

Abstract
Numerous inbred mouse strains comprise models for human diseases and diversity, but the molecular differences between them are mostly unknown. Several mammalian genomes have been assembled, providing a framework for identifying structural variations. To identify variants between inbred mouse strains at a single nucleotide resolution, we aligned 26 million individual sequence traces from four laboratory mouse strains to the C57BL/6J reference genome. We discovered and analyzed over ten thousand intermediate-length genomic variants (from 100 nucleotides to 10 kilobases) distinguishing these strains from the C57BL/6J reference. Approximately 85% of such variants are due to recent mobilization of endogenous retrotransposons, predominantly L1 elements, greatly exceeding that reported in humans. Many genes’ structure and expression are altered directly by polymorphic L1 retrotransposons, including Drosha, Parp8, Scn1a, Arhgap15 and others including novel genes. L1 polymorphisms are distributed non-randomly across the genome, as they are excluded significantly from the X chromosome and from genes associated with the cell cycle, but are enriched in receptor genes. Thus, recent endogenous L1 retrotransposition has diversified genomic structures and transcripts significantly, distinguishing mouse lineages and driving a major portion of natural genetic variation.
INTRODUCTION
Inbred mouse strains form a foundation for mammalian genetics research. Hundreds of distinct lineages including well-known laboratory strains were generated from limited founders by repetitive crosses of highly related animals within the past 100–300 years. Individuals of a given strain are both virtually homozygous at all autosomal loci and isogenic (Beck et al. 2000). The power of mouse genetics research in part comes from naturally occurring genetic variation between different strains. Phenotypic differences between mouse lineages, such as disease susceptibility traits, behavioral differences and many other characteristics, are widely used to model human developmental and metabolic disorders, cancers, and many other diseases and traits (Beck et al. 2000).
Genome sequence assemblies have been completed recently for the mouse and other mammalian species. Large-scale resequencing projects have focused upon identification of certain forms of sequence variation, especially short variants such as single nucleotide polymorphisms (SNPs) (International HapMap Consortium 2005), that might account for functional differences between mammalian individuals or lineages. Such work has helped map a small number of quantitative trait loci, tabulated common variants associated with cancers and other diseases, and facilitated analysis of mammalian evolution (Conrad et al. 2006; Frazer et al. 2007; Wade and Daly 2005). More recently, longer structural variants have been identified, distinguishing human individuals and mouse sub-strains (Egan et al. 2007; Korbel et al. 2007; Mills et al. 2006). Several recent studies on human structural variation revealed that non-homologous end joining and endogenous transposition of retroelements have contributed mechanistically to most insertion or deletion (indel) changes between human genomes (Korbel et al. 2007; Mills et al. 2006).
Various classes of repetitive elements, mostly transposons, make up nearly half of the mammalian genomes assembled (Lander et al. 2001; Waterston et al. 2002). While some retrotransposon families are actively mobilized in mouse and human genomes (Kazazian 2004), occasionally resulting in disease-causing mutations (Chen et al. 2005) and various forms of genomic instability (Symer et al. 2002), their contributions to structural variation are largely unknown. Since transposons can introduce promoters, terminators, and alternative splice sites, and affect local chromatin structures (Belancio et al. 2006; Chen et al. 2006; Roy-Engel et al. 2005; Wheelan et al. 2005; Whitelaw and Martin 2001), their active mobilization in genomes is a likely determinant of transcriptional variation (Horie et al. 2007), and therefore at least some cases of phenotypic variation.
A comprehensive analysis of structural variation between classical inbred mouse strains has not been conducted to date, except for SNPs and certain copy number variants (CNVs). In this study, to identify intermediate-length structural variants between inbred mouse strains at extremely high resolution, i.e. single nucleotide resolution, we aligned individual sequence traces to the reference mouse genome using a fast and accurate new method. Virtually all sampled predictions were validated by specific polymerase chain reaction (PCR) assays. Surprisingly, most of the identified genomic variants between mouse strains were caused by recent mobilization of endogenous transposable elements, of which L1 retrotransposons were most active. Additionally, as described here, we found that a substantial number of these polymorphic transposons directly altered transcript structures and expression levels in corresponding mouse strains.
RESULTS
Most intermediate-size mouse structural variants are due to transposition
High-resolution data from whole genome shotgun (WGS) sequencing of four inbred mouse strains, A/J, DBA/2J, 129S1/SvImJ (henceforth, 129S1), and 129X1/SvJ (129X1) (Mural et al. 2002), whose genomes remain unassembled, were deposited recently at the National Center for Biotechnology Information (NCBI) trace archive (Mural et al. 2002; Wade and Daly 2005). To identify genomic variants distinguishing these strains, we downloaded ~26 million WGS sequence traces (cumulative length ~ 18 billion nucleotides, nt) and aligned them individually to the reference C57BL6/J (C57) genome assembly using GMAP. This software application was developed to map exons and therefore is well-suited to align genomic fragments with intervening breaks. It appeared to speed alignments over other applications such as Blat by 10–100-fold (R. M. Stephens, N. Volfovsky, unpublished data). We found that 73% of the individual sequence traces align unambiguously to the C57 reference genome with minimal or no variation (Fig. 1, Table 1, Supplementary Fig. 1). Many traces validate known SNPs and/or identify new ones, and show that significant portions of the compared strains’ genomes are non-polymorphic in pairwise comparisons (Wade et al. 2002). By contrast, others align to multiple repetitive elements or to no unique locus, identify short tandem repeat (STR) polymorphisms (N. Volfovsky et al., manuscript in preparation), and/or identify indel variants.
Figure 1. Discovery of structural variation between inbred mouse strains by WGS trace alignment.
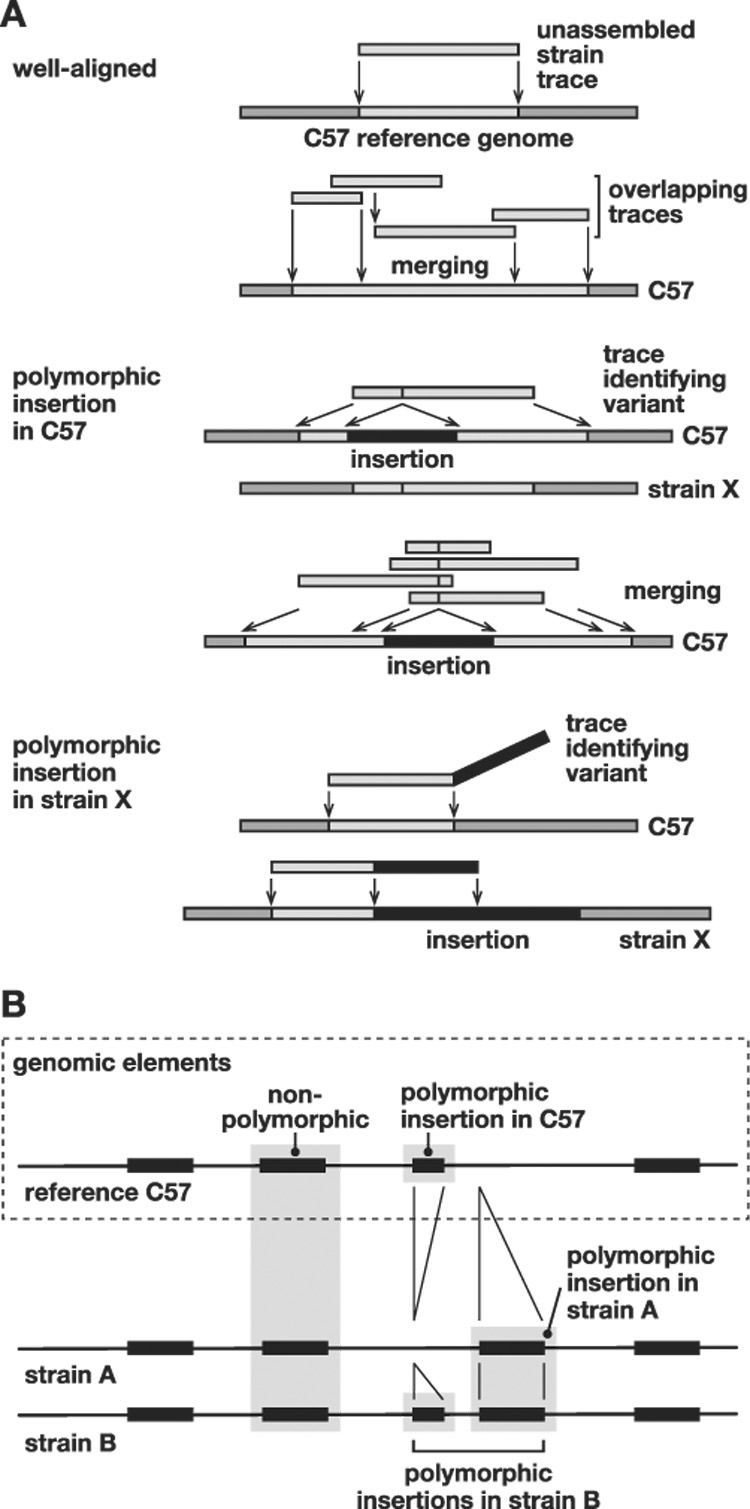
(A) Presented here is a schematic illustrating several types of alignments of WGS traces against the reference genome (Supp. Fig. 1). (Top) “Well-aligned” traces aligned almost completely at unique locations in the C57 reference genome (light gray). Overlapping sequences were merged into a contig. (Middle) “Polymorphic insertion in C57” traces identify an indel present in the C57 reference genome but absent from the genome of the trace’s source, unassembled strain X. The insertion (black) interrupts the trace’s alignment (light gray) with the reference sequence. Overlapping traces were merged, identifying a unique indel. (Bottom) “Polymorphic insertion in strain X” represents an indel present in that strain (black) but absent from the reference genome. The sequence trace from strain X aligns well, but only partially, to the reference genome; another contiguous part of the trace does not align to the reference genome, identifying an indel variant (Supp. Fig. 1).
(B) An example of aligned genomic features in reference C57 and unassembled strains A and B genomic sequences, identifying various intermediate-length elements including polymorphic, non-polymorphic and reference sequences such as L1 retrotransposons. A sequence called an insertion in one strain might alternatively be considered a deletion from another.
Table 1. Categorization and coverage of 26 million WGS sequence traces.
(A) WGS traces from four unassembled mouse strain genomes were aligned to the C57 reference using GMAP and a genome variation discovery pipeline. Resulting categories of alignment were tabulated for each alternative strain (see Supplementary Information). (B) A summary of the coverage by clustered sequence traces mapped to the reference assembly, whose size in mm8 release is 2,644,077,689 nt.
| A. Alignment categories of WGS sequence traces | |||||||
| Number of traces | |||||||
| Strain | 129S1 | 129X1 | A/J | DBA/2J | total | % total | |
| total | 1,461,249 | 5,621,095 | 11,094,880 | 7,667,299 | 25,844,523 | 100 | |
| minimal variation | 1,118,796 | 4,031,241 | 7,953,029 | 5,812,166 | 18,915,232 | 73.19 | |
| polymorphism in C57 | 2,376 | 7,731 | 16,742 | 12,465 | 39,314 | 0.15 | |
| polymorphism in str X | 67,263 | 619,002 | 1,075,807 | 401,187 | 2,163,259 | 8.37 | |
| B. Coverage by clustered sequence traces. | |||||||
| Strain | 129S1 | 129X1 | A/J | DBA/2J | C57 (reference) | ||
| % coverage | 27.19 | 64 | 82.08 | 75.33 | 98.6 | ||
| clustered coverage (nt) | 718,816,791 | 1,692,267,619 | 2,170,166,237 | 1,991,719,990 | 2,607,156,572 | ||
| gap size (nt) | 1,925,260,898 | 951,810,070 | 473,911,452 | 652,357,699 | 36,921,117 | ||
| cluster number | 758,235 | 1,063,806 | 688,928 | 897,162 | 1,324 | ||
| average cluster (nt) | 948 | 1,591 | 3,150 | 2,220 | 1,969,151 | ||
| average gap (nt) | 2,539 | 895 | 688 | 727 | 27,886 | ||
Upon merging overlapping individual WGS traces (Fig. 1a and Supp. Fig. 1), more than ten thousand intermediate-sized variants, ranging from 100 nt to 10 kb, are predicted by this analysis to be present in the C57 reference but absent from at least one of the other strain(s) (Fig. 1, Fig. 2 and Supplementary Table 1). We call such indel variants a “polymorphic insertion in C57” since they are present in the reference genome (Fig. 1) but absent from another strain. Even more variants were found present in at least one of the four unassembled strains but absent from the reference (“polymorphic insertion in strain X”). These latter variants are difficult to characterize without full genome assemblies, precluding their detailed analysis here. We do not wish to imply by this nomenclature that the polymorphisms’ mechanism of formation is known in all cases; an indel variant that we call an insertion in a given strain could alternatively have been deleted from another strain. All polymorphisms identified here were determined from comparisons with the reference C57 mouse genome. Our alignment procedures, categorization of WGS traces, and resulting sequence coverage for each strain are described in Fig. 1, Table 1, Supplementary Fig. 1, Supplementary Tables 1–2 and Supplementary Materials and Methods. Comprehensive data about the genomic variants distinguishing mouse strains, as discovered in this study, are available using PolyBrowse, our new genomic polymorphism query and display website at http://polybrowse.abcc.ncifcrf.gov/ (Stephens et al. 2008).
Figure 2. Intermediate-sized structural variation between mouse strains due to endogenous retrotransposition.
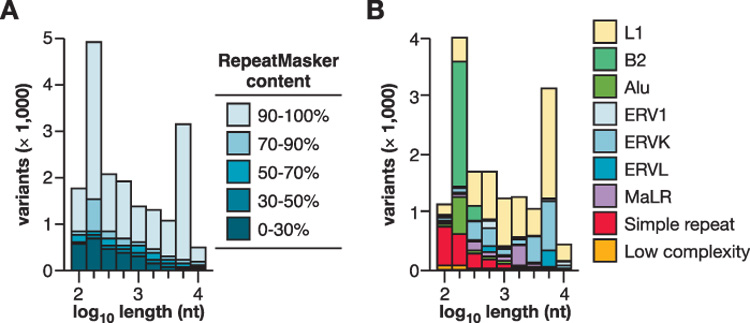
(A) Distribution of lengths of all variants identified here (ranging from 100 nt to 10 kb), predicted from WGS trace alignments. Each polymorphic integrant is present in C57 reference genome and absent from at least one of the unassembled strain(s). (Legend) The percentages indicate the relative composition of each variant, identified by RepeatMasker as repetitive element sequences.
(B) Classes of repeats in variants. Polymorphisms present in C57, containing >70% RepeatMasker content and ranging in length from 100 nt to 10 kb include: Alu, 5.4% of the total number of such variants; B2 SINEs, 19.2%; ERV1, 1.8%; ERV-K, 16.4%; ERV-L, 3.5%; L1, 38.8%; MaLR, 4.0%; simple repeats, 8.8%; and other, 2.0%. L1 retrotransposition is the most numerous cause of intermediate length variation between the strains.
Almost all such variants include at least 70% sequence content from various classes of repetitive elements (Fig. 2a), as identified by RepeatMasker (Smit et al. 2007). A large majority contains >90% transposon sequences per variant. Their length distribution is strikingly bimodal, matching transposons’ known structures in the mouse genome (Fig. 2). Of these transposon indels, L1 (LINE, Long Interspersed Element) retrotransposons are the most numerous. L1 integrants are frequently truncated from the 5’ end, but many others are full length (Symer et al. 2002). L1 polymorphisms contributed the most variant nucleotides to the strains’ genomes overall; their mean +/− standard deviation (SD) length is 1,130 +/− 590 nucleotides (nt). Other classes of active transposable elements, including short interspersed elements (SINEs, mostly B2 elements) and long terminal repeat-containing retrotransposons (e.g. ERV-K and MaLR elements), are also very frequently polymorphic between strains (Fig. 2b).
We tabulated a total of 666,328 “reference L1s” (each > 100 nt) in the haploid C57 reference genome using RepeatMasker (Smit et al. 2007), based on their evolutionary ages and structures (Fig. 1b and Supplementary Table 3). These counts are likely to be inexact because gaps remain in the reference genome assembly, currently 98.6% complete (Table 1). Remaining gaps frequently include highly repetitive sequences. Mouse Y chromosome sequences have not been assembled, and some transposons are “compound” elements contiguous to one another that cannot be counted unambiguously.
At least 127,803 L1 elements (19.2% of the total) are present in all four strains’ unassembled genomes and in C57, so we call them “non-polymorphic” (Fig. 1b). Notably, some of these may be fixed in all mouse lineages, but their presence has been determined only for the five inbred strains here. By contrast, at least 6,723 (1%) distinct elements are L1 polymorphisms in C57, i.e. present in the C57 reference and possibly other strains, but absent from at least one strain. We compared the absent or present status (A/P call) for all five inbred strains in 1,861 fully predicted cases out of 6,723 L1 polymorphisms. These pairwise comparisons confirmed that 129S1 and 129X1 strains are most similar, while A/J and DBA/2J are most divergent (Supplementary Table 4). These results corroborate both earlier phylogenetic analyses using SNPs and other genomic markers, and strains’ known breeding histories (Wade et al. 2002).
If a similar proportion of all reference L1s were polymorphic, then up to ~33,000 L1s would be absent from at least one of the four unassembled strains. Additionally, many thousands of other currently unknown L1 integrants, absent from the reference genome, are likely to be present in one or more of the unassembled mouse strains. Thus the analysis presented here substantially under-estimates structural variation including transposition-mediated variation between the strains.
To validate predictions of L1s present or absent in the strains, we arbitrarily selected a set of 31 L1 integrants for validation by polymerase chain reaction (PCR) (Table 2). This collection is an arbitrary sample of mouse L1s genome-wide, as we included 22 independent polymorphic L1s present in the C57 reference but absent from at least one of the other strains. Of these, 11 were chosen from several regions of chromosome 10, and others were picked at a frequency of approximately one per chromosome. The remaining 9 elements were chosen for validation based upon their activity in a screen for fusion transcripts (see below). PCR assays were run both across left and right junctions between L1s and flanking genomic sequences, and across empty and/or occupied genomic target sites. We required results from the three PCR tests to be self-consistent. Predictions from all but one of 78 individual WGS traces (99%) identifying empty target sites (where reference L1s are absent from a strain) were validated (Supplementary Table 5), suggesting very low error rates in trace sequencing and alignments, and minimal confounding by other forms of genomic variation such as copy number variants. A predicted integrant on chr. 17 could not be assayed in any strain, probably because its target site lies within an ancient element repeated in many genomic locations (Table 2).
Table 2.
Validation of L1 polymorphisms in classical and wild mouse strains.
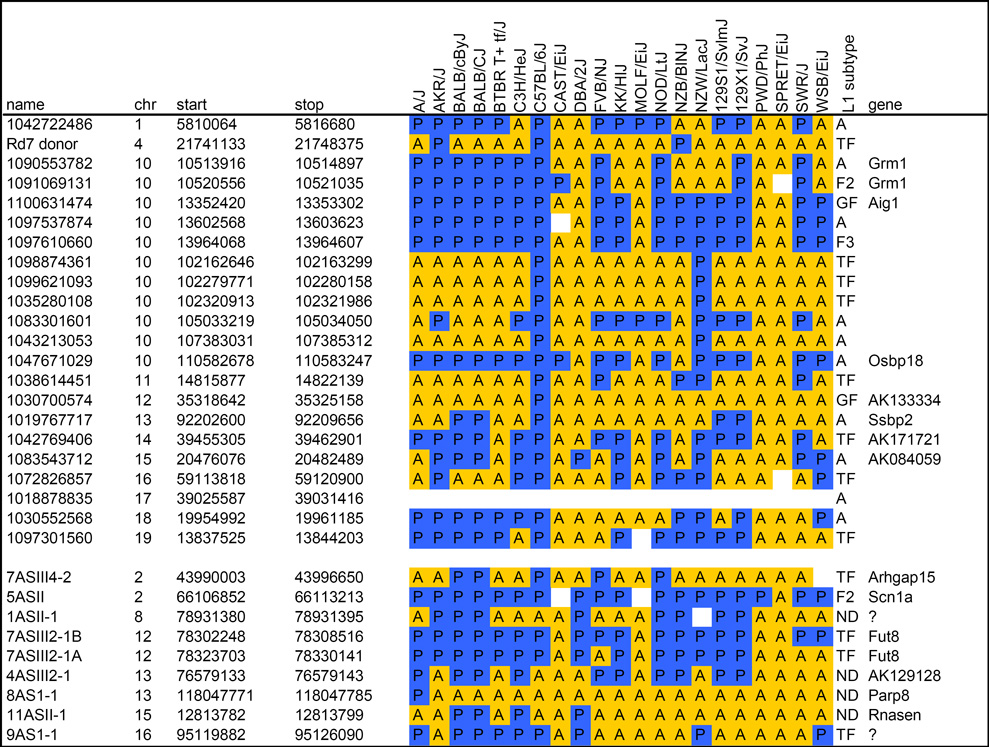 |
Top: Candidate L1 polymorphisms from chromosomes 10 and others were arbitrarily selected as described in the text for validation by PCR. Bottom: Nine putative L1 integrants from a screen of fusion transcripts were identified in unassembled strains by chromosome walking. PCR reactions across left and right genomic junctions and empty target sites validated presence (blue, P) or absence (yellow, A) of individual integrants, as predicted by WGS trace alignments for four unassembled strains. In a few cases, no PCR product was obtained (white), suggesting additional genetic variation in a strain, or suboptimal PCR design. Trace ID or cDNA clone names, chromosomal coordinates, spanning gene names, and L1 subtypes from RepeatMasker classification and Cross_Match re-classification are indicated (see Supplementary Materials and Methods).
We wanted to determine if more extensive genomic variation distinguishes other lineages. Therefore, the same L1 integrants were assayed by PCR in 16 additional mouse strains and related species that have been studied in large-scale SNP discovery and analysis projects (Table 2) (Frazer et al. 2007; Wade and Daly 2005; Yang et al. 2007). Strikingly, none of the 31 L1s assayed (0%) is present in SPRET/EiJ, although Mus spretus diverged from ancestors of the classical inbred strains approximately one million years ago, and our collection emphasized integrants known to be polymorphic among those laboratory strains. If we had assayed mostly non-polymorphic L1s, presumably some would be present at conserved loci in Mus spretus. Only 2/28 (7%) each are present in CAST/EiJ (Mus castaneus) and MOLF/EiJ (Mus molossinus), respectively, and 1/30 (3%) is in PWD/PhJ. For comparison, the overall contribution from the genomes of these ancestral strains to classical inbred mouse strains has been estimated to be 3% from CAST/EiJ, 10% from MOLF/EiJ and 6% from PWD/PhJ, illustrating that our collection approximates the genome-wide contributions of these ancestors estimated by SNP analysis (Frazer et al. 2007). However, in WSB/EiJ, a strain most closely related to Mus musculus domesticus (the common ancestor for a majority of classical mouse strain genomes) (Wade et al. 2002), only a small minority (10 out of 29; 34%) of the assayed L1 integrants is present. This value deviates substantially from expected contribution (68%) from Mus musculus domesticus to the classical inbred mouse strains (Frazer et al. 2007), but might be explained by the small sample size and non-random distribution of L1s assayed here (Table 2).
Although most of the integrants chosen for validation are polymorphic, three of the 31 validated integrants are non-polymorphic in the five strains. Of these, none are fixed in all 21 lineages (Table 2). Several integrants are present only in a few strains, suggesting that they integrated very recently in evolutionary time, quite possibly within the past few hundred years or less. This relatively rapid rate of genomic change is comparable with that reported for copy number variants, which have emerged within several hundred generations of inbreeding of C57BL6 sub-strains (Egan et al. 2007). While more than 19% of reference L1 elements are non-polymorphic in the five strains, a substantially smaller fraction likely will be non-polymorphic in all strains. These results are consistent with a recent analysis of SNPs in classical inbred mice, supporting their intrasubspecific origin (Yang et al. 2007). Additional WGS sequencing of divergent mouse species such as Mus spretus and Mus castaneus likely would identify fundamentally different patterns of transposon integrants and resulting differences in chromosome structures.
The chromosomal distributions of reference and polymorphic L1 retrotransposons were compared to genes and G/C-rich regions (Fig. 3). As expected, L1s are not uniformly distributed genome-wide, but tend to be located in gene-poor regions (Ostertag and Kazazian 2001). Strikingly, the mouse genome contains many more reference L1 elements than exons. Polymorphic L1s and exons contribute to similar extents (Fig. 3a). L1s are also enriched in A/T-rich genomic regions (Gasior et al. 2007). Variation in L1 polymorphism densities along chromosomes is not due simply to differences in WGS trace coverage (Supplementary Fig. 2, Supplementary Tables 2–3). We cannot analyze the Y chromosome since its coverage is minimal due to its composition of arrayed Huge Repeats.
Figure 3. Chromosomal distribution of mouse L1s and SNPs.
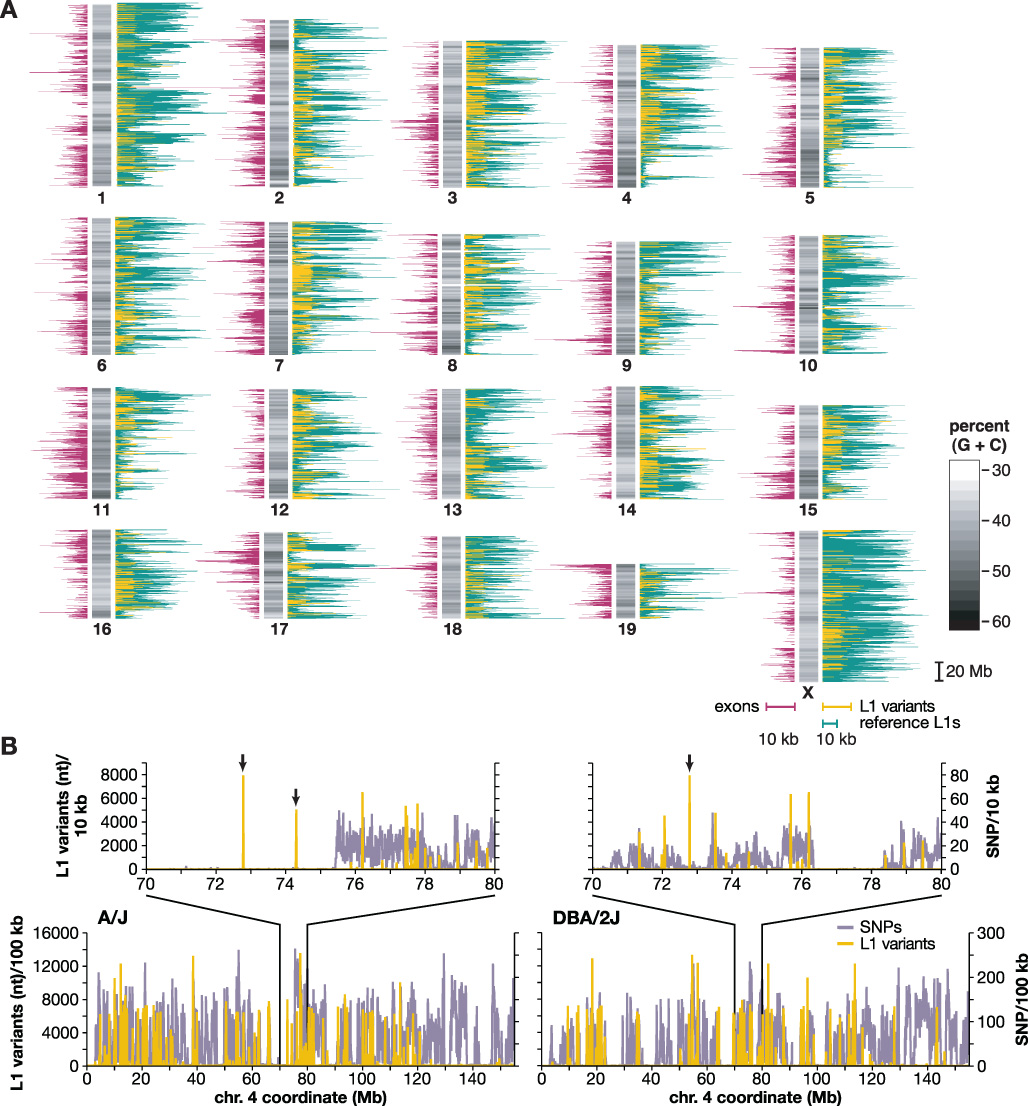
(A) A schematic mouse karyotype containing 19 autosomes and the X chromosome (vertical bars, center), indicates variable G:C content (grayscale). Darker shades indicate (G+C)-rich regions. Histograms display exon content (left, maroon), reference C57 strain L1 retrotransposons (right, green) and polymorphic L1s absent from an unassembled strain(s) (right, yellow), as nt per 100 kb genomic sequence (scale bars, 10 kb per 100 kb genomic sequence, below X chromosome).
(B) Densities of SNPs (lavender) and L1 variants (yellow) are compared between two strains each along chromosome 4 and (inset) at its coordinates 70–80 Mb. (left) A/J vs. C57 reference; (right) DBA/2J vs. C57, nt per 10 kb. Note that the nt scale differs between L1 (y-axis, left) and SNPs (right) by a factor of 100x. Polymorphic L1 integrants in chromosomal regions lacking SNPs in these pairwise comparisons are marked (arrows).
Compared with autosomes, the X chromosome has a significantly higher density of reference L1s (Fig. 3a and Table 3; p = 0), as expected (Ostertag and Kazazian 2001). Less purifying selection on the sex chromosomes would allow accumulation of deleterious L1s on chromosome X (Boissinot et al. 2001). Chromosome 11 contains a substantially lower density of reference L1s (Table 3a; p = 0).
Table 3. Non-random distribution of L1 retrotransposons on chromosomes and within genes.
(A) Chromosomal distribution. A total of 3,361,500 simulated “insertion events” were distributed randomly genome-wide, proportionally matching the relative lengths of chromosomes as expected. A total of 666,328 reference L1s were identified, of which 600,486 are on autosomes. A total of 6,723 polymorphic L1s were found, of which 6,484 are autosomal. Particularly significant enrichments or exclusions of reference or polymorphic L1 elements are highlighted (light gray). Numbers indicated for the Y chromosome are not reliable, due to its poor sequence coverage (dark gray).
(B) Distribution within annotated genes. Simulation again had 3,361,500 “insertion events” distributed randomly genome-wide. The p-value for the comparison of reference L1s inside genes, vs. simulated events inside genes, is < 1 × 10−100. The p-value for polymorphic L1s inside genes vs. simulated events inside genes is 2.13 × 10−83 (highlighted gray).
| (A) The chromosomal distribution of reference and polymorphic L1s is non-random. | ||||||||
| chromosome length | reference L1s | polymorphic L1s | ||||||
| chr. | length (nt) | % total | no. events, % total | fold change vs chr. length | p-value | no. events, % total | fold change vs reference | p-value |
| 1 | 197,069,962 | 7.45% | 7.63% | 1.02 | 3.25E-07 | 10.53% | 1.38 | 0.00E+00 |
| 2 | 181,976,762 | 6.88% | 6.38% | 0.93 | 4.96E-59 | 6.57% | 1.03 | 1.00E+00 |
| 3 | 159,872,112 | 6.05% | 6.65% | 1.10 | 1.41E-09 | 8.98% | 1.35 | 0.00E+00 |
| 4 | 155,029,701 | 5.86% | 5.75% | 0.98 | 4.22E-04 | 6.25% | 1.09 | 1.00E+00 |
| 5 | 152,003,063 | 5.75% | 5.18% | 0.90 | 1.23E-91 | 5.15% | 0.99 | 3.54E-01 |
| 6 | 149,525,685 | 5.66% | 5.82% | 1.03 | 3.38E-08 | 5.93% | 1.02 | 1.00E+00 |
| 7 | 145,134,094 | 5.49% | 5.34% | 0.97 | 2.19E-06 | 7.02% | 1.31 | 1.36E-06 |
| 8 | 132,085,098 | 5.00% | 4.48% | 0.90 | 2.92E-83 | 6.41% | 1.43 | 3.65E-06 |
| 9 | 124,000,669 | 4.69% | 4.11% | 0.88 | 3.63E-116 | 3.30% | 0.80 | 1.89E-07 |
| 10 | 129,959,148 | 4.92% | 4.74% | 0.96 | 1.63E-10 | 2.74% | 0.58 | 3.98E-18 |
| 11 | 121,798,632 | 4.61% | 3.47% | 0.75 | 0.00E+00 | 3.90% | 1.12 | 5.24E-02 |
| 12 | 120,463,159 | 4.56% | 4.53% | 0.99 | 1.00E+00 | 5.07% | 1.12 | 5.07E-01 |
| 13 | 120,614,378 | 4.56% | 4.47% | 0.98 | 5.47E-03 | 4.19% | 0.94 | 1.00E+00 |
| 14 | 123,978,870 | 4.69% | 5.03% | 1.07 | 4.72E-09 | 6.04% | 1.20 | 5.89E-06 |
| 15 | 103,492,577 | 3.91% | 3.93% | 1.00 | 1.00E+00 | 4.18% | 1.06 | 1.00E+00 |
| 16 | 98,252,459 | 3.72% | 3.82% | 1.03 | 1.07E-04 | 3.17% | 0.83 | 1.77E-01 |
| 17 | 95,177,420 | 3.60% | 3.31% | 0.92 | 7.21E-37 | 2.96% | 0.89 | 4.49E-02 |
| 18 | 90,736,837 | 3.43% | 3.41% | 0.99 | 1.00E+00 | 2.41% | 0.71 | 1.60E-05 |
| 19 | 61,321,190 | 2.32% | 2.07% | 0.89 | 1.26E-43 | 1.64% | 0.79 | 1.16E-03 |
| X | 165,556,469 | 6.26% | 9.76% | 1.56 | 0.00E+00 | 3.53% | 0.36 | 1.47E-22 |
| (Y) | 16,029,404 | 0.61% | 0.12% | 0.20 | 0.00E+00 | 0.03% | 0.25 | 3.26E-14 |
| total | 2,644,077,689 | 100.00% | 100.00% | 100.00% | ||||
| (B) The distribution of reference and polymorphic L1s inside genes is non-random. | ||||||||
| simulation | reference L1s | polymorphic L1s | ||||||
| location | no. events | % total | no. integrants | % total | no. integrants | % total | ||
| no gene | 1,268,578 | 37.74% | 314,532 | 47.20% | 3,358 | 49.95% | ||
| inside | 1,031,092 | 30.67% | 133,963 | 20.10% | 1,351 | 20.10% | ||
| 3 prime | 487,983 | 14.52% | 101,543 | 15.24% | 950 | 14.13% | ||
| 5 prime | 573,847 | 17.07% | 116,290 | 17.45% | 1,064 | 15.83% | ||
| Total | 3,361,500 | 100.00% | 666,328 | 100.00% | 6,723 | 100.00% | ||
By contrast, there are many fewer L1 polymorphisms on the X chromosome and chromosome 10, and increased numbers of L1 polymorphisms on chromosomes 1 and 3. Out of 600,486 autosomal L1s, 6,484 (1.08%) are polymorphic, while only 237 out of 65,038 L1s on the X chromosome (0.36%) are polymorphic (p = 1.47 × 10−22; Fig. 3a and Table 3a). The high density of L1s on the X chromosome, together with its paradoxical lack of L1 polymorphisms, could be due to prevention of or strong selection against new insertions, or selection for older ones. This apparent contradiction suggests that non-polymorphic L1s may play an important biological role there, perhaps in X inactivation (Lyon 1998).
We compared L1 variants and SNPs pairwise between the reference genome and A/J or DBA/2J, respectively. Such pairwise comparisons revealed that most polymorphic L1 integration sites coincide with SNP-dense regions (p < 1E-10; Fig. 3b and Supplementary Materials and Methods). A plausible explanation for this concordance between a large majority of L1 variants and SNP-dense regions is that most polymorphic transposon integration sites and flanking genomic sequences, co-inherited from distant ancestors, then diverged with a subsequent accumulation of SNPs. Alternatively, these two forms of genomic variation might be expected to coincide in those chromosomal regions where such changes can be tolerated. While independent polymorphic L1s are substantially less numerous than SNPs (Frazer et al. 2007), they contain at least a thousand-fold more nucleotides per variant (Fig. 3b).
Importantly, occasional L1 variants integrated into genomic regions without apparent SNPs, so-called “identical by descent” (IBD) (insets, Fig. 3b). However, such transposon integrants clearly have caused substantial local variation despite lack of SNPs. Screening for polymorphic transposons might provide a powerful new way to genotype mouse strains and other mammalian species, particularly in IBD regions with few or no SNPs available (2005; Yang et al. 2007).
Several structural features of polymorphic L1s are consistent with their young evolutionary ages. In contrast with both reference and non-polymorphic elements, polymorphic L1s have a bimodal length distribution with a significantly increased number of long, full-length elements (Fig. 4). They also more frequently have target site duplications (TSDs) and poly(A) tails, and when present, their TSDs and poly(A) tails are significantly longer than those of reference or non-polymorphic L1s (Supplementary Fig. 3). Polymorphic L1s also have a canonical target site preference, lower nucleotide substitution rate, and more frequently are classified as young, active L1 subfamily members (Supplementary Table 6). These results strongly suggest that such genomic integrants are bona fide products of recent retrotransposition (Symer et al. 2002).
Figure 4. Polymorphic L1s are bona fide products of recent retrotransposition.
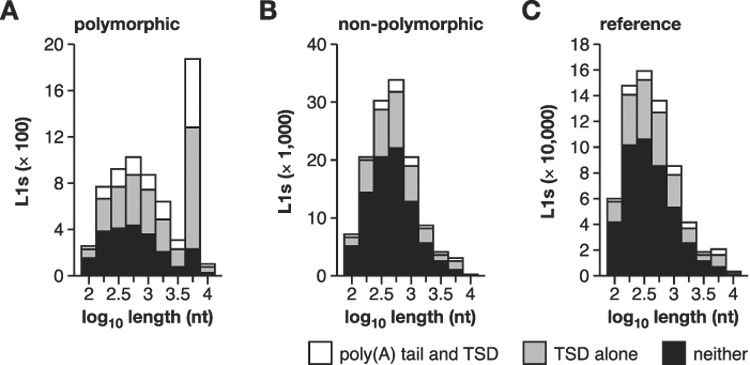
The length distribution of (A) polymorphic L1s (absent from at least one of the unassembled strains); (B) non-polymorphic L1s (present in all five strains); and (C) reference L1s (present in the C57 genome) is presented for elements with both a poly(A) tail and TSD (white); TSD alone (light gray); or neither (black). Polymorphic L1s are much more likely to be full-length and to have both a poly(A) tail and TSD. See Supp. Fig. 3 and Supp. Table 6.
Three young L1 subfamilies are currently active in mouse; some members of these active subfamilies have caused murine diseases by insertional mutagenesis. Ranked by their occurrence in the reference genome, these are TF, A and GF (Goodier et al. 2001; Naas et al. 1998; Ostertag and Kazazian 2001; Saxton and Martin 1998). Similarly, a majority (59%) of polymorphic L1s are products of retrotransposition by young, active donors, i.e. TF (28%), A (23%) and GF (8%) subfamily members (Supplementary Table 3).
These results collectively show that polymorphic L1s are substantially younger than other L1s in the mouse genome. However, L1 polymorphisms typically are localized in high-density SNP regions (Fig. 3b), suggesting their localization and co-inheritance within divergent ancestral blocks (Wade et al. 2002). Clearly, determination of the ages and evolutionary relationships of individual transposon integrants and other genomic variants along chromosomes in different strains will require further investigation.
Multiple forms of transcriptional variation have been linked previously with transposons, which may contribute cryptic or alternative promoters, terminators and/or splice sites, affect RNA polymerase processivity, trigger altered chromatin conformations, mediate homologous recombination and/or template small RNA expression (Belancio et al. 2006; Ostertag and Kazazian 2001; Speek 2001; Wheelan et al. 2005; Yang and Kazazian 2006). However, the extent of transcriptional variation due to endogenous transposition is not known.
Nearly half (53%) of both non-polymorphic and polymorphic L1s are located within 100 kb of annotated RefSeq genes. Approximately 20% of both reference L1s and L1 variants occur inside transcription units, representing a significant bias against L1 integrants within genes, since 28–30% of the mouse genome is comprised of annotated RefSeq genes including introns (An et al. 2006) (Table 3b). Presumably this relative exclusion of L1 elements from genes reflects selection against them, or less likely, their non-random integration into intergenic regions.
Of the non-polymorphic L1s within introns, approximately 68% are oriented antisense to the open reading frame (ORF; Supplementary Table 7). A smaller majority (58%) of polymorphic L1s are antisense within genes. An antisense orientation bias also was observed for de novo L1 integrants within genes in cultured human cells (Symer et al. 2002). By contrast, both non-polymorphic and polymorphic L1s within an interval of 100 kb upstream or downstream of genes occur in both orientations (Supplementary Table 7), suggesting a neutral orientation preference during retrotransposon integration per se, as expected (Gilbert et al. 2005). Presumably the observed orientation bias within genes is due to positive selection upon antisense elements or negative selection upon sense integrants (Boissinot et al. 2001). The smaller majority of antisense polymorphic L1s within genes may reflect selection over a shorter period of time upon these evolutionarily younger integrants.
To find L1s associated with transcriptional variation in mouse strains, we screened pooled testis cDNA libraries for fragments of L1 TF sequences. This approach allowed us to discover a new antisense promoter active within many full-length, young L1s (Li et al. 2008). In an initial survey, a diverse collection of spliced, polyadenylated L1-gene fusion cDNAs, initiated by L1 elements in various gene introns or in intergenic regions, was identified (Supplementary Table 8). Their corresponding antisense L1 templates are polymorphic, but non-polymorphic elements also can be expressed (Li et al. 2008). Approximately half are present in the C57 genome, while others are absent (Table 2 and Supplementary Table 8). The latter putative L1 integrants were identified in other strains’ genomic DNA by chromosome walking from expressed exons into adjacent introns. Each unknown L1 integrant’s genomic flanks were sequenced, revealing canonical TSDs and a poly(A) tail. Once identified, the presence or absence of each L1 template was determined by PCR in all 21 lineages. In one case, a polymorphic L1 is present exclusively in the A/J lineage but none of the others, suggesting that it integrated very recently (Table 2).
To verify that fusion transcripts are present exclusively in strains containing a putative genomic L1 template, we analyzed total RNAs isolated from adult male testes from the five strains. For example, fusion transcripts of L1-Drosha, L1-Parp8 and an L1-novel gene were identified only in strains with relevant antisense L1 polymorphisms present (Fig. 5). Similarly, other fusion transcripts were detected only in strains with corresponding L1 templates, including a chimeric transcript from the L1-ArhGAP15 locus (Table 2, Supplementary Fig. 2, Supplementary Table 8).
Figure 5. Transcriptional variation due to L1 variants.
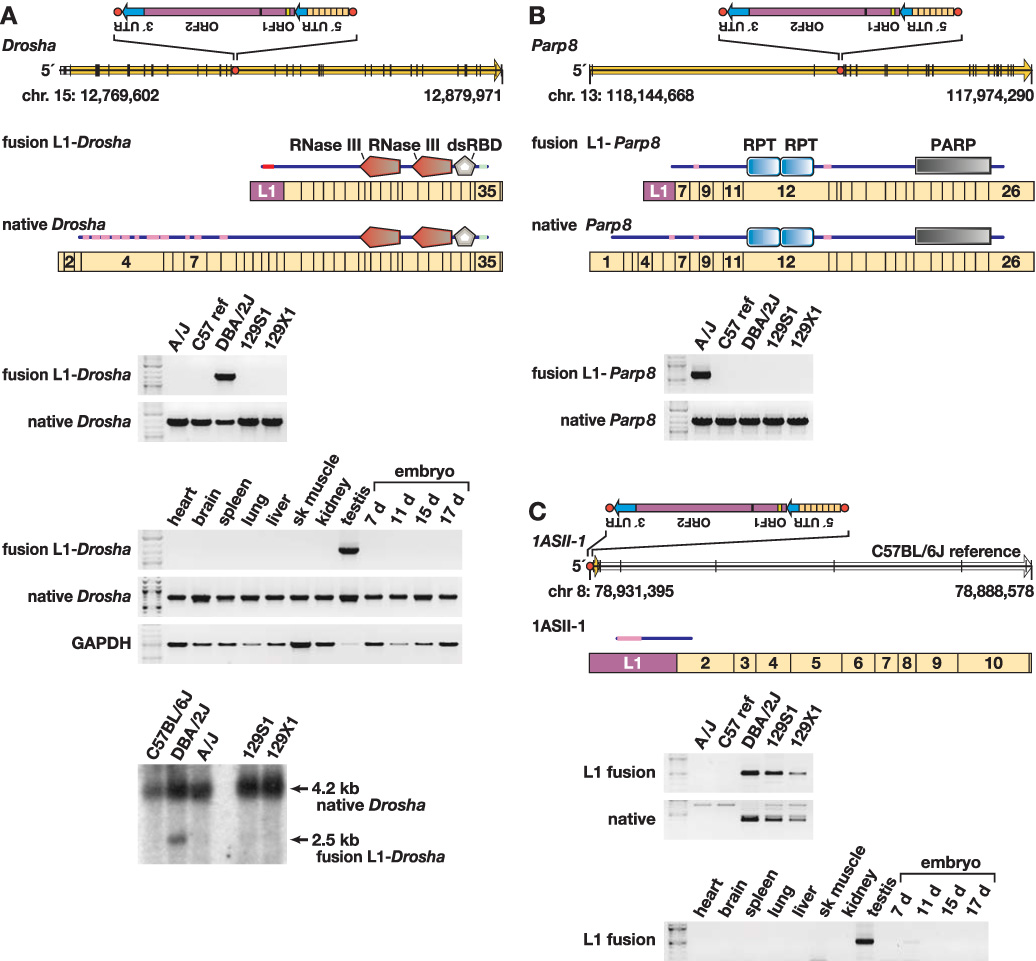
(A) Top: Genomic structure of Drosha (Rnasen) on mouse chromosome 15 (Feb., 2006 assembly), presented left to right (5’ to 3’), with exons (black vertical lines); open reading frame (ORF) (yellow arrow); intronic, antisense L1 polymorphism including its 5’ untranslated region (5’ UTR), ORF-1 and ORF-2, and 3’ UTR (inset); and L1 target site (red dot) as indicated. The L1 target site sequence, presented in the orientation of Drosha, is 5’-TCGCGCTTTGGCTTCTTT. Also presented are fusion L1-Drosha and native Drosha spliced, poly(A)+ transcripts structures including relative lengths and numbers of Drosha (tan rectangle) and antisense L1 (purple) exons. Above each transcript is a schematic indicating predicted translation products (from start to stop codons) including RNaseIII and double stranded RNA binding domains, and low complexity (pink) and coiled-coil (light blue) domains (annotated by SMART program). Middle: Reverse transcriptase-mediated PCR (RT-PCR) assay for fusion L1-Drosha and native transcripts in total RNA from five mouse strain testes, and assay for fusion L1-Drosha transcript from Balb/cJ tissues as indicated. Bottom: Northern blot probed for Drosha transcripts, indicating fusion L1-Drosha expression only in DBA/2J mice.
(B) Top: Genomic structure of Parp8 on the minus strand of chromosome 13, including genomic features as in (A). The L1 target site sequence, in the orientation of Parp8, is 5’-CCTCCGACGTTAAAG. Also presented are fusion L1-Parp8 and native Parp8 spliced, poly(A)+ transcripts, including relative exon lengths (tan rectangles), numbers and the antisense L1 exon (purple). Above each transcript is a schematic indicating predicted translation products including internally repeated (RPT) and poly(ADP-ribose) polymerase (PARP) catalytic domains, and low complexity (pink) domains (SMART). Bottom: RT-PCR assay for fusion L1-Parp8 and native transcripts.
(C) Top: Genomic and transcript structures for 1ASII-1, a novel, spliced transcript initiated by a polymorphic L1 on the minus strand of chromosome 8, including genomic features as in (A). The L1 target site sequence, presented in the sense orientation of 1ASII-1, is 5’-GACGTATAGACAAGAA. Also presented is poly(A)+ transcript 1ASII-1 (open arrow), including its relative exon lengths (tan rectangles), numbers and the antisense L1 exon (purple). Above it is a schematic indicating predicted translation products with low complexity (pink) domain as indicated (SMART program). Bottom: RT-PCR assay for fusion L1-1ASII-1 and native (lacking the L1 exon) transcripts and for the fusion L1 transcript in Balb/cJ tissues as indicated. This L1 variant initiates transcription in testis and 11 d embryo, only in strains containing the variant.
Fusion L1 transcripts are exemplified by the L1-Drosha fusion transcript, which is expressed at ~30% of the level of native Drosha in testis (Fig. 5a). This transcript contains both translation start and splice donor sites from L1, and is spliced in-frame with downstream exons encoding catalytic domains of Drosha (also called Rnasen), an RNaseIII gene centrally involved in microRNA biosynthesis (Murchison and Hannon 2004). Similarly, an L1-Parp8 fusion transcript also is predicted to be in-frame, and its open reading frame contains most functional domains of Parp8 (Fig. 5b). As a control, an assay for readthrough transcripts for the canonical genes, from which L1 polymorphisms are spliced out with usual introns, showed comparable expression levels. Remarkably, a novel, spliced transcript 1ASII-1 is promoted by a polymorphic L1 (Fig. 5c) in a genomic region where no cDNA or expressed sequence tag (EST) had been reported previously.
No appreciable fusion L1-Drosha transcript was identified by reverse transcriptase-mediated (RT-) PCR in non-gonadal tissues (Fig. 5a). By contrast, the novel fusion transcript 1ASII-1 was detected both in testis and 11 day-embryo tissues (Fig. 5c). We speculate that mechanisms such as transcriptional or post-transcriptional gene silencing, position effects, and/or availability of tissue-specific transcription factors may contribute to variable expression and control of particular transposon integrants in different developmental states (Whitelaw and Martin 2001). These and other fusion transcripts may encode protein variants or noncoding RNAs with regulatory or other functions.
We asked what proportion of endogenous L1 variants might contribute to transcriptional variation in the strains. Therefore, we screened adult testis total RNA samples for more L1 fusion transcripts. Out of 205 full-length, antisense L1 polymorphisms predicted inside RefSeq genes in the C57 genome, an arbitrary sample of 68 was screened. Of these, 13 (19%) drive fusion L1-gene transcripts, including 40% of the TF polymorphisms tested (Supplementary Table 9) (Li et al. 2008). Additionally, fusion L1-Arhgap15 transcription was identified in another screen (Table 2, Supplementary Table 9, and Supplementary Fig. 2b). Notably, two distinct intronic L1 polymorphisms occur in Grid2 in different strains, but only one drives expression of a fusion L1-Grid2 transcript while the other does not (Supplementary Table 9). Thus, we speculate that both polymorphic and non-polymorphic L1s may initiate additional transcripts in testes or other tissues, developmental stages and/or disease states such as cancers.
Another way by which L1 variants can affect tissue-specific gene structure and expression (Fig. 6) is illustrated by the rd7 mouse model of retinal degeneration (Chen et al. 2006). A de novo insertion of a full-length antisense L1 into exon 5 of Nr2e3 disrupts that gene’s normal transcription and splicing. Its donor itself is polymorphic, present only in C57, NZB/BinJ, and AKR/J out of the 21 strains tested (Table 2), thereby providing the first example of a “hot” endogenous mouse L1 that actively retrotransposed from its chromosomal location (Brouha et al. 2003). Thus, other full-length, polymorphic L1s also may be highly active donors in vivo.
Figure 6. Genomic and transcriptional variation due to endogenous transposition.
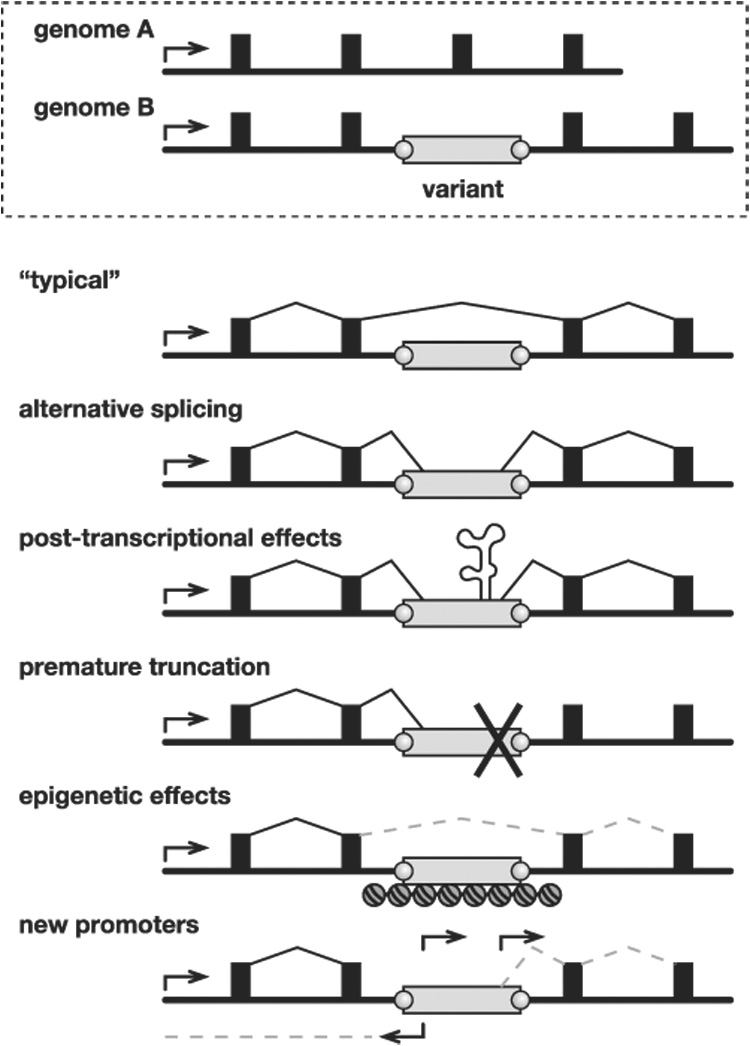
Top: Schematic of allelic variants A and B at a genomic locus including a promoter (arrow), exons (filled boxes), introns (underlying black line), and a polymorphic transposon integrant (open rectangle, genome B) with target site duplications (grey circles). Bottom: Possible forms of transcriptional variation due to a transposon integrant. (“Typical” transcript) Because transposons are ubiquitous, a typical transcript might lack an intronic integrant by splicing between exons. (Alternative splicing) Transcripts might include portions of transposon integrants due to their internal splice donor and splice acceptor sites. (Post-transcriptional effects) Transposon sequences may introduce autoregulatory elements affecting RNA stability, intracellular compartmentalization, etc. (Premature truncation) Similar to alternative splicing, except that transcripts end prematurely due to a transcription terminator in the transposon. (Epigenetic effects) Read-through transcription may be repressed by heterochromatin, DNA methylation and/or other epigenetic controls at transposon integrants. (New promoters) Gene expression and structure may be altered by introduction of new sense and/or antisense promoters in transposon integrants. This is the main form of transcriptional variation described in this report.
Ontology analysis (Mi et al. 2005) of annotated genes containing L1 polymorphisms showed a significant exclusion from certain categories of genes, including genes associated with cell cycle, nucleic acid metabolism and oncogenesis (Table 4, Supplementary Table 10). By contrast, L1 polymorphisms are significantly enriched in the receptors category of molecular functions, suggesting that these genes generally may tolerate added structural or transcriptional variability mediated by transposon integration events. Non-polymorphic L1s and reference L1s were enriched significantly in brain-associated genes along with other ontological categories (Supplementary Table 10). A recent, high resolution analysis of copy number variation between mouse strains revealed that these structural variants also are excluded from similar groups of mouse genes required in fundamental cellular processes, e.g. those involved in cell cycle and nucleic acid metabolism (Cutler et al. 2007).
Table 4.
Exclusion or enrichment of polymorphic L1s within genes in various ontological categories.
| (A) Polymorphic L1s vs random simulation: biological processes | ||||
| biological process | intronic L1 polymorphism, % | random simulation, % | fold-change | p-value |
| Nucleoside, nucleotide and nucleic acid metabolism | 8.44 | 13.42 | 0.63 | 3.34E-07 |
| Cell cycle | 1.88 | 4.38 | 0.43 | 1.63E-05 |
| Oncogenesis | 1.13 | 2.57 | 0.44 | 5.39E-03 |
| (B) Polymorphic L1s vs reference L1s: biological processes | ||||
| biological process | intronic L1 polymorphism, % | reference L1, % | fold-change | p-value |
| Cell cycle | 1.88 | 3.96 | 0.47 | 4.32E-04 |
| (C) Polymorphic L1s vs random simulation: molecular functions | ||||
| molecular function | intronic L1 polymorphism, % | random simulation, % | fold-change | p-value |
| Receptor | 16.14 | 10.71 | 1.51 | 3.55E-08 |
| Nucleic acid binding | 6.26 | 10.10 | 0.62 | 1.41E-05 |
| (D) Polymorphic L1s vs reference L1s: molecular functions | ||||
| molecular function | intronic L1 polymorphism, % | reference L1, % | fold-change | p-value |
| Receptor | 16.14 | 12.28 | 1.31 | 6.54E-04 |
Annotated genes containing 1,327 distinct intronic L1 polymorphisms were identified. They were assigned to top-level ontological categories (including biological processes and molecular functions) using Gene Ontology (GO) Panther software. Because many genes are included in more than one ontological category, a total of 2,184 assignments were made for these L1 polymorphisms. Only significant differences in ontological categories are listed. Additional information about non-polymorphic and reference L1 elements is presented in Supplementary Table 10.
(A), (B) In silico simulations resulted in 2,045,793 “integrants” that are distributed randomly across the reference mouse genome, within annotated genes. As expected, they are distributed proportionally according to gene and chromosome lengths. Their annotated (A) biological processes and (B) molecular functions were determined. The frequency of integrants within each category was calculated as the ratio of the count of integrants divided by the total number of integrants (1,327 polymorphic L1s or 2,045,793 simulated integrants, respectively). Because more than one ontological category can be assigned to a given gene, the sum of these frequencies for all top-level ontological categories exceeds 100%. P-values were calculated using the binomial statistic and are adjusted based upon the Bonferroni correction. Only statistically significant differences in ontological categories (corrected p-values < 0.01) are listed here.
(C), (D) Since reference L1s are non-randomly distributed in the genome, and as they comprised the basis for identification of most polymorphic L1s described here, we compared the ontological categories of polymorphic L1 genes against reference L1 genes. Their annotated (C) biological processes and (D) molecular functions were determined.
DISCUSSION
In this comprehensive study of intermediate length structural variants that distinguish different inbred mouse strains, we found that a large majority was caused by endogenous retrotransposition, predominantly by L1 retrotransposons. Other classes of active retrotransposons, including LTR elements and SINEs, also have caused substantial variation between the strains (Fig. 2). These variants, which could become a useful adjunct to SNPs and STRs in genotyping studies, can be accessed in detail using the mouse PolyBrowse website (Stephens et al. 2008). While we identified over ten thousand independent variants (Fig. 2), their total numbers do not remotely approximate 8.3 million SNPs identified to date in 16 classical and wild strains (Frazer et al. 2007). Nevertheless, summation of their cumulative lengths (Fig. 2) strongly suggests that these variants have altered millions of nucleotides genome-wide, affecting the structures of perhaps hundreds of genes. Recently, a similar scope of structural variation has been attributed to copy number variation between mouse strains (Cutler et al. 2007).
The extent of recent endogenous transposition in causing structural variation between mouse strains also appears to be substantially larger than that in humans, where non-homologous end joining appears to have been a predominant mechanism for generating variation (Korbel et al. 2007; Levy et al. 2007; Mills et al. 2006). The reasons for this striking difference are unclear, since human L1 retrotransposons (which mobilize LINEs, SINEs and SVA elements) paradoxically are more active than mouse L1s in tissue culture assays (Han and Boeke 2004). Moreover, their overall content in the human genome exceeds that in mouse (Lander et al. 2001; Waterston et al. 2002). Determination and comparison of the rates of structural variation by endogenous retrotransposition and by other mechanisms (Egan et al. 2007; Korbel et al. 2007) in mouse, man and other species will require additional study.
In this study, we used GMAP (Wu and Watanabe 2005) in a new way to align individual sequence traces to the C57 reference genome assembly (Fig. 1 and Supplementary Fig. 1). It is important to note that this alignment procedure, while fast and accurate, is also very stringent, as many additional polymorphisms are likely to remain uncounted. For example, variants in genomic regions with low sequence trace coverage were not counted here. If by chance single sequence traces did not span a variant substantially on both sides, that variant would not be counted. Moreover, polymorphisms that are present in an unassembled genome but absent from the C57 reference genome were not fully identified here. In an effort to describe the complete extent of variants existing between strains, we currently are comparing classes of variants that can be identified by different methods including mate pair alignments (Dew et al. 2005), and documenting many more novel variants present in strains with unassembled genomes.
The genomes of more distantly related mouse species such as Mus spretus are likely to be even more distinct from the classical strains analyzed here, due in large part to consequences of active endogenous transposition. As shown in Table 2, not a single one of the arbitrary, polymorphic L1 retrotransposons that we assayed is present in the Mus spretus genomic DNA, suggesting that a major component of its genomic architecture (likely corresponding to many thousands of elements, on average approximately 1 kb long) is fundamentally different from that in its relatives. It is possible that such non-coding genomic compartments, outside of conserved exons, have been shaped differentially by endogenous transposition, but might contribute nevertheless to important biological differences between species, since their coding exons are expected to be extremely similar.
A substantial fraction of L1 variants directly affect neighboring gene expression and structures in a range of tissues, possibly contributing to functional differences between strains (Muotri et al. 2005). However, we presume that a majority of both polymorphic and non-polymorphic L1s still do not significantly affect expression of overlapping or nearby genes in most tissues (Supplementary Table 9), as we do not anticipate large differences between strains in the structure or expression of most genes. We cannot exclude the possibility that polymorphic transposons, in many cases, may cause subtle differences in the expression and structures of many genes (Han et al. 2004). It will be of great interest to compare transcriptomes in various mouse species with very distinctive genome structures, for example using gene expression microarrays or ultra-high-throughput sequencing, to elucidate the relationship between structural variation and transcriptional variation more fully (Stranger et al. 2007).
Many of the novel fusion L1 transcripts that we identified reflect altered gene structures. For example, the L1-Drosha and L1-Parp8 fusion transcripts (Fig. 5a and b, Supp. Table 8) are predicted to encode many of the catalytic domains of the native gene products, together with short domains from the antisense L1 elements. Others, such as the novel spliced transcript 1ASII-1 (Fig. 5c), also demonstrate that transcription levels can be altered dramatically at a genomic locus previously thought to be devoid of exons. As the biological significance of such fusion transcripts remains unclear, we currently are evaluating whether such transcripts, initiated by certain polymorphic transposons, could rescue upstream promoter traps or affect tissue-specific gene expression levels. At least some of the variant fusion transcripts resulting directly from L1 retrotransposon polymorphisms may be noncoding RNAs with possible regulatory roles.
It is entirely possible that other structural variants, including those caused by other classes of retrotransposon polymorphisms (Fig. 2), may exert even larger effects upon transcriptional variation. For example, LTR retrotransposons may contain stronger promoters active in additional tissues and in other genomic contexts (Horie et al. 2007). Thus the functional consequences of transposon-mediated genomic variation upon transcripts may be variable themselves (Han et al. 2004). Variable transcription or added regulation mediated by polymorphic transposon promoters could provide a selective advantage that helps explain how mammalian hosts tolerate huge numbers of transposons in their genomes, despite the negative burden that their dispersal and maintenance engenders (Bestor 2003; Boissinot et al. 2001; Han et al. 2004; Yoder et al. 1997).
The generation of diversity between and within very recently separated mouse lineages by active mobilization of L1 retrotransposons emphasizes in detail that these elements are a built-in, active, dynamic engine for evolutionary changes – driving genetic variation and providing a substrate for natural selection – that operates even now (Kazazian 2004). As we documented here, the resulting changes caused by endogenous transposons are not merely structural, genomic variants: they can bring about direct changes in expressed transcripts, and quite likely phenotypic variation, as well.
METHODS
Identification of mouse genomic sequence variants
Approximately 26 million sequence traces (~18 billion nucleotides) from four inbred mouse strains (A/J, DBA2/J, 129S1/SvImJ, and 129X1/SvJ) were downloaded from the tracedb archive, National Center for Biotechnology Information (NCBI, NIH). Only high quality (>300 nt with Phred score >Q20) sequence traces were included, thereby excluding a very small percentage of traces. GMAP was used to align each individual trace to the C57 genome assembly (Stephens et al. 2008; Wu and Watanabe 2005). Possible alignment categories included no best alignment; polymorphism in C57; polymorphism in strain X; almost perfect alignment; and others (Fig. 1 and Supplementary Fig. 1). Candidate indels’ boundaries were determined by merging traces.
Databases/ public graphical display browser
PolyBrowse, a query tool and graphical browser at http://polybrowse.abcc.ncifcrf.gov/ based on GBrowse (Stein et al. 2002), was developed to display all indels described here together with other available genomic variants and annotated features (Stephens et al. 2008). C57 reference genomic data were downloaded from UCSC website, http://genome.ucsc.edu/, Feb. 2006 release. Protein domains were predicted using the SMART database, http://smart.embl-heidelberg. de/ (Letunic et al. 2006).
Bioinformatic identification of polymorphic transposons
Procedures are described in Supplementary Materials and Methods.
Mouse tissues’ total RNA isolation
Total RNA was isolated from grossly dissected adult testes (fasted, 72–75 day old males, harvested at same time of day), frozen in RNALater (Ambion), and homogenized in Trizol (Invitrogen) following standard protocols.
Validation of genomic polymorphisms
Genomic DNA from C57, 129S1, 129X1, A/J and DBA/2J mice was purchased from The Jackson Laboratory (Bar Harbor, ME). A locus-specific PCR amplicon was designed across the empty target site of each polymorphic repetitive element (Table 2 and Supplementary Table 5). Occasionally the same PCR reaction detected smaller integrants (<500 nt), while both left and/or right junctions of larger integrants were assayed using unique locus-specific primers in flanking genomic sequences paired with primers within the repetitive element (sequences available upon request). PCR products were assessed by agarose gel electrophoresis using standard methods.
Identification of L1 fusion transcripts
Screens of commercial phage libraries and online EST libraries were performed as described in Supplementary Materials and Methods.
cDNA sequencing
Synthesis of cDNAs was performed using SuperScript II (Invitrogen) with oligo-dT and gene-specific primers. Sequencing was performed as described in Supplementary Methods.
Correlation with SNPs
SNP reference genome coordinates were downloaded from NIEHS Perlegen and Celera databases (stored at tracedb, NCBI website) and compared to polymorphic transposon coordinates as described (Stephens et al. 2008).
Simulations and ontology analysis
To test various hypotheses about the genome-wide distribution of the 6,723 independent polymorphic L1s identified here, we generated lists of simulated integrants using a random number generator to assign chromosomal coordinates. To approximate genomic or intragenic distributions, 6,723 integrant locations were simulated 500 times, resulting in 3,361,500 simulated L1 insertions. Intronic integrants were identified by comparison with a database of RefSeq genes (NCBI). P-values were calculated using the binomial statistic and were adjusted by applying the Bonferroni correction (SPSS software) (Slonim 2002).
To sample gene categories randomly for ontology analysis, based on their relative lengths, the simulation was performed 1,000 times, resulting in 6,723,000 simulated integrants. To investigate whether genes are involved in a biological process affected by polymorphisms, we used the GeneID associated with each accession to query the PANTHER database (Mi et al. 2005) at http://www.panther.org. Simulated integrants or reference L1s were used alternatively as reference groups, as indicated. Biological process or molecular function categories were deemed significant if, upon applying the Bonferroni correction, their p-values are less than 0.01 as determined by the binominal statistic (Mi et al. 2005).
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Maxine Singer, Michael Kuehn, Beverly Mock, Maura Gillison, and Berton Zbar for helpful comments on drafts of this manuscript, and members of the Symer lab for constructive discussions. This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH and in part was funded by NCI contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government.
NCI-Frederick is accredited by Association for Assessment and Accreditation of Laboratory Animal Care International and follows the U.S. Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the "Guide for Care and Use of Laboratory Animals" (National Research Council, 1996, National Academy Press, Washington, D.C.). Mouse studies were performed following a protocol approved by the Animal Care and Use Committee, NCI Frederick.
Footnotes
This manuscript is accompanied by Supplementary Information. GenBank accession numbers EF591871 – EF591883 are included in tables with novel sequences. No unpublished results from individuals other than the authors have been referenced in this manuscript.
REFERENCES
- An W, Han JS, Wheelan SJ, Davis ES, Coombes CE, Ye P, Triplett C, Boeke JD. Active retrotransposition by a synthetic L1 element in mice. Proc Natl Acad Sci U S A. 2006;103:18662–18667. doi: 10.1073/pnas.0605300103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM. Genealogies of mouse inbred strains. Nat Genet. 2000;24:23–25. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- Belancio VP, Hedges DJ, Deininger P. LINE-1 RNA splicing and influences on mammalian gene expression. Nucleic Acids Res. 2006;34:1512–1521. doi: 10.1093/nar/gkl027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH. Cytosine methylation mediates sexual conflict. Trends Genet. 2003;19:185–190. doi: 10.1016/S0168-9525(03)00049-0. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Furano AV. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol. 2001;18:926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Rattner A, Nathans J. Effects of L1 retrotransposon insertion on transcript processing, localization and accumulation: lessons from the retinal degeneration 7 mouse and implications for the genomic ecology of L1 elements. Hum Mol Genet. 2006;15:2146–2156. doi: 10.1093/hmg/ddl138. [DOI] [PubMed] [Google Scholar]
- Chen JM, Stenson PD, Cooper DN, Ferec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum Genet. 2005;117:411–427. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, Rosenberg NA, Pritchard JK. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- Cutler G, Marshall LA, Chin N, Baribault H, Kassner PD. Significant gene content variation characterizes the genomes of inbred mouse strains. Genome Res. 2007;17:1743–1754. doi: 10.1101/gr.6754607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dew IM, Walenz B, Sutton G. A tool for analyzing mate pairs in assemblies (TAMPA) J Comput Biol. 2005;12:497–513. doi: 10.1089/cmb.2005.12.497. [DOI] [PubMed] [Google Scholar]
- Egan CM, Sridhar S, Wigler M, Hall IM. Recurrent DNA copy number variation in the laboratory mouse. Nat Genet. 2007;39:1384–1389. doi: 10.1038/ng.2007.19. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- Gasior SL, Preston G, Hedges DJ, Gilbert N, Moran JV, Deininger PL. Characterization of pre-insertion loci of de novo L1 insertions. Gene. 2007;390:190–198. doi: 10.1016/j.gene.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, Moran JV. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol. 2005;25:7780–7795. doi: 10.1128/MCB.25.17.7780-7795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Ostertag EM, Du K, Kazazian HH., Jr A novel active L1 retrotransposon subfamily in the mouse. Genome Res. 2001;11:1677–1685. doi: 10.1101/gr.198301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Boeke JD. A highly active synthetic mammalian retrotransposon. Nature. 2004;429:314–318. doi: 10.1038/nature02535. [DOI] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- Horie K, Saito ES, Keng VW, Ikeda R, Ishihara H, Takeda J. Retrotransposons influence the mouse transcriptome: implication for the divergence of genetic traits. Genetics. 2007;176:815–827. doi: 10.1534/genetics.107.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34:D257–D260. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PloS Biology. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Kannan M, Symer DE. Diverse fusion transcripts are initiated by a novel antisense promoter in mouse L1 retrotransposons. 2008 doi: 10.1093/nar/gku091. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. X-chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet. 1998;80:133–137. doi: 10.1159/000014969. [DOI] [PubMed] [Google Scholar]
- Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, Guo N, Muruganujan A, Doremieux O, Campbell MJ, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33:D284–D288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- Mural RJ, Adams MD, Myers EW, Smith HO, Miklos GL, Wides R, Halpern A, Li PW, Sutton GG, Nadeau J, et al. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science. 2002;296:1661–1671. doi: 10.1126/science.1069193. [DOI] [PubMed] [Google Scholar]
- Murchison EP, Hannon GJ. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr Opin Cell Biol. 2004;16:223–229. doi: 10.1016/j.ceb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Naas TP, DeBerardinis RJ, Moran JV, Ostertag EM, Kingsmore SF, Seldin MF, Hayashizaki Y, Martin SL, Kazazian HH. An actively retrotransposing, novel subfamily of mouse L1 elements. Embo J. 1998;17:590–597. doi: 10.1093/emboj/17.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Roy-Engel AM, El-Sawy M, Farooq L, Odom GL, Perepelitsa-Belancio V, Bruch H, Oyeniran OO, Deininger PL. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet Genome Res. 2005;110:365–371. doi: 10.1159/000084968. [DOI] [PubMed] [Google Scholar]
- Saxton JA, Martin SL. Recombination between subtypes creates a mosaic lineage of LINE-1 that is expressed and actively retrotransposing in the mouse genome. J Mol Biol. 1998;280:611–622. doi: 10.1006/jmbi.1998.1899. [DOI] [PubMed] [Google Scholar]
- Slonim DK. From patterns to pathways: gene expression data analysis comes of age. Nat Genet. 2002;32 Suppl:502–508. doi: 10.1038/ng1033. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker. 2007 [Google Scholar]
- Speek M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol Cell Biol. 2001;21:1973–1985. doi: 10.1128/MCB.21.6.1973-1985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens RM, Akagi K, Collins JR, Neelam B, McCullough D, Volfovsky N, Symer DE. PolyBrowse: An interface to access, query and display mouse genomic variation. 2008 Submitted for publication. [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human L1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- Wade CM, Daly MJ. Genetic variation in laboratory mice. Nat Genet. 2005;37:1175–1180. doi: 10.1038/ng1666. [DOI] [PubMed] [Google Scholar]
- Wade CM, Kulbokas EJ, 3rd, Kirby AW, Zody MC, Mullikin JC, Lander ES, Lindblad-Toh K, Daly MJ. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420:574–578. doi: 10.1038/nature01252. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wheelan SJ, Aizawa Y, Han JS, Boeke JD. Gene-breaking: a new paradigm for human retrotransposon-mediated gene evolution. Genome Res. 2005;15:1073–1078. doi: 10.1101/gr.3688905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelaw E, Martin DI. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat Genet. 2001;27:361–365. doi: 10.1038/86850. [DOI] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F. On the subspecific origin of the laboratory mouse. Nat Genet. 2007;39:1100–1107. doi: 10.1038/ng2087. [DOI] [PubMed] [Google Scholar]
- Yang N, Kazazian HH., Jr L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.