothelin-1--induced enhancement of coronary smooth muscle contraction via MAPK-dependent and MAPK-independent [Ca(2+)](i) sensitization pathways. Hypertension. 2002;39:543–549. doi: 10.1161/hy0202.103129. [DOI] [PubMed] [Google Scholar]
- 128.Khalil RA. Modulators of the vascular endothelin receptor in blood pressure regulation and hypertension. Curr Mol Pharmacol. 2011;4:176–186. doi: 10.2174/1874467211104030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schiffrin EL. Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension. 1995;25:1135–1143. doi: 10.1161/01.hyp.25.6.1135. [DOI] [PubMed] [Google Scholar]
- 130.Kanayama Y, Negoro N, Okamura M, Konishi Y, Nishimura M, Umetani N, Inoue T, Takeda T. Modulation of protein kinase C in aorta of spontaneously hypertensive rats with enalapril treatment. Osaka City Med J. 1994;40:83–97. [PubMed] [Google Scholar]
- 131.Bell PD, Mashburn N, Unlap MT. Renal sodium/calcium exchange; a vasodilator that is defective in salt-sensitive hypertension. Acta Physiol Scand. 2000;168:209–214. doi: 10.1046/j.1365-201x.2000.00671.x. [DOI] [PubMed] [Google Scholar]
- 132.Nowicki S, Kruse MS, Brismar H, Aperia A. Dopamine-induced translocation of protein kinase C isoforms visualized in renal epithelial cells. Am J Physiol Cell Physiol. 2000;279:C1812–1818. doi: 10.1152/ajpcell.2000.279.6.C1812. [DOI] [PubMed] [Google Scholar]
- 133.Ridge KM, Dada L, Lecuona E, Bertorello AM, Katz AI, Mochly-Rosen D, Sznajder JI. Dopamine-induced exocytosis of Na,K-ATPase is dependent on activation of protein kinase C-epsilon and -delta. Mol Biol Cell. 2002;13:1381–1389. doi: 10.1091/mbc.01-07-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Banday AA, Fazili FR, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor-kappaB and protein kinase C. J Am Soc Nephrol. 2007;18:1446–1457. doi: 10.1681/ASN.2006121373. [DOI] [PubMed] [Google Scholar]
- 135.Hughes-Darden CA, Wachira SJ, Denaro FJ, Taylor CV, Brunson KJ, Ochillo R, Robinson TJ. Expression and distribution of protein kinase C isozymes in brain tissue of spontaneous hypertensive rats. Cell Mol Biol (Noisy-le-grand) 2001;47:1077–1088. [PubMed] [Google Scholar]
- 136.Rao F, Zhang L, Wessel J, Zhang K, Wen G, Kennedy BP, Rana BK, Das M, Rodriguez-Flores JL, Smith DW, Cadman PE, Salem RM, Mahata SK, Schork NJ, Taupenot L, Ziegler MG, O’Connor DT. Tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis: discovery of common human genetic variants governing transcription, autonomic activity, and blood pressure in vivo. Circulation. 2007;116:993–1006. doi: 10.1161/CIRCULATIONAHA.106.682302. [DOI] [PubMed] [Google Scholar]
- 137.Hempel A, Maasch C, Heintze U, Lindschau C, Dietz R, Luft FC, Haller H. High glucose concentrations increase endothelial cell permeability via activation of protein kinase C alpha. Circ Res. 1997;81:363–371. doi: 10.1161/01.res.81.3.363. [DOI] [PubMed] [Google Scholar]
- 138.Williams B, Howard RL. Glucose-induced changes in Na+/H+ antiport activity and gene expression in cultured vascular smooth muscle cells. Role of protein kinase C. J Clin Invest. 1994;93:2623–2631. doi: 10.1172/JCI117275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Farese RV, Standaert ML, Ishizuka T, Yu B, Hernandez H, Waldron C, Watson J, Farese JP, Cooper DR, Wickstrom E. Antisense DNA downregulates protein kinase C isozymes (beta and alpha) and insulin-stimulated 2-deoxyglucose uptake in rat adipocytes. Antisense Res Dev. 1991;1:35–42. doi: 10.1089/ard.1991.1.35. [DOI] [PubMed] [Google Scholar]
- 140.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 141.Young JL, Libby P, Schonbeck U. Cytokines in the pathogenesis of atherosclerosis. Thromb Haemost. 2002;88:554–567. [PubMed] [Google Scholar]
- 142.Waehre T, Yndestad A, Smith C, Haug T, Tunheim SH, Gullestad L, Froland SS, Semb AG, Aukrust P, Damas JK. Increased expression of interleukin-1 in coronary artery disease with downregulatory effects of HMG-CoA reductase inhibitors. Circulation. 2004;109:1966–1972. doi: 10.1161/01.CIR.0000125700.33637.B1. [DOI] [PubMed] [Google Scholar]
- 143.Sardella G, Mariani P, D’Alessandro M, De Luca L, Pierro M, Mancone M, Porretta A, Accapezzato D, Fedele F, Paroli M. Early elevation of interleukin-1beta and interleukin-6 levels after bare or drug-eluting stent implantation in patients with stable angina. Thromb Res. 2006;117:659–664. doi: 10.1016/j.thromres.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 144.Lubrano V, Cocci F, Battaglia D, Papa A, Marraccini P, Zucchelli GC. Usefulness of high-sensitivity IL-6 measurement for clinical characterization of patients with coronary artery disease. J Clin Lab Anal. 2005;19:110–114. doi: 10.1002/jcla.20061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Funayama H, Ishikawa SE, Kubo N, Katayama T, Yasu T, Saito M, Kawakami M. Increases in interleukin-6 and matrix metalloproteinase-9 in the infarct-related coronary artery of acute myocardial infarction. Circ J. 2004;68:451–454. doi: 10.1253/circj.68.451. [DOI] [PubMed] [Google Scholar]
- 146.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–940. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 147.Das M, Dempsey EC, Bouchey D, Reyland ME, Stenmark KR. Chronic hypoxia induces exaggerated growth responses in pulmonary artery adventitial fibroblasts: potential contribution of specific protein kinase c isozymes. Am J Respir Cell Mol Biol. 2000;22:15–25. doi: 10.1165/ajrcmb.22.1.3536. [DOI] [PubMed] [Google Scholar]
- 148.Littler CM, Morris KG, Jr, Fagan KA, McMurtry IF, Messing RO, Dempsey EC. Protein kinase C-epsilon-null mice have decreased hypoxic pulmonary vasoconstriction. Am J Physiol Heart Circ Physiol. 2003;284:H1321–1331. doi: 10.1152/ajpheart.00795.2002. [DOI] [PubMed] [Google Scholar]
- 149.Ito T, Ozawa K, Shimada K. Current drug targets and future therapy of pulmonary arterial hypertension. Curr Med Chem. 2007;14:719–733. doi: 10.2174/092986707780059562. [DOI] [PubMed] [Google Scholar]
- 150.Puri A, McGoon MD, Kushwaha SS. Pulmonary arterial hypertension: current therapeutic strategies. Nat Clin Pract Cardiovasc Med. 2007;4:319–329. doi: 10.1038/ncpcardio0890. [DOI] [PubMed] [Google Scholar]
- 151.Barman SA. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muscle involves Rho-kinase and protein kinase C. Am J Physiol Lung Cell Mol Physiol. 2007;293:L472–479. doi: 10.1152/ajplung.00101.2006. [DOI] [PubMed] [Google Scholar]
- 152.Khalil RA, Granger JP. Vascular mechanisms of increased arterial pressure in preeclampsia: lessons from animal models. Am J Physiol Regul Integr Comp Physiol. 2002;283:R29–45. doi: 10.1152/ajpregu.00762.2001. [DOI] [PubMed] [Google Scholar]
- 153.Sheppard SJ, Khalil RA. Risk factors and mediators of the vascular dysfunction associated with hypertension in pregnancy. Cardiovasc Hematol Disord Drug Targets. 2010;10:33–52. doi: 10.2174/187152910790780096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Magness RR, Rosenfeld CR, Carr BR. Protein kinase C in uterine and systemic arteries during ovarian cycle and pregnancy. Am J Physiol. 1991;260:E464–470. doi: 10.1152/ajpendo.1991.260.3.E464. [DOI] [PubMed] [Google Scholar]
- 155.Kanashiro CA, Cockrell KL, Alexander BT, Granger JP, Khalil RA. Pregnancy-associated reduction in vascular protein kinase C activity rebounds during inhibition of NO synthesis. Am J Physiol Regul Integr Comp Physiol. 2000;278:R295–303. doi: 10.1152/ajpregu.2000.278.2.R295. [DOI] [PubMed] [Google Scholar]
- 156.Kanashiro CA, Alexander BT, Granger JP, Khalil RA. Ca(2+)-insensitive vascular protein kinase C during pregnancy and NOS inhibition. Hypertension. 1999;34:924–930. doi: 10.1161/01.hyp.34.4.924. [DOI] [PubMed] [Google Scholar]
- 157.Khalil RA, Crews JK, Novak J, Kassab S, Granger JP. Enhanced vascular reactivity during inhibition of nitric oxide synthesis in pregnant rats. Hypertension. 1998;31:1065–1069. doi: 10.1161/01.hyp.31.5.1065. [DOI] [PubMed] [Google Scholar]
- 158.Crews JK, Novak J, Granger JP, Khalil RA. Stimulated mechanisms of Ca2+ entry into vascular smooth muscle during NO synthesis inhibition in pregnant rats. Am J Physiol. 1999;276:R530–538. doi: 10.1152/ajpregu.1999.276.2.R530. [DOI] [PubMed] [Google Scholar]
- 159.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kupferminc MJ, Peaceman AM, Wigton TR, Rehnberg KA, Socol ML. Tumor necrosis factor-alpha is elevated in plasma and amniotic fluid of patients with severe preeclampsia. Am J Obstet Gynecol. 1994;170:1752–1757. discussion 1757–1759. [PubMed] [Google Scholar]
- 161.Vince GS, Starkey PM, Austgulen R, Kwiatkowski D, Redman CW. Interleukin-6, tumour necrosis factor and soluble tumour necrosis factor receptors in women with pre-eclampsia. Br J Obstet Gynaecol. 1995;102:20–25. doi: 10.1111/j.1471-0528.1995.tb09020.x. [DOI] [PubMed] [Google Scholar]
- 162.Conrad KP, Benyo DF. Placental cytokines and the pathogenesis of preeclampsia. Am J Reprod Immunol. 1997;37:240–249. doi: 10.1111/j.1600-0897.1997.tb00222.x. [DOI] [PubMed] [Google Scholar]
- 163.Williams MA, Mahomed K, Farrand A, Woelk GB, Mudzamiri S, Madzime S, King IB, McDonald GB. Plasma tumor necrosis factor-alpha soluble receptor p55 (sTNFp55) concentrations in eclamptic, preeclamptic and normotensive pregnant Zimbabwean women. J Reprod Immunol. 1998;40:159–173. doi: 10.1016/s0165-0378(98)00074-6. [DOI] [PubMed] [Google Scholar]
- 164.LaMarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr Hypertens Rep. 2007;9:480–485. doi: 10.1007/s11906-007-0088-1. [DOI] [PubMed] [Google Scholar]
- 165.Benyo DF, Smarason A, Redman CW, Sims C, Conrad KP. Expression of inflammatory cytokines in placentas from women with preeclampsia. J Clin Endocrinol Metab. 2001;86:2505–2512. doi: 10.1210/jcem.86.6.7585. [DOI] [PubMed] [Google Scholar]
- 166.Davis JR, Giardina JB, Green GM, Alexander BT, Granger JP, Khalil RA. Reduced endothelial NO-cGMP vascular relaxation pathway during TNF-alpha-induced hypertension in pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2002;282:R390–399. doi: 10.1152/ajpregu.00270.2001. [DOI] [PubMed] [Google Scholar]
- 167.Orshal JM, Khalil RA. Reduced endothelial NO-cGMP-mediated vascular relaxation and hypertension in IL-6-infused pregnant rats. Hypertension. 2004;43:434–444. doi: 10.1161/01.HYP.0000113044.46326.98. [DOI] [PubMed] [Google Scholar]
- 168.Giardina JB, Green GM, Cockrell KL, Granger JP, Khalil RA. TNF-alpha enhances contraction and inhibits endothelial NO-cGMP relaxation in systemic vessels of pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R130–143. doi: 10.1152/ajpregu.00704.2001. [DOI] [PubMed] [Google Scholar]
- 169.Orshal JM, Khalil RA. Interleukin-6 impairs endothelium-dependent NO-cGMP-mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1013–1023. doi: 10.1152/ajpregu.00729.2003. [DOI] [PubMed] [Google Scholar]
- 170.Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension. 2007;50:1142–1147. doi: 10.1161/HYPERTENSIONAHA.107.096594. [DOI] [PubMed] [Google Scholar]
- 171.Gilbert JS, Gilbert SA, Arany M, Granger JP. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension. 2009;53:399–403. doi: 10.1161/HYPERTENSIONAHA.108.123513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bagrov AY, Dmitrieva RI, Dorofeeva NA, Fedorova OV, Lopatin DA, Lakatta EG, Droy-Lefaix MT. Cicletanine reverses vasoconstriction induced by the endogenous sodium pump ligand, marinobufagenin, via a protein kinase C dependent mechanism. J Hypertens. 2000;18:209–215. doi: 10.1097/00004872-200018020-00012. [DOI] [PubMed] [Google Scholar]
- 173.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 174.McCarty MF. Up-regulation of intracellular signalling pathways may play a central pathogenic role in hypertension, atherogenesis, insulin resistance, and cancer promotion--the ‘PKC syndrome’. Med Hypotheses. 1996;46:191–221. doi: 10.1016/s0306-9877(96)90243-1. [DOI] [PubMed] [Google Scholar]
- 175.Davis MD, Sheetz MJ, Aiello LP, Milton RC, Danis RP, Zhi X, Girach A, Jimenez MC, Vignati L. Effect of ruboxistaurin on the visual acuity decline associated with long-standing diabetic macular edema. Invest Ophthalmol Vis Sci. 2009;50:1–4. doi: 10.1167/iovs.08-2473. [DOI] [PubMed] [Google Scholar]
- 176.Aiello LP, Vignati L, Sheetz MJ, Zhi X, Girach A, Davis MD, Wolka AM, Shahri N, Milton RC. Oral protein kinase c beta inhibition using ruboxistaurin: efficacy, safety, and causes of vision loss among 813 patients (1,392 eyes) with diabetic retinopathy in the Protein Kinase C beta Inhibitor-Diabetic Retinopathy Study and the Protein Kinase C beta Inhibitor-Diabetic Retinopathy Study 2. Retina. 2011;31:2084–2094. doi: 10.1097/IAE.0b013e3182111669. [DOI] [PubMed] [Google Scholar]
- 177.Joy SV, Scates AC, Bearelly S, Dar M, Taulien CA, Goebel JA, Cooney MJ. Ruboxistaurin, a protein kinase C beta inhibitor, as an emerging treatment for diabetes microvascular complications. Ann Pharmacother. 2005;39:1693–1699. doi: 10.1345/aph.1E572. [DOI] [PubMed] [Google Scholar]
- 178.Haller H, Quass P, Lindschau C, Luft FC, Distler A. Platelet-derived growth factor and angiotensin II induce different spatial distribution of protein kinase C-alpha and -beta in vascular smooth muscle cells. Hypertension. 1994;23:848–852. doi: 10.1161/01.hyp.23.6.848. [DOI] [PubMed] [Google Scholar]
- 179.Singer HA. Phorbol ester-induced stress and myosin light chain phosphorylation in swine carotid medial smooth muscle. J Pharmacol Exp Ther. 1990;252:1068–1074. [PubMed] [Google Scholar]
- 180.Ohanian V, Ohanian J, Shaw L, Scarth S, Parker PJ, Heagerty AM. Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circ Res. 1996;78:806–812. doi: 10.1161/01.res.78.5.806. [DOI] [PubMed] [Google Scholar]
- 181.Watanabe M, Hachiya T, Hagiwara M, Hidaka H. Identification of type III protein kinase C in bovine aortic tissue. Arch Biochem Biophys. 1989;273:165–169. doi: 10.1016/0003-9861(89)90175-6. [DOI] [PubMed] [Google Scholar]
- 182.Goodnight JA, Mischak H, Kolch W, Mushinski JF. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J Biol Chem. 1995;270:9991–10001. doi: 10.1074/jbc.270.17.9991. [DOI] [PubMed] [Google Scholar]
- 183.Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. J Physiol. 1997;500(Pt 1):95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]