

Abstract
Castrate-resistant prostate cancer (CRPC) is a fatal, metastatic form of prostate cancer. CRPC is characterized by reactivation of the androgen axis due to changes in androgen receptor signaling and/or adaptive intratumoral androgen biosynthesis. AKR1C3 is upregulated in CRPC where it catalyzes the formation of potent androgens. This makes AKR1C3 a target for the treatment of CRPC. AKR1C3 inhibitors should not inhibit AKR1C1/AKR1C2, which inactivate 5α-dihydrotestosterone. Indomethacin, used to inhibit cyclooxygenase, also inhibits AKR1C3 and displays selectivity over AKR1C1/AKR1C2. Parallel synthetic strategies were used to generate libraries of indomethacin analogs, which exhibit reduced cyclooxygenase inhibitory activity but retain AKR1C3 inhibitory potency and selectivity. The lead compounds inhibited AKR1C3 with nanomolar potency, displayed >100-fold selectivity over AKR1C1/AKR1C2, and blocked testosterone formation in LNCaP-AKR1C3 cells. The AKR1C3·NADP+ ·2′-des-methylindomethacin crystal structure was determined and it revealed a unique inhibitor binding mode. The compounds reported are promising agents for the development of therapeutics for CRPC.
INTRODUCTION
Aldo-keto reductase 1C3, also known as type 5 17β-hydroxysteroid dehydrogenase, is a critical enzyme in androgen biosynthesis within the prostate and has been implicated in the development of castrate-resistant prostate cancer (CRPC). CRPC is characterized by the intratumoral reactivation of androgen receptor signaling caused by adaptive intratumoral androgen biosynthesis and/or androgen receptor (AR) amplification leading to resistance to androgen deprivation therapy.1–3 Elevated androgen levels within the tumor occur due to the upregulation of key enzymes involved in androgen biosynthesis.4–6 AKR1C3 catalyzes the conversion of the weak androgen precursors 4-androstene-3,17-dione (Δ4AD) and 5α-androstane-3,17-dione to the potent androgens testosterone and 5α-dihydrotestosterone (DHT), respectively, and is one of the most highly upregulated genes in CRPC, Figure 1.4, 5
Figure 1.

Representative reactions catalyzed by AKR1C3. AR = androgen receptor, FP = F prostanoid receptor, PG = prostaglandin.
The success in phase III clinical trials of abiraterone acetate (Zytiga®), a CYP17α–hydroxylase/17,20-lyase inhibitor, and its subsequent approval by the FDA support the involvement of adaptive androgen biosynthesis within the tumor in CRPC.7–10 However, abiraterone blocks glucocorticoid (cortisol) biosynthesis. Abiraterone therefore needs to be co-administered with the glucocorticoid, prednisone to limit adverse effects. Since AKR1C3 acts further downstream of CYP17, it does not play a role in glucocorticoid biosynthesis, which in addition to its intratumoral localization makes it an appealing therapeutic target in CRPC.
AKR1C3 through its prostaglandin (PG) F2α synthase activity also re-directs the metabolism of PGD2 into 11β-PGF2α and can suppress the activation of PPARγ by putative PGJ2 ligands, which would favor differentiation.11–15 Thus, AKR1C3 inhibition might be beneficial in either hormone-dependent or hormone-independent prostate cancer.
AKR1C1 and AKR1C2 are isoforms closely related to AKR1C3 and are required to inactivate DHT in the prostate.16–19 It is therefore imperative that AKR1C3 be inhibited selectively. Several selective AKR1C3 inhibitors have recently been reported. These compounds include 3-(phenylamino)benzoic acids,20–22 baccharin,23 3-(3,4-dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids,24 N-benzoylanthranilic acids,25 and a thiazolidinedione 26 that was discovered by virtual screening (Figure 2).
Figure 2.

Structures of other known selective AKR1C3 inhibitors.
Non-steroidal anti-inflammatory drugs (NSAIDs) are potent inhibitors of the AKR1C enzymes at therapeutically relevant concentrations.12,27–29 In contrast to most NSAIDs, indomethacin inhibits AKR1C3 with little or no effect on AKR1C1 and AKR1C2. Indomethacin has been shown to inhibit AKR1C3 dependent processes in human cell lines and murine xenografts.14, 30. However, the potential therapeutic usefulness of indomethacin in the context of CRPC is limited because of the undesired side effects associated with chronic COX inhibition. Despite this, indomethacin represents a vital lead compound that can be used to develop selective AKR1C3 inhibitors that are stripped of COX inhibitory activity.
Extensive SAR studies on indomethacin (Figure 3) have shown that the p-chlorobenzoyl group, the 2′-methyl group, and the 3′-acetic acid side chain are essential for COX inhibition. Previous efforts in our lab led to the discovery of N-(4-chlorobenzoyl)-melatonin, which inhibited AKR1C3 with potency comparable to that seen with indomethacin; moreover it was selective over AKR1C1 and AKR1C2, and it did not inhibit COX.31, 32 However, the therapeutic usefulness of this compound is limited due to its poor solubility and bioavailability.
Figure 3.

The structure of indomethacin (1) is shown. Groups shown in red are essential for COX-inhibition and were modified to yield Class I analogs; Class II compounds are 2′-des-methyl-indomethacin analogs; and Class III compounds are 3′-alkyl analogs.
In this study, we report the synthesis and structure-activity relationship studies of three series of indomethacin analogs. These compounds have been classified based on the presence and position of the 2′-alkyl group and the 3′-aliphatic acid group on the indole ring.33 Class I analogs have the 2′-alkyl, 3′-aliphatic acid arrangement on the indole ring of indomethacin preserved, class II compounds are based on 2-des-methyl-indomethacin and retain the 3′-aliphatic acid group on the indole ring but lack the 2′-methyl group, and class III compounds have this arrangement reversed to give an aliphatic acid group on the 2′-position and an alkyl group on the 3′-positon of the indole ring (Figure 3).
This study led to the discovery of potent and selective AKR1C3 inhibitors that are devoid of inhibitory activity against AKR1C1, AKR1C2, and COX. Additionally, these compounds block androgen synthesis in a prostate cancer cell line that over-expresses AKR1C3 and have no agonist effect on the AR. The crystal structure of AKR1C3 in complex with 2′-des-methylindomethacin is also reported, showing that this class of compounds occupies a unique binding pose in the enzyme active site.
RESULTS
Chemistry
Our search for novel AKR1C3 inhibitors was aided by the availability of high-throughput chemistry and analysis technologies. All three classes of compounds were prepared using parallel synthesis approaches based on well-established Fischer–indole chemistry. We reported previously an efficient three-step reaction sequence that is amenable to rapid and high-yielding analog production (outlined in Scheme 1).34 The optimized route used indole alkanoic acid methyl- or allyl esters as intermediates, featured maximum synthetical flexibility (e.g. variation of the indole 5′-substituents or late-stage N-acylation) and was more sustainable than the conventional pathway shown in Scheme 2 for quite a few of the envisioned analogs, including 2′-des-methyl-indomethacin (20). Compounds are characterized by either a 5′-methoxy or 5′-flouro group on the indole ring, alternating 2′-/3′-(cyclo)aliphatic acid substituents and variable N1-acyl moieties. Utilization of Fischer indolization/N-acylation/(ester cleavage) with microwave heating led to assembly of novel N-acyl indole alkanoic acids or esters 2 through 13, 17 and 19 (Table 1; class I analogs with 2′-alkyl and 3′-aliphatic acid groups), the 2′-des-methyl derivatives 20 through 25, 42 and 43 (Table 2; class II: 3′-aliphatic acid group) as well as compounds 44 and 62 (Table 3; class III: ‘reversed’ 2′-aliphatic acid/3′-alkyl or 2′-/3′-cyclized acid substituents). For technical details please refer to the cited literature. Moreover, the procedure enabled simple scale-up from milligram to multi-gram quantities (e.g. for lead compounds 20 and 50), which is desirable for testing efficacy in murine xenograft models of CRPC.30
Scheme 1.
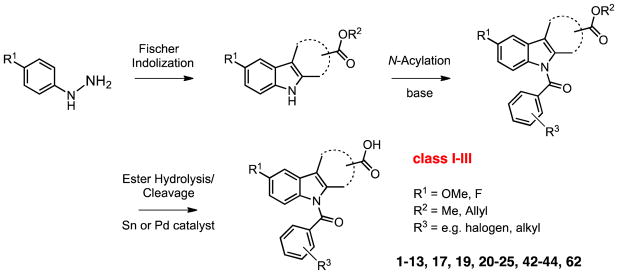
Optimized Fischer indolization/N-acylation/ester cleavage approach for expedient production of different N-acyl indole alkanoic acid analogs.34
Scheme 2.

Classical synthetic strategy for the preparation of N-(4-chlorobenzoyl) indole alkanoic acid analogs and sulfonamide derivatives as COOH bioisosteres. Reagents and conditions: (i) 1. Et3N, 2. CH3CHO, toluene, 0 to 25 °C, 3h; (ii) 4-chloro-benzoyl chloride, pyridine, 10 to 25 °C, 2h; (iii) HCl (g), CH2Cl2, 0°C to rt, 1h; (iv), (cyclo)aliphatic keto acid, AcOH, 70–80 °C, 3h; (v) 1,1′-carbonyldiimidazole (CDI), CH2Cl2, 0–5 °C, 2h, then alkyl-/arylsulfonamide, diazabicyclo[5.4.0]undec-7-ene (DBU), rt, 4–6h; (vi) 1. oxalyl chloride, CH2Cl2, 0 °C to rt, 8h, 2. alkyl-/arylsulfonamide, 1,2-DCE or CH2Cl2, pyridine, rt, overnight.
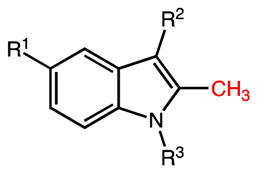
Table 1.
Inhibitory Properties of Indomethacin Analogs on AKR1C3 and AKR1C2
| ||||||
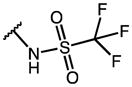
|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | AKR1C3 IC50 (μM) | AKR1C2 IC50 (μM) | IC50 Ratio 1C2/1C3 |
| IMN (1) | OMe |
|
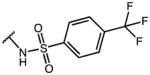
|
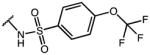
0.10 | 36.53 | 365 |
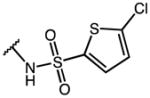
| 2 | OMe |
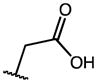
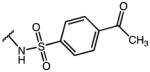
|
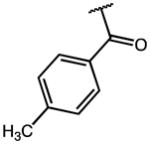
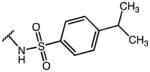
|
0.16 | 54.50 | 336 |
| 3 | OMe |
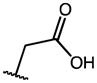
|
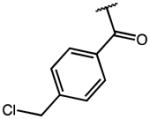
|
0.12 | 40.74 | 329 |
| 4 | OMe |
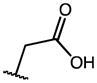
|
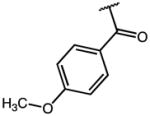
|
0.14 | 45.14 | 332 |
| 5 | OMe |
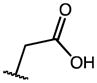
|
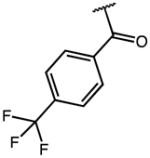
|
0.27 | 35.73 | 134 |
| 6 | OMe |
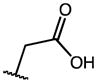
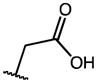
|
|
2.44 | 19.51 | 8 |
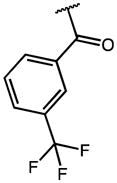
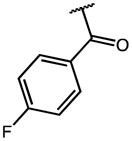
| 7 | OMe |
|
|
0.71 | 60.97 | 86 |
| 8 | F |
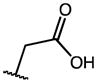
|
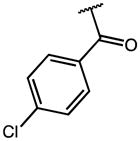
|
0.25 | 6.19 | 25 |
| 9 | F |
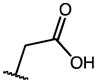
|
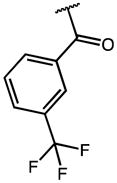
|
2.23 | 7.84 | 4 |
| 10 | OMe |
|
|
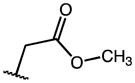
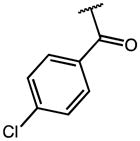
5.67 | >100 (56% Enz. activity at 100 μM) | >18 |
| 11 | OMe |
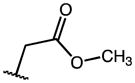
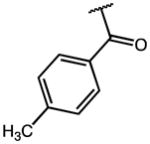
|
|
7.54 | 43.59 | 6 |
| 12 | OMe |
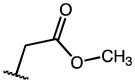
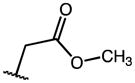
|
|
15.71 | >100 (79% Enz. activity at 100 μM) | >6 |
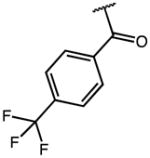
| 13 | OMe |
|
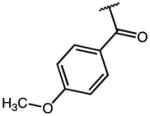
|
7.59 | >100 (53% Enz. activity at 100 μM) | >12 |
| 14 | OMe |
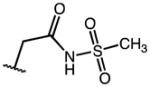
|
|
2.24 | 65.39 | 29 |
| 15 | OMe |
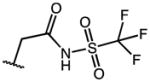
|
|
0.74 | 80.95 | 108 |
| 16 | OMe |
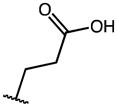
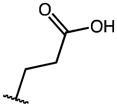
|
|
0.22 | 56.53 | 257 |
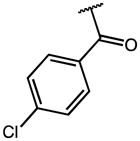
| 17 | F |
|
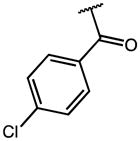
|
0.29 | 13.55 | 47 |
| 18 | OMe |
|
H | >100 | 100 | NA |
| 19 | OMe | H |
|
3.56 | 23.99 | 7 |
Table 2.
Inhibitory properties of 2-Des-Methyl Indomethacin Analogs on AKR1C3 and AKR1C2
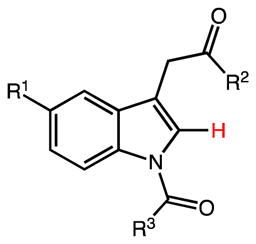
| ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | AKR1C3 IC50 (μM) | AKR1C2 IC50 (μM) | IC50 Ratio 1C2/1C3 |
| 20 | OMe | OH | p-Cl-Phe | 0.96 | 100 | 100 |
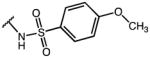
| 21 | OMe | OMe | p-Cl-Phe | 11.34 | >100 (74% Enz. activity at 100 μM) | >8 |
| 22 | OMe | OH | p-F-Phe | 4.96 | >100 | >20 |
| 23 | OMe | OH | p-CF3-Phe | 1.08 | >100 | >100 |
| 24 | OMe | OH | m-CF3-Phe | 5.21 | 100 | 19 |
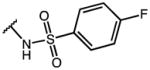
| 25 | F | OH | p-Cl-Phe | 0.50 | 23.25 | 47 |
| 26 | OMe |
|
p-Cl-Phe | 12.0 | 16.68 | 1.4 |
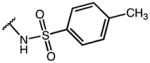
| 27 | OMe |
|
p-Cl-Phe | 0.21 | 50.13 | 240 |
| 28 | OMe |
|
p-Cl-Phe | 12.4 | 42.74 | 3.5 |
| 29 | OMe |
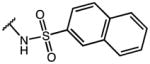
|
p-Cl-Phe | 1.19 | 16.83 | 14.1 |
| 30 | OMe |
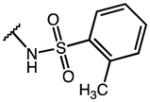
|
p-Cl-Phe | 8.82 | 15.27 | 1.7 |
| 31 | OMe |
|
p-Cl-Phe | 2.07 | 38.22 | 18.5 |
| 32 | OMe |
|
p-Cl-Phe | 2.65 | 59.39 | 22.4 |
| 33 | OMe |
|
p-Cl-Phe | 3.73 | 15.1 | 4.0 |
| 34 | OMe |
|
p-Cl-Phe | 2.09 | 17.31 | 8.3 |
| 35 | OMe |
|
p-Cl-Phe | 6.34 | 7.9 | 1.2 |
| 36 | OMe |
|
p-Cl-Phe | 5.80 | 39.9 | 6.9 |
| 37 | OMe |
|
p-Cl-Phe | 7.86 | 39.91 | 5.1 |
| 38 | OMe |
|
p-Cl-Phe | 2.52 | *65.1% activity @ 10μM | NA |
| 39 | OMe |
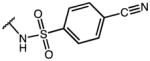
|
p-Cl-Phe | 1.11 | 32.99 | 29.7 |
| 40 | OMe |
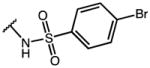
|
p-Cl-Phe | 3.97 | 39.91 | 10.1 |
| 41 | OMe |
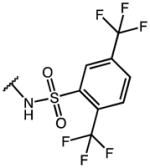
|
p-Cl-Phe | 1.94 | 19.25 | 9.9 |
| 42 | OMe | Et | OH | 2.4 | 100 | 42 |
| 43 | OMe | Et | OMe | 22.78 | >100 μM | >4 |
Table 3.
Inhibitory properties of 3-Alkyl Indomethacin Analogs on AKR1C3 and AKR1C2
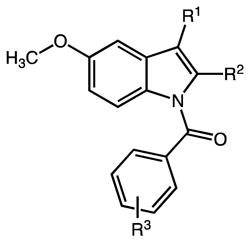
| ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | AKR1C3 IC50 (μM) | AKR1C2 IC50 (μM) | IC50 Ratio 1C2/1C3 |
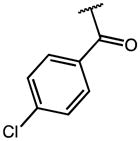
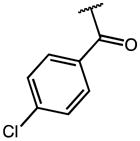
| 44 | Me |
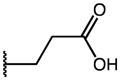
|
p-Cl | 0.13 | 14.45 | 111 |
|
| ||||||
| 45 (mix of isomers A:B=3:1) |
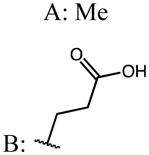
|
|
p-Cl | 0.15 | 18.31 | 119 |
|
| ||||||
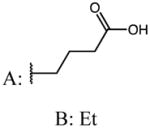
| 46 (mix of isomers A:B=3:1) |
|
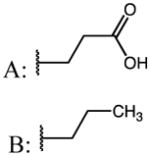
|
p-Cl | 0.07 | 20.70 | 279 |
|
| ||||||
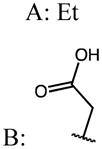
| 47 | Et |
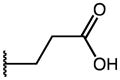
|
p-Cl | 0.09 | 49.57 | 539 |
| 48 | Me |
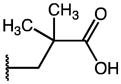
|
p-Cl | 0.12 | 9.82 | 85 |
| 49 | Me |
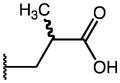
|
p-Cl | 0.29 | 12.28 | 42 |
| 50 | Me |
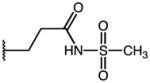
|
p-Cl | 0.34 | 93.0 | 275 |
| 51 | Me |
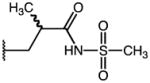
|
p-Cl | 5.53 | 115.8 | 21 |
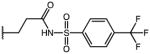
| 52 | Me |
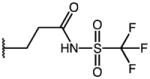
|
p-Cl | 1.44 | *56.8% activity @ 10μM | NA |
| 53 | Me |
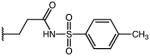
|
p-Cl | 3.11 | 12.26 | 3.9 |
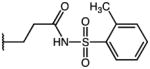
| 54 | Me |
|
p-Cl | 2.54 | 72.3 | 28.5 |
| 55 | Me |
|
p-Cl | 3.25 | 21.13 | 6.5 |
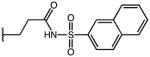
| 56 | Me |
|
p-Cl | 1.11 | 33.74 | 30.4 |
| 57 | Me |
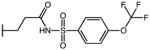
|
p-Cl | 2.50 | 40.43 | 16.2 |
| 58 | Me |
|
p-Cl | ND | ND | NA |
| 59 | Me |
|
p-Cl | 2.65 | 27.35 | 10.3 |
| 60 | Me |
|
p-Cl | 4.69 | 55.34 | 11.8 |
| 61 |
|
p-Cl | 0.16 | 53.65 | 331 | |
| 62 |
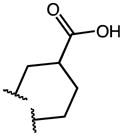
|
m-CF3 | 0.279 | 100 | 358 | |
| 63 |
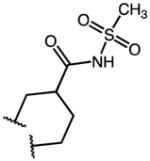
|
p-Cl | 5.07 | >100 | > 20 | |
| 64 |
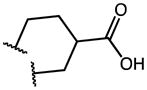
|
p-Cl | 0.34 | 48.2 | 143 | |
Indomethacin analogs with conserved 5′-methoxy groups and 4-chlorobenzoyl tails at their indole N1 were (also) synthesized following the conventional reaction scheme, Scheme 2, which was primarily adopted by Yamamoto 35 for assembly of 1. The key reagent for the underlying Fischer indolization step, 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (D), was prepared according to the reported literature procedures from the aryl hydrazine precursor A via intermediates B and C.36 Target compounds 16 (Table 1; 3′-propionic acid group), and 20 (Table 2, 2′-des-methyl-indomethacin) were readily obtained from D by refluxing with slight excess of 5-oxohexanoic acid E (here: R1=Me, n=2), and 4-oxobutanoic acid E (R1=H, n=1) in AcOH, respectively. Application of 4-oxohexanoic acid F (R1=Me, n=1) as cyclization reagent quantitatively yielded the ‘reversed’ 2′-propionic acid/3′-alkyl indole derivative 44 as the only isolable product.37 The purity of compounds 16, 20, and 44 after a simple filtration and elutriation with water was 99% based on LC-MS analysis. In contrast, reaction of D with either 5-oxoheptanoic acid F (R1=Me, n=2) or 4-oxoheptanoic acid F (R1=Et, n=1) in a trial to further extend the aliphatic acid chain in position 2′ (for 45) or the 3′-alkyl substituent (for 46) of the indole, led to isomer mixtures containing only about 75% of the envisaged target structure (by 1H NMR; compare Supporting Information). Mixtures of 45 and 46 of known composition were evaluated in the biochemical screens as such. We later successfully isolated the major indole regioisomer 47 (2′-propionic acid/3′-ethyl) from mixture 46 with an acceptable sample/isomeric purity of 95% by repeated trituration of the crude product precipitate with diethyl ether.
Analogs 48 and 49 structurally derive from compound 44, and bear an additional α,α-dimethyl (48) or α-methyl group (49) in their aliphatic acid chain. Their synthesis could be accomplished by converting D with appropriately pre-built keto acids. However, these cyclization reagents, namely 2,2-dimethyl-4-oxohexanoic acid, and 2-methyl-4-oxohexanoic acid, were not commercially available, and needed to be prepared beforehand following existing protocols (for details see Supporting Information).37, 38 Whereas analog 49 could be accumulated as solid and filtered pure from the watery reaction solution, compound 48 needed to be extracted into dichloromethane and purified on silica gel. The purities of 48 (after flash chromatography) and 49 (dried filter residue) were judged to be 95%, and >97%, respectively.
In compounds 61 and 64, the 2′- and 3′-positions are bridged by a fused six-membered aliphatic ring; the two structures only vary in the position of the carboxylic acid functionality attached to the cyclohexane ring. Although compounds 61 and 64 formally represent 2,3,4,9-tetrahydro-1H-carbazole-carboxylic acid derivatives, they were considered possible AKR1C3 inhibitor candidates as well. The synthesis followed the above-mentioned universal procedure in Scheme 2, except that different cyclic keto acids G (for 61: p-keto; for 64: m-keto) were used for the ring-closure with the hydrazine hydrochloride precursor D (Scheme 2). Both the symmetrical 4-oxocyclohexanecarboxylic acid, and more surprisingly the unsymmetrical 3-oxocyclohexanecarboxylic acid yielded a single and clean reaction product (filterable precipitate) in high yield (>82%; purity ~99% by LC-MS).39
In order to enhance the desired biological (and physical) properties of the compounds, (bio)isosteric replacements of the free carboxylic acid functions were accomplished on indomethacin (1), 2′-des-methyl-indomethacin (20), the 2′-propionic acid/3′-methyl indole derivatives 44 and 51, and the carbazole carboxylic acid analog 61. Different isostere options were considered and finally the sulfonamide group was chosen as it retained the essential acidic character of the former COOH group. Moreover, this moiety facilitated the introduction of further (sterically demanding) substituents at the sulfone tail end, which might in turn affect the size, shape, conformation, inductive and mesomeric effects, polarizability, H-bond-formation capacity, lipophilicity or pKa of the new analogs. A library of initially 30 sulfonamide derivatives was generated as specified in Scheme 2 (right hand side) applying state-of-the-art methods of combinatorial chemistry: entries 14 and 15 (Table 1) represent class I analogs, entries 26–41 (Table 2) belong to class II, and finally compounds 50, 51, 52–60 and 63 (Table 3), are members of structural class III (see above). For the standard (one-pot) two-step amide coupling reactions the acid groups of starting compounds 1, 20, 44, 51 and 61 were activated with carbonyldiimidazole or infrequently as acid chlorides (to improve the efficiency of the reaction e.g. for 15 and 27) prior to sulfonylamine addition.40–42 After acidic workup (to minimize amide hydrolysis) and evaporation of the organic solvents, the residues were combined with DMSO (1 mL each) and subjected to automated mass-directed HPLC purification with a set purity threshold of ≥95%.
The chemical structures of all intermediates and target compounds were confirmed and fully characterized by NMR spectroscopy and mass spectrometry. The purity of the test compounds was determined using liquid chromatography [UV detection at 215 and 254 nm along with ELSD detection] and was ≥95%, if not otherwise denoted (compare Experimental Section and/or Supporting Information).
Strategy
The synthesized compounds were compared for their ability to inhibit AKR1C3 over AKR1C2. AKR1C2 was used in this comparison due to its similarity in amino acid sequence and structure, and because its inhibition is undesirable. The ratio of the IC50 values for the two enzymes was computed to generate a selectivity ratio. The higher the ratio the more selective the compound is in its ability to inhibit AKR1C3.
Class I Analogs
These compounds retain the 2′-alkyl, 3′-aliphatic acid substitution on the indole ring of indomethacin and possess one or more of the following modifications: substitution on the p-chlorobenzoyl group, replacement of the 5′-OCH3 group, esterification/amidation of the free acid, and modification of the acetic acid side chain. Results from the AKR1C3/AKR1C2 screen of these compounds are shown in Table 1. Indomethacin is a potent and selective inhibitor of AKR1C3. It inhibited AKR1C3 with an IC50 value of 100 nM and was over 300-fold selective over AKR1C2. To evaluate the importance of substitution on the phenyl ring, the p-Cl substituent of indomethacin was replaced with substituents of varying electronic properties and lipophilicity. Introduction of either p-CH3 (2), p-CH2Cl (3), or p-OCH3 (4) on the phenyl ring did not significantly alter either AKR1C3 potency or selectivity. These compounds displayed IC50 values between 120–160 nM for AKR1C3 and were over 300-fold selective for AKR1C3 over AKR1C2. However, when the p-Cl group was substituted with a p-CF3 group (5), the inhibitor lost both potency and selectivity and was only 130-fold selective for AKR1C3. The reduction in potency and selectivity was even more pronounced with a smaller p-F (7) substitution and was reduced further when the CF3 group was moved to the meta-position (6: AKR1C3-IC50 > 2 μM). Removal of the p-chlorobenzoyl group altogether to yield des-chlorobenzoyl-indomethacin 18 (a major metabolite and decomposition product of indomethacin) showed that inhibition potency and selectivity was completely eliminated. These findings underscored the need for the N-benzoyl ring as well as a suitable substitution at the para position of the phenyl ring for AKR1C3 inhibition. Interchanging the 5′-OCH3 groups in compounds 1 and 6 with fluorines at the same indole position in analogs 8 and 9 was detrimental to AKR1C3 selectivity as AKR1C3 inhibition was mostly unchanged whilst undesired AKR1C2 inhibition was enhanced.
The free carboxylic acid, which exists predominantly as the negatively charged carboxylate at physiological pH, is thought to contribute to the general inhibition of AKR1C enzymes by NSAIDs. Consistent with this, esterification of indomethacin to give the methyl ester (10) led to a 57-fold loss of inhibitory potency for AKR1C3 and a reduction in selectivity. This trend was also seen with all other methyl esters 11–13 when compared to their corresponding free acid derivatives (2, 4 and 5). Despite this loss of potency, the rank order of inhibitory potency for the methyl esters was similar to the free acids. The requirement for an acid functionality for optimal AKR1C3 inhibition was likewise apparent with compound 19. This indomethacin fragment lacks the complete carboxyalkyl chain in position 3′ of the indole, and revealed a 36-fold lower inhibitory activity on the target enzyme in comparison to 1. However, the interactions of 19 and 1 with AKR1C2 were still comparable clearly indicating differences in the way the compounds bind to AKR1C3 and AKR1C2. The AKR1C inhibition data for compound 18 reconfirm the need for an intact N-acyl (here: p-Cl-benzoyl) substituent. The decrease in inhibitor potency (and selectivity) observed for AKR1C3 appeared to be less with 19 than with 18 (see above).
Further modification of the acetic acid group of indomethacin yielded the N-(sulfonyl)acetamide analogs 14 and 15. While the N-(methylsulfonyl)acetamide analog, 14, led to a 22-fold reduction in inhibitory potency for AKR1C3 (IC50 value for AKR1C3 = 2.24 μM), potency was partially restored by the formation of the N-(trifluoromethylsulfonyl)acetamide analog 15, a compound with an IC50 value of 0.74 μM and 108-fold selectivity for AKR1C3.
Increasing the length of the aliphatic side chain of indomethacin from -ethyl to -propyl (16) led to a twofold reduction in AKR1C3 potency but the compound retained 257-fold selectivity for AKR1C3 over AKR1C2 (Table 1). Replacement of the 5′-OCH3 group of this compound with a fluorine atom (17) did not change the AKR1C3 inhibitory potency but this compound was a more potent AKR1C2 inhibitor than 16 leading to a loss of AKR1C3 selectivity.
Class II : 2-Des-methyl-indomethacin Analogs
Structurally, these compounds have a 3′aliphatic acid on the indole ring but the 2′-methyl group on the indole ring has been eliminated. Compared to indomethacin, the parent compound in this class, 2′-des-methyl-indomethacin (20) was a less potent and less selective AKR1C3 inhibitor (Table 2). It displayed an IC50 value of 960 nM for AKR1C3 and an IC50 value of 100 μM for AKR1C2, which translated to 100-fold selectivity for AKR1C3. The loss in AKR1C3 potency with removal of the 2′-CH3 group was seen across most of the des-methyl-indomethacin analogs when compared to their respective class I analogs (e.g. 21 vs. 10, 23 vs. 5). Within the triad of class II compounds 22–24 with alternating substituents at the benzoyl moiety, the potency and selectivity decreased in the same rank order as discovered for the indomethacin counterparts 5–7 in Table 1 from p-CF3 > p-F > m-CF3. One exception with quite promising inhibitory qualities was the N-(trifluoromethylsulfonyl)acetamide derivative, 27 which gave an IC50 value of 210 nM for AKR1C3 and was 240-fold selective for AKR1C3 over AKR1C2, while the corresponding indomethacin analog 15 was less potent and less selective. The presence of a trifluoromethyl group on the acetamide seems critical for AKR1C3 inhibition since the N-(methylsulfonyl)acetamide analog of des-methyl-indomethacin (26) displayed weak AKR1C3 inhibition and a total loss of selectivity for AKR1C3. Further substitution on the N-(sulfonyl)acetamide group with substituted phenyl groups or heterocycles (28–41) did not significantly improve AKR1C3 potency or selectivity, although analogs with bulky aromatic rings and/or aromatic rings with substituents capable of forming electrostatic interactions generally gave better AKR1C3 inhibitors. Introduction of a 5′-F on the indole ring of des-methyl-indomethacin to give 25 led to little or no improvement in AKR1C3 potency and a 4-fold increase in AKR1C2 inhibitory potency. This translated to a reduction in AKR1C3 selectivity.
Finally, compounds 42 and 43 display structurally edited versions of the parent molecules, des-methyl-indomethacin 20 and its methyl ester 21, respectively, whose terminal aryl rings of the original N-4-Cl-benzoyl substituents were truncated leaving a linear propanoyl group in each case. Both derivatives 42 and 43 were about 2 to 2.5-fold less potent than their Nbenzoyl analogs 20 and 21 in inhibiting AKR1C3. The methyl ester 43 again was clearly inferior with respect to AKR1C3 inhibition and selectivity.
Class III: 3-Alkyl Indomethacin Analogs
The compounds in this class have the acetic acid side chain of indomethacin 1 replaced with a 3′-alkyl group and the 2′-CH3 group substituted with an aliphatic acid or a substituted N-(sulfonyl)acetamide, in effect reversing the position of the aliphatic acid and alkyl group on the indole ring of 1. Relative to indomethacin and the Class I inhibitor 16, compound 44, with a 3′-CH3 group and a 2′-propionic acid substituent on the indole ring, retained AKR1C3 potency with IC50 value of 130 nM (Table 3). This result indicated that AKR1C3 will tolerate flexibility in the position of the aliphatic acid side chain and alkyl group on the indole ring. However, 44 was less selective for AKR1C3 than both 1 and 16, being 111-fold selective for AKR1C3 over AKR1C2. Introduction of an α-CH3 group on the aliphatic acid side chain of 44 introduced a chiral center and the racemate of this compound, 49, displayed twofold lower potency for AKR1C3 and an unchanged AKR1C2 potency relative to 44. Removal of the chiral center by formation of the geminal di-methyl analog, 48 restored AKR1C3 potency. Synthesis of the N-(methylsulfonyl)acetamide analog, 50, led to a 3-fold loss of AKR1C3 potency (340 nM) but an increase in AKR1C3 selectivity (275-fold). However, its α-CH3 analog, 51, exhibited poor inhibition of AKR1C3. Replacement of the methylsulfonyl group of 50 with a trifluoromethylsulfonyl group (52) also did not improve AKR1C3 potency and selectivity in contrast to class I and II analogs, neither did a series of further sulfonamides 53–60 with bulkier (hetero)aryl substituents attached to their sulfonyl tail end.
The library also produced mixtures of 45 in which the 3′ substituent is methyl and the 2′ substituent is butyric acid or the 3′ substituent is propionic acid and the 2′ substituent is a propyl group. This mixture produced an IC50 value = 0.15 μM for AKR1C3 and was a 119-fold selective for the inhibition of AKR1C3 over AKR1C2. Additionally, the library produced mixtures of 46 in which either the 3′ substituent is ethyl and the 2′ substituent is propionic acid or the 3′ substituent is acetic acid and the 2′ substituent is a propyl group. This mixture gave an IC50 value of 70 nM for AKR1C3 that was 279-fold selective for the inhibition of AKR1C3 over AKR1C2. The latter mixture was then resolved and the active component identified as compound 47.
Compound 47 was the most potent and selective compound in the study, with an IC50 value of 90 nM for AKR1C3 and was 540-fold selective for AKR1C3 over AKR1C2. Cyclization of the side chains at the 2′- and 3′-position on the indole ring of 47 to form the racemic tetrahydrocarbazole analog with a 3′-carboxylic acid group (61) did not produce a pronounced change in either AKR1C3 potency or selectivity. This compound gave an IC50 value of 160 nM for AKR1C3 and showed a 330-fold selectivity for AKR1C3 over AKR1C2. Changing the p-chloro substituent on the benzoyl ring of 61 with a m-CF3 group to give 62 fractionally improved the selectivity to a factor of 360 by raising the IC50 value for AKR1C2 inhibition more than the IC50 value for AKR1C3 inhibition. In contrast, replacement of the carboxylic acid function of 61 by the isosteric N-(methylsulfonyl)acetamide decreased the potency of 63 for AKR1C3 and drastically reduced the selectivity to a factor of only 20. Compound 64, a close analog of the 2,3,4,9-tetrahydro-1H-carbazole-carboxylic acid 61, but with the −COOH group in position 2 in the six-membered cyclohexene ring did not exhibit improved potency and selectivity over the primary lead compound 44 within this series (IC50: 340 nM).
Inhibition of Other AKR1C Isoforms and COX (secondary screens)
Lead compounds were subsequently counterscreened against other human AKR1C enzymes (AKR1C1 and AKR1C4). A complete profile of the inhibitory properties of these compounds on all human AKR1C enzymes is shown in Table 4. All the compounds tested had IC50 values for AKR1C1 in the high micromolar range and showed between 100–800 fold selectivity for AKR1C3 over AKR1C1.
Table 4.
Inhibitory effects of compounds on other AKR1C enzymes.
| IC50 Values (μM)
| ||||
|---|---|---|---|---|
| a Compds | AKR1C3 | AKR1C1 | AKR1C2 | AKR1C4 |
| 2 | 0.16 | >100 ( > 625) | 54.50 (336) | >100 ( > 625) |
| 3 | 0.12 | 100 (833) | 40.74 (329) | 49.75 (415) |
| 5 | 0.27 | >100 ( > 370) | 35.73 (134) | >100 ( > 370) |
| 15 | 0.74 | >100 ( > 135) | 81.0 (108) | >100 ( > 135) |
| 16 | 0.22 | >100 ( > 455) | 57.0 (257) | >100 ( > 455) |
| 20 | 0.96 | >100 (> 100) | 100 (100) | 48.7 (357) |
| 27 | 0.21 | >100 (> 478) | 50.13 (240) | >100 (> 478) |
| 44 | 0.13 | 17.73 (136) | 14.45 (111) | 3.51 (27) |
| 47 | 0.09 | 30.71 (341) | 49.57 (538) | 1.95 (22) |
| 50 | 0.34 | 100 (296) | 93.0 (275) | 12.64 (37) |
| 61 | 0.16 | 76.25 (477) | 53.50 (331) | 3.15 (20) |
Also, all of the compounds tested had IC50 values between 50 μM to 100 μM for AKR1C4 and had over 130–600 fold selectivity for AKR1C3 over AKR1C4 with the exception of the class III compounds, 44, 47, 50 and 61, which showed less than 50-fold selectivity.
Secondary screens also involved a screen against COX-1 and COX-2 to reaffirm that compounds will not act as NSAIDs. Initially we used a continuous colorimetric COX-1 assay that monitored the oxidation of N, N, N, N-tetramethyl-1,4-phenylenediamine (TMPD) when it is coupled to the formation of PGH2 from PGG2 employing arachidonic acid (AA) as substrate.
The activity of the lead compounds on COX-1 is shown in Table 5. Indomethacin (1) potently inhibited COX-1 with an IC50 of 20 nM. This value was consistent with reported inhibitory potency of 1 for COX-1.43 All the other lead compounds were at least 37-fold less potent than 1 on COX-1. Compound 61 with IC50 value for COX-1 of 750 nM was the most potent COX-1 inhibitor among the lead compounds. Surprisingly, 16, which contains a propyl side chain instead of an ethyl side chain, was 2000-fold weaker as a COX-1 inhibitor than 1. The 3′-alkyl analogs 44 and 50 did not display significant inhibitory activity on COX-1 at the highest inhibitor concentration (100 μM) tested. Compared to their respective AKR1C3 inhibitory potency, all the compounds with the exception of 27 and 61, displayed over 200-fold selectivity for AKR1C3 over COX-1.
Table 5.
COX-1 inhibitory activity of lead compounds. Data derived from the continuous colorimetric assay.
| Compound | AKR1C3 IC50 (μM) | COX 1 IC50 (μM) | COX 1 IC50: AKR1C3 IC50 |
|---|---|---|---|
| Indomethacin (1) | 0.10 | 0.02 | 0.2 |
| 16 | 0.22 | 48.8 | 222 |
| 27 | 0.21 | 2.19 | 10 |
| 44 | 0.13 | 100 | 770 |
| 50 | 0.34 | >100 | >294 |
| 61 | 0.16 | 0.75 | 5 |
To further evaluate the residual COX-1 and COX-2 inhibitory activity of a subset of AKR1C3 inhibitors, we also used a discontinuous 14C-AA TLC-based COX assay, where samples were taken, the reaction stopped and the concentration of radioactive substrates/products determined on a plate reader (for details see Supporting Information). Compounds were tested at four distinct concentrations, 250 nM, 1000 nM, 4 μM and 25 μM on either ovine COX-1 or mouse COX-2 and IC50 values (if applicable) were detected graphically from the resulting dose-response curves. Results are compiled in Table 6. The AA concentration of 5 μM used in the latter assay represents the Km for COX-1 and COX-2, which enables rapid, reversible, and slow, tight-binding inhibitors to be detected. Both assays are well established in our laboratories and have been described earlier in literature.20, 44
Table 6.
COX-1 and COX-2 inhibitory activities of selected AKR1C3 inhibitors Data derived from the discontinuous radioactive TLC assay.
| Compound | oCOX 1 IC50 (μM) | wt mCOX 2 IC50 (μM) | COX 1 IC50: COX 2 IC50 |
|---|---|---|---|
| Indomethacin (1) | 0.05 | 0.20 | 0.25 |
| 8 | 0.49 | 0.09 | 5.44 |
| 20 | 15.27 (P)a | 3.27 (P) | 4.67 |
| 25 | 42.7 % inhib. @ 25 μM | 35.6 % inhib. @ 25 μM | n/a |
| 44 | 11 % inhib. @ 25 μM | 17.6 % inhib. @ 4 μM | n/a |
| 47 | 15.6 % inhib. @ 25 μM (P) | 7.0 | n/a |
| 48 | 24.2 % inhib. @ 25 μM (P) | 33.7 % inhib. @ 25 μM (P) | n/a |
| 50 | 30.8 % inhib. @ 25 μM (P) | 32.4 % inhib. @ 25 μM (P) | n/a |
| 61 | 1.0 | 3.02 (P @ 61.4%) | 0.33 |
| 64 | 2.4 (P @ 56.1%) | 1.48 (P @ 62.6%) | 1.62 |
P = plateau; oCOX-1 = ovineCOX-1; mCOX-2 = murineCOX-2
Unlike 1, its 5′-fluoro equivalent 8 (Table 1), and to some degree the Class III inhibitors 61 and 64 (Table 3), the other indole-based lead AKR1C3 inhibitors listed in Table 6 do not strongly inhibit COX-1 or COX-2. As reported earlier, 2′-des-methyl-indomethacin 20 is only a very weak competitive inhibitor of both COX enzymes with a preference for COX-2.45
The same proved true for its close analog 25 (here: 5′-OCH3 replaced by 5′-F), but without preference for either COX enzyme. The best results from the standpoint of minimal COX interaction were achieved with the 3′-alkyl indole derivatives 44, 47, 48 (free acids), and 50 (sulfonamide), whose percent COX-1/-2 inhibition did not exceed 34% at 25 μM. In particular, for compounds 44 (3′-methyl) and 47 (3′-ethyl), with an unbranched 2′-aliphatic acid chain, the maximum inhibition achieved was even lower. Compound 44 inhibited oCOX-1 at 11% (25 μM) and mCOX-2 at <18% (4 μM). The homo analog 47 demonstrated 16% inhibition of oCOX-1 at 25 μM and a poor IC50 of 7 for mCOX-2. In many cases the formation of a plateau at higher inhibitor concentrations suggests that inhibition is readily reversible. Collectively, compounds 16 (Class I), compounds 20, 27 (Class II) and compounds 44–47 and 50 (Class III) are among the most promising lead compounds not to inhibit COX-1 and COX-2.
Inhibition of Testosterone Formation in LNCaP-AKR1C3 cells
To evaluate the efficacy of the lead compounds in a prostate cancer cell model, we tested their ability to block testosterone formation in LNCaP cells stably transfected with AKR1C3.46 Cells were incubated with 0.1 μM Δ4-AD and their ability to block the formation of testosterone-17β-glucuronide was measured (Figure 4). In the absence of the inhibitor, Δ4-AD is converted to testosterone and androsterone glucuronides. We found that there was robust inhibition of testosterone formation when the cells were treated with 0.1 μM Δ4-AD in the presence of 30 μM 1, 44, and 50 (Figure 4).
Figure 4.

Inhibitory effect of indomethacin and indomethacin analogs 44 and 50 on testosterone formation in LNCaP-AKR1C3 cells measured by radiochromatography
Effects of Lead Compounds on AR Reporter gene Assay
Since AR activation is involved in progression of CRPC, we next evaluated the effect of these compounds on AR signaling using a luciferase reporter gene assay. Agonist activity by these compounds would be deleterious in the context of CRPC. When HeLa-AR3A-PSA-(ARE)4-Luc13 cells (a kind gift from Dr. Elizabeth Wilson) were treated with the compounds in the absence of DHT, none of the compounds displayed agonist activity on the AR in the absence of DHT.
The compounds also did not enhance DHT-mediated AR signaling (Figure 5). The EC50 value for DHT induction of the luciferase reporter in the presence and absence of the indomethacin analogs was not significantly different. The EC50 value for DHT induction of the luciferase reporter was 0.13 ± 0.02 nM, while the values in the presence of 1, 27, 44 and 50 were 0.14 ± 0.02 nM, 0.10 ± 0.02 nM, 0.15 ± 0.03 nM, and 0.14 ± 0.05 nM, respectively (Figure 5). This data also indicates that these compounds did not act as AR antagonists.
Figure 5.

Dose-response curve of 5α-dihydrotestosterone (5α-DHT) in HeLa-AR3A-PSA-(ARE)4-Luc13 cells in the presence and absence of 10 μM of lead compounds.
Crystal Structure of the AKR1C3•NADP+•20 (2′-des-methyl-indomethacin) complex
The structure of AKR1C3 in complex with NADP+ and 2′-des-methyl-indomethacin was determined at a resolution of 1.8 A by molecular replacement (Table 7 and Figure 6).47 We found that there are two monomers of AKR1C3 in each unit cell. Monomer A exhibited a lower average B-factor compared to monomer B, but the two monomers showed almost identical conformation with a rmsd of 0.23 A. The cofactor binding cavity of AKR1C3 is not perturbed upon binding of the inhibitor. NADP+ maintains the same binding position and conformation as in the other AKR1C3 structures determined.48–51 The steroid/inhibitor binding cavity of AKR1C3 is composed of five compartments: oxyanion site (formed by Tyr55, His117, and NADP+), steroid channel (Trp227 and Leu54), and three sub-pockets, SP1 (Ser118, Asn167, Phe306, Phe311, and Tyr319), SP2 (Trp86, Leu122, Ser129, and Phe311), and SP3 (Tyr24, Glu192, Ser221, and Tyr305).52 Two molecules of compound 20 are found in each steroid/inhibitor binding cavity (Figure 6A). One molecule interacts with the oxyanion site through hydrogen bonding between the carboxylate group and the active site residues Tyr55 and His117. The carboxylate group of this molecule also lies on the face of the positively-charged nicotinamide ring of NADP+ and makes a favorable charge-charge interaction. Finally, the indole ring of this molecule is bound between the two gate keepers of the steroid channel, Trp227 and Leu54, and the p-chorobenzoyl ring of the inhibitor projects into the SP1 subpocket. This molecule is likely the first molecule of 20 to enter the active site considering its deeper penetration in the cavity. The second molecule of 20 stacks on the first molecule through offset stacked aromatic interactions between the p-chorobenzoyl rings and edge-to-face aromatic interactions between the indole rings. The indole ring of the second molecule of 20 projects to the steroid channel. In contrast to the crystal structure, the inhibition dose-response curve of 20 for AKR1C3 exhibits a slope factor of 1, which disagrees with multisite binding. Non-specific binding or occupancy of low affinity binding sites by more than one inhibitor molecule per enzyme active site can occur in the presence of excess inhibitor used under the crystallization conditions.
Table 7.
Data collection and refinement statistics for the AKR1C3•NADP+• 20 complex (PDB ID : 4DBW).
| Data collection | |
| Resolution range (Å) | 50.0–1.80 |
| Unique reflections measured | 60585 (5607)b |
| Rmerge a | 0.050 (0.28) b |
| I/σ (I) | 19.8 (3.3)b |
| Completeness (%) | 96.7 (89.8)b |
|
| |
| Refinement statistics | |
| Reflections used in refinement/test set | 56940/2893 |
| R/Rfree c | 0.178/0.233 |
| Protein atoms d | 5050 |
| Water moleculesd | 611 |
| NADP+ moleculesd | 2 |
| inhibitor moleculesd | 4 |
| r.m.s. deviations | |
| Bond lengths (A) | 0.005 |
| Bond angles (°) | 1.0 |
| Overall B-factor (A2) | 28 |
| Cofactor B-factor (A2) | 21 |
| Inhibitor B-factor (A2) | 34 |
| Ramachandran statisticse | |
| Allowed (%) | 92.9 |
| Additionally allowed (%) | 6.8 |
| Generously allowed (%) | 0.2 |
| Disallowed (%) | 0.2 |
Rmerge = Σ |I − 〈I〉|/Σ I, where I is the observed intensity and 〈I〉 is the average intensity calculated for replicate data.
The number in parentheses refers to the outer 0.1-A shell of data.
Crystallographic R-factor, R = Σ||Fo|−|Fc||/Σ|Fo| for reflections contained in the working set. Free R-factor, Rfree = Σ| |Fo|−|Fc||/Σ|Fo| for reflections contained in the test set excluded from refinement. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Per asymmetric unit.
Ramachandran statistics were calculated with PROCHECK 53.
Figure 6.
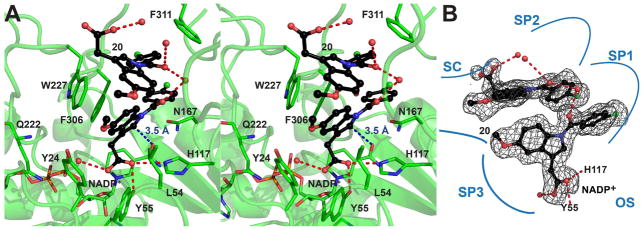
Stereoview (A) and schematic (B) illustrate the occupancy of the AKR1C3 active site by compound 2′-des-methyl-indomethacin (20) (PDB ID: 4DBW). The first molecule of 20 is anchored to the active site through hydrogen bonding to Tyr55 and His117. The second molecule of 20 stacks on the first molecule. Both molecules project their p-chorobenzoyl rings into the SP1 pocket. The distance between the 2′-position on the indole ring of the first molecule of 20 and the carboxamide group of the nicotinamide cofactor is only 3.5 A (blue dash, panel A), prohibiting indomethacin from assuming this binding pose. The simulated-annealing omit map of 20 is contoured at 2.3σ. Compound 20 is shown in ball-and-stick representation with carbon atoms colored in black. The non-carbon atoms are colored: oxygen = red, nitrogen = blue, chlorine = green,, phosphorus = orange. Water molecules are shown as red spheres. Hydrogen bonds are indicated by red dashes.
DISCUSSION
AKR1C3 is a critical enzyme in the intraprostatic biosynthesis of androgens and has been implicated in the progression of CRPC.4–6 Therefore, AKR1C3 inhibition is expected to be an effective therapy in CRPC. We describe the synthesis and structure-activity relationship studies of indomethacin-based AKR1C3 inhibitors. Based on the known SAR for COX inhibition, three classes of compounds were synthesized: (I) analogs in which groups essential for COXinhibition were substituted; (II) 2′-des-methyl-indomethacin analogs in which the 2′-CH3 group essential for tight binding to COX 54 was removed from the structure; and (III) 3′-alkyl analogs in which the acetic acid side chain essential for COX inhibition was replaced with an alkyl group and the acid-side chain was moved to the 2′-position.
This effort led to the discovery of potent AKR1C3 inhibitors that retain selectivity for AKR1C3 over other AKR1C enzymes and have been stripped of COX inhibitory activity. These compounds inhibit testosterone formation in a prostate cancer cell line and display no agonist or antagonist activity at the androgen receptor. The crystal structure of a lead compound, 2′-des-methyl-indomethacin (20), in complex with AKR1C3 was also obtained to elucidate enzyme-inhibitor interactions. Of the compounds synthesized and tested, seven class I and class II analogs had the required AKR1C3 selectivity and potency. Many of the class III analogs had desirable AKR1C3 potency but were less selective for AKR1C3 over AKR1C4, which plays a critical role in bile-acid biosynthesis.
Structural Consideration of AKR1C3 Inhibitor Potency and Selectivity
Five crystal structures exist for AKR1C3 inhibitor complexes that are relevant to the present study: AKR1C3•NADP+•indomethacin,49, 55 AKR1C3•NADP+•2′-des-methyl-indomethacin (20),47 and AKR1C3•NADP+•Z-sulindac55 (Figure 7), which can be used to explain the binding and selectivity of class I – class III inhibitors. These structures show that the active site of AKR1C3 contains three subpockets (SP1–SP3) and depending on the inhibitor class, different subpockets may be occupied. At the base of the active site, an oxyanion site exists and consists of Tyr55, His117, and Lys84, which for most NSAID derivatives provides a counterion to bind the carboxylic acid on these drugs.55 The steroid channel extends from the oxyanion hole and is flanked by Trp227 and Leu54 on either side. Surprisingly, few NSAID derivatives fully occupy the steroid channel.
Figure 7.

Binding poses of indomethacin analogs in AKR1C3. (A) indomethacin (IMN, black) at pH 6 (PDB ID: 1S2A), where the carboxylate group anchors the indole ring in the SP3 pocket by interacting with Q222 and a phosphate group of the pyrophosphate moiety of the cofactor. 2′-Des-methyl-indomethacin (20, green, PDB ID: 4DBW) is superimposed as a reference showing how the oxyanion site is occupied by the carboxylate group and the SP1 pocket is occupied by the p-chorobenzoyl ring. (B) indomethacin (black) at pH 7.5 (PDB ID: 3UG8), where the carboxylate group is tethered to the oxyanion site and the p-chorobenzoyl ring penetrates the SP1 pocket. Two alternative conformations exist for Trp227, each with 50% occupancy. Note that the 5′-methoxy group on the indole ring points into SP3 so that the indole ring of indomethacin assumes a ~120° angle with the indole ring of 2′-des-methyl-indomethacin. (C) Z-sulindac (black, PDB ID: 3R7M), where the carboxylate group is anchored to the oxyanion site and the 4-methylsulfinyl group approaches the SP2 pocket. (D) the proposed fifth binding pose illustrated by compound 27 (black, generated by AutoDock Vina), where the N-(sulfonyl)acetamide group occupies the SP3 pocket, the p-chorobenzoyl ring occupies the SP1 pocket, and the indole ring almost overlaps with the indole ring of 2′-des-methyl-indomethacin. Non-carbon atoms, water, and hydrogen bonds are presented as described in Figure 6.
Three AKR1C3•NADP+•indomethacin structures have been reported in the Protein Data Bank.49, 55 The structure obtained at pH 6.0 (PDB ID: 1S2A) shows a unique binding pose, in which the carboxylic acid, presumably ionized as the negatively charged carboxylate, is not anchored in the oxyanion site but instead projects into SP3, where it interacts with Gln222 and with the pyrophosphate bridge of the cofactor (Figure 7A). As the pH of the crystallization condition is increased to 7.5 (PDB ID: 3UG8), deprotonation of the pyrophosphate bridge weakened the interactions in SP3 so that the indole ring rotates by ~40° to permit the carboxylate group to interact with the oxyanion site. In this binding pose, the 5′-OCH3 group still remains in SP3 while the p-chlorobenzoyl ring enters SP1 (Figure 7B). In the AKR1C3•NADP+•2′-des-methyl-indomethacin complex (PDB ID: 4DBW), the carboxylate remains anchored by the oxyanion hole but the 5′-OCH3 group no longer interacts with SP3. In addition, the orientation of the p-chlorobenzoyl ring is altered so that it lies perpendicular to this ring in the indomethacin structure determined at pH 7.5 (Figure 7B). Indomethacin cannot assume the binding pose of 2′-des-methyl-indomethacin since the 2′-methyl group of indomethacin would clash with the nicotinamide head group of the cofactor. In the AKR1C3•NADP+•Z-sulindac structure (PDB ID: 3R7M), the indene ring is flipped by 180° relative to the indole ring in the 2′-des-methylindomethacin structure and the 4-methylsulfinyl group approaches SP2 (Figure 7C).
Most of the class I compounds would adopt the binding conformations exemplified by indomethacin. Since our inhibition experiments were performed at pH 7.0, it is likely that both pH 6 and pH 7.5 binding poses for indomethacin will coexist in solution. The unsubstituted free acids inhibit AKR1C3 with the highest potency and selectivity indicating that the interaction between the binding sites and the carboxylate is essential for inhibitor anchoring. Analogs with an extended acid side chain at the 3′-position, i.e. replacement of the acetic acid chain by a propionic acid side chain (compound 16), is also well tolerated suggesting there is space available in the binding cavity to accommodate the additional carbon atom. But larger subsitituents such as the N-(sulfonyl)acetamide derivatives cannot be accommodated by either of the two indomethacin binding poses observed in the available crystal structures.. Upon inspection of the AKR1C3 structures, we propose a fifth binding pose for these bulky analogs, in which inhibitors extend their p-chorobenzoyl ring into the SP1 and the bulky 3-subsitituents would occupy the SP3 (Figure 7D, generated by AutoDock Vina56). The SP3 pocket is lined by polar/charged residues including Tyr24, Gln222, Asp224, and Arg226, which supports the observation that polar substituents are generally favored and the very bulky side chains may be exposed through an opening in the SP3 pocket into bulk solvent. This new binding pose is supported by docking studies and can be applied to all the bulky analogs in all three classes of inhibitors.
Only two Class II compounds based on 2′-des-methyl-indomethacin (20) are likely to adopt the binding pose of 20, namely compounds 21 and 25. Interaction with the oxyanion site is again supported since the corresponding methyl esters are weak inhibitors of AKR1C3. The N-(sulfonyl)-acetamide of 2′-des-methyl-indomethacin is too bulky to interact with the oxyanion site and likely assumes the fifth binding pose proposed.
Class III compounds that have a bulky side chain at the 2′-position cannot be easily superimposed on the indomethacin or 2′-des-methyl-indomethacin binding poses presented in any of the crystal structures. But all these compounds could assume the fifth binding pose proposed, By contrast Class III compounds containing a 3′-methyl/ethyl group may adopt the Z-sulindac binding conformation since this conformation imposes least restriction on the size of the 2′ substitution. Structural information of a AKR1C3•NADP+•class III inhibitor would be invaluable to confirm that this new binding pose exists.
Promising Lead Compounds
Eleven compounds were selected for further evaluation. The selection criteria was AKR1C3 potency < 1 μM and greater than 100-fold selectivity for AKR1C3 over AKR1C2. On this list, five were class I analogs, two were class II analogs and four were class III analogs. These compounds were all greater than 100-fold selective for AKR1C3 over AKR1C1, ensuring that there would be no interference with DHT inactivation within the prostate. All the compounds were also selective for AKR1C3 over AKR1C4. While the 3′-alkyl analogs were still over 20-fold selective for AKR1C3 over AKR1C4, they displayed lower AKR1C3 selectivity relative to AKR1C4 in comparison to the class I and II analogs. AKR1C4 is primarily involved in bile acid biosynthesis and steroid hormone biotransformation in the liver, where it is predominantly expressed.17, 57, 58 The consequence, if any, of the low selectivity of the 3-alkyl analogs for AKR1C3 over AKR1C4 will need to be explored.
The absence of intrinsic agonist activity or positive modulation of DHT-mediated AR transcription in the reporter gene assay by all the lead compounds is important since either effect on the AR would counteract the effect of AKR1C3 inhibition.
The compounds were also efficacious in a prostate cancer cell model where they produced robust inhibition of testosterone formation when cells were treated with the AKR1C3 substrate 4Δ-AD. This indicates the compounds possess adequate physicochemical properties to traverse the cell membrane and more importantly, are able to target AKR1C3 in a cellular context. This is in agreement with the report of Cai et al., which showed that indomethacin was effective in vivo, in a castrate-resistant prostate cancer setting at inhibiting tumor cell proliferation.30 These findings support the potential therapeutic usefulness of AKR1C3 inhibitors in CRPC. It equally indicates that the indomethacin analogs and importantly those with little or no COX activity such as the lead compounds reported in this study should block the adaptive intratumoral biosynthesis that drives CRPC, thereby inhibiting cancer progression without any of the adverse effects secondary to chronic COX inhibition. Since the compounds identified in this study are expected to have pharmacokinetic profiles comparable to that of indomethacin, we anticipate that the lead compounds will be well-tolerated for CRPC treatment.
In conclusion, we have discovered indomethacin-based inhibitors of AKR1C3 that are devoid of activity against other AKR1C isoforms, do not display COX inhibitory activity or AR agonist activity, and are effective in a prostate cancer cell model. These compounds are useful therapeutic leads in the management of CRPC. These compounds have the potential to be superior therapeutic agents compared to abiraterone in the management of CRPC because they do not interfere with glucocorticoid biosynthesis and therefore would not require co-administration with a glucocorticoid.
EXPERIMENTAL
Chemistry
General
All commercially available reagents and anhydrous solvents were used as received. Microwave reactions were carried out using a Biotage Initiator 8-EXP Microwave Synthesizer. Analytical thin-layer chromatography was carried out using glass-backed plates coated with fluorescent silica gel 60 F254 from Whatman (Partisil LK6D). Spots were visualized under natural light, and UV illumination at λ = 254 and 365 nm. Flash chromatography was conducted on a Biotage SP1 automated flash chromatography system equipped with a fixed wavelength UV detector (λ = 254 nm). Samples were preabsorbed onto ready-made silica gel samplets and then applied on normal-phase flash chromatography cartridges (Biotage KP-SIL, size according to requirements) eluting with a 0–100% EtOAc/hexane (0.5% acetic acid) gradient. Where stated, purification was performed utilizing a custom high-throughput mass-directed preparative (RP-) HPLC platform using an appropriate acetonitrile/water gradient with TFA modifier.59 1H, and 13C NMR spectra were recorded at 400, and 100 MHz, respectively, using a Bruker AV-400 with sample changer (BACS 60). 19F NMR spectra were measured at 282 MHz using a Bruker AV-300. MS samples were analyzed on an Agilent 1200 LCMS system operating in single MS mode with electrospray ionization. Samples were introduced into the mass spectrometer using chromatography. A purity of ≥95% (unless otherwise indicated) for the final compounds in this study was confirmed by analytical HPLC on either a Waters HPLC system with PDA detector (set at λ=254 nm) equipped with a Supelco Supelcosil LC-18 reverse-phase column (15 cm × 3 mm, 5/m) or the aforementioned Agilent 1200 analytical LCMS with UV detection at λ=215 and 254 nm along with ELCD detection (Stationary phase: YMC J’sphere H-80 S-4 column (3.0 × 50 mm)), using two different gradient methods. The mobile phases were MeOH/0.01 M KH2PO4, and ACN/H2O (plus 0.1% TFA), respectively (for further details see Supporting Information).
Preparation and Analytical Data of Exemplified Test Compounds
The general synthetic procedures A–D are specified in the Supporting Information.
2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)-N-(methylsulfonyl)acetamide (14)
According to general procedure B, the title compound was obtained from 2-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)acetic acid (IMN) (60 mg, 0.17 mmol) and methansulfonamide (16 mg, 0.17 mmol) after 20 h at room temperature. The crude product was purified by flash chromatography (SiO2, ethyl acetate/hexane gradient). Yield: 55 mg (75%), a white solid: C20H19ClN2O5S, Mr = 434.89; 1H NMR (400 MHz, DMSO-d6) δ: 2.23 (s, 3H), 3.24 (s, 3H), 3.72 (s, 2H), 3.76 (s, 3H), 6.71 (dd, J=2.4/8.8 Hz, 1H), 6.91 (d, J=8.8 Hz, 1H), 7.08 (d, J=2.8 Hz, 1H), 7.63–7.69 (m, 4H); LCMS (ESI) (method 2) tR: 2.55 min (>99%, UV220, UV254), m/z: 435.0 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl)propanoic acid (16)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (50 mg, 0.16 mmol), 5-oxohexanoic acid (25 mg, 0.19 mmol) in AcOH (0.5 mL) after 3 h at 80 °C in 69% yield (41 mg) as an off-white solid. C20H18ClNO4, Mr = 371.81; 1H NMR (400 MHz, DMSO-d6) δ: 2.19 (s, 3H, C2′-CH3), 2.49 (t, J=7.4 Hz, 2H, overlayed by DMSO-signal), 2.88 (t, J=7.4 Hz, 2H), 3.77 (s, 3H, −OCH3), 6.69 (dd, J=2.6 Hz, 1H), 6.93 (d, J=8.8 Hz, 1H), 7.06 (d, J=2.4 Hz, 1H), 7.61–7.64 (m, 4H); LCMS (ESI) (method 2) tR: 2.69 min (>99%, UV220, UV254), m/z: 372.0 [M+H]+.
2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetic acid (20)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (200 mg, 0.64 mmol), 4-oxobutanoic acid (succinic semialdehyde, 15% solution in H2O; 565 mg, 0.83 mmol) in AcOH (1.5 mL) after 3 h at 80°C in 45% yield (99 mg) as an off-white solid. C18H14ClNO4, Mr = 343.76; 1H NMR (400 MHz, DMSO-d6) δ: 3.67 (s, 2H), 3.81 (s, 3H), 6.99 (dd, J=2.4/9.2 Hz, 1H), 7.12 (d, J=2.4 Hz, 1H), 7.32 (s, 1H), 7.65–7.77 (m, 4H), 8.17 (d, J=9.2 Hz, 1H); LCMS (ESI) (method 2) tR: 2.60 min (>99%, ELSD), m/z: 344.0 [M+H]+.
2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetyl chloride
In accordance with general procedure C: Oxalyl chloride (30 XL, 0.35 mmol) was added dropwise to a solution of 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetic acid 20 (100 mg, 0.29 mmol) in 2 mL of dry CH2Cl2 under argon. The reaction mixture was stirred overnight at room temperature. The solvent was evaporated and the crude product was washed with dry hexane (3×1 mL) and dried in vacuo to give the title compound (pale white solid) in 95% yield (100 mg). C18H13Cl2NO3, Mr = 362.21; 1H NMR (400 MHz, CDCl3) δ: 3.90 (s, 3H), 4.21 (s, 2H), 6.95 (d, J=2.4 Hz, 1H), 7.04 (dd, J=2.4/9.0 Hz, 1H), 7.29 (s, 1H), 7.52–7.55 (m, 2H), 7.67–7.71 (m, 2H), 8.29 (d, J=9.2 Hz, 1H); LCMS (ESI) (method 2) tR: 2.72 min (>99%, UV254), m/z: 358.2 [M+H]+.
2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)-N-((trifluoromethyl)sulfonyl) acetamide (27)
In accordance with general procedure D: 80 mg (0.22 mmol) 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetyl chloride and 49.4 mg (0.33 mmol) trifluoromethanesulfonamide were dissolved in 1.8 mL 1,2-dichloroethane (DCE) under stirring. Then 17.5 mg (0.22 mmol) pyridine was added and the reaction was allowed to run at ambient temperature until the staring material was consumed (~ 4 h). After the addition of 13 XL of AcOH, the organic solution was washed with H2O (3×2 mL), dried over Na2SO4, filtered and the concentrated in vacuo. The crude product was purified by flash chromatography (SiO2, ethyl acetate/hexane, 0.5% AcOH gradient) to afford the title compound in 70% yield (73 mg). C19H14ClF3N2O5S, Mr = 474.84; 1H NMR (400 MHz, DMSO-d6) δ: 3.44 (s, 2H), 3.79 (s, 3H), 6.95 (dd, J=2.6/9.0 Hz, 1H), 7.14 9d, J=2.4 Hz, 1H), 7.23 (s, 1H), 7.64–7.66 (m, 2H), 7.72–7.74 (m, 2H), 8.15 (d, J=8.8 Hz, 1H, C7′-H); 19F NMR (282 MHz, DMSO-d6) δ: −75.58 (s, −CF3); LCMS (ESI) (method 2) tR: 2.37 min (>99%, UV254, ELSD), m/z: 475.0 [M+H]+.
2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)-N-(naphthalen-2-ylsulfonyl)acetamide (29)
According to general procedure B, the title compound was obtained from 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetic acid 20 (30 mg, 0.087 mmol) and naphthalene-2-sulfonamide (18.1 mg, 0.087 mmol) after 20 h at room temperature. The crude product was purified by automated mass-directed HPLC (RP-18, ACN/H2O gradient). Yield: 21 mg (45%). C28H21ClN2O5S, Mr = 532.99; 1H NMR (400 MHz, DMSO-d6) δ: 3.64 (s, 3H), 3.65 (s, 2H), 6.87 (d, J=2.4 Hz, 1H), 6.92 (dd, J=2.6/9.0 Hz, 1H), 7.21 (s, 1H), 7.61–7.75 (m, 6H), 7.82 (dd, J=2.0/8.8 Hz, 1H), 8.02–8.16 (m, 4H), 8.55 (d, J=1.6 Hz, 1H); LCMS (ESI) (method 2) tR: 3.01 min (98%, UV254, ELSD), m/z: 533.1 [M+H]+.
2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)-N-((4-(trifluoromethoxy)phenyl)sulfonyl)-acetamide (32)
According to general procedure B, the title compound was obtained from 2-(1-(4-chlorobenzoyl)-5-methoxy-1H-indol-3-yl)acetic acid 20 (30 mg, 0.087 mmol) and 4-(trifluoromethoxy)benzene-sulfonamide (21 mg, 0.087 mmol) after 20 h at room temperature. The crude product was purified by automated mass-directed HPLC (RP-18, ACN/H2O gradient). Yield: 25.4 mg (51%). C25H18ClF3N2O6S, Mr = 566.93; 1H NMR (400 MHz, DMSO-d6) δ: 3.66 (s, 2H), 3.74 (s, 3H), 6.89 (d, J=2.4 Hz, 1H), 6.97 (dd, J=2.6/9.0 Hz, 1H), 7.24 (s, 1H), 7.55 (dd, J=0.8/8.8 Hz, 1H), 7.64–7.75 (m, 4H), 7.99–8.02 (m, 2H), 8.14 (d, J=9.2 Hz, 1H); 19F NMR (282 MHz, DMSO-d6) δ: −54.88 (s, OCF3); LCMS (ESI) (method 2) tR: 3.08 min (98%, UV254, ELSD), m/z: 567.0 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid (44)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (400 mg, 1.28 mmol), 4-oxohexanoic acid (200 mg, 1.54 mmol) in AcOH (4 mL) after 3 h at 80 °C in 75% yield (368 mg) as an off-white solid. C20H18ClNO4, Mr = 371.81; 1H NMR (400 MHz, DMSO-d6) δ: 2.20 (s, 3H), 2.46 (t, J=7.2 Hz, 2H), 3.08 (t, J=7.6 Hz, 2H), 3.75 (s, 3H), 6.44 (d, J=8.8 Hz), 6.63 (dd, J=2.4/9.0 Hz, 1H), 7.01 (d, J=2.4 Hz, 1H), 7.63–7.68 (m, 4H); 13C NMR (100 MHz, DMSO-d6) δ: 8.78 (s, −CH3), 21.61 (s, −CH2-), 34.12 (s, −CH2CO), 55.81 (s, −OCH3), 101.93 (s, C4′ indole), 111.97 (s, C6′ indole), 114.76 (s, C7′ indole), 116.11 (s, C3′ indole), 129.53 (s, C3′/C5′ 4-Cl-benzoyl-), 130.63 (s), 131.58 (s), 131.62 (s, C2′/C6′ 4-Cl-benzoyl-), 134.42 (s), 137.09 (s), 138.14 (s), 155.83 (s, C5′ indole), 168.14 (s, >C=O), 173.77 (s, −C=O(OH)); LCMS (ESI) (method 2) tR: 2.68 min (99%, UV254, ELSD), m/z: 372.1 [M+H]+.
3-(1-(4-chlorobenzoyl)-3-ethyl-5-methoxy-1H-indol-2-yl)propanoic acid (47)
The title compound was synthesized via general procedure A as described for isomer mixture 46 (see SI). H2O was added. The watery reaction mixture was extracted with methylene chloride (2x) and the combined organic phases dried over Na2SO4. The solvent was removed under reduced pressure and the residual beige solid repeatedly triturated with diethyl ether. The resulting white precipitation was collected by filtration (ether wash phases abolished) and dried in vacuo to afford 47 as a single product! Yield: 36 mg, 29%. C21H20ClNO4, Mr = 385.84; 1H NMR (400 MHz, DMSO-d6) δ: 1.18 (t, J=7.6 Hz, 3H), 2.46 (t, J=7.6 Hz, 2H), 2.69 (q, J=7.6 Hz, 2H), 3.09 (t, J=7.6 Hz, 2H), 3.75 (s, 3H), 6.42 (d, J=9.2 Hz, 1H), 6.63 (dd, J=2.4/9.0 Hz, 1H), 7.03 (d, J=2.4 Hz, 1H), 7.63 (m, 4H); LCMS (ESI) (method 2) tR: 2.82 min (95%, UV220, UV254), m/z: 386.2 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)-2,2-dimethylpropanoic acid (48)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (50 mg, 0.16 mmol), 2,2-dimethyl-4-oxohexanoic acid (30 mg, 0.19 mmol) in AcOH (0.5 mL) after 3 h at 80 °C, extraction into hot hexane and subsequent purification by flash chromatography in 36% yield (23 mg) as a yellow oil, which crystallized constantly upon drying at high vacuum. C22H22ClNO4, Mr = 399.87; 1H NMR (400 MHz, DMSO-d6) δ: 1.03 (s, 6H), 2.21 (s, 3H), 3.25 (s, 2H), 3.76 (s, 3H), 6.52 (d, J=9.2 Hz, 1H), 6.67 (dd, J=2.6/9.0 Hz, 1H), 7.03 (d, J=2.4 Hz, 1H), 7.64–7.68 (m, 4H); LCMS (ESI) (method 2) tR: 2.89 min (95%, UV220, UV254), m/z: 400.0 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)-2-methylpropanoic acid (49)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (50 mg, 0.16 mmol), 2-methyl-4-oxohexanoic acid (28 mg, 0.19 mmol) in AcOH (0.5 mL) after 3 h at 80 °C in 49% yield (30 mg) as a beige solid. C21H20ClNO4, Mr = 385.84; 1H NMR (400 MHz, DMSO-d6) δ: 1.01 (d, J=6.8 Hz, 3H, alpha-CH3), 2.19 (s, 3H, C3-CH3), 2.54 (sex, J=7.2 Hz, 1H, methin-H), 2.91–3.19 (m, 2H, −CH2-), 3.76 (s, 3H, −OCH3), 6.46 (d, J=9.2 Hz, 1H, C7-H), 6.64 (dd, J=2.4/9.0 Hz, 1H, C6-H), 7.02 (d, J=2.4 Hz, 1H, C4-H); LCMS (ESI) (method 2) tR: 2.75 min (>97%, UV220, UV254, MSD), m/z: 386.1 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)-N-(methylsulfonyl)propanamide (50)
According to general procedure B, the title compound was obtained from 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid 44 (40 mg, 0.11 mmol) and methansulfonamide (10 mg, 0.11 mmol) after 20 h at room temperature. The crude product was purified by flash chromatography (SiO2, EtOAc:Hexane gradient = 1+1 (0.5% AcOH). Yield: 32 mg (66%), a bright yellow solid: C21H21ClN2O5S, Mr = 448.92; 1H NMR (400 MHz, DMSO-d6) δ: 2.21 (s, 3H), 2.55 (t, J=7.2 Hz, 2H), 3.11 (t, J=7.2 Hz, 2H), 3.16 (s, 3H), 3.75 (s, 3H), 6.43 (d, J=9.2 Hz, 1H), 6.64 (dd, J=9.0 Hz, 1H), 7.02 (d, J=2.4 Hz, 1H), 7.64–7.69 (m, 4H); LCMS (ESI) (method 2) tR: 2.74 min (>96%, UV254, ELSD), m/z: 249.1 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)-N-tosylpropanamide (53)
According to general procedure B, the title compound was obtained from 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid 44 (30 mg, 0.081 mmol) and 4-methylbenzenesulfonamide (15.2 mg, 0.089 mmol) after 20 h at room temperature. The crude product was purified by automated mass-directed HPLC (RP-18, ACN/H2O gradient). Yield: 23.1 mg (55%). C27H25ClN2O5S, Mr = 525.02; 1H NMR (400 MHz, DMSO-d6) δ: 2.07 (s, 3H), 2.35 (s, 3H), 2.49 (t, J=7.2 Hz, 2H, partially overlayed by solvent signal), 2.99 (t, J=7.2 Hz, 2H), 3.76 (s, 3H), 6.37 (d, J=9.2 Hz, 1H), 6.63 (dd, J=2.6/9.0 Hz, 1H), 6.96 (d, J=2.4 Hz, 1H), 7.31 (d, J=8.0 Hz, 2H), 7.57–7.64 (m, 4H), 7.69 (d, J=8.4 Hz, 2H); LCMS (ESI) (method 2) tR: 3.09 min (95%, UV254, ELSD), m/z: 525.1 [M+H]+.
3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)-N-((5-chlorothiophen-2-yl)sulfonyl)-propanamide (58)
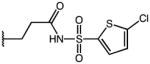
According to general procedure B, the title compound was obtained from 3-(1-(4-chlorobenzoyl)-5-methoxy-3-methyl-1H-indol-2-yl)propanoic acid 44 (30 mg, 0.081 mmol) and 5-chlorothiophene-2-sulfonamide (17.5 mg, 0.089 mmol) after 20 h at room temperature. The crude product was purified by automated mass-directed HPLC (RP-18, ACN/H2O gradient). Yield: 12.3 mg (28%). C24H20Cl2N2O5S2, Mr = 551.46; 1H NMR (400 MHz, DMSO-d6) δ: 2.10 (s, 3H), 2.54 (t, J=7.2 Hz, 2H), 3.06 (t, J=7.2 Hz, 2H), 3.76 (s, 3H), 6.40 (d, J=9.2 hz, 1H), 6.63 (dd, J=2.6/9.0 Hz, 1H), 6.96 (d, J=2.4 Hz, 1H), 7.21 (d, J=4.0 Hz, 1H), 7.60 (d, J=4.0 Hz, 1H), 7.64 (pseudo-s, 4H); LCMS (ESI) (method 2) tR: 3.19 min (99%, UV254, ELSD), m/z: 551.0 [M+H]+.
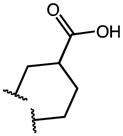
9-(4-chlorobenzoyl)-6-methoxy-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylic acid (61)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (50 mg, 0.16 mmol), 4-oxocyclohexanecarboxylic acid (27.2 mg, 0.19 mmol) in AcOH (0.5 mL) after 3 h at 80 °C in 83% yield (51 mg) as an off-white solid. C21H18ClNO4, Mr = 383.82; 1H NMR (400 MHz, DMSO-d6) d: 1.66–1.76 (m, 1H), 2.02–2.08 (m, 1H), 2.52 (m, 2H, partially overlaid by DMSO signal), 2.66–2.75 (m, 2H), 2.87–2.95 (m, 1H, methin-H), 3.77 (s, 3H, −OCH3), 6.74 (dd, J=2.6/9.0 Hz, 1H), 7.02 (d, J=2.8 Hz, 1H), 7.10 (d, J=8.8 Hz, 1H), 7.61–7.67 (m, 4H, ar-H 4-Clbenzoyl); LCMS (ESI) (method 2) tR: 2.71 min (>99%, ELSD), m/z: 384.1 [M+H]+.
9-(4-chlorobenzoyl)-6-methoxy-N-(methylsulfonyl)-2,3,4,9-tetrahydro-1H-carbazole-3-carboxamide (63)
According to general procedure B, the title compound was obtained from 9-(4-chlorobenzoyl)-6-methoxy-2,3,4,9-tetrahydro-1H-carbazole-3-carboxylic acid 61 (20 mg, 0.052 mmol) and methansulfonamide (5 mg, 0.052 mmol) after 20 h at room temperature. The crude product was purified by flash chromatography (SiO2, ethyl acetate/hexane gradient). Yield: 19 mg (79%), a white solid: C22H21ClN2O5S, Mr = 460.93; 1H NMR (400 MHz, DMSO-d6) δ: 1.63–1.73 (m, 1H), 2.03–2.06 (m, 1H), 2.51–2.57 (m, 2H, partially overlayed by DMSO-signal), 2.65–2.76 (m, 2H), 2.88–2.91 (m, 1H), 3.25 (s, 3H, −SO2-CH3), 3.77 (s, 3H, −OCH3), 6.74 (dd, J=2.6/9.0 Hz, 1H), 7.03 (d, J=2.8 Hz, 1H), 7.09 (d, J=8.8 Hz, 1H), 7.61–7.68 (m, 4H); LCMS (ESI) (method 2) tR: 2.65 min (>99%, UV220, UV254), m/z: 461.0 [M+H]+.
9-(4-chlorobenzoyl)-6-methoxy-2,3,4,9-tetrahydro-1H-carbazole-2-carboxylic acid (64)
According to general procedure A, the title compound was obtained from 4-chloro-N-(4-methoxyphenyl)benzohydrazide hydrochloride (50 mg, 0.16 mmol), 3-oxocyclohexanecarboxylic acid (27.2 mg, 0.19 mmol) in AcOH (0.5 mL) after 3 h at 80 °C in 82% yield (50 mg) as an off-white solid. C21H18ClNO4, Mr = 383.82; 1H NMR (400 MHz, DMSO-d6) δ: 1.79–1.87 (m, 1H), 2.11–2.14 (m, 1H), 2.57–2.75 (m, 5H), 6.73 (dd, J=2.8/9.0 Hz, 1H), 6.97 (d, J=2.8 Hz, 1H), 7.06 (d, J=9.2 Hz, 1H), 7.62–7.69 (m, 4H); LCMS (ESI) (method 2) tR: 2.82 min (>99%, ELSD), m/z: 384.0 [M+H]+.
Enzyme purification
Homogenous recombinant enzymes (AKR1C-4, COX 1 and COX 2) used in this study were prepared and purified as previously described.16, 17,60–62
AKR1C Isoforms
The potency of the compounds on all AKR enzymes was determined by their ability to inhibit the NADP+ dependent oxidation of S-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) catalyzed by AKR1C isoforms, as previously described.20, 21 The concentration of S-tetralol used in the assays for each AKR1C enzyme was equal to the Km value for the respective enzyme so that the IC50 values can be directly compared.
COX 1 and 2
The ability of compounds to inhibit COX activity was determined by two different test systems: a continuous colorimetric assay that monitored the oxidation of N, N, N, N-tetramethyl-1,4-phenylenediamine (TMPD) when it is coupled to the formation of PGH2 from PGG2 using arachidonic acid (AA) as substrate, and a discontinuous 14C-AA TLC-based COX assay, where samples were taken, the reaction stopped and the concentration of radioactive substrates/products determined on a plate reader. Both assays have been described earlier in literature.20, 44
IC50 Determination (except for the discontinuous COX assay, see above)
Initial velocities of an enzyme reaction measured in the presence of varying concentrations of inhibitor were compared to a solvent control to give percent inhibition values. IC50 values of compounds was determined by fitting the inhibition data using Grafit 5.0 software[y _ (range)/[1 _ (I/IC50)S] _ background] and obtained from a single experiment with each inhibitor concentration run in quadruplicate with the exception of COX assays that were conducted in duplicate. Substrate, cofactor and enzyme were titrated for each experiment. Selectivity of a compound for AKR1C3 relative to another tested enzyme is defined as the ratio of the IC50 values for the tested enzyme: IC50 value for AKR1C3; the higher the ratio, the greater the selectivity for AKR1C3.
Androgen receptor Reporter Gene Assay
HeLa cells that are stably transfected with the AR and a luciferase reporter gene (HeLa-AR3A-PSA-(ARE)4-Luc13 cells- a kind gift from Dr. Elizabeth Wilson) were used for this assay. These cells are plated in a 96 -well plate at 1.5 × 104 cell/100Xl/well in phenol red free MEM supplemented with 5% charcoal stripped FBS, 1% Pen/Strep, 2mM L-Glutamine, geneticin (500 g/ml) and hygromycin (200 g/ml) (CSS media) to maintain selection. The cells are incubated for 6–8 h after which time the media is aspirated and fresh CSS media containing 0.1 nM -100 nM DHT in the presence and absence of 10 μM lead compound was added to the cells. The cells were then incubated for 20 h at 37 °C and 5% CO2 after which luciferase expression in the cells is measured using the BrightGlo luciferase assay kit according to the manufacturer’s instructions. Briefly, after the incubation time, the cells were allowed to equilibrate to room temperature. The luciferase substrate was then added to the cells and the luminescence in each well was measured on a BIOTEK Synergy 2 plate reader. The maximal luciferase activity was seen with 100 nM DHT.
LNCaP-AKR1C3 Mediated Formation of Testosterone
Androgen dependent prostate cancer cells (LNCaP) were genetically engineered to stably express AKR1C3 (LNCaP-AKR1C3).46 The cell line was used to determine the ability of lead compounds to block the conversion of Δ4-AD to testosterone. LNCaP-AKR1C3 cells were plated in RPMI-1640 supplemented with 5% charcoal stripped serum, 1% Pen/strep, 2 mM L-glutamine and 500 μg/ml G418 (CSS media). After 24 h incubation, the media was aspirated and fresh CSS media containing 0.1 μM [14C]-Δ4-AD with or without inhibitor was added. The media was collected after 48 h treatment and analyzed for testosterone levels. Briefly, the collected media was extracted with ethyl acetate. The aqueous fraction was then treated with β-glucuronidase (200 units/ml at pH 6.6) for 24 h at 37 °C to liberate conjugated testosterone and extracted with ethyl acetate. The organic extracts were pooled and dried under vacuo. Steroid reference standards and extracts were dissolved in 100 Xl ethyl acetate were applied to LK6D Silica TLC plates (Whatman Inc., Clifton, NJ). TLC plates were developed using a dichloromethane/ethyl acetate (80:20 v/v) solution and were counted with a Bioscan System 200 plate reader (Washington, DC).
Crystallography
Crystals of the AKR1C3•NADP+•20 complex were obtained using the hanging drop vapor diffusion method at 4 °C. Drops typically contained 3.0 μL of protein solution (9.3 mg/mL AKR1C3, 10 mM potassium phosphate (pH 7.0), 1.0 mM BME, 1.0 mM EDTA, 7% (v/v) DMSO, 1.33 mM 20, and 2.0 mM NADP+) and 3.0 μL of reservoir solution (0.1 M 2-(Nmorpholino) ethanesulfonic acid (pH 6.0), 15% (w/v) polyethylene glycol 8000). Crystals appeared and grew to a suitable size for diffraction in approximately 3 days. Prior to flash-cooling, crystals were soaked for 5 min in a cryoprotective solution containing 16% (w/v) Jeffamine ED-2001. Crystals yielded diffraction data to 1.80 A at beamline X29 (λ = 0.9795 A) of the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY). Crystals belong to space group P1 with unit cell parameters a = 47.35 A, b = 49.09 A, c = 83.44 A, α = 74.30°, β = 87.04°, and γ = 69.95°. The unit cell contains two monomers of AKR1C3. Data were integrated and scaled with HKL2000 and Scalepack 63. Data collection and reduction statistics are reported in Table 7.
The structure of the AKR1C3•NADP+•20 complex was solved by molecular replacement performed with PHASER 64 from the CCP4 suite of programs using the coordinates of the AKR1C3•NADP+•indomethacin complex (PDB: 1S2A) 49 less ligand and solvent molecules as a search probe. The programs CNS 65, PHENIX 66, and COOT 67 were used for refinement and model fitting. NADP+ and 20 were built into the electron density map at the final stage of refinement. The correct modeling of the ligands was confirmed by simulated annealing omit maps. The quality of the model was verified with Molprobity and PROCHECK. The refinement statistics are reported in Table 7.
Supplementary Material
Acknowledgments
Supported by grant R01-CA89450 from the National Institutes of Health (NIH) to L.J.M, by R01-CA90744, P30-ES013508 from the NIH and a Prostate Cancer Foundation Challenge grant to T.M.P., by grant GM-056838 from the NIH to D.W.C., and by grant F32 (F32-DK089827) awarded by the NIH to M.C. A.J.L. was awarded a postdoctoral fellowship by the German Research Foundation, DFG, for a research stay in the United States (LI2019/1-1, Einzelprojekt). The X-ray crystallographic studies are based upon research conducted at beamline X25 and X29 of the National Synchrotron Light Source. Financial support for the National Synchrotron Light Source comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the NCRR of the NIH grant number P41RR012408. We thank Ms. Ling Duan for help with the metabolism studies and Mr. Kebreab Ghebreselasie for assistance with the ‘sensitive’ COX-1 and COX-2 activity screens. We are also grateful to Mr. Nathan Kett and Mr. Sichen Chang (Craig Lindsley Research Group) for the purification of compounds via the mass-directed HPLC system. The compounds disclosed in the abstract are protected by a US Provisional Patent Application No 61/548,004 filed on October 21, 2011 and converted to an International Patent Application No PCT/US1242-96 on October 17, 2012.
ABBREVIATION
- AA
arachidonic acid
- Δ4-AD
4-androstene-3-17dione
- ADT
androgen deprivation therapy
- AKR
aldo-keto reductase
- AR
androgen receptor
- BME
β-mercaptoethanol
- CRPC
castrate resistant prostate cancer
- COX
cyclooxygenases (PGH2-synthase I and II)
- CRPC
castrate resistant prostate cancer
- DHEA
dehydroepiandrosterone
- DHT
5α-dihydrotestosterone
- DMSO
dimethyl sulfoxide
- FLU
flufenamic acid
- IMN
indomethacin
- LC-MS
liquid chromatography-mass spectrometry
- N-PA
N-phenylanthranilates
- NSAIDs
non-steroidal anti-inflammatory drugs
- PSA
prostate specific antigen
- TLC
thin-layer chromatography
Footnotes
Supporting Information Available: Details of the synthesis and physicochemical characterization of reported compounds are available as Supporting Information. This material is available free of charge on the internet at http://pubs.acs.org.
References
- 1.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgenindependent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 5.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–64. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 7.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M. Hormonal impact of the 17α-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90:2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, de Bono JS. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–3748. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, Molife LR, Hunt J, Messiou C, Parker C, Dearnaley D, Swennenhuis JF, Terstappen LW, Lee G, Kheoh T, Molina A, Ryan CJ, Small E, Scher HI, de Bono JS. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–1495. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. Abiraterone increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–12. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 12.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3α-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124:940–946. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki-Yamamoto T, Nishizawa M, Fukui M, Okuda-Ashitaka E, Nakajima T, Ito S, Watanabe K. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 1999;462:335–40. doi: 10.1016/s0014-5793(99)01551-3. [DOI] [PubMed] [Google Scholar]
- 14.Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, Bunce CM. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003;63:505–512. [PubMed] [Google Scholar]
- 15.Wang T, Xu J, Yu X, Yang R, Han ZC. Peroxisome proliferator-activated receptor gamma in malignant diseases. Crit Rev Oncol Hematol. 2006;58:1–14. doi: 10.1016/j.critrevonc.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Burczynski ME, Harvey RG, Penning TM. Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry. 1998;37:6781–6790. doi: 10.1021/bi972725u. [DOI] [PubMed] [Google Scholar]
- 17.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizner TL, Lin HK, Peehl DM, Steckelbroeck S, Bauman DR, Penning TM. Human type 3 3alpha-hydroxysteroid dehydrogenase (aldo-keto reductase 1C2) and androgen metabolism in prostate cells. Endocrinology. 2003;144:2922–2932. doi: 10.1210/en.2002-0032. [DOI] [PubMed] [Google Scholar]
- 19.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 20.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, Penning TM. Development of potent and selective inhibitors of Aldo-Keto Reductase 1C3 (Type 5 17β-Hydroxysteroid Dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J Med Chem. 2012;55:2311–23. doi: 10.1021/jm201547v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, Penning TM. Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Bioorg Med Chem Lett. 2011;21:1464–8. doi: 10.1016/j.bmcl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett. 2012;22:3492–7. doi: 10.1016/j.bmcl.2012.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endo S, Matsunaga T, Kanamori A, Otsuji Y, Nagai H, Sundaram K, El-Kabbani O, Toyooka N, Ohta S, Hara A. Selective inhibition of human type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J Nat Prod. 2012;75:716–21. doi: 10.1021/np201002x. [DOI] [PubMed] [Google Scholar]
- 24.Jamieson SM, Brooke DG, Heinrich D, Atwell GJ, Silva S, Hamilton EJ, Turnbull AP, Rigoreau LJ, Trivier E, Soudy C, Samlal SS, Owen PJ, Schroeder E, Raynham T, Flanagan JU, Denny WA. 3-(3,4-Dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids: highly potent and selective inhibitors of the type 5 17β-hydroxysteroid dehydrogenase AKR1C3. J Med Chem. 2012;55:7746–58. doi: 10.1021/jm3007867. [DOI] [PubMed] [Google Scholar]
- 25.Sinreih M, Sosic I, Beranic N, Turk S, Adeniji AO, Penning TM, Rizner TL, Gobec S. N-Benzoyl anthranilic acid derivatives as selective inhibitors of aldo-keto reductase AKR1C3. Bioorg Med Chem Lett. 2012;22:5948–51. doi: 10.1016/j.bmcl.2012.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brozic P, Turk S, Adeniji AO, Konc J, Janezic D, Penning TM, Lanisnik Rizner T, Gobec S. Selective inhibitors of aldo-keto reductases AKR1C1 and AKR1C3 discovered by virtual screening of a fragment library. J Med Chem. 2012;55:7417–24. doi: 10.1021/jm300841n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 28.Pawlowski J, Huizinga M, Penning TM. Isolation and partial characterization of a full-length cDNA clone for 3 α-hydroxysteroid dehydrogenase: a potential target enzyme for nonsteroidal anti-inflammatory drugs. Agents Actions. 1991;34:289–293. doi: 10.1007/BF01993305. [DOI] [PubMed] [Google Scholar]
- 29.Penning TM, Talalay P. Inhibition of a major NAD(P)+-linked oxidoreductase from rat liver cytosol by steroidal and nonsteroidal anti-inflammatory agents and by prostaglandins. Proc Natl Acad Sci U S A. 1983;80:4504–4508. doi: 10.1073/pnas.80.14.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalgutkar AS, Crews BC, Saleh S, Prudhomme D, Marnett LJ. Indolyl esters and amides related to indomethacin are selective COX-2 inhibitors. Bioorg Med Chem. 2005;13:6810–22. doi: 10.1016/j.bmc.2005.07.073. [DOI] [PubMed] [Google Scholar]
- 33.Adeniji AO, Liedtke A, Byrns MC, Jin Y, Marnett LJ, Penning TM. Development of Potent and Selective Indomethacin Analogs for the Inhibition of AKR1C3 in Castrate-Resistant Prostate Cancer. Endocrine Reviews. 2012;33:SAT-537. [Google Scholar]
- 34.Liedtke AJ, Kim K, Stec DF, Sulikowski GA, Marnett LJ. Straightforward protocol for the efficient synthesis of varied N1-acylated (aza)indole 2-/3-alkanoic acids and esters: optimization and scale-up. Tetrahedron. 2012;68:10049–10058. doi: 10.1016/j.tet.2012.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto H. 1-Acyl-indoles. II. A new syntheses of 1-(ion-chlorobenzoyl)-5-methoxy-3-indolylacetic acid and its polymorphism. Chem Pharm Bull (Tokyo) 1968;16:17–19. doi: 10.1248/cpb.16.17. [DOI] [PubMed] [Google Scholar]
- 36.Bartolini W, Cali BM, Chen B, Chien Y-T, Currie MG, Milne GT, Pearson JP, Talley JJ, Zimmerman C. 2004–979794 20050234244, 20041101. Synthesis of acylindole COX-2 and FAAH inhibitors. 2005
- 37.Conn RSE, Douglas AW, Karady S, Corley EG, Lovell AV, Shinkai I. An unusual Fischer indole synthesis with 4-keto acids: an indole incorporating the terminal hydrazine nitrogen. J Org Chem. 1990;55:2908–13. [Google Scholar]
- 38.Kato I, Higashimoto M, Tamura O, Ishibashi H. Total Synthesis of mappicine ketone (Nothapodytine B) by means of sulfur-directed 5-exo-selective aryl radical cyclization onto enamides. J Org Chem. 2003;68:7983–7989. doi: 10.1021/jo030177m. [DOI] [PubMed] [Google Scholar]
- 39.Allen GR., Jr Selectivity in the Fischer indolization of phenylhydrazones derived from 3-ketocyclohexanecarboxylic acid. Journal of Heterocyclic Chemistry. 1970;7:239–41. [Google Scholar]
- 40.Allegretti M, Bertini R, Cesta MC, Bizzarri C, Di Bitondo R, Di Cioccio V, Galliera E, Berdini V, Topai A, Zampella G, Russo V, Di Bello N, Nano G, Nicolini L, Locati M, Fantucci P, Florio S, Colotta F. 2-Arylpropionic CXC Chemokine Receptor 1 (CXCR1) Ligands as Novel Noncompetitive CXCL8 Inhibitors. J Med Chem. 2005;48:4312–4331. doi: 10.1021/jm049082i. [DOI] [PubMed] [Google Scholar]
- 41.Bikzhanova GA, Toulokhonova IS, Gately S, West R. Novel silicon-containing drugs derived from the indomethacin scaffold: Synthesis, characterization and evaluation of biological activity. Silicon Chemistry. 2007;3:209–217. [Google Scholar]
- 42.Halder S, Satyam A. Accidental discovery of a ‘longer-range’ vinylogous Pummerer-type lactonization: Formation of sulindac sulfide lactone from sulindac. Tetrahedron Letters. 2011;52:1179–1182. [Google Scholar]
- 43.Riendeau D, Percival MD, Boyce S, Brideau C, Charleson S, Cromlish W, Ethier D, Evans J, Falgueyret JP, Ford-Hutchinson AW, Gordon R, Greig G, Gresser M, Guay J, Kargman S, Leger S, Mancini JA, O’Neill G, Ouellet M, Rodger IW, Therien M, Wang Z, Webb JK, Wong E, Chan CC, et al. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br J Pharmacol. 1997;121:105–17. doi: 10.1038/sj.bjp.0701076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liedtke AJ, Crews BC, Daniel CM, Blobaum AL, Kingsley PJ, Ghebreselasie K, Marnett LJ. Cyclooxygenase-1-selective inhibitors based on the (E)-2′-des-methyl-sulindac sulfide scaffold. J Med Chem. 2012;55:2287–2300. doi: 10.1021/jm201528b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Felts AS, Ji C, Stafford JB, Crews BC, Kingsley PJ, Rouzer CA, Washington MK, Subbaramaiah K, Siegel BS, Young SM, Dannenberg AJ, Marnett LJ. Desmethyl derivatives of indomethacin and sulindac as probes for cyclooxygenase-dependent biology. ACS Chem Biol. 2007;2:479–483. doi: 10.1021/cb700077z. [DOI] [PubMed] [Google Scholar]
- 46.Byrns MC, Mindnich R, Duan L, Penning TM. Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5α-reductase inhibitor finasteride. J Steroid Biochem Mol Biol. 2012;130:7–15. doi: 10.1016/j.jsbmb.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen M, Adeniji AO, Twenter BM, Liedtke AJ, Winkler JD, Marnett LJ, Christianson DW, Penning TM. Crystal structures of human 17β-hydroxysteroid dehydrogenase Type 5 (AKR1C3) in complex with N-phenylanthranilic acid and indomethacin-based selective inhibitors. Endocrine Reviews. 2012;33:SAT-536. [Google Scholar]
- 48.Komoto J, Yamada T, Watanabe K, Takusagawa F. Crystal structure of human prostaglandin F synthase (AKR1C3) Biochemistry. 2004;43:2188–2198. doi: 10.1021/bi036046x. [DOI] [PubMed] [Google Scholar]
- 49.Lovering AL, Ride JP, Bunce CM, Desmond JC, Cummings SM, White SA. Crystal structures of prostaglandin D2 11-ketoreductase (AKR1C3) in complex with the nonsteroidal anti-inflammatory drugs flufenamic acid and indomethacin. Cancer Res. 2004;64:1802–1810. doi: 10.1158/0008-5472.can-03-2847. [DOI] [PubMed] [Google Scholar]
- 50.Qiu W, Zhou M, Labrie F, Lin SX. Crystal structures of the multispecific 17β-hydroxysteroid dehydrogenase type 5: critical androgen regulation in human peripheral tissues. Molecular Endocrinology. 2004;18:1798–1807. doi: 10.1210/me.2004-0032. [DOI] [PubMed] [Google Scholar]
- 51.Yamada T, Watanabe K, Takusagawa F. Crystal Structure of Human Prostaglandin F Synthase (AKR1C3) Biochemistry. 2004;43:2188–2198. doi: 10.1021/bi036046x. [DOI] [PubMed] [Google Scholar]
- 52.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. Journal of Steroid Biochemistry and Molecular Biology. 2011;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laskowski R, MacArthur A, Moss D, Thornton J. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
- 54.Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Molecular basis of the time-dependent inhibition of cyclooxygenases by indomethacin. Biochemistry. 2004;43:15439–45. doi: 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- 55.Flanagan JU, Yosaatmadja Y, Teague RM, Chai MZ, Turnbull AP, Squire CJ. Crystal structures of three classes of non-steroidal anti-inflammatory drugs in complex with aldo-keto reductase 1C3. PLoS ONE. 2012;7:e43965. doi: 10.1371/journal.pone.0043965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. Journal of Computational Chemistry. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Penning TM, Ma H, Jez JM. Engineering steroid hormone specificity into aldo-keto reductases. Chem Biol Interact. 2001;130–132:659–671. doi: 10.1016/s0009-2797(00)00257-x. [DOI] [PubMed] [Google Scholar]
- 58.Jin Y. Activities of aldo-keto reductase 1 enzymes on two inhaled corticosteroids: implications for the pharmacological effects of inhaled corticosteroids. Chem Biol Interact. 2011;191:234–238. doi: 10.1016/j.cbi.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C. Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries. Journal of Combinatorial Chemistry. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 60.Quinn AM, Harvey RG, Penning TM. Oxidation of PAH trans-dihydrodiols by human aldo-keto reductase AKR1B10. Chem Res Toxicol. 2008;21:2207–2215. doi: 10.1021/tx8002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.George HJ, Van Dyk DE, Straney RA, Trzaskos JM, Copeland RA. Expression purification and characterization of recombinant human inducible prostaglandin G/H synthase from baculovirus-infected insect cells. Protein Expr Purif. 1996;7:19–26. doi: 10.1006/prep.1996.0003. [DOI] [PubMed] [Google Scholar]
- 62.Smith T, Leipprandt J, DeWitt D. Purification and characterization of the human recombinant histidine-tagged prostaglandin endoperoxide H synthases-1 and -2. Arch Biochem Biophys. 2000;375:195–200. doi: 10.1006/abbi.1999.1659. [DOI] [PubMed] [Google Scholar]
- 63.Otwinowski Z, Minor M. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 64.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software system for macromolecular structure determination. Acta Cryst. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 66.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.