

Abstract
Retrograde synaptic signaling by endogenous cannabinoids (endocannabinoids) is a recently discovered form of neuromodulation in the brain. In the basolateral amygdala (BLA), endocannabinoid signaling has been implicated in learning and memory, specifically in extinction of aversive memories. To examine retrograde endocannabinoid signaling in this brain region, BLA neurons were freshly isolated using an enzyme-free procedure. These isolated neurons retain attached functional excitatory and inhibitory synaptic boutons. Spontaneous GABAergic IPSCs (sIPSCs) were isolated from these freshly isolated neurons and a 4 s step of depolarization from –60 to 0 mV produced suppression of sIPSC frequency and amplitude. A similar depolarization-induced suppression of inhibition (DSI) was observed in neurons in BLA slices. DSI in the single-cell preparation was abolished by the CB1 receptor antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide, and DSI duration was shortened in the presence of 2-methyl-6-(phenylethynyl) pyridine, an mGluR5 (metabotropic glutamate receptor 5) antagonist. The initial decrease in sIPSCs induced by the DSI procedure was greatly attenuated in recordings with 20 mM BAPTA containing postsynaptic internal solution, but a delayed-onset decrease was observed under this recording condition. A CB1 agonist decreased sIPSC frequency and amplitude, whereas CB1 antagonists increased these responses. The antagonist-induced increase was abolished in 20 mM BAPTA-filled cells. These data provide solid evidence for retrograde endocannabinoid signaling in the BLA and also indicate that this retrograde signaling requires only a postsynaptic neuron and attached synaptic boutons.
Keywords: mGluR5, GABAergic transmission, CB1 receptors, depolarization-induced suppression of inhibition, IPSC, basolateral amygdala
Introduction
Cannabinoids are the psychoactive ingredients of preparations derived from Cannabis sativa. These compounds alter a variety of brain functions mainly via activation of the CB1 cannabinoid receptor (Matsuda et al., 1990; Freund et al., 2003). The endogenous agonists for this receptor, the so-called endocannabinoids, participate in recently discovered, important neuromodulatory mechanisms. Endocannabinoid release from postsynaptic neurons functions as a rapid, retrograde signal that activates presynaptic CB1 cannabinoid receptors and inhibits transmitter release (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). For example, depolarization of hippocampal CA1 pyramidal neurons produces endocannabinoid-mediated, depolarization-induced suppression of inhibition (DSI) at GABAergic synapses (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). DSI may serve to facilitate memory and synaptic plasticity (Carlson et al., 2002).
The basolateral amygdala (BLA) is a brain region with critical roles in emotional control and fear conditioning (Davis et al., 1994). Endocannabinoid signaling in BLA has been implicated in the extinction of aversive memories (Marsicano et al., 2002). Furthermore, endocannabinoids and CB1 receptors play a critical role in long-term synaptic depression of GABAergic synapses in BLA and glutamatergic synapses in lateral amygdala (Marsicano et al., 2002; Huang et al., 2003). However, many aspects of endocannabinoid signaling in this brain region, including the presence or absence of DSI and mechanisms controlling endocannabinoid signaling, remain to be examined. To explore these issues under well controlled experimental conditions, we took advantage of a highly reduced preparation consisting of an isolated postsynaptic neuron from BLA and attached GABAergic presynaptic terminals to demonstrate DSI and examine the time course of presynaptic cannabinoid modulation.
Materials and Methods
Brain slices (400 μm thickness) containing the amygdala were prepared from Sprague Dawley rats [postnatal day 14 (P14)–P17]. Sections were cut with a Vibratome in cold buffer oxygenated with 95% O2/5% CO2. The composition of the slicing buffer was the following (in mM): 124 NaCl, 3 KCl, 1.3 Mg2SO4, 2 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 10 glucose. Slices were maintained at room temperature for at least 1 h before the dissociation.
Mechanical dissociation. Single BLA neurons were isolated from BLA slices using an enzyme-free mechanical dissociation procedure, as described by Akaike and Moorhouse (2003). Briefly, BLA slices were transferred to a 35 mm dish (coated with poly-d-lysine) with an external recording buffer containing (in mM) 140 NaCl, 5 KCl, 10 HEPES, 2 MgCl2, 2 CaCl2, 10 glucose, 0.005 2,3-dihydroxy-6-nitro-7-sulfonylbenzo[f] quinoxaline (NBQX), and 0.025 dl-2-amino-5-phosphonopentanoic acid (AP-5), with pH adjusted to 7.4 using NaOH and osmolarity of ∼300 mOsm. A fire-polished glass micropipette was placed on the surface of the BLA. The tip of the pipette was vibrated horizontally at 6 — 8 Hz for ∼2 min. The isolated neurons were allowed to settle to the bottom of the dish for 10 —15 min. Neurons were placed on the stage of an inverted microscope for visualization and performance of electrophysiological recordings. For the experiments examining the effects of CB1 agonists and antagonists on action potential-independent IPSCs, tetrodotoxin (TTX) was added during mechanical dissociation of BLA neurons and remained in the perfusion solution throughout the experiment.
Electrophysiology. Whole-cell patch-clamp recordings were made at room temperature using conventional patch-clamp techniques and an Axopatch 200B amplifier (Molecular Devices, Union City, CA). Neurons were held at –60 or –90 mV during voltage-clamp recordings. The extracellular recording solution was the same as that used during neuronal isolation. Patch pipettes had resistances of 4 — 6 MΩ after filling with a solution containing (in mM) 140 CsCl, 10 HEPES, 0.2 or 20 BAPTA, 2.0 MgCl2, 2.0 Mg-ATP, 0.3 GTP, 5 5-N-(2,6-dimethyl-phenylcarbamoylmethyl)-triethylammonium bromide, pH 7.20 —7.25, with CsOH and 295 mOsm. In a few neurons, we examined input resistance using a solution in which CsCl was replaced by KMeSO4. We also examined glutamatergic transmission in a small subset of cells using this KMeSO4-based internal solution. An array of extracellular superfusion pipettes was placed ∼100 μm from the recorded neuron. External solution change was achieved by rapid lateral movement of three-port square glass pipettes driven by a Warner Instruments (Hamden, CT) Perfusion Fast-Step system (solution exchange time in whole-cell mode averaged ∼200 ms). In experiments examining GABA-activated current, we applied an approximately EC50 concentration of GABA (5 μM) so that we could observe either increases or decreases in current amplitude. This concentration was determined from concentration–response curves generated in the mechanically dissociated BLA pyramidal neurons (data not shown). The [K+]o was increased to 9 mM only during selected DSI tests and remained at 5 mM in other experiments. Spontaneous synaptic currents were initially detected using Mini Analysis software (Synaptosoft, Decatur, GA) with a threshold criterion of 20 pA, and smaller events (down to 10 pA) were manually detected during visual inspection of traces.
In the majority of experiments in isolated neurons, 100 s traces were sampled for DSI analysis, and a 4 s depolarization pulse (from –60 to 0 mV) was inserted after 20 s of sampling. For brain slice experiments, whole-cell recordings were made at room temperature using the same buffer as that used for slice preparation. Monosynaptic GABAergic currents [evoked IPSCs (eIPSCs)] were evoked with bipolar electrodes (Teflon-coated platinum/iridium wire, 75 μm in diameter) placed in the BLA in the presence of NBQX (5 μM) and AP-5 (25 μM). DSI was tested every 4 min and consisted of 33 stimuli at 0.33 Hz with depolarization from –60 to 0 mV for 4 s inserted after the seventh stimulus. For DSI analysis, the means of the five eIPSCs evoked just before depolarization and the three eIPSCs just after the depolarization were used as ampbaseline and amptest, respectively. DSI magnitude (%) = 100 (1 – (amptest/ampbaseline). Analysis of spontaneous IPSCs (sIPSCs) in brain slice experiments was performed as described for experiments on isolated neurons.
Data were filtered at 2 kHz, digitized at 5 kHz using pClamp8 software through a Digidata 1200 interface (Molecular Devices), and plotted with SigmaPlot (SPSS, Chicago, IL). BAPTA, bicuculline, AP-5, GABA, and TTX were purchased from Sigma (St. Louis, MO). N-piperidino-5-(4-chloro-phenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole carboxamide (SR141716) was from the National Institute of Mental Health chemical synthesis program. Cyclothiazide, 2-methyl-6-(phenylethynyl) pyridine (MPEP), NBQX, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1 H-pyrazole-3-carboxamide (AM251), and R-(+)-(2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrol[1,2,3-de]-1,4-benzoxazin-6-yl)(1-naphthalenyl) methanone monomethanesulfonate (WIN55212-2; WIN) were obtained from Tocris Cookson (Ellisville, MO). Averaged data are expressed as mean ± SE; data significance was analyzed by t test unless indicated otherwise. The criterion for statistical significance was p < 0.05.
Results
Freshly mechanically isolated neurons from BLA retain attached functional GABAergic synaptic boutons (Fig. 1A) Drewe et al., 1988; Akaike and Moorhouse, 2003). The postsynaptic elements of neurons isolated in this manner consist of a soma and processes of either short or medium (Fig. 1A) length. When whole-cell patch recordings were made with a KMeSO4-based internal solution, we found that resting potential was –50 ± 2 mV (n = 7), and the input resistance in these neurons was 1.0 ± 0.03 GΩ (n = 12). Injection of positive current produced a pattern of action potential firing, as described by Washburn and Moises (1992), and injection of a negative current led to a small amplitude “sag” during hyperpolarization (Fig. 1B). In the present study, the majority of whole-cell patch recordings were made with a CsCl-based internal solution, and neurons examined under this condition also showed a high input resistance (936 ± 105 MΩ; n = 6). We observed evidence that both GABAergic and glutamatergic boutons remain attached to these isolated neurons. Figure 1C shows spontaneous inward postsynaptic currents recorded using a KMeSO4-based internal solution in the presence of 20 μM bicuculline and 50 μM cyclothiazide. Similar currents observed in three neurons were abolished by AP-5/NBQX, suggesting that they are EPSCs generated by glutamate release from excitatory presynaptic terminals.
Figure 1.
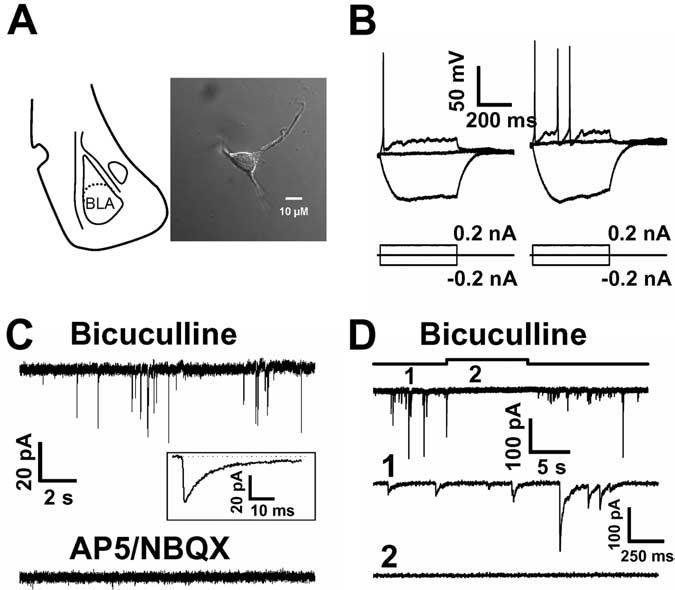
Excitability and synaptic responses in freshly mechanically isolated BLA neurons. A, Schematic representation of the location of the BLA in a coronal brain slice (left). A typical freshly mechanically isolated BLA neuron is shown on the right [image acquired using differential interference contrast optics on an Olympus (Tokyo, Japan) IX-71 inverted microscope, with a 60× 1.45 numerical aperture objective, a Qimaging (Barnaby, British Columbia, Canada) Retiga EXi 1394 camera, and Improvision (Lexington, MA) Openlab software]. B, Top, Traces recorded with KMeSO4-based internal solution; injection of positive current produced action potential firing, whereas negative current injection led to a hyperpolarization sag. C, Top, Spontaneous inward currents, recorded with KMeSO4-based internal solution in the presence of bicuculline, were abolished by AP-5/NBQX (bottom). C, Inset, Single sEPSC trace (average of 75 events).All traces were recorded in the presence of 50μM cyclothiazide. D, Traces recorded with CsCl-based internal solution from an isolated BLA neuron, illustrating spontaneous GABAergic currents that are reversibly suppressed by the GABAA antagonist bicuculline (20 μM). D, Expanded traces recorded before and during bicuculline application are shown in the middle and bottom traces.
Spontaneous GABAergic IPSCs were isolated in the presence of NBQX/AP-5 (5 μM/25 μM) (Fig. 1D). A total of 90 neurons were examined, with recordings that lasted for up to 1 h. The mean sIPSC frequency in a given neuron varied from 40 to 480 events per min (mean for all cells = 127 ± 15). The number of sIPSCs per minute decreased from 118 ± 21 to 17 ± 5 (n = 9) after the addition of 1 μM TTX, indicating that events recorded under our standard recording conditions are mainly sIPSCs driven by action potentials. Application of 20 μM bicuculline produced fast, reversible inhibition of the sIPSCs (Fig. 1D), demonstrating that they are mediated by GABA release and activation of postsynaptic GABAA receptors.
If endocannabinoids function as rapid retrograde signaling molecules in this simple neuron/bouton preparation, we should be able to elicit DSI (Pitler and Alger, 1992; Vincent and Marty, 1993). To examine DSI under conditions in which release probability was high, we increased external [K+] to 9 mM. Under the high [K+]o condition (280 ± 33 events/min; n = 16), depolarization from –60 to 0 mV for 4 s produced a profound reduction in sIPSC frequency and amplitude (Fig. 2A, B). Comparing 20 s periods before and just after depolarization, the number of events after depolarization was decreased by 50 ± 4% from the prepulse level of 162 ± 18 events (mean ± SE; p < 0.01; t test; n = 10). Depression of sIPSC frequency was evident immediately after the offset of the depolarizing pulse, and maximal depression was at 16 s after the end of the depolarization (31 ± 8% of baseline values). The sIPSC frequency recovered to 50% of baseline levels at 46 ± 3 s after the end of depolarization and maintained this plateau level of depression (50 ± 20% of baseline) for up to 80 s (the time point at which the majority of experiments were terminated). Subsequent, longer-lasting recordings revealed that the decrease in sIPSC frequency did not fully recover to baseline levels even when examined >15 min after depolarization in the high K solution (n = 3).
Figure 2.
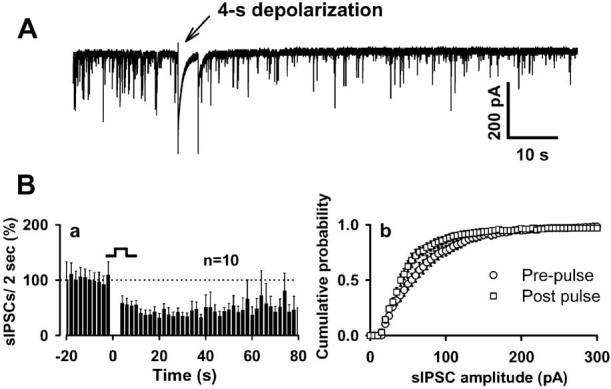
DSI in the neuron/bouton preparation. A, A 4s depolarizing step (–60 to 0 mV) produces robust reduction in sIPSC frequency. Ba, Time course of DSI from 10 neurons. sIPSC amplitude was also inhibited by depolarization, as indicated in the cumulative sIPSC amplitude distribution (Bb). Events from 20 s recording periods before and just after depolarization were pooled to create this cumulative amplitude distribution. Error bars represent SEM.
DSI in this preparation was abolished in the presence of the CB1 receptor antagonist AM251 (1 μM) (Fig. 3Aa). The rapid decrease in sIPSCs produced by the DSI protocol, which was normally evident after cessation of depolarization, was not observed in recordings with 20 mM BAPTA-containing postsynaptic internal solution (Fig. 3Ab). However, a slowly developing decrease in sIPSC frequency was retained in the BAPTA-filled cells. The latency to onset of the decrease in sIPSC frequency (defined as the occurrence of two successive bins in which frequency was significantly decreased; p < 0.05; t test) was 32 s after the end of the depolarizing pulse, and sIPSC frequency remained depressed at 70 s after the end of the depolarization (60 ± 18% of baseline; p < 0.05; t test; compared with the baseline). MPEP (10 μM), a metabotropic glutamate receptor 5 (mGluR5) antagonist, did not block DSI but shortened DSI duration (Fig. 3Ac). The time to observe the recovery to 50% of the baseline sIPSC frequency was 24 ± 3 s after depolarization in the presence of MPEP, and full recovery was first observed within ∼42 s (time at which there are two successive nonsignificant bins). sIPSC frequency was 95 ± 23% of the baseline at 70 s after depolarization (p > 0.2; t test; compared with control baseline; n = 6). To determine whether there was any “rundown” of sIPSCs during high K+ application, we applied high K+ alone for the same period as that used in the standard DSI experiment. As shown in Figure 3B, high K+ alone did not produce any clear time-dependent change in the frequency of sIPSCs. The sIPSC frequency 40 s after the beginning of the baseline period was significantly lower in the neurons given depolarization compared with neurons given high K+ alone (p < 0.01; unpaired t test). As noted above, the DSI evoked in the neuron/bouton preparation lasted at least 80 s, a duration longer than DSI observed in hippocampal slices or cultured hippocampal neurons (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). The observation that MPEP decreased the duration of DSI indicates that the longer-lasting component of depression is attributable to mGluR5 prolongation of DSI, a phenomenon that has also been observed in the hippocampus (Varma et al., 2001).
Figure 3.
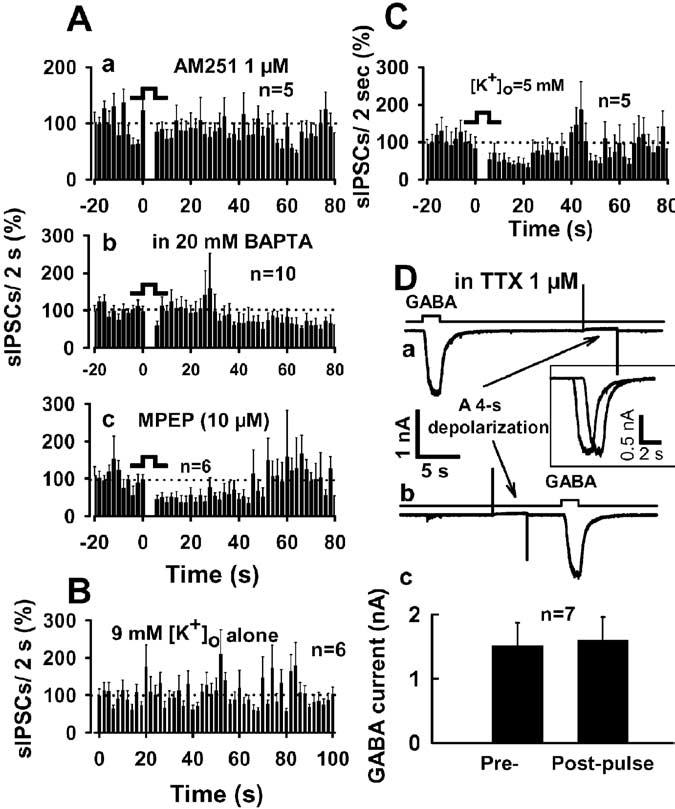
Characterization of DSI in the neuron/bouton preparation. Aa, Ab, Inhibition of sIPSC frequency by depolarization was abolished in the presence of the CB1 antagonist 1 μM AM251 (Aa) and was delayed and attenuated by a 20 mM BAPTA-containing postsynaptic internal solution(Ab). Ac, MPEP(10μM), the mGluR5 antagonist, shortened DSI duration (but did not prevent DSI) in acutely isolated BLA neurons. B, High [K +]o alone did not produce any clear time-dependent decrease in the frequency of sIPSCs. C, Short-lasting DSI was also observed in an external solution containing 5 mM [K +]o. D, Depolarizing voltage step did not affect GABA-activated currents. Da, Db, Exogenous GABA (5 μM) was applied every 4 min before (Da) or after (Db) a 4 s depolarization in the presence of TTX (1 μ). Dc, Summary of exogenous GABA-activated currents. All DSI tests were performed in high [K +]o, except C and D. Error bars represent SEM.
We also examined DSI using an external solution containing 5 mM [K+]o in those neurons in which we observed an especially high basal frequency of sIPSCs (226 ± 38 events/min; n = 5). Figure 3C shows that a 4 s depolarization caused suppression of sIPSCs. Interestingly, DSI observed under this condition was shorter in duration than that observed under the higher extracellular K+ condition (compare with Fig. 2Ba). The time to 50% recovery was 20 ± 3 s after depolarization, and full recovery was observed at ∼36 s, defined by two successive nonsignificant bins. At 70 s after depolarization, sIPSC frequency was 110 ± 25% of the baseline (p > 0.5; t test; compared with control baseline; n = 5).
It is possible that DSI could involve decreased GABAA receptor function. Use of the isolated neuron/bouton preparation allowed us to directly test this idea by applying GABA to neurons using rapid solution-exchange techniques. We applied 5 μM GABA to neurons before and 4 s after the 4 s depolarization used to induce DSI (Fig. 3Da, Db). The peak amplitude of GABA-activated current was not altered in the postdepolarization time period relative to predepolarization values. GABA produced a 1599 ± 365 pA current, and current amplitude decreased by only 2 ± 7% (p > 0.5; n = 7) after depolarization (Fig. 3Dc).
WIN55212-2, a selective CB1 agonist, decreased sIPSC frequency in the neuron/bouton preparation (Fig. 4A), confirming the presence of CB1 receptors in the neuron/bouton preparation. WIN55212-2 (500 nM) also reduced sIPSC amplitude (Fig. 4B). On average, for a period of 1 min during drug application, WIN55212-2 produced a 58 ± 3% (p < 0.01; n = 5; t test) reduction in the number of sIPSCs from the control level of 102 ± 11 events observed during the last minute before agonist application. The time course of WIN55211-2 inhibition of sIPSC frequency is shown in Fig. 4C. The rapidly reversible inhibition by WIN55212-2 illustrates a distinct advantage of this preparation over brain slices, in which it is often difficult or impossible to remove synthetic cannabinoid agonists on the time scale of a standard experiment. There was a significant increase in the frequency of sIPSC events after washing out WIN55212-2, and the peak of this “rebound” increase (52 ± 11% above pre-WIN baseline levels; p < 0.05; t test) was observed at 120 s after returning to control buffer. The sIPSC frequency subsequently returned to baseline levels within ∼50 s of the peak rebound increase.
Figure 4.
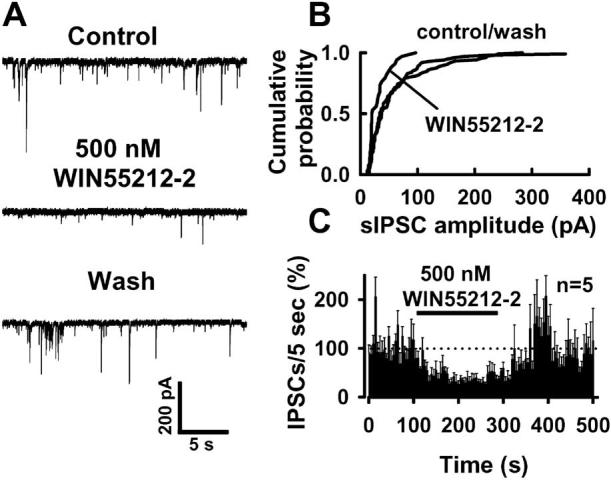
Activation of CB1 receptors inhibits spontaneous GABAergic current. A, B, WIN55212-2 (500 nM), a CB1 agonist, suppressed sIPSCs in mechanically isolated BLA neurons by increasing the interevent intervals (A) and decreasing sIPSC amplitudes (B). C, Time course of the WIN55212-2 effect on the frequency of the spontaneous GABAergic events. All tests were performed in 5 mM [K]o.
We next performed experiments in the presence of 1 μM TTX to determine whether CB1 agonist effects are dependent on pre-synaptic action potentials. On average, the number of IPSCs per minute in this experiment was 28 ± 3 (n = 12), and mean amplitude of IPSCs was 46 ± 4.7 pA (n = 12). As shown in Figure 5A, WIN55212-2 reversibly decreased the frequency of IPSCs. On average, WIN produced a 48 ± 5% inhibition of the frequency of IPSCs and a 40 ± 8% inhibition of IPSC amplitude (p < 0.01; n = 6) in the presence of TTX. We also consistently observed a postdrug rebound increase (110 ± 12% increase) in event frequency (p < 0.01; n = 6) under this condition. The time course of the post-WIN peak increase in frequency and the return to baseline levels were similar to those observed in the absence of TTX. The rise and decay times were 0.64 ± 0.1 and 20 ± 2.4 ms in control and 0.60 ± 0.1 and 18 ± 3.0 ms in the presence of WIN55212-2 (p > 0.05; n = 6). Histograms of event amplitudes (Fig. 5B, C) showed that, in the presence of WIN555212-2, the most prominent change in IPSC amplitude distribution was the reduction in events with amplitudes >100 pA, with much less change observed at the lower end of the distribution.
Figure 5.
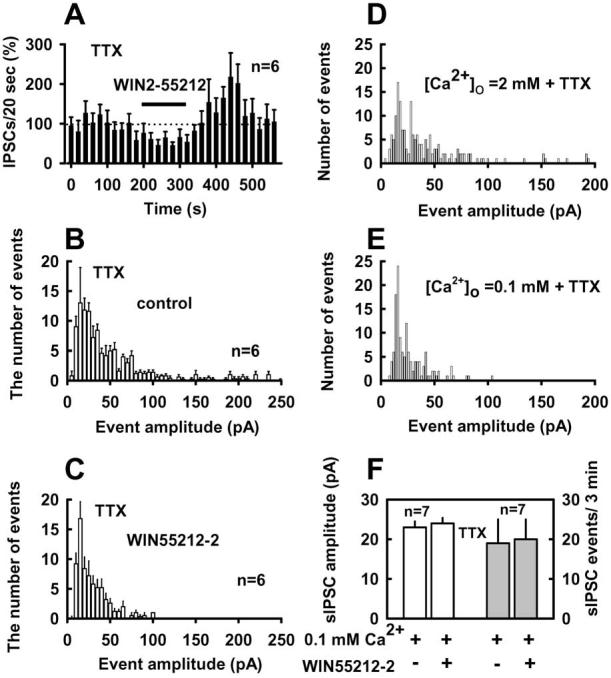
Effects of WIN55212-2 on IPSCs are calcium dependent. A, WIN55212-2 suppression of IPSCs in the present of TTX (1μM). A–C, Under this condition, activation of CB1 receptors by WIN55212-2 decreased the frequency and amplitude of action potential-independent IPSCs. GABAergic events were collected in 5 pA bins for plotting histograms of event amplitude, as shown in B and C. Note that the bin size in A has been increased to 20 s. E, [Ca2+]o (0.1 mM) plus TTX prevented WIN55212-2 effects on IPSCs. D, E, Histograms of GABAergic event amplitudes from a representative experiment; bin size is 2 pA. Recordings (3 min) were collected for [Ca2+]o = 2 mM and 10 min recordings for [Ca2+]o = 0.1 mM. F, GABAergic sIPSC frequency and amplitude were not altered by WIN55212-2 in the presence of 0.1 mM [Ca2+]o plus TTX. [K +]o was 5 mM in all tests.
To exclude the possibility that the WIN55212-2-induced decrease in event frequency is a secondary consequence of the reduction of sIPSC amplitude, we examined the effect of WIN55212-2 on IPSCs in the presence of TTX under more negative holding potential (–90 mV). This condition increases the driving force for Cl- efflux during the IPSC and thus increases average sIPSC amplitude. In five neurons tested, the number of events per minute was 30 ± 4 and 34 ± 8 at Vh = –60 and –90 mV, respectively (Δ = 12 ± 20%; p > 0.5; paired t test). Mean IPSC amplitude increased from 43 ± 4 to 63 ± 5 pA when Vh was increased from –60 to –90 mV (p < 0.01). WIN55212-2 produced a 43 ± 6% decrease in the number of IPSCs (p < 0.01) per minute and a 30 ± 6% reduction in mean IPSC amplitude (p < 0.05) at Vh = –90 mV. At Vh = –90 mV, we did not observe sIPSCs with amplitudes <10 pA in either the absence or presence of WIN55212-2. These findings indicate that the CB1 agonist does not decrease event amplitudes to levels below the threshold for detection.
The observation of large amplitude (>100 pA) IPSCs in the presence of TTX indicated that action potential blockade did not eliminate all multiquantal events. To determine whether the occurrence of these large events was attributable to calcium entry in the presence of TTX, we decreased extracellular Ca2+ to 0.1 mM. Reducing [Ca2+]o in the presence of TTX decreased the mean amplitude of IPSCs and eliminated almost all IPSCs with amplitudes >100 pA (Fig. 5D, E). In seven neurons tested, mean IPSC amplitude was 41 ± 3 pA in 2 mM [Ca2+]o and 23 ± 2 pA in 0.1 mM [Ca2+]o. Under the TTX plus reduced [Ca2+]o, WIN55212-2 had no effect on either the frequency or amplitude of IPSCs. The mean amplitude and the frequency of events averaged 23 ± 2 and 19 ± 5 pA per 3 min in the control condition and 24 ± 1 and 20 ± 5 pA per 3 min in the presence of WIN55212-2 (Fig. 5F)(p > 0.5; paired t test).
Interestingly, we noted that the application of 1 μM AM251 for 2 min caused a 118 ± 19% increase in the number of sISPCs. AM251 also increased event amplitudes (Fig. 6A, B). The average sIPSC frequency (n = 5) was reversibly increased by AM251 with a rapid onset, and this effect reversed over a 1–2 min time course (Fig. 6C). Application of SR141716 (1 μM), another CB1 antagonist, for 1.5–2 min produced a 144 ± 19% (n = 3) increase in sIPSC frequency. As shown in Figure 6D, in recordings with 20 mM BAPTA-containing postsynaptic internal solution, AM251 failed to increase sIPSC frequency. AM251 also had no significant effect on GABAergic event amplitude in these BAPTA-filled cells (Fig. 6E). Figure 6F shows the effects of AM251 on average sIPSC frequency in pooled data from six cells with 20 mM BAPTA in the recording pipette.
Figure 6.
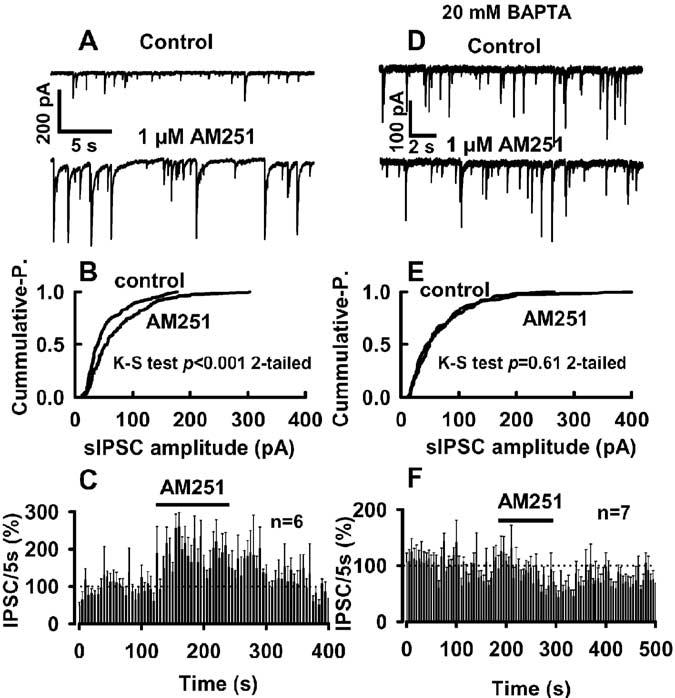
Ongoing endocannabinoid signaling in the neuron/bouton preparation. A–C, Application of 1 μM AM251 enhances spontaneous GABAergic transmission by decreasing the interevent intervals (A) and increasing sIPSC amplitudes [B; p < 0.001; Kolmogorov–Smirnov (K-S) test]. The average magnitude and time course of AM251 effects on GABAergic event frequency are shown in C(n=6). D–F, Traces(D) show that AM251 had no significant effecton GABAergic transmission in cells filled with 20 mM BAPTA. E, AM251 had also no effect on event amplitude distribution under the high buffering condition (p > 0.6; K-S test). F, The time course of the effect of AM251 on event frequency. A, B and D, E are from the same neuron, respectively. [K]o was 5 mM in all tests.
The CB1 antagonist effect on IPSCs was also TTX independent. AM251 (1 μM) produced a 344 ± 111% increase in the frequency of IPSCs and a 42 ± 14% increase in IPSC amplitude in the presence of TTX (p < 0.05; n = 5) (Fig. 7A, C). AM251 had no significant effect on the rise and decay times of IPSCs (0.80 ± 0.1 and 21 ± 1.2 ms for control and 0.80 ± 0.1 and 22 ± 1.2 ms in the presence of AM251; p > 0.05; n = 5). However, reduction of extracellular Ca2+ to 0.1 mM prevented AM251 effects on IPSCs (Fig. 7A). The mean frequency of the events was 44 ± 10 per 5 min in TTX plus reduced [Ca2+]o and 46 ± 11 per 5 min in the presence of 1 μM AM251 (p > 0.2; n = 5; paired-t test). Under TTX plus reduced [Ca2+]o, AM251 had no effect on the cumulative distributions of either event intervals or amplitudes (Fig. 7 E, F).
Figure 7.
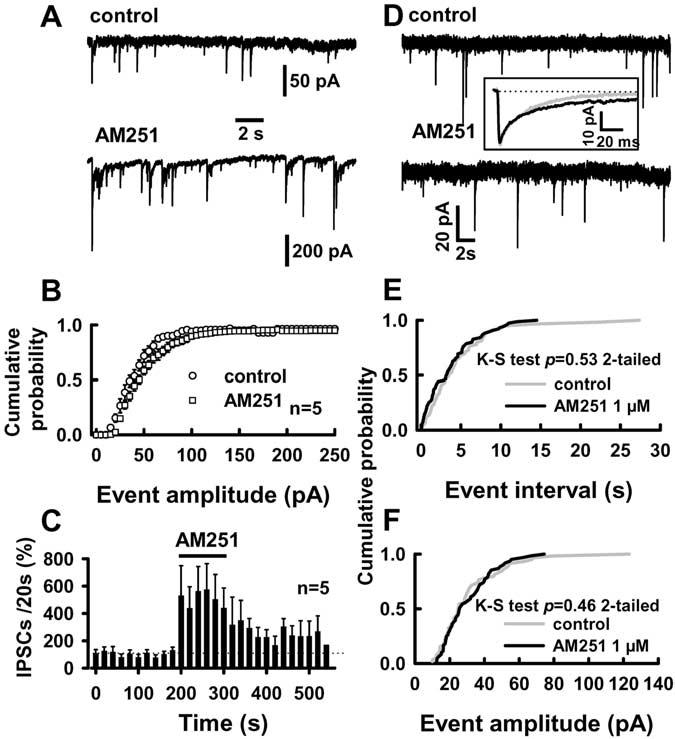
Reduced [Ca2+]o plus TTX prevents AM251 effects on IPSCs. A, AM251 effects on IPSCs in TTX alone. Traces in A are from a representative experiment. TTX (1μM) was present in all experiments. B, The events were collected in 5 pA bins for cumulative distributions. AM251 leftward shifted the distribution of event amplitudes [p < 0.05; Kolmogorov–Smirnov (K-S) test] and increased the frequency of the events (C). D–F, AM251 failed to affect IPSCs in reduced [Ca2+]o plus TTX. D, Traces from a representative experiment. Inset, Traces are averages of 59 and 61 events for control (5 min) and 1 μM AM251 (5 min), respectively. E, F, The cumulative distributions were constructed from 5 min recordings. [K +]o was 5 mM in all tests.
It is possible that the agonist- and antagonist-induced alterations in IPSC amplitude in the presence TTX are attributable to CB1-mediated alterations of postsynaptic responses to GABAA or direct effects of WIN55212-2 and AM251 on postsynaptic GABAA receptors. Thus, agonist and antagonist effects on exogenous GABA-activated current were examined. These experiments were performed using rapid superfusion of the whole cell at an approximately EC50 GABA concentration (5 μM). WIN55212-2 did not alter the peak amplitude of current activated by exogenous GABA (Fig. 8A). Peak GABA-activated current averaged 773 ± 237 pA and was decreased by only 3 ± 5% (p > 0.5; n = 7) in the presence of 500 nM WIN55212-2 (Fig. 8B). To our surprise, AM251 (1 μM) produced a 31 ± 6% reduction of the amplitude of GABA-activated current from the control level of 727 ± 146 pA (p < 0.01; n = 7) (Fig. 8A, B). This effect was opposite of the AM251 effect on sIPSC amplitude.
Figure 8.
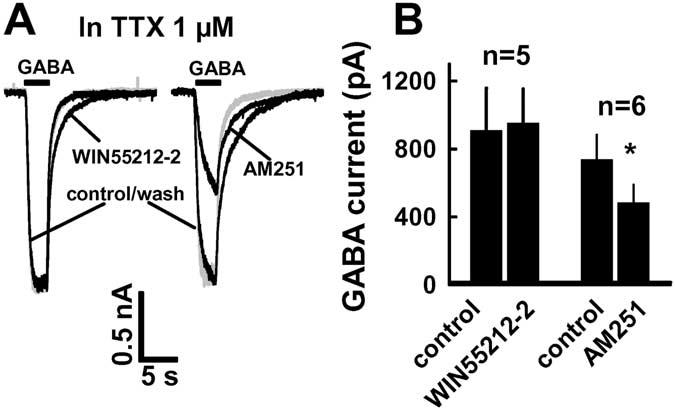
WIN55212-2 and AM251 effects on currents activated by exogenous GABA (5μM). A, Left, WIN55212-2 did not suppress GABA-activated current but slowed down recovery of GABA current after the end of GABA application; right, AM251 reversibly suppressed GABA-activated currents. B, Summaries of WIN55212-2 and AM251. *p < 0.05 (t test) compared with control. The traces in A are from different neurons. All experiments were performed in the presence of TTX and [K +]o (5 mM).
Finally, we wanted to determine whether retrograde endocannabinoid signaling seen in the dissociated neuron/bouton preparation could also be observed in neurons in the BLA slice preparation, because DSI had not been reported previously in BLA slices. DSI was examined in BLA neurons in brain slices, using an extracellular solution that contained 3 mM [K+]o. When we examined the effects of postsynaptic depolarization on sIPSCs, we chose P16 –P17 rats for recordings, because sIPSC frequency was sufficiently high (175 ± 13 events/min; n = 7) to allow evaluation before and after depolarization. A 4 s depolarization produced a 44 ± 6% decrease in sIPSC frequency (p < 0.05; n = 7) and a 34 ± 7% decrease in event amplitude (p < 0.05; n = 7) in recordings from BLA principal neurons in the slice preparation (Fig. 9A, B). The magnitude of these decreases is similar to those observed in the neuron/bouton preparation. The duration of DSI in the slices was comparable with that observed under the low [K+]o condition in the neuron/bouton preparation (sIPSC frequency returned to baseline levels at ∼30 s). We also examined the effect of a 4 s depolarization on evoked IPSCs in the BLA slice preparation. Figure 9C shows that depolarization produced a short-term suppression of eIPSCs similar to that observed in the hippocampal CA1 region (Pitler and Alger, 1992). On average, a 4 s depolarization caused a 37 ± 8% suppression of eIPSCs (p < 0.01; n = 6), which was prevented in the presence of AM251 (1 μM), a CB1 antagonist (Fig. 9C, D). Application of AM251 alone produced an increase in sIPSC frequency by 13 and 22% in two of five neurons examined. The remainder of the cells showed no change in sIPSC frequency after AM251 application.
Figure 9.
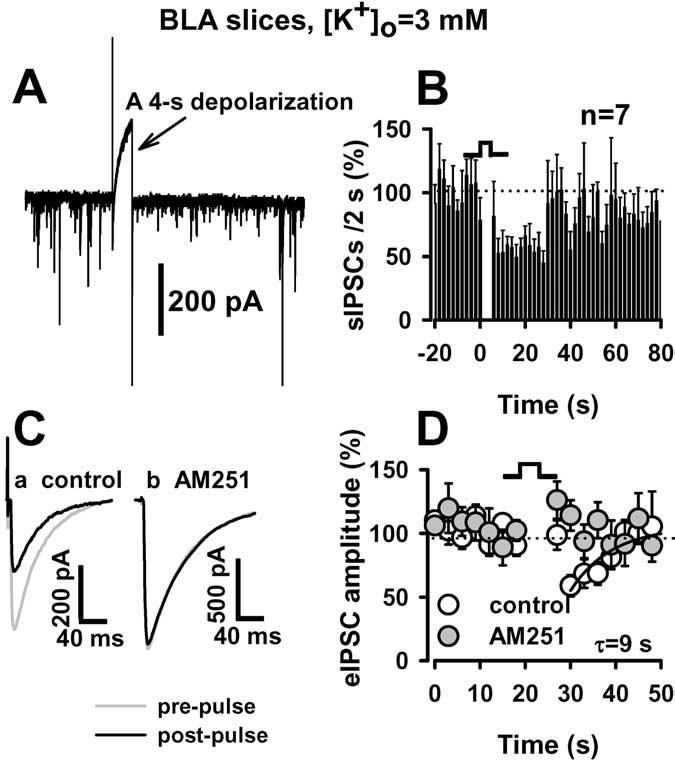
DSI in the BLA brain slice preparation. A, A 4 s depolarization led to inhibition of spontaneous GABAergic current. B, Summary of depolarization effects on sIPSC frequency. C, Evoked IPSCs were recorded from BLA neurons in brain slices. eIPSCs before (gray line) and after (dark line) depolarization are superimposed. Depolarization-induced suppression of eIPSCs was abolished in the presence of AM251 (1 μM). D, Time course of the DSI. Note that Ca and Cb are from different neurons. The time course of recovery was best fit by the single exponential function of y=y0+k×exp(-X/τ), where y is the percentage or control amplitude at a given time point, y0 is the relative amplitude at t = 0, x is a given time point, and τ is the time constant of DSI recovery. [K +]o was 3 mM in all slice experiments.
Discussion
Endocannabinoid-dependent DSI and presynaptic cannabinoid modulation were observed in a preparation consisting of a single mechanically isolated BLA neuron with attached GABAergic terminals. This retrograde signaling appears to be independent of supporting cells and other cellular elements that are found in the brain slice and neuronal culture preparations used previously for performing such experiments (Llano et al., 1991; Pitler and Alger, 1992; Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001).
The use of this simple preparation allows for examination of retrograde signaling with greatly improved pharmacological and physiological control. We can now gain precise information about the onset and duration of CB1 receptor agonist and antagonist effects. The fact that this preparation contains few postsynaptic processes minimizes space-clamp errors that could alter quantification of synaptic modulation and plasticity. The observation that CB1 agonist-induced synaptic depression was completely reversible in this preparation allowed us to accurately determine the time course of the effects of receptor activation alone and will facilitate comparison with studies of CB1 modulation of ion channels in single-cell preparations (Mackie and Hille, 1992; Guo and Ikeda, 2004). We were also able to examine GABAA receptor function by applying exogenous GABA in this preparation, which is difficult to accomplish under well controlled pharmacological conditions in brain slices. These experiments indicate that changes in GABAA receptor function do not contribute to DSI or to changes in IPSCs produced by CB1 ligands. This enhanced pharmacological control should allow us to define the magnitude and duration of effects of receptor activation alone in relation to both short-and long-lasting changes in synaptic efficacy.
Modulation of sIPSCs by WIN55212-2 was action potential independent, indicating that presynaptic firing is not necessary for this CB1 receptor-mediated effect. Thus, synaptic depression produced by WIN55212-2 is predominantly, if not exclusively, attributable to presynaptic modulation. IPSC amplitude in the presence of TTX was decreased by WIN55212-2, and this finding could be construed as evidence that the agonist has a postsynaptic action. However, the finding that IPSC amplitude was not decreased in TTX and low extra-cellular calcium suggests that this is not the case. Furthermore, the WIN55212-2 did not alter postsynaptic current activated by exogenous GABA, indicating that CB1 activation does not decrease postsynaptic GABAA receptor function. The most likely explanation for the WIN55212-2 effects on sIPSC amplitude in the presence of TTX is that events observed under this condition are mainly multiquantal, and CB1 activation reduces the amplitude of large, presumably multiquantal IPSCs under this condition. To obtain single quantal mIPSCs, it is necessary to decrease presynaptic calcium influx both in the neuron/bouton and slice preparations (Katona et al., 2001). This observation is in agreement with previous evidence that multiquantal event frequency increases with increasing release probability (Tong and Jahr, 1994). We do not yet know whether these events arise from the release of multiple quanta per synaptic bouton or simultaneous release from multiple boutons. We observed sIPSCs of >100 pA amplitude in BLA slices in the presence of TTX (data not shown), but only events with much smaller amplitudes, likely single quantal events, have been observed in TTX and cadmium (Katona et al., 2001). The lack of a WIN55212-2 effect in the TTX and low extracellular calcium condition is in agreement with the observation by Katona et al. (2001) that CB1-mediated inhibition in BLA slices was absent when single quantal events were examined. Thus, the cannabinoid effect on IPSCs appears to depend on calcium influx, as suggested previously in hippocampus (Hoffman and Lupica, 2000).
Because WIN55212-2-induced reduction of IPSC frequency and amplitude was similar in magnitude both at Vh = –60 or –90 mV, the decreased frequency is not attributable to an inability to detect small events during an amplitude decrease. The amplitude increase produced by the change in driving force is greater than the decrease produced by WIN55212-2 (see Results) and thus would offset any apparent loss of events resulting from decreases in amplitude below the detection threshold.
Examination of DSI in the presence of elevated [K+]o in this preparation revealed what appear to be two separable components of endocannabinoid-mediated synaptic depression. Depression immediately after depolarization depends on increased postsynaptic calcium but is independent of mGluR5 activation. In contrast, the later-onset, more persistent component requires mGluR5 activation but is resistant to strong postsynaptic calcium buffering with BAPTA. This is reminiscent of mGluR-induced retrograde signaling described in previous studies (Maejima et al., 2001; Chevaleyre and Castillo, 2003). This mGluR-dependent component of DSI may be similar to endocannabinoid-dependent long-term depression (LTD) (Gerdeman et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003; Azad et al., 2004), but additional studies examining the duration of this synaptic depression will be needed to determine whether this is the case. A recent study emphasizes the role of mGluR1 in endocannabinoid actions in the BLA (Azad et al., 2004). Thus, both mGluR1 and mGluR5 could play important roles in endocannabinoid signaling in the BLA. The glutamatergic transmission shown in Figure 1C indicates that intact glutamate-releasing synapses are present in the neuron/bouton preparation. These synapses are a likely source of the glutamate that activates mGluRs under the elevated [K+]o condition in this preparation. However, we do not have direct evidence at present that mGluRs are activated in this preparation.
DSI can also be induced in the presence of relatively low [K+]o, indicating that elevated [K+]o is not necessary for DSI. However, the DSI is shorter in duration under the 5 mM [K+]o condition than it is when 9 mM [K+]o is present. This finding further supports the idea that two DSI components can be separated in the isolated cell preparation. The observation that only short-duration DSI is observed when [K+]o is lowered is consistent with our findings in the brain slice preparation in which experiments were performed with 3 mM [K+]o.
These findings extend our knowledge of endocannabinoid signaling in the BLA, a brain region rich in endocannabinoids and CB1 receptors (Katona et al., 2001; Marsicano et al., 2002). Manifestation of DSI in the BLA slice further indicates the potential physiological relevance of endocannabinoid signaling in the BLA. Endocannabinoid-dependent LTD has been observed at BLA GABAergic synapses (Marsicano et al., 2002), and there is emerging evidence that a retrograde signal is involved in this form of plasticity (Azad et al., 2004). However, DSI has not been reported previously at GABAergic synapses onto BLA. Our findings provide evidence for retrograde endocannabinoid signaling in the BLA.
The effects of CB1 antagonists suggest that there is ongoing endocannabinoid signaling in isolated BLA neurons. The observed rebound increase in sIPSC frequency after WIN55212-2 application might also result from such a tone, because there is likely to be receptor desensitization after prolonged agonist application that would disrupt the endocannabinoid effect. However, caution should be exercised in generalizing these findings to the intact BLA. Although we did observe inconsistent AM251-induced potentiation of sIPSC frequency in the BLA slice, as yet there are no data indicating whether an endocannabinoid tone exists in the BLA in vivo. It is possible that the release of endocannabinoids, or their local extracellular concentration or diffusion, is tightly regulated in the slice preparation by adjacent cellular elements and/or transporters. Transient intracellular calcium release may also be larger in magnitude or produce larger local calcium increases in isolated BLA neurons than in neurons within slices. Differences in tonic endocannabinoid signaling may be a consequence of the experimental conditions in isolated neurons versus brain slices. Thus, it is premature to draw conclusions about the relevance of the endocannabinoid tone. AM251 also increased the amplitude of sIPSCs in the presence of TTX. However, AM251 decreased exogenous GABA-activated postsynaptic current. It is therefore unlikely that the AM251-elicited increase sIPSC amplitude is mediated by effects on GABAA receptors.
The BLA has a critical role in fear conditioning (Davis et al., 1994) and appears to provide the anatomical substrate for fear memory storage (LaLumiere et al., 2003; Gale et al., 2004). GABAergic control of projection neuron activity is thought to play a crucial role in information processing in the amygdala (Lang and Paré, 1998). CB1 immunoreactivity was detected in a distinct subset of GABA-containing interneurons in the BLA (Katona et al., 2001), the nucleus in which aversive memories might be formed and stored (Medina et al., 2002). Furthermore, endocannabinoid signaling in BLA has been implicated in extinction of aversive memories (Marsicano et al., 2002). Examination of the detailed mechanisms of endocannabinoid signaling using the neuron/bouton preparation should help us to understand the molecular underpinnings of cannabinoid effects on emotion and endocannabinoid roles in emotional memory. Use of this preparation may also allow us to examine molecular mechanisms by which stress and fear conditioning affect endocannabinoid signaling at a single set of synapses in BLA. This work not only demonstrates endocannabinoid signaling in BLA neurons but also provides a powerful tool for pharmacological and physiological studies of endocannabinoid signaling in this brain region.
Footnotes
This work was supported by the National Institute on Alcohol Abuse and Alcoholism Division of Intramural Clinical and Basic Research. We thank Dr. Susumu Koyama for assistance with implementation of the neuron/bouton preparation and Dr. Stephen Ikeda for assistance in photographing isolated neurons.
References
- Akaike N, Moorhouse AJ. Techniques: applications of the nerve-bouton preparation in neuropharmacology. Trends Pharmacol Sci. 2003;24:44–47. doi: 10.1016/s0165-6147(02)00010-x. [DOI] [PubMed] [Google Scholar]
- Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Drewe JA, Childs GV, Kunze DL. Synaptic transmission between dissociated adult mammalian neurons and attached synaptic boutons. Science. 1988;241:1810–1813. doi: 10.1126/science.2459774. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, Wilt-gen B, Fanselow MS. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Guo J, Ikeda SR. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol. 2004;65:665–674. doi: 10.1124/mol.65.3.665. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, Wang SJ, Chiou LC, Gean PW. Mediation of amphetamine-induced long-term depression of synaptic transmission by CB1 cannabinoid receptors in the rat amygdala. J Neurosci. 2003;23:10311–10320. doi: 10.1523/JNEUROSCI.23-32-10311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- LaLumiere RT, Buen TV, McGaugh JL. Post-training intrabasolateral amygdala infusions of norepinephrine enhance consolidation of memory for contextual fear conditioning. J Neurosci. 2003;23:6754–6758. doi: 10.1523/JNEUROSCI.23-17-06754.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang EJ, Paré D. Synaptic responsiveness of interneurons of the cat lateral amygdaloid nucleus. Neuroscience. 1998;83:877–889. doi: 10.1016/s0306-4522(97)00420-x. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Mackie OK, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di M, V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Medina JF, Christopher RJ, Mauk MD, LeDoux JE. Parallels between cerebellum- and amygdala-dependent conditioning. Nat Rev Neurosci. 2002;3:122–131. doi: 10.1038/nrn728. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to pre-synaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE.Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus J Neurosci 200121RC1881–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P, Marty A. Neighboring cerebellar Purkinje cells communicate via retrograde inhibition of common presynaptic interneurons. Neuron. 1993;11:885–893. doi: 10.1016/0896-6273(93)90118-b. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Muscarinic responses of rat basolateral amygdaloid neurons recorded in vitro. J Physiol (Lond) 1992;449:121–154. doi: 10.1113/jphysiol.1992.sp019078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]