

Abstract
Trophic support and myelination of axons by Schwann cells in the PNS are essential for normal nerve function. Herein, we show that deletion of the LDL receptor-related protein-1 (LRP1) gene in Schwann cells (scLRP1−/−) induces abnormalities in axon myelination and in ensheathment of axons by non-myelinating Schwann cells in Remak bundles. These anatomical changes in the PNS were associated with mechanical allodynia, even in the absence of nerve injury. In response to crush injury, sciatic nerves in scLRP1−/− mice showed accelerated degeneration and Schwann cell death. Remyelinated axons were evident 20 days after crush injury in control mice, yet were largely absent in scLRP1−/− mice. In the partial nerve ligation model, scLRP1−/− mice demonstrated significantly increased and sustained mechanical allodynia and loss of motor function. Evidence for central sensitization in pain processing included increased p38MAPK activation and activation of microglia in the spinal cord. These studies identify LRP1 as an essential mediator of normal Schwann cell-axonal interactions and as a pivotal regulator of the Schwann cell response to PNS injury in vivo. Mice in which LRP1 is deficient in Schwann cells represent a model for studying how abnormalities in Schwann cell physiology may facilitate and sustain chronic pain.
Introduction
Schwann cells originate in the neural crest and differentiate by two alternative pathways. Non-myelinating Schwann cells ensheath multiple small-caliber, C-fibers to form Remak bundles. Schwann cells, which associate with larger axons, segregate these axons and myelinate them at a one-to-one ratio. Schwann cell-axonal interactions are critical in controlling the physiology of both cell types (Jessen and Mirsky, 1999; Witt and Brady, 2000). Although several receptors have been implicated in axonal-Schwann cell interactions (Feltri et al., 2002; Taveggia and Salzer, 2007; Yu et al., 2007), this process remains incompletely understood.
In injury to the peripheral nervous system (PNS), Schwann cells are activated and play a critical role in the programmed series of steps that lead to eventual nerve regeneration (Grinspan et al., 1996; Meier et al., 1999). Activated Schwann cells at the injury site de-differentiate, proliferate, migrate, and participate in phagocytosis of debris (Chen et al., 2007). Extracellular matrix (ECM) proteins, which are secreted by Schwann cells, assemble into a scaffold that facilitates axonal regeneration (Bunge and Pearse, 2003; Chen et al., 2007). When the response to PNS injury is abnormal, as in end-bulb neuroma formation or denervation hyperalgesia, chronic neuropathic pain may result (Rogawski and Loscher, 2004). Chronic neuropathic pain is a highly prevalent health problem and there is a profound need to identify novel targets for therapeutics development (Goldberg and McGee, 2011).
The endocytic, transmembrane receptor, LDL receptor-related protein (LRP1), is a potent regulator of Schwann cell physiology in vitro (Campana et al., 2006a). LRP1 binds diverse proteins produced in the injured PNS, including proteases such as MMP-9 and ECM proteins (Strickland et al., 1990; La Fleur et al., 1996; Akassoglou et al., 2000; Strickland et al., 2002). Ligand-binding to LRP1 activates pro-survival signaling, including ERK/MAP kinase, the PI3K–Akt pathway (Campana et al., 2006a; Mantuano et al., 2008). LRP1 also promotes Schwann cell survival by antagonizing the unfolded-protein response (Mantuano et al., 2011). By regulating Rho family GTPases, LRP1 promotes Schwann cell migration (Mantuano et al., 2010). Thus, Schwann cell LRP1 expresses multiple activities that may be important in the response to PNS injury.
LRP1 gene deletion in the mouse is embryonic-lethal (Herz et al., 1992), precluding the use of this mouse model system to characterize Schwann cell LRP1. Furthermore, other cell types present in the injured peripheral nerve, including neurons and macrophages, express LRP1 (Lillis et al., 2008). Thus, results obtained using reagents such as receptor-associated protein (RAP), which antagonize LRP1 in all cell types, may be difficult to interpret. To address this problem, we developed a unique mouse model in which LRP1 is deleted under the control of the P0 promoter, which is active selectively in Schwann cells (Feltri et al., 1999).
Herein, we show that LRP1 gene deletion in Schwann cells affects the structure of uninjured nerve fibers, including myelinated fibers and C-fibers in Remak bundles. These changes are associated with altered pain processing even in the absence of injury. LRP1 deficiency in Schwann cells also substantially compromises the response to injury. Accelerated degeneration Schwann cell death and reduced regeneration are observed in association with robust and sustained neuropathic pain. We conclude that Schwann cell LRP1 is required for normal Schwann cell-axonal interactions and as a pivotal regulator of the response to PNS injury.
Material and Methods
Animals
Transgenic mice carrying LRP1 alleles with LoxP sites, so that Cre-mediated recombination eliminates part of the promoter and the first two exons (LRP1flox/flox mice), were originally generated by Rohlmann et al. (Rohlmann et al., 1996). We obtained mice which carry two copies of the floxed LRP1 gene and Cre recombinase expressed under the control of the Lysozyme M promoter (Overton et al., 2007). These mice were crossed with C57BL/6 mice to regenerate LRP1flox/flox mice, without LysM-Cre, in the C57BL/6 genetic background. PCR analysis was performed on genomic DNA isolated from tail clips (BioPioneer, Inc). Homozygous floxed lrp1 alleles were identified by a 350bp fragment amplified by PCR using forward 5’CATACCCTCTTCAAACCCCTTC3’ and reverse 5’GCAAGCTCTCCTGGTCAG-ACC3’ primers (see Fig. 1). P0-Cre mice, in which Cre is expressed selectively in Schwann cells, are previously described (Feltri et al., 1999; Feltri et al., 2002). For our studies, P0-Cre mice in the C57BL/6 genetic background were crossed with LRP1flox/flox mice. Progeny that were heterozygous for the LRP1floxed gene and P0-Cre-positive were bred with LRP1flox/flox mice. Approximately 25% of the resulting pups were homozygous for the LRP1floxed gene and P0-Cre-positive, as anticipated for a system in which there is no embryonic lethality. These mice were bred with LRP1flox/flox mice to generate experimental animals. Male littermates that were used in experiments were LRP1flox/flox and either P0-Cre-positive or P0-Cre–negative. Hemizygous P0-Cre mice were identified by a 492 bp fragment amplified in PCR reactions using forward 5’CCACCACCTCTCCATTG-CAC3’ and reverse 5’GCTGGCCCAAATGTTCGTGG3’ primers. Mice that are deficient in Schwann cell LRP1 are called scLRP1−/− mice and littermate controls containing Schwann cell LRP1 are called scLRP1+/+ mice. All breeding procedures were performed according to the protocols approved by the University of California, San Diego Committee on Animal Research, and conform to NIH Guidelines for Animal Use. All mice were housed with a 12 h:12 h light: dark cycle with ad libitum access to food and water.
Figure 1.

LRP1 inactivation in Schwann cells. (A) Double-label immunofluorescence microscopy of LRP1 (green) in an adult myelinated sciatic nerve fiber. Nuclei are identified with DAPI (blue). Note some residual LRP1 immunoreactivity in axoplasm of scLRP1−/− nerves not associated with Schwann cells. Images represent n=4 mice/group. Scale bar, 4 µm. (B) Quantification of LRP1 immunoreactive Schwann cells and axons in uninjured sciatic nerves. Immunoreactive cells were counted in 4 separate sections from each mouse (*p<0.01; n=4 mice/group). (C) Immunofluorescence microscopy of LRP1 (red) in DRGs. Note similar intensity and distribution of immunoreactivity in DRG neuron cell bodies. Nuclei are identified with Dapi (blue). Scale bar, 45 µm.
Mouse surgery
In crush injury experiments, mice were anesthetized with 3% isoflurane (IsoSol; VedCo, St. Joseph MO) and maintained with 2% isoflurane. An incision was made along the long axis of the femur. The sciatic nerve was exposed at mid-thigh level by separating the biceps femoris and the gluteus superficialis and then carefully cleared of surrounding connective tissue. The sciatic nerve was crushed twice for 30 sec with flat forceps (Azzouz et al., 1996). The site of crush injury was marked with an epineural suture on the muscle surface. The muscle and skin layers were closed using 6.0 silk sutures. In partial nerve ligation (PNL, Seltzer model) studies, adult mice were subjected to PNL of the left sciatic nerve (Seltzer et al., 1990) under anesthesia using 3% isoflurane initially, as described above. The sciatic nerve was exposed at mid-thigh level as described above. A 9-0 silk suture was inserted into the nerve with a curved, reversed-cutting mini-needle, and ligated so that the dorsal one-third to one-half of the nerve thickness was held within the ligature. The wound was closed with a single muscle suture and the skin was tightly sutured with 4-0 nylon suture. In sham-operated animals, the nerve was exposed at mid-thigh level but not ligated. Some animals received minocycline (Sigma, St. Louis, MO). Minocycline was prepared and dissolved in sterile water. Mice received daily intraperitoneal injections (40 mg/kg) for 7 days, with the first injection administered 4 hours prior to surgery. The dosage of minocycline was determined based on the previous reports regarding the therapeutic efficacy of minocycline (Ledeboer et al., 2005).
Motor and sensory function
Male mice were randomized and acclimated to the test chamber for 1 h prior to performing behavioral tests. Baseline thresholds were obtained for three days prior to surgery. Following surgery, tests were performed every day until euthanasia. Mechanical allodynia was assessed by probing the hindpaw with non-noxious graded stimuli (von Frey filaments ranging from 0.008 to 6 g of force) applied perpendicularly to the plantar surface of the paw (between the footpads) through wire-mesh observation cages. The paw withdrawal threshold (PWT) was determined by sequentially increasing and decreasing the stimulus strength and analyzing withdrawal data using a Dixon nonparametric test (Dixon, 1980), as described by Chaplan et al. (Chaplan et al., 1994). Motor testing was performed using a rotarod. Mice were randomized and placed on a rotating cylinder that accelerated from 4 to 40 rotations per minute (rpm) over 5 min (Ugo Basile, Comerio, Italy). Mice were allowed to remain on the cylinder for a maximum of 700 s and the time to failure was measured in different trials (two trials with four replicates per day). Any mice remaining on the apparatus after 700 s were removed and their times were scored as 700 s (Bolis et al., 2005). In all behavior studies, experimentalists were blinded to group identity.
TUNEL staining
TUNEL assays were performed as described by Grinspan et al. (Grinspan et al., 1996), with a few modifications. Briefly, adult sciatic nerves immediately distal to injury sites (3 mm) were dissected, fixed in 4% paraformaldehyde in PBS for 2 h, and then cryopreserved. Cryosections were prepared and mounted on glass slides. TUNEL-staining was performed using the ApopTag® Fluorescein in situ Apoptosis Detection Kit (Millipore). Labeled sections were mounted with coverslips using Pro-long® Gold and nuclei were counterstained by DAPI. Images were captured on a Leica DM IRE2 fluorescent microscope.
Immunoblot analysis
Immunoblot analysis was performed as previously described (Campana et al., 2006a; Mantuano et al., 2008). Briefly, extracts of sciatic nerve 3 mm distal from an injury site and spinal dorsal horn (ipsi and contralateral from the L4-L6 lumbar enlargement) were prepared in RIPA buffer. The protein content of each extract was determined by bicinchoninic acid assay (BCA) assay. An equivalent amount of protein (20 µg per lane) was subjected to 10% SDS-PAGE and electrotransferred to nitrocellulose membranes. Blots were blocked with 5% nonfat dry milk in Tris-HCl buffered saline, pH 7.4, with Tween-20 and subsequently incubated with primary polyclonal antibodies to cleaved caspase-3 (1:500; Cell Signaling), LRP1 (1:1000; C-terminal, Sigma), phospho-p38 MAPK, total-p38 MAPK (1:1000; Cell signaling), myelin basic protein (MBP) (1:1000; Abcam) or monoclonal antibody to β-actin (1:2000; Sigma) overnight at 4°C. Antibody binding was detected by HRP-conjugated species-specific secondary antibodies (1:2000; Cell Signaling) followed by enhanced chemiluminescence (GE Healthcare). Blots were scanned (Cannoscan) and densitometry was performed using Image J software.
Immunofluorescence microscopy and immunohistochemistry
Mice were anesthetized with 3% isoflurane (IsoSol; VedCo, St. Joseph MO) and subjected to intracardiac perfusion with fresh 4% paraformaldehyde in 0.1 M sodium phosphate buffer. Tissue (sciatic nerve, DRGs) was dissected, cryoprotected, and cut into 10–20 µm sections. Spinal cord tissue from the lumbar region (L4-L6) was sectioned as transverse free-floating spinal sections (35 µm). A minimum of four sections per animal were analyzed. Nonspecific binding sites were blocked with 10% goat or horse-serum in 0.1% Triton X-100 and PBS for 60 min at room temperature, and then incubated with primary antibodies diluted in blocking solution overnight at 4°C. The following primary antibodies were used: LRP1 (C-terminal) (Sigma-Aldrich, 1:1000); P0 (Millipore, 1:100); p-p38 MAPK (Cell Signaling, 1:200); OX-42 (Serotec, 1:800); and IgG at appropriate matched dilution. Tissue was washed in PBS and incubated with the appropriate fluorescent secondary antibodies (Alexa Fluor 488- or 594-conjugated antibodies, 1:1000; Molecular Probes) for 1 h at room temperature. For dual-labeling studies, a second set of primary and secondary antibodies was added. Preparations were mounted on slides using Pro-long Gold with DAPI for nuclear labeling (Invitrogen). Images were captured on an Olympus Fluoview1000 confocal microscope.
In some cases, nerves were processed for paraffin embedding as described previously (Campana and Myers, 2001; Mantuano et al., 2011). Nerve sections were cut (4 µm) and incubated with Tris-EDTA (Ventana Discovery Ultra Medical Systems, Tucson, AZ) for 4 min and 8 min cycles at 95°C. Three sections from each nerve were sampled. Non-specific binding was blocked with 10% nonfat milk. Primary antibodies (S100 and MBP; Millipore) were subsequently incubated in antibody diluent (Dako, Carpenteria, CA) for 1 h at 22°C. Next, sections were incubated with anti-mouse antibodies conjugated to HRP (Ventana Discovery Ultra Medical Systems, Tucson, AZ) and developed with 3’3 diaminobenzidine (DAB). Some sections were treated with secondary antibody only, as a control. Imaging was performed using a Leica DCF2500 microscope with Leica Imaging Software 2.8.1 (Leica Microsystems, Inc. Buffalo Grove, IL).
Quantitative analysis of S100-positive Schwann cells
The number of S100-positive cells was determined in the mouse sciatic nerve 1 day after crush lesion. Counting was performed 3 mm distal to the crush injury site and in the contralateral (non-lesioned) nerve. S100 immunostaining was located in Schwann cell cytoplasm and only S100-positive cytoplasm in crescents was counted. Counting was performed using a light microscope (Leitz Wetzlar Dialux 20) with the 40× objective and an ocular grid containing 10×10 squares. One square was 25×25 µm (625 µm2). Counting of S100 Schwann cells in the distal nerve sections was performed in a total of 20 squares per section, representing approximately 10% of the total nerve bundle area.
Transmission electron microscopy
Sciatic nerves from scLRP1−/− and littermate control mice were processed for plastic embedding as described elsewhere (Campana et al., 2006a). Glutaraldehyde (2.5%)/0.1 M sodium phosphate, 150 mM NaCl, pH 7.4 (Fixation Buffer) was applied directly onto mouse sciatic nerves prior to removal. Resected tissue was further incubated in Fixation Buffer for an additional 72 h at 4°C, washed, chilled in 0.1 M Cacodylate Buffer, pH 7.4, and then secondarily fixed in 1% Osmium Tetroxide/CB for 30 min. Sciatic nerve samples were further washed and then dehydrated in serially increasing concentrations of ethanol. Specimens were embedded in Durcupan resin (Sigma-Aldrich). Semi-thin sections (50–60 nm) were applied to copper grids (300 mesh). The grids were viewed using a Tecnai G2 Spirit BioTWIN transmission electron microscope (TEM) equipped with an Eagle 4k HS digital camera (FEI, Hilsboro, OR) or with a JOEL FX1200 Transmission Electron Microscope.
Quantitative TEM analysis
Ultrathin sections (50–60 nm) of sciatic nerve were used to count myelinated nerve fibers and Remak bundles in scLRP1−/− and scLRP1+/+ mice. Nerves were imaged at 1900X. Myelinated fibers, C-fibers and Remak bundles were counted in a 500 µm2 surface area using OpenLab software (Improvision, Conventry, UK). The number of myelinating and nonmyelinating Schwann cells is presented as the average per cross-section. For g-ratios, mean axonal and fiber (axon including myelin sheath) diameters were measured. The g-ratio was determined by dividing the mean axon diameter by the mean fiber diameter. Myelin area was determined by subtracting the axonal area from the total fiber area. Approximately 100–120 nerve fibers were counted from three individual animals per genotype. Axon diameters from both genotypes were divided into three groups: 0–3, 3.1–6 and >6.1 µm and the frequency of g-ratio was recorded and averaged for each group. The criteria used for defining abnormal non-myelinating Schwann cells was 1) the presence of an axon greater than >2 µm in the Remak bundle and 2) lack of Schwann cell cytoplasm between axons so that multiple axons are within one cytoplasmic domain.
Statistical analysis
Behavioral and functional data are presented as the mean ± SEM. Results were analyzed by repeated-measures ANOVA and Scheffe’s post hoc test. For quantitative immunohistochemistry and TEM, immunoblotting, and qPCR data, we performed a one-way ANOVA followed by a Tukey’s post hoc test when more than two groups of results were compared. A student’s t-test was used when two groups were compared. For non-parametric data, a Kolmogorov-Smirnov test was used to measure and compare frequency distributions. In all statistical analysis, a significance criterion of p<0.05 was used.
Results
Targeted disruption of LRP1 in Schwann cells
Mice harboring loxP recognition sites in the LRP1 gene (Rohlmann et al., 1996) were crossed with mice in which Cre recombinase is expressed under the control of the P0 promoter (Feltri et al., 1999). The P0 promoter becomes active specifically in Schwann cell precursors at embryonic day 13.5–14.5, including cells that develop into myelinating and non-myelinating Schwann cells; the P0 promoter is inactive in other glial cells and dorsal root ganglion neurons (Feltri et al., 1999; Feltri et al., 2002). Mice that are homozygous for the LRP1floxed gene and carry the P0-Cre recombinase transgene (scLRP1−/−) were grossly normal. These animals were fertile and born at the expected Mendelian frequency, without evidence for embryonic lethality. Adult mice were the same size and weight as mice that were homozygous for the LRP1floxed gene and negative for P0-Cre (scLRP1+/+). Gait and motor function were qualitatively intact, and there was no evidence of overt neurological abnormalities.
scLRP1−/− and scLRP1+/+ mice were euthanized at 10 weeks. mRNA was harvested from uninjured sciatic nerves. LRP1 mRNA levels were determined by qPCR (n=8 mice/group). LRP1 mRNA was reduced by 52±5% in nerves from scLRP1−/− mice, compared with nerves isolated from scLRP1+/+ mice (p<0.01), suggesting that the LRP1 gene was indeed deleted in a sub-population of cells present in the sciatic nerve. Immunofluorescence microscopy, using antibody specific for LRP1, showed loss of antigen (green fluorescence) in association with the nucleated cytoplasmic crescents of myelinating Schwann cells in scLRP1−/− mice (Fig. 1A). Axonal immunoreactivity was still observed in sections of nerve from scLRP1−/− mice, as anticipated (Campana et al., 2006a). By visual examination, greater than 95% of the myelinating Schwann cells were LRP1-immunonegative in scLRP1−/− mice, compared with Schwann cells from scLRP1+/+ mice, which were uniformly LRP1-immunopositive (Fig. 1B). As a further control, we examined dorsal root ganglia (DRGs) from scLRP1+/+ and scLRP1−/− mice. LRP1 was detected in the neuronal cell bodies in DRGs of control animals, as previously reported (Shi et al., 2009). Conditional LRP1 gene deletion in Schwann cells did not affect LRP1 expression in DRG neuronal cell bodies (Fig. 1C).
LRP1 gene deletion in Schwann cells affects the phenotype of myelinating and non-myelinating cells
Non-lesioned sciatic nerves were examined histologically by preparing semi-thin plastic sections, which were stained with methylene blue/Azure II. By light microscopy, the morphology of the sciatic nerves from scLRP1−/− mice appeared normal (Fig. 2A). Axons of various diameters were present at proportions that appeared similar to those observed in scLRP1+/+ mice. The number of myelinated axons per field (500 µm2) was not significantly different in scLRP1−/− (19±1) compared with scLRP1+/+ (21±2) mice.
Figure 2.

Effects of LRP1 deletion on the structure of myelinating Schwann cells. (A) Semi-thin transverse sections of uninjured sciatic nerves from scLRP1+/+ and scLRP1−/− mice were stained with methylene blue/Azure II. Scale bar, 12 µm. (B) TEM analysis of a myelinated axon and quantification of axon and myelin area in scLRP1+/+ or scLRP1−/− sciatic nerves (*p<0.05 compared to scLRP1+/+ nerve). Scale bar, 3.0 µm. (C) Scatter plots of g-ratio versus axon diameter for myelinated axons from scLRP1+/+ and scLRP1−/− mice (n=100–120 axons/genotype). Higher g-ratios were observed in scLRP1−/− nerves compared to scLRP1+/+ (**p<0.01).
When uninjured sciatic nerves from scLRP1−/− mice were examined by TEM, we noticed that the thickness of the myelin surrounding many axons appeared decreased (Fig. 2A). To explore this observation, we selected, at random, 47 myelinated axons from scLRP1−/− mice, with diameters ranging from 3.1–6 µm, and 48 equivalently-sized axons from scLRP1+/+ nerves. Axonal and myelin areas were determined. The mean axonal area for the cohort of axons from scLRP1−/− mice was actually slightly greater than that of the wild-type cohort. Nevertheless, the mean myelin area for the cohort of axons from scLRP1−/− nerves was significantly decreased (p<0.01; Fig. 2B).
Next, we conducted a more comprehensive analysis of TEM images, calculating g-ratios, which report mean axonal diameters divided by mean fiber diameters. Fig. 2C shows that the g-ratios appeared increased in nerves from scLRP1−/− mice across the continuum of axon size (p<0.05; Kolmogorov-Smirnov). We confirmed this observation by applying statistical analysis to axon populations that were stratified according to size. For axons ranging from 0–3 µm, the g-ratios were 0.54 ± 0.02 for scLRP1+/+ mice and 0.64 ± 0.02 for scLRP1−/− mice (mean±SEM, p<0.001). For axons ranging from 3–6 µm, the g-ratios were 0.58 ± 0.01 for scLRP1+/+ nerves and 0.68 ± 0.01 for scLRP1−/− nerves (p<0.001). For axons ranging from 6–8 µm, the g-ratios were 0.62 ± 0.01 for scLRP1+/+ nerves and 0.72 ± 0.02 for scLRP1−/− nerves (p<0.001).
In normal peripheral nerves, non-myelinating Schwann cells ensheath multiple small pain transmitting axons (C-fibers) to form Remak bundles. Fig. 3A shows TEM images of Remak bundles from scLRP1+/+ and scLRP1−/− mice. In scLRP1+/+ mice, C-fibers ensheathed by non-myelinating Schwann cells were well-organized and of normal size (<1.2 µm). Distinct Schwann cell cytoplasm separated each individual axon. In contrast, nerves from scLRP1−/− mice, had axons in Remak bundles that appeared abnormally shaped and ensheathed (Fig. 3B). The percentage of axons that was greater than 2 µm in diameter was significantly increased (p<0.01) and the percentage of axons that appeared improperly separated from neighboring axons by Schwann cell cytoplasm was also significantly increased (p<0.01). The density of non-myelinating Schwann cells in nerve sections from scLRP1−/− mice (5.1±0.5 per 500 µm2 field) was not significantly changed compared with littermate control mice (5.5±0.8 per 500 µm2); however, we observed a significant increase in the number of Remak bundles containing more than 21 axons in scLRP1−/− mice (p<0.05; Fig. 3C). Collectively, these findings suggest that there are abnormalities in Schwann-cell-axonal interactions, in scLRP1−/− mice, involving myelinating and non-myelinating Schwann cells.
Figure 3.
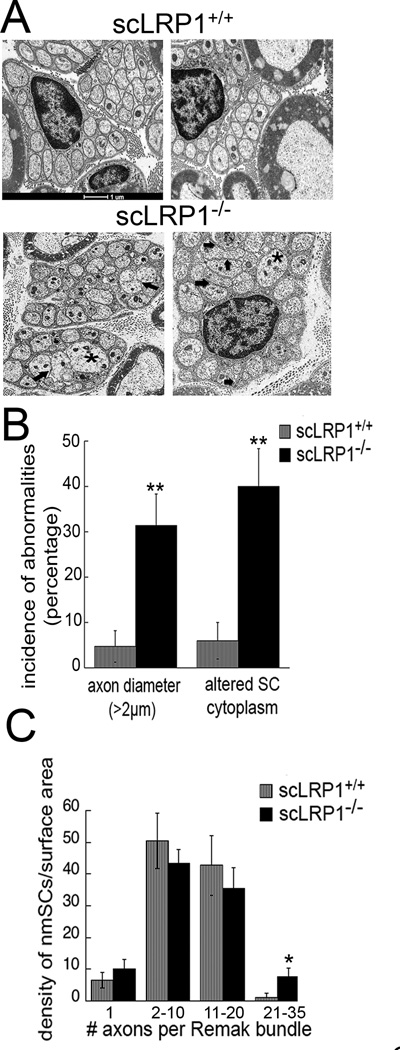
LRP1 deletion in Schwann cells alters Remak bundle properties. (A) TEM of transverse thin sections of sciatic nerves from scLRP1+/+ and scLRP1−/− mice. Asterisks identify axons with >2 µm diameter. Thick arrows demarcate areas in which the amount of Schwann cell cytoplasm between axons is decreased/absent. (B) Quantification of abnormal Remak bundles per surface area (500 µm2) in scLRP1+/+ and scLRP1−/− mice. Abnormal Remak bundles are defined as (and see Material and Methods): 1) the presence of axons greater than >2 µm in the Remak bundle; and 2) lack of Schwann cell cytoplasm between individual axons so that multiple axons are within one cytoplasmic domain. Abnormal Remak bundles were counted and expressed as a percentage of total number of Remak bundles. Data are presented as the mean ±SEM (**p<0.01). (C) The number of Remak bundles per surface area (500 µm2) in scLRP1+/+ and scLRP1−/− sciatic nerves in each of four categories was counted. Categories are distinguished by the number of ensheathed axons. Data represent the mean±SEM for 4 mice/genotype (*p<0.05 compared to littermate controls).
Given the abnormalities observed in Schwann cell-axonal ultrastructure in uninjured scLRP1−/− mice, we performed evoked sensory and motor testing experiments. Mechanical allodynia was measured by non-noxious probing of the hindpaw using von Frey filaments in adult mice. A significant reduction in sensory threshold was observed in scLRP1−/− mice (1.3 ± 0.15 g), compared with 2.3 ± 0.3 g, in scLRP1+/+ mice, which indicates that these scLRP1−/− mice have mechanical allodynia in the absence of injury (p<0.05; see Fig. 8). Motor function was measured by Rotarod testing. No change in motor function was evident (108 ± 3 seconds in scLRP1 −/− mice and 107 ± 4 in scLRP1+/+ mice; and see Fig. 8).
Figure 8.

Sensory and motor deficits are exacerbated and sustained in scLRP1−/− mice following PNL. (A) Mechanical allodynia was measured by non-noxious probing of the hindpaw using von Frey filaments before and after PNL. Data are expressed as means±SEM (N=10–12/cohort). Differences in treatment groups were compared by Scheffe’s post hoc test (* p<0.05 comparing genotypes at baseline; *p<0.05 compared with sham-operated mice in the same genotype; **p<0.01 comparing scLRP1−/− mice with all groups). (B) Rotarod-testing of scLRP1+/+ and scLRP1−/− mice prior to and after PNL. The time until failure is plotted (*p<0.05 compared with sham-operated mice, n=12/cohort).
Nerve degeneration is accelerated after injury in scLRP1−/− mice
To determine whether LRP1 deficiency in Schwann cells affects the response to injury, sciatic nerves were crush-injured in scLRP1−/− and scLRP1+/+ mice. Distal nerve sections (3 mm from the crush injury site) were examined by immunohistochemistry (IHC), using an antibody that detects MBP. We chose to image MBP to highlight myelin structure. In uninjured nerves, MBP immunoreactivity was relatively similar in both genotypes (Fig. 4A, B). The immunostain clearly demarcated intact myelinated axons of variable size.
Figure 4.

Degeneration is accelerated following crush injury of sciatic nerves in scLRP1−/− mice. Immunohistochemistry of MBP in sciatic nerves from scLRP1+/+ and scLRP1−/− mice is shown in (A,B) uninjured sciatic nerves; (C,D) in distal nerve sections (3 mm distal from the crush site) recovered one day after injury; and (E,F) and distal nerve sections recovered five days after injury. Scale bar, 25 µm. Note the loss of MBP and accelerated degeneration in injured scLRP1−/− nerves. Images are representative of those obtained with 4 mice per group. (G) MBP was determined by immunoblot analysis in extracts on nerves isolated 5 days after nerve injury. (H) Quantification of MBP ratios by densitometry 5 days after sciatic nerve crush injury. Data represent the mean±SEM for 4 mice/genotype (*p<0.05 compared to littermate controls).
One day after crush injury, nerves from scLRP1−/− mice showed profound loss of MBP immunoreactivity, reflecting degeneration of myelin sheaths, together with large areas of edema and apparent loss of nerve fiber architecture (Fig. 4D). In the control scLRP1+/+ mice, 24 h after crush injury, the structure of the nerve remained more intact (Fig. 4C), suggesting that degeneration was accelerated in scLRP1−/− mice. Five days after crush injury, degeneration of myelin sheath structure was still more advanced in scLRP1−/− mice; however, at this time point, nerve degeneration was obvious in the control scLRP1+/+ mice as well (Fig. 4E, F).
To confirm the results of our IHC studies, we performed immunoblotting experiments to detect MBP. In uninjured (contralateral) nerves, we did not detect significant differences in the total level of MBP, despite the ultrastructural changes in myelin sheaths in nerves from scLRP1−/− mice observed by TEM (Fig. 4G, H). There are many possible explanations for this result, including a change in the composition of PNS myelin in scLRP1−/− mice; however, the major focus of this experiment was to confirm more rapid degradation of myelin following injury in scLRP1−/− mice. Five days after nerve injury, when degeneration was apparent in control nerves, the level of MBP was significantly decreased in nerve extracts from scLRP1−/− mice. We interpret these IHC and immunoblottting results to indicate more rapid and robust degeneration of myelin in crush-injured nerves from scLRP1−/− mice. When immunoblots were performed using equivalent amounts of extracted sciatic nerve protein, β-neuronal tubulin was not significantly decreased 5 days after crush-injury in nerves from scLRP1−/− and scLRP1+/+ mice, suggesting that axonal cytoskeleton does not degenerate at the same rate as myelin in crush-injured nerves.
Schwann cell survival is compromised after nerve injury in vivo by LRP1 deficiency
In Schwann cells, LRP1 functions as a cell-signaling receptor, activating pro-survival pathways and antagonizing endoplasmic reticulum stress signaling (Campana et al., 2006a; Mantuano et al., 2011). To identify pathways that may be responsible for the accelerated degeneration of crush-injured sciatic nerves in scLRP1−/− mice, we performed TUNEL-labeling experiments. In control, uninjured contralateral nerves, TUNEL-positive cells were rare in both scLRP1−/− and scLRP1+/+ mice (Fig. 5A, B). Following crush injury, TUNEL positivity increased selectively in scLRP1−/− nerves (Fig. 5C, D). TUNEL-positivity was detected in 36±9% of the DAPI-labeled cells in injured nerves from scLRP1−/− mice, compared with <3% in injured nerves from scLRP1+/+ mice (p<0.05).
Figure 5.

Schwann cell viability is compromised in scLRP1−/− mice. (A–D) Sections of crush-injured and control, contralateral sciatic nerves were analyzed by TUNEL-staining (green). Nuclei are stained blue with DAPI. Images are representative of those observed with 3 mice/cohort. Scale bar, 15 µm. (E) Immunoblot analysis of LRP1 (85 kDa) and cleaved caspase-3 in sciatic nerve distal to a crush injury site. Nerves were isolated 24 h after injury. Equal amounts of nerve protein (30 µg) were loaded in each lane and subjected to SDS-PAGE. β-actin was measured as a loading control. Each lane represents an individual animal (n=3/group). (F) Quantification of LRP1 in sciatic nerve (SN) and cleaved caspase-3 by densitometry. Quantification of LRP1 in spinal cord by densitometry. Data are expressed as mean±SEM (*p<0.05).
Next, we tested whether apoptotic cell signaling is increased in scLRP1−/− nerves compared with scLRP1+/+ nerves. Nerve extracts were isolated 24 h after crush injury and subjected to immunoblot analysis. The total level of LRP1 (85 kDa light chain) was decreased in scLRP1−/− mice (Fig. 5E). This was anticipated because, after nerve injury, LRP1 expression in axons rapidly declines and Schwann cells are responsible for an increased fraction of total LRP1 in the nerve (Campana et al., 2006a). Cleaved caspase-3 was significantly increased in sciatic nerves from scLRP1−/− mice, providing further evidence that LRP1 deficiency in Schwann cells compromises cell viability after nerve injury.
We subjected our immunoblotting results to densitometry analysis with standardization against β-actin (Fig. 5F). LRP1 light chain was significantly decreased in injured sciatic nerves from scLRP1−/− mice (p<0.05) and cleaved caspase-3 was significantly increased (p<0.05). As a control, we showed that LRP1 was not regulated in spinal cords isolated from scLRP1−/− animals.
To further test the hypothesis that LRP1 deficiency in Schwann cells compromises cell viability after nerve injury, we subjected sections of sciatic nerve to S100 IHC. S100 is a well-defined Schwann cell marker (Jessen and Mirsky, 2005). In uninjured nerves from scLRP1+/+ and scLRP1−/− mice, the IHC images were similar. Representative low magnification and high magnification images of scLRP1+/+ nerve are shown in Figs. 6A and 6B, respectively. 24 h after crush-injury, activated Schwann cells with plump, robustly S100-immunopositive cytoplasmic crescents were abundant in nerves from scLRP1+/+ mice (Fig. 6C, D). In nerves from scLRP1−/− mice, activated Schwann cells were more difficult to identify (Fig. 6E, F). Interestingly, the residual Schwann cells in these nerves demonstrated contracted cytoplasm or appeared degenerated (see panel F). Large areas devoid of immunostain suggested extensive nerve edema. We quantitated the number of S100-immunopositive Schwann cell crescents and demonstrated a significant decrease in crescents in injured nerves from scLRP1−/− mice (p<0.01; Fig. 6G). The number of Schwann cell crescents was not decreased in contralateral, uninjured nerves.
Figure 6.

Loss of Schwann cells and failed regeneration in crush injured sciatic nerves from scLRP1−/− mice. (A–F) S100 immunohistochemistry of uninjured and crush-injured sciatic nerves. Note the intense immunoreactivity of Schwann cell crescents in the injured (much more activated) control nerves (arrowheads). In scLRP1−/− nerves (panel F), fewer intact S100-immunopositive crescents are present and degenerating Schwann cells are visible (astericks). Images are representative of studies with 4 mice per group. Scale bar, 12 µm. (E) Quantification of S100-immunoreactive Schwann cell crescents in tissue sections (**p<0.01; n=16–20 sections/group). (F) TEM analysis of crush-injured sciatic nerves after 20 days in scLRP1+/+ and scLRP1−/− mice. Arrow points to onion bulb formation.
Schwann cells are known to be essential for peripheral nerve regeneration (Ide et al., 1996). Because Schwann cell survival is compromised in scLRP1−/− mice, we subjected scLRP1−/− and scLRP1+/+ mice to crush injury and studied recovered nerves by TEM, 20 days after injury. Myelinated axons were evident in nerves isolated from scLRP1+/+ mice (Fig. 6H). Because axonal deterioration downstream of crush injury sites is typically complete, we interpreted the presence of myelinated axons 20 days post-injury as evidence of nerve regeneration. By contrast, sections of scLRP1−/− nerves showed no evidence of successfully re-myelinated axons. Instead, degenerated myelin swirls, electron-opaque structures resembling vacuoles and numerous onion bulb formations, which form from layers of Schwann cell cytoplasm, were present, suggestive of failed regeneration (Thomas, 1970; Ohara et al., 1988).
scLRP1−/− mice develop severe and sustained neuropathic pain
The relationship between abnormal Schwann cell-axonal interactions, demyelination, and pain has been previously reported (Fujita et al., 2007; Ueda, 2008). Thus, we hypothesized that abnormal Schwann cell-axonal interactions in scLRP1−/− mice would result in altered pain responses following injury. To test this hypothesis, mice were subjected to Seltzer injury (also called partial nerve ligation, PNL), which is a well-established neuropathic pain model (Shir and Seltzer, 1990). In this model system, one-third to one-half of the sciatic nerve is ligated. Heterogeneity in damage to different areas of a single nerve is apparent in semi-thin sections of nerve from scLRP1−/− and scLRP+/+ mice, isolated 20 days after PNL and stained with methylene blue Azure II (Fig. 7A, B). When the most highly damaged areas of the nerves were examined at high power, some of the major observations from the crush injury model system were conserved. In these areas of scLRP1−/− nerves, degenerating axons and inflammatory cells were abundant. Similar areas from the nerves of scLRP1+/+ mice showed substantial evidence of regeneration, including thinly myelinated axons with small diameters (Fig. 7C, D). The myelinated axons are most likely axons coming through or around surgical ligations. These differences also were apparent by TEM (Fig. 7E, F). The onion bulb formations in sections of nerve from scLRP1−/− mice, subjected to PNL 20 days prior to nerve harvest, provide suggestive evidence for failed regeneration (Thomas, 1970).
Figure 7.
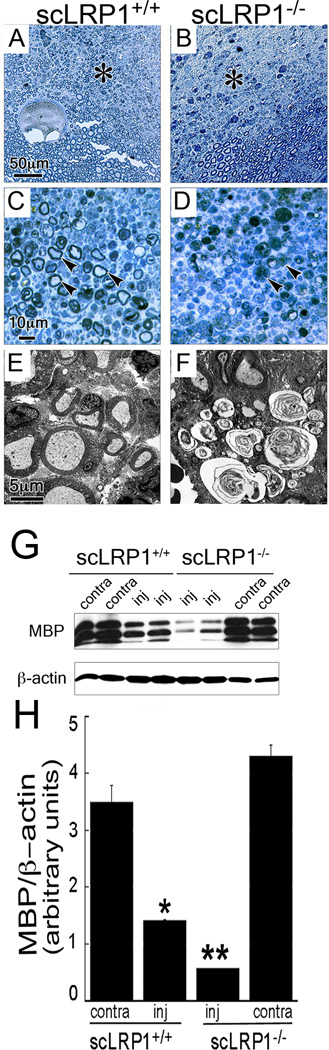
Characterization of structural changes following PNL of sciatic nerves in scLRP1−/− mice. (A–D) Transverse semi-thin plastic sections of PNL-injured sciatic nerves isolated 20 days after injury and stained with methylene blue/Azure II. Asterisks in panels A and B identify areas shown in panels C and D at higher magnifcation. Arrowheads in panel C identify myelinated axons. Arrowheads in panel D show degenerated nerve fibers. (E, F) TEM analysis of PNL-injured sciatic nerves isolated 20 days after injury. The images shown are representative of studies conducted with 3–4 mice/group. (G) Immunoblot analysis of MBP levels in PNL-injured nerves isolated 20 days after injury. Each lane represents an individual mouse. (H) Quantification of immunoblots by densitometry (*p<0.05; n=4/group).
To confirm that the abundance of myelinated fibers is decreased in scLRP1−/− mice, we performed immunoblotting experiments to detect MBP in sciatic nerves 20 days after PNL (Fig. 7G). MBP levels were not significantly different in uninjured contralateral nerves isolated from scLRP1+/+ and scLRP1−/− mice, confirming the results shown in Fig. 4G. Twenty days after PNL, MBP was significantly decreased in distal nerve extracts from scLRP1−/− and scLRP1+/+ mice, compared with levels observed in uninjured nerves. Furthermore, the level of MBP was decreased in injured nerves isolated from scLRP1−/− mice, compared with injured nerves from scLRP1+/+ mice. Densitometry analysis confirmed that these results were significant (Fig. 7H). The greater decrease in MBP in injured nerves from scLRP1−/− mice may be multifactorial, and include the fact that axonal degeneration is more robust in scLRP1−/− nerves. However, at 20 days, the lower level of MBP observed in nerves isolated from scLRP1−/− mice also is interpreted as resulting from failure of regenerating myelinated axons to penetrate into the distal nerve segment.
Next, mechanical allodynia was measured using von Frey hairs for up to 19 days following PNL. scLRP1−/− mice exhibited statistically significant (p<0.05) allodynia during baseline testing (Fig. 8A). After nerve injury, scLRP1−/− mice developed robust mechanical allodynia, characterized by significantly lower paw withdrawal thresholds, compared with scLRP1+/+ mice (p<0.01). Mechanical allodynia was sustained in scLRP1−/− mice for 19 days, when the experiments were terminated. By contrast, scLRP1+/+ began to recover 15 days post-injury, as anticipated (Ulmann et al., 2008). By 19 days, thresholds in injured scLRP1+/+ mice were nearly equivalent to those recorded in uninjured and sham-operated scLRP1+/+ animals. Sham-operated scLRP1−/− mice had significant allodynia initially (the first week) but recovered to baseline levels 11 days after surgery.
Motor function was assessed by Rotarod testing. Prior to nerve injury, scLRP1−/− and scLRP1+/+ mice demonstrated equivalent motor function (Fig. 8B). PNL transiently decreased motor function in scLRP1+/+ mice; however, in scLRP1−/− mice, loss of motor function was more substantial and sustained (p<0.01). Sham-operated scLRP1+/+ and scLRP1−/− mice also initially demonstrated decreased motor function, but function was completely restored by 17 days (p<0.01; Fig. 8B). PNL typically does not induce sustained motor loss. These results suggest that abnormal Schwann cell-axonal interactions in scLRP1−/− mice may affect the function of diverse neuronal cells types in sciatic nerve injury.
scLRP1−/− mice demonstrate increased p38MAPK phosphorylation and activation of microglia in the spinal cord in response to PNL
p38 MAPK phosphorylation and microglial activation in the spinal dorsal horn have been identified as key biomarkers of neuropathic pain processing (Ji and Suter, 2007). To assess these biomarkers, we subjected scLRP1−/− and scLRP1+/+ mice to PNL. Spinal cords were harvested seven days later. Phospho-p38 MAPK (P-p38MAPK) and the microglial cell marker, CD11b/OX-42, were analyzed by immunofluorescence microscopy. In the uninjured spinal cords from scLRP1+/+ and scLRP1−/− mice, phospho-p38 MAPK was barely detectable (Fig. 9A, B), as anticipated. In the injured spinal cords from scLRP1+/+ mice, phospho-p38 MAPK was increased on the ipsilateral side, relative to the contralateral side, in lamina I through III; however, increased antigen also was observed in the contralateral dorsal horn (Fig. 9C). Phospho-p38 MAPK was substantially increased in both magnitude and distribution in the ipsilateral dorsal horn of injured scLRP1−/− mice (Fig. 9D). Robust signal extended from lamina I all the way to lamina VI, with extension into the contralateral spinal dorsal horn.
Figure 9.

Sustained pain-related behavior in scLRP1−/− mice is associated with phosphorylation of p38 MAPK (P-p38MAPK) and activation of microglia in the spinal dorsal horn. (A–D) Immunofluorescence for P-p38MAPK in the spinal dorsal horn of mice in uninjured (A,B) and injured (C,D) seven days after PNL. (E,F) Immunofluorescence of cd11b/OX-42 in the spinal dorsal horn after seven days of PNL injury. Images are representative of those obtained in studies with 3–4 mice per cohort. Each pair of images shown in panels A-F (A and B, C and D, E and F) is matched for exposure. Scale bar, 400 µm (G, H) Quantification by densitometry of P-p38MAPK and OX-42 in the ipsilateral and contralateral spinal dorsal horn. Signal intensity in the ipsilateral horn was significantly increased in scLRP1−/− mice compared with scLRP1+/+ mice (*p<0.05, n=4/group). Signal intensity also was significantly increased in the ipsilateral compared with the contralateral horn for each biomarker (*p<0.05). (I) Immunoblot analysis of P-p38 MAPK in spinal cord dorsal horn isolated seven days after PNL. Equal amounts of protein (50 µg) were loaded in each lane and subjected to SDS-PAGE. Total p38 MAPK was determined as a loading control. (J) Quantification of P-p38MAPK to T-p38MAPK ratio by densitometry in uninjured and injured spinal cord (n=3–4 mice) *p<0.05 comparing injury in each genotype.
OX-42 immunofluorescence also was increased in the ipsilateral dorsal horn of scLRP1−/− mice, 7 days after PNL, compared with scLRP1+/+ mice (Fig. 9E, F), suggesting extensive microglial cell proliferation, recruitment, and/or activation. Immunofluorescence intensity was quantitated by densitometry and is shown for phospho-p38 MAPK and OX-42 in Figs. 9G and 9H, respectively. Both markers were significantly increased in intensity in the ipsilateral horn in scLRP1−/− mice, compared with scLRP1+/+ mice (p<0.05).
To confirm the results of our immunofluorescence microscopy studies, tissue from the spinal dorsal horn was harvested 7 days after PNL and subjected to immunoblot analysis. Phospho-p38 MAPK was occasionally detected in uninjured spinal dorsal horn, in scLRP1−/− mice; however, the signal was extremely weak. Following PNL, phospho-p38 MAPK was readily detected and signal was significantly greater in scLRP1−/− mice, compared with scLRP1+/+ mice. Total p38 MAPK was unchanged (Fig. 9I). Immunoblots showing phopho-p38 MAPK and total MAPK were subjected to densitometry. The results are quantified and summarized in Fig. 9J.
To determine whether p38 MAPK was activated in microglia in the spinal cords of scLRP1−/− mice that were subjected to PNL, we performed dual-labeling immunofluorescence microscopy studies. Areas of phospho-p38 MAPK immunostaining (green) co-localized with microglial cell marker, OX-42 (red), suggesting that p38 MAPK was activated in microglia (Fig. 10A). However, phospho-p38MAPK immunoreactivity also occasionally co-localized with NeuN, a neuronal marker, as well (results not shown). These findings are consistent with previous observations regarding p38MAPK activation in the spinal cord (Svensson et al., 2008; Zhang et al., 2010).
Figure 10.

P-p38MAPK localizes to microglia in injured mice. (A) Double-label immunofluorescence microscopy of OX-42 (red) and P-p38MAPK (green) in the ipsilateral spinal dorsal horn in scLRP1+/+ and scLRP1−/− mice seven days after PNL. Co-localization of p38MAPK and OX-42 is shown in yellow. Cell nuclei were identified by Dapi (blue). Scale bar, 5 µm. (B) Mechanical allodynia was measured in mice prior to and after PNL by non-noxious probing of the hind paws with von Frey filaments. Mice were treated systemically with minocycline (40 mg/kg) or vehicle 4 h prior to PNL and daily for seven days after PNL (*p<0.05 comparing scLRP1+/+ mice treated with minocycline to scLRP1+/+ mice treated with vehicle; no significant differences between scLRP1−/− mice with and without minocycline; n=6/cohort).
Minocycline is an antibiotic known to inhibit microglial activation in injured spinal cords (Raghavendra et al., 2004). To demonstrate that microglial activation contributes to the sustained mechanical allodynia observed in scLRP1−/− mice, we treated scLRP1+/+ and scLRP1−/− mice with minocycline (40 mg/kg) by intraperitoneal injection 4 hours prior to PNL and then daily. Cohorts of scLRP1+/+ and scLRP1−/− mice were injected with vehicle, as a control. Mechanical allodynia was monitored daily prior to surgery and from 4–8 days after PNL. At day 4, both scLRP1+/+ and scLRP1−/− mice, which were treated with vehicle, demonstrated allodynia (Fig. 10B). Control scLRP1+/+ mice treated with minocycline showed significantly less allodynia, as anticipated. Although scLRP1−/− mice, which were treated with minocycline demonstrated a trend towards reduced allodynia, the drug was minimally effective at best and extensive allodynia remained throughout the testing period. We interpret these results to suggest that the more extensive damage associated PNL in the sciatic nerves of scLRP1−/− mice overwhelms the activity of this drug.
Discussion
Based mainly on in vitro studies, we have previously shown that membrane-anchored LRP1 orchestrates many Schwann cell activities that are observed in vivo, when the PNS is injured and Schwann cells become activated. These include activation of pro-survival cell-signaling pathways (Campana et al., 2006a) and promotion of cell migration (Mantuano et al., 2008; Mantuano et al., 2011). In this study, we have shown that in the uninjured rodent peripheral nerve, LRP1 deficiency in Schwann cells is associated with ultrastructural changes in both myelinating and non-myelinating Schwann cells. After peripheral nerve injury, nerve degeneration is accelerated and associated with substantial loss of Schwann cell viability, as might have been predicted by previous in vitro cell-signaling studies. Accelerated degeneration is associated wtih increased and sustained allodynia, more extensive loss of motor function, and apparently failed regeneration. scLRP1−/− mice demonstrate increased central sensitization in response to PNL, as evidenced by activation of p38 MAPK and microglial activation in the dorsal horn of the spinal cord. Overall, these studies indicate a pervasive role for LRP1 in regulating Schwann cell physiology and the response to peripheral nerve injury.
A striking abnormality was noted in the ultrastructure of non-myelinating Schwann cells and their interaction with pain-transmitting axons in uninjured nerves isolated from scLRP1−/− mice. Abnormal Remak bundles were characterized by 1) axons with mean diameters exceeding 2 µm; 2) improper separation of axons by Schwann cell cytoplasm; and 3) a greater frequency of Remak bundles in which a high number of axons are contained. The presence of large caliber axons in Remak bundles in nerves from scLRP1−/- mice may reflect incorporation of axons that are typically myelinated in wild-type nerves or axonal swelling due to improper ensheathment. That LRP1 deficiency regulates axonal sorting is plausible because LRP1 regulates β1 integrin levels on the cell surface of diverse cell types (Salicioni et al., 2004). Conditional deletion of β1 integrin in Schwann cells has profound effects on axonal sorting and myelination (Feltri et al., 2002). Dilation of axons within Remak bundles may be observed when there is insufficient Schwann cell trophic support (Chen et al., 2003). The presence of an increased number of axons in association with LRP1-deficient Schwann cells has not been explained; however, we hypothesize that the previously demonstrated activity of LRP1 as a regulator of Rho family GTPases may be involved (Mantuano et al., 2010). Rho GTPases are known to control Schwann cell cytoskeletal and plasma membrane dynamics required for development of correct axonal interactions and myelination (Melendez-Vasquez et al., 2004).
Schwann cells in Remak bundles are termed “first responders” to injury or disease in the PNS microenvironment (Griffin and Thompson, 2008). The altered Remak bundle structure we observed may underlie the presence of allodynia in scLRP1−/− mice in the absence of injury. Small C-fibers ensheathed by non-myelinating Schwann cells are high-threshold pain-producing neurons. If axons are improperly ensheathed by Schwann cells, then adjacent axons come into contact. This provides a mechanism for impulse conduction through the nerve membrane in either direction, allowing neighboring axons to “cross-talk” and engage in spontaneous ephaptic transmission (Sadjadpour, 1975; Ueda, 2008). Ultrastructural abnormalities in myelinated Schwann cells also may have contributed to the baseline changes in sensory processing in uninjured scLRP1−/− mice. Aδ fibers, which are myelinated, mediate a large proportion of withdrawal reactions to light-touch (Shir and Seltzer, 1990) and contribute to excessive sensitivity in human peripheral demyelinating and degenerative diseases (Pentland and Donald, 1994).
Following nerve injury, in each of the model systems we studied, evidence for more rapid and/or severe nerve degeneration was observed in scLRP1−/− mice. These anatomic changes were associated with more severe loss of nerve function. We recognize at least three candidate mechanisms by which LRP1 deficiency in Schwann cells may exacerbate responses to nerve injury and neuropathic pain. First, sciatic nerves in scLRP1−/− mice already exhibit lower thresholds prior to injury and therefore may respond to noxious stimuli in an exaggerated manner. Second, the accelerated degeneration of myelinated nerve fibers observed in scLRP1−/− mice may cause extensive modifications in neurolemma architecture. These changes increase sodium channel activity and turnover in the neural membranes, thereby contributing to sustained afferent hyperexcitability in injured nerves (Devor et al., 1993). Moreover, demyelination and subsequent physical crosstalk between noxious and innocuous fibers may lead to pathological pain-generating synapses involving Aβ-fibers (Ueda, 2006; Ueda, 2011). Third, indirect effects of myelinated fiber degeneration on neighboring Schwann cells in Remak bundles may explain the rapid onset and sustaining of exacerbated mechanical allodynia. Previously, it has been shown that Schwann cells in Remak bundles transition to a proliferating phenotype when exposed to degenerating myelinated fibers (Murinson et al., 2005), which coincides with induction of spontaneous c-fiber activity (Wu et al., 2002). Myelin and lipid products in the injured nerves of scLRP1−/− mice also may indirectly exacerbate the activity of C-fibers that are abnormally supported by LRP1-deficient non-myelinating Schwann cells.
LRP1 is released from cell-surfaces by α-secretases (Liu et al., 2009). The shed form of LRP1 contains the intact 515-kDa LRP1 α-chain and part of the ectodomain region of the 85 kDa β chain. Shed LRP1 is biologically active as a regulator of the function of both Schwann cells (Gaultier et al., 2008) and macrophages (Gorovoy et al., 2010). In the injured PNS, shed LRP1 counteracts the activity of pro-inflammatory cytokines known to activate nociceptors, including TNF-α and IL1-β (Gaultier et al., 2008). Loss of LRP1 in scLRP1−/− mice would be expected to decrease the pool of available shed LRP1 in the injured PNS, thereby de-repressing TNF-α and IL1-β from inhibition. These cytokines are known to induce central sensitization in the spinal dorsal horn (Schafers et al., 2003), which may underpin some of the allodynia associated with chronic neuropathic pain (Hathway et al., 2009). Evidence of central sensitization observed in scLRP1−/− mice included robust and sustained activation of p38 MAPK in the spinal dorsal horn and activation of spinal microglia. Thus, loss of shed LRP1 from the injured PNS may have contributed to hyperactivity of spinal dorsal horn neurons and may have modulated spinal cord synaptic transmission that is characterized by pain hypersensitivity. Lack of shed LRP1 also may have de-repress factors that induce demyelination in the periphery, such as lysophosphatidic acid, which has been shown to initiate sustained microglia activation (Fujita et al., 2007; Ueda et al., 2008).
Our ultrastructural studies of sciatic nerves that were injured for 20 days provided additional clues regarding sustained pain states in scLRP1−/− mice. In both the PNL and crush injury model systems, the presence of prominent onion bulb formations suggested that axonal regeneration was initiated but then failed in these mice (Thomas, 1970). Onion bulb formation is typically observed after repeated injury to the peripheral nerve of wild-type mice (O’Hara et al., 1986). Increased ectopic activity from aborted regenerating sprouts can maintain allodynia (Nachemson and Bennett, 1993). Aberrant axonal regeneration may contribute to allodynic states and spontaneous pain observed in chemotherapy-induced neuropathies and peripheral neuropathies, such as Charcot-Marie-Tooth disease type 1 and type 4 (Carter et al., 1998) and Guillain-Barré syndrome (Pentland and Donald, 1994). Because abnormalities in sciatic nerve function are sustained in scLRP1−/− mice, these mice may provide a model of chronic neuropathic pain states in humans.
Deletion of LRP1 selectively in Schwann cells represents a “loss of function” model system. However, based on our results, we offer some novel hypotheses regarding Schwann cell LRP1 as a target for therapeutics development in neuropathic pain. We propose that it may be possible to enhance LRP1 receptor activation as a pathway to further improve Schwann cell viability, capacity for migration, and secretion of mediators involved in the response to PNS injury. We previously described recombinant products, derived from α2-macroglobulin and MMP-9, which function as LRP1 agonists, activating cell signaling in Schwann cells (Mantuano et al., 2008; Mantuano et al., 2011). Similarly, derivatives of shed LRP1, by their capacity to regulate TNF-α and IL-1β (Gaultier et al., 2008), may limit central sensitization. A recent population-based genome-wide association study showed that an LRP1 SNP is associated with common migraine headache, linking LRP1 to migraine pathophysiology (Chasman et al., 2011). The molecular mechanism underlying this genetic association remains to be determined. We propose that the ability of LRP1 to couple endocytosis and phagocytosis of extracellular ligands and debris with cell signaling responses underlies many of the activities of LRP1 in Schwann cells, which may be exploited for therapeutics development to improve the PNS response to injury.
In summary, we have shown that LRP1 is a major determinant of normal structure of both non-myelinating and myelinating Schwann cell phenotypes. When LRP1 is deleted, Schwann cell physiology is altered and this is evident in PNS injury. Wallerian degeneration is accelerated, regeneration is aborted and pain states are exacerbated and sustained. Overall, these studies demonstrate a pervasive relationship between normal Schwann cell physiology and preventing of neuropathic pain.
Acknowledgements
This work was supported by NINDS R01 NS-057456, R01 NS-054671, P30 NS047101, NCRR 5P41RR004050-24 and NIGMS P41GM103412-24. The authors would like to thank Donna Hacelrode, Timo Meerloo and Ying Jones for excellent technical assistance.
Abbreviations List
- DAB
diaminobenzidine
- DRG
dorsal root ganglion
- ECM
extracellular matrix
- LRP1
LDL receptor-related protein-1
- MBP
myelin basic protein
- nm
non-myelinating
- PNL
partial nerve ligation
- PNS
peripheral nervous system
- PWT
paw withdrawal threshold
- TEM
transmission electron microscopy
Footnotes
Author contributions: All authors designed research and analyzed data; S.O., K.H., E.M., A.D., A.G.,T.I., M.P., and W.M.C. performed research; S.O., S.L.G., and W.M.C. wrote the paper; all authors reviewed and approved content.
The authors declare no competing financial interests.
References
- Akassoglou K, Kombrinck KW, Degen JL, Strickland S. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol. 2000;149:1157–1166. doi: 10.1083/jcb.149.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzouz M, Kennel PF, Warter JM, Poindron P, Borg J. Enhnacement of mouse sciatic nerve regeneration by the long chain fatty alcohol, N-Hexacosanol. Expt Neurol. 1996;138:189–197. doi: 10.1006/exnr.1996.0057. [DOI] [PubMed] [Google Scholar]
- Bolis A, Coviello S, Bussini Dina G, Pardini C, Prvitali SC, Malaguti M, Morana P, Del Carro U, Feltri ML, Quattrini A, Wrabetz L, Bolino A. Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Cahrcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. J Neurosci. 2005;25:8567–8577. doi: 10.1523/JNEUROSCI.2493-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge MB, Pearse DD. Transplantation strategies to promote repair of the injured spinal cord. J Rehabil Res Dev. 2003;40:55–62. doi: 10.1682/jrrd.2003.08.0055. [DOI] [PubMed] [Google Scholar]
- Campana WM, Myers RR. Erythropoietin and erythropoietin receptors in the peripheral nervous system: changes after nerve injury. Faseb J. 2001;15:1804–1806. doi: 10.1096/fj.00-0857fje. [DOI] [PubMed] [Google Scholar]
- Campana WM, Li X, Dragojlovic N, Janes J, Gaultier A, Gonias SL. The low-density lipoprotein receptor-related protein is a pro-survival receptor in Schwann cells: possible implications in peripheral nerve injury. J Neurosci. 2006a;26:11197–11207. doi: 10.1523/JNEUROSCI.2709-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana WM, Li X, Shubayev VI, Angert M, Cai K, Myers RR. Erythropoietin reduces Schwann cell TNF-alpha, Wallerian degeneration and pain-related behaviors after peripheral nerve injury. Eur J Neurosci. 2006b;23:617–626. doi: 10.1111/j.1460-9568.2006.04606.x. [DOI] [PubMed] [Google Scholar]
- Carter GT, Jensen MP, Galer BS, Kraft GH, Crabtree LD, Beardsley RM, Abresch RT, Bird TD. Neuropathic pain in Charcot-Marie-Tooth disease. Arch Phys Med Rehabil. 1998;79:1560–1564. doi: 10.1016/s0003-9993(98)90421-x. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chasman DI, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43:695–698. doi: 10.1038/ng.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G. Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci. 2003;6:1186–1193. doi: 10.1038/nn1139. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Yu WM, Strickland S. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209–233. doi: 10.1146/annurev.neuro.30.051606.094337. [DOI] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R, Angelides K. Na+ channel immunolocalization in peripheral mammalian axons and changes following nerve injury and neuroma formation. J Neurosci. 1993;13:1976–1992. doi: 10.1523/JNEUROSCI.13-05-01976.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Feltri ML, D’Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–123. [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita R, Kiguchi N, Ueda H. LPA-mediated demyelination in ex vivo culture of dorsal root. Neurochem Internat. 2007;50:351–355. doi: 10.1016/j.neuint.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Gaultier A, Wu X, Le Moan N, Takimoto S, Mukandala G, Akassoglou K, Campana WM, Gonias SL. Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J Cell Sci. 2009;122:1155–1162. doi: 10.1242/jcs.040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaultier A, Arandjelovic S, Li X, Janes J, Dragojlovic N, Zhou GP, Dolkas J, Myers RR, Gonias SL, Campana WM. A shed form of LDL receptor-related protein-1 regulates peripheral nerve injury and neuropathic pain in rodents. J Clin Invest. 2008;118:161–172. doi: 10.1172/JCI32371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg DS, McGee SJ. Pain as a global public health priority. BMC Public Health. 2011;11:770–775. doi: 10.1186/1471-2458-11-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovoy M, Gaultier A, Campana WM, Firestein GS, Gonias SL. Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J Leukoc Biol. 2010;88:769–778. doi: 10.1189/jlb.0410220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Thompson WJ. Biology and pathology of nonmyelinating Schwann cells. Glia. 2008;56:1518–1531. doi: 10.1002/glia.20778. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Marchionni MA, Reeves M, Coulaloglou M, Scherer SS. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci. 1996;16:6107–6118. doi: 10.1523/JNEUROSCI.16-19-06107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathway GJ, Vega-Avelaira D, Moss A, Ingram R, Fitzgerald M. Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain. 2009;144:110–118. doi: 10.1016/j.pain.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Clouthier DE, Hammer RE. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell. 1992;71:411–421. doi: 10.1016/0092-8674(92)90511-a. [DOI] [PubMed] [Google Scholar]
- Ide C. Peripheral Nerve Regeneration. J Neurosci Res. 1996;25:101–121. doi: 10.1016/0168-0102(96)01042-5. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci. 1999;22:402–410. doi: 10.1016/s0166-2236(98)01391-5. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Fleur M, Underwood JL, Rappolee DA, Werb Z. Basement membrane and repair of injury to peripheral nerve: defining a potential role for macrophages matrix metalloproteinases and tissue inhibitor of metalloproteinases-1. J Exp Med. 1996;184:2311–2326. doi: 10.1084/jem.184.6.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor related protein 1: unique tissue specific functions revealed by seledtive gene knoxckout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhang J, Tran H, Verbeek MM, Reiss K, Estus S, Bu G. LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol Neurodegener. 2009;4:17. doi: 10.1186/1750-1326-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantuano E, Inoue G, Li X, Takahashi K, Gaultier A, Gonias SL, Campana WM. The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of schwann cells by binding to low-density lipoprotein receptor-related protein. J Neurosci. 2008;28:11571–11582. doi: 10.1523/JNEUROSCI.3053-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantuano E, Jo M, Gonias SL, Campana WM. Low density lipoprotein receptor-related protein (LRP1) regulats Rac1 and RhoA recipricoally to control Schwann cell adhesion and migration. J Biol Chem. 2010;285:14259–14266. doi: 10.1074/jbc.M109.085126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantuano E, Henry K, Yamauchi T, Hiramatsu N, Yamauchi K, Orita S, Takahashi K, Lin JH, Gonias SL, Campana WM. The unfolded protein response is a major mechanism by which LRP1 regulates Schwann cell survival after injury. J Neurosci. 2011;31:13376–13385. doi: 10.1523/JNEUROSCI.2850-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier C, Parmantier E, Brennan A, Mirsky R, Jessen KR. Developing Schwann cells acquire the ability to survive without axons by establishing an autocrine circuit involving insulin-like growth factor, neurotrophin-3, and platelet-derived growth factor-BB. J Neurosci. 1999;19:3847–3859. doi: 10.1523/JNEUROSCI.19-10-03847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez-Vasquez CV, Einheber S, Salzer JL. Rho kinase regulates schwann cell myelination and formation of associated axonal domains. J Neurosci. 2004;24:3953–3963. doi: 10.1523/JNEUROSCI.4920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murinson BB, Archer DR, Li Y, Griffin JW. Degeneration of myelinated efferent fibers prompts mitosis in Remak Schwann cells of uninjured C-fiber afferents. J Neurosci. 2005;25:1179–1187. doi: 10.1523/JNEUROSCI.1372-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachemson AK, Bennett GJ. Does pain damage spinal cord neurons? Transsynaptic degeneration in rat following a surgical incision. Neurosci Lett. 1993;162:78–80. doi: 10.1016/0304-3940(93)90564-2. [DOI] [PubMed] [Google Scholar]
- Ohara S, Ikuta F. Schwann cell resonses during regeneration after one or more crush injuries to myelinated nerve fibres. Neuropath Appl Neurobiol. 1988;14:229–245. doi: 10.1111/j.1365-2990.1988.tb00884.x. [DOI] [PubMed] [Google Scholar]
- Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res. 2007;100:670–677. doi: 10.1161/01.RES.0000260204.40510.aa. [DOI] [PubMed] [Google Scholar]
- Pentland B, Donald SM. Pain in the Guillain-Barre syndrome: a clinical review. Pain. 1994;59:159–164. doi: 10.1016/0304-3959(94)90068-X. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–334. doi: 10.1038/sj.npp.1300315. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Loscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10:685–692. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- Rohlmann A, Gotthardt M, Willnow TE, Hammer RE, Herz J. Sustained somatic gene inactivation by viral transfer of Cre recombinase. Nat Biotechnol. 1996;14:1562–1565. doi: 10.1038/nbt1196-1562. [DOI] [PubMed] [Google Scholar]
- Sadjadpour K. Postfacial palsy phenomena: faulty nerve regeneration or ephaptic transmission? Brain Res. 1975;95:403–406. doi: 10.1016/0006-8993(75)90117-1. [DOI] [PubMed] [Google Scholar]
- Salicioni AM, Gaultier A, Brownlee C, Cheezum MK, Gonias SL. Low density lipoprotein receptor-related protein-1 promotes beta1 integrin maturation and transport to the cell surface. J Biol Chem. 2004;279:10005–10012. doi: 10.1074/jbc.M306625200. [DOI] [PubMed] [Google Scholar]
- Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003;23:3028–3038. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Shi Y, Mantuano E, Inoue G, Campana WM, Gonias SL. Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci Signal. 2009;2:ra18. doi: 10.1126/scisignal.2000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shir Y, Seltzer Z. A-fibers mediate mechanical hyperesthesia and allodynia and C-fibers mediate thermal hyperalgesia in a new model of causalgiform pain disorders in rats. Neurosci Lett. 1990;115:62–67. doi: 10.1016/0304-3940(90)90518-e. [DOI] [PubMed] [Google Scholar]
- Strickland DK, Gonias SL, Argraves WS. Diverse roles for the LDL receptor family. Trends Endocrinol Metab. 2002;13:66–74. doi: 10.1016/s1043-2760(01)00526-4. [DOI] [PubMed] [Google Scholar]
- Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS. Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J Biol Chem. 1990;265:17401–17404. [PubMed] [Google Scholar]
- Svensson C, Medicherla S, Malkmus S, Jiang Y. Ma JYRole of p38 mitogen activated protein kinase in a model of osteosarcoma-induced pain. Pharmacol Biochem Behavior. 90:664–675. doi: 10.1016/j.pbb.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Salzer JL. PARsing the events of myelination. Nat Neurosci. 2007;10:17–18. doi: 10.1038/nn0107-17. [DOI] [PubMed] [Google Scholar]
- Thomas PK. The cellular responses to nerve injury. J Anat. 1970;106:463–470. [PMC free article] [PubMed] [Google Scholar]
- Ueda H. Lysophosphadic acid as the initiator of neuropathic pain. Biol Pharm Bull. 2011;34:1154–1158. doi: 10.1248/bpb.34.1154. [DOI] [PubMed] [Google Scholar]
- Ueda H. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Therap. 2006;109:57–77. doi: 10.1016/j.pharmthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Ueda H. Peripheral mechanisms of neuropathic pain- involvement of lysophosphatidic acid receptor-mediated demyelination. Molec Pain. 2008;4:11. doi: 10.1186/1744-8069-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2×4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt A, Brady ST. Unwrapping new layers of complexity in axon/glial relationships. Glia. 2000;29:112–117. doi: 10.1002/(sici)1098-1136(20000115)29:2<112::aid-glia3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Wu G, Ringkamp M, Murinson BB, Pogatzki EM, Hartke TV, Weerahandi HM, Campbell JN, Griffin JW, Meyer RA. Degeneration of myelinated efferent fibers induces spontaneous activity in uninjured C-fiber afferents. J Neurosci. 2002;22:7746–7753. doi: 10.1523/JNEUROSCI.22-17-07746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WM, Yu H, Chen ZL. Laminins in peripheral nerve development and muscular dystrophy. Mol Neurobiol. 2007;35:288–297. doi: 10.1007/s12035-007-0026-x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Nei H, Dougherty PM. A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-alpha. J Neurosci. 2010;30:12844–12855. doi: 10.1523/JNEUROSCI.2437-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]