

Abstract
Upon entering a presynaptic terminal, an action potential opens Ca2+-channels, and transiently increases the local Ca2+-concentration at the presynaptic active zone. Ca2+ then triggers neurotransmitter release within a few hundred microseconds by activating synaptotagmin Ca2+-sensors. Synaptotagmins bind Ca2+ via two C2-domains, and transduce the Ca2+-signal into a mechanical activation of the membrane fusion machinery; this activation is mediated by the Ca2+-dependent interaction of the synaptotagmin C2-domains with phospholipids and SNARE proteins. In triggering exocytosis, synaptotagmins do not act alone, but require an obligatory co-factor called complexin, a small protein that binds to SNARE complexes and simultaneously activates and clamps the SNARE complexes, thereby positioning the SNARE complexes for subsequent synaptotagmin action. The conserved function of synaptotagmins and complexins operates generally in most, if not all, Ca2+-regulated forms of exocytosis throughout the body, including degranulation of mast-cells, acrosome exocytosis in sperm cells, hormone secretion from endocrine cells, and neuropeptide release.
Introduction
Synaptic transmission is initiated when an action potential invades a nerve terminal, opening Ca2+-channels which gate a highly localized, transient increase in intracellular Ca2+ at the active zone (Fig. 1A). Ca2+ triggers synaptic vesicle exocytosis, thereby releasing the neurotransmitters contained in the vesicles and initiating synaptic transmission. This fundamental mechanism was discovered in pioneering work on the neuromuscular junction by Katz and Miledi (1967). Its precise time course was studied in many ‘model’ synapses, including the giant squid axon synapse (Augustine et al., 1985) and rat cerebellar parallel fiber synapses (Sabatini and Regehr, 1996), but characterized in greatest detail in the calyx of Held synapse in the brainstem (Fig. 1B; reviewed in Meinrenken et al., 2003). Overall, the high-resolution electrophysiological studies on neurotransmitter release revealed that a presynaptic action potential is tightly coupled to Ca2+-influx and synaptic vesicle fusion, such that under physiological conditions, Ca2+ triggers fusion in a few hundred microseconds, or possibly even in less than a 100 microseconds (Sabatini and Regehr, 1996).
Figure 1. Principle and time course of Ca2+-induced initiation of synaptic transmission.

A. Schematic diagram of a synapse illustrating the localized influx of Ca2+ at the active zone (red = secreted neurotransmitters).
B. Schematic illustration of the sequence and time course of synaptic transmission as measured by simultaneous pre- and postsynaptic patch-clamp recordings at the calyx of Held synapse (modified from Südhof (2004) and Meinrenken et al. (2003); note that Ca2+-currents and EPSC are shown inverted).
The amazing speed and precision of Ca2+-triggered neurotransmitter release raised the question of how such speed might be possible – how can Ca2+ induce exocytosis in a few hundred microseconds, on the same timescale as the gating of an ion channel? As we will describe in this review, the speed and precision of release are mediated, at least in part, by the properties of the Ca2+-sensor synaptotagmin (Syt) and its co-factor complexin. Moreover, increasing evidence indicates that the principal mechanism of Ca2+-triggered exocytosis, although not the organization of this mechanism, is conserved in other, slower forms of Ca2+-induced exocytosis. Thus, the Syt-based fusion apparatus emerges as a general paradigm that accounts for most Ca2+-regulated exocytosis in eukaryotic cells.
In addition to normal action potential-evoked neurotransmitter release, synapses exhibit two additional forms of Ca2+-dependent synaptic exocytosis: spontaneous ‘mini’ release and asynchronous release (Pang and Südhof, 2010). Spontaneous mini release was discovered by Katz (Fatt and Katz, 1952), and is thought to represent the background activity of a nerve terminal that is triggered by spontaneous Ca2+-fluctuations. Although the biological significance of spontaneous release remains debated, its presence can be observed in all neurons. When calculated on a per synapse basis, spontaneous release occurs only once every 2–3 hours at an individual excitatory synapse of a pyramidal neuron in the CA1 region of the hippocampus, and approximately once every 3 minutes at an individual inhibitory synapses. Asynchronous release, finally, is undetectable in most synapses under physiological conditions, but becomes apparent when the Syt Ca2+-sensor for regular release is ablated (Geppert et al., 1994). Some inhibitory synapses exhibit a slow form of release upon repetitive stimulation (Hefft and Jonas, 2005) that may represent either genuine physiological asynchronous release or simply synaptotagmin-dependent delayed release.
Structure and biochemical properties of synaptotagmins
Syts are defined as proteins containing a short N-terminal non-cytoplasmic sequence followed by a transmembrane region, a central linker sequence of variable length, and two C-terminal C2-domains that account for the majority of the protein (Fig. 2A; Perin et al., 1990). Sixteen genes encoding canonical Syts are expressed in mammals; in addition, mammals contain a Syt-related gene encoding a similar protein (the so-called B/K-protein) that lacks a transmembrane region, and is occasionally called Syt17. Synaptotagmins are evolutionarily conserved, and invertebrates also express multiple Syt isoforms. However, synaptotagmins are absent from plants and unicellular eukaryotes, suggesting that they evolved at the same time as animals. Moreover, a large family of Syt-like proteins is expressed in animals; these proteins also contain two C2-domains, but will not discussed here further (see Pang and Südhof, 2010). Syt1, Syt2, Syt9, and Syt12 are present on synaptic vesicles and on secretory granules in neuroendocrine cells; the latter also contain Syt7. In addition, Syt10 was recently found on secretory vesicles containing IGF-1 in olfactory mitral neurons (Cao et al., 2011), Syt4 was localized to the trans-Golgi complex, synaptic vesicles, and postsynaptic organelles, and may be widely distributed in neuronal organelles (reviewed in Südhof, 2004), and Syt7 was additionally localized to ‘secretory lysosomes’ in non-neuronal and non-endocrine cells (Andrews and Chakrabarti, 2005). Other Syts appear to be primarily localized to transport vesicles, although some Syt isoforms are enriched on the plasma membrane (Butz et al., 1999), and the identity of many of the Syt-containing vesicles remains unknown.
Figure 2. Structures and Ca2+-binding properties of synaptotagmins.
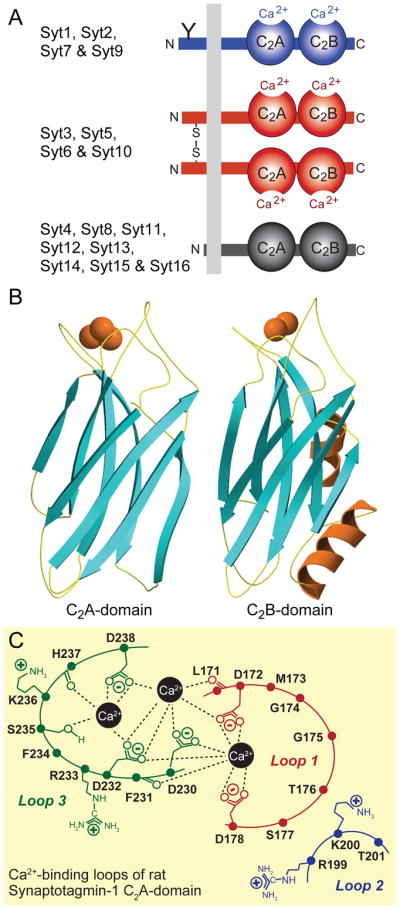
A. Canonical domain structures and classification of synaptotagmins. Mammals express 16 synaptotagmins composed of an N-terminal transmembrane region preceded by a short non-cytoplasmic sequence and followed by a variable linker sequence and two C2-domains; in addition, a 17th related protein called B/K-protein contains the same domain structure but an N-terminal lipid anchor instead of the transmembrane region (see Pang and Südhof, 2010, for a discussion of additional synaptotagmin-related proteins). Eight synaptotagmins bind Ca2+ (Syt1, 2, 3, 5, 6, 7, 9, and 10; blue and red); the remaining synaptotagmins do not (black). The eight Ca2+-binding synaptotagmins fall into two broad classes that differ in the absence (Syt1, 2, 7 and 9; blue) or presence (Syt3, 5, 6, and 10; red) of disulfide-bonded cysteine residues in their N-terminal sequences. Note that Syt1 and 2 include an N-glycosylated sequence at the N-terminus (indicated by a ‘Y’), and that Syt7 is extensively alternatively spliced in the linker sequence (Han et al., 2004).
B. Atomic structures of the C2A- and C2B-domains of Syt1 containing bound Ca2+-ions (red spheres) bound to flexible loops formed by an eight-stranded β-sandwich (from Sutton et al., 1995; Ubach et al., 1999; Fernandez et al., 2001).
C. Structure of the Ca2+-binding sites of the Syt1 C2A domain (modified from Fernandez-Chacon et al., 2001). The Syt1 C2A-domain contains three Ca2+-binding sites, of which the central two are canonical for all Ca2+-binding C2-domains, whereas the third Ca2+-site on the left is variably observed in C2-domains, and is absent from the Syt1 C2B-domain. A further Ca2+-binding site that localizes to loop1 on the right is observed in some C2-domains, but is absent from Syt1 and is not shown. Loops refer to the flexible sequences on the top of the Syt1 C2-domain sandwich; the structures of some of the side chains are indicated.
C2-domains were identified as sequence motifs in protein kinase C isoforms (Coussens et al., 1986), but their functional activity as autonomously folded Ca2+-binding domains was first identified in Syt1 (Davletov and Südhof, 1993). The atomic structures of the C2A-domain of Syt1, the first C2-domain structure determined (Sutton et al., 1995), revealed that C2-domains are composed of a stable β-sandwich with flexible loops emerging from the top and bottom (Fig. 2B); all subsequently determined C2-domain structures described a similar fold. Ca2+ binds exclusively to the top loops of the C2-dmains, which form binding sites for 2–3 Ca2+-ions in close vicinity (Shao et al., 1996). Although not all C2-domains bind Ca2+ (see below), all of those that do and have been studied in detail contain the same Ca2+-binding site architecture, with the Ca2+-ions ligated primarily by aspartate side chains (Fig. 2C). C2-domains are now recognized as general Ca2+-binding modules that are found in a large number of signal transduction and membrane trafficking proteins.
Approximately half of all C2-domains lack the canonical aspartate residues required for Ca2+-binding, including the C2A-domains of Syt4 and Syt11, and all C2-domains of Syt8 and Syt12-Syt16, which are thus thought to be Ca2+-independent. Even the C2B-domains of Syt4 and Syt11 that include all of the requisite Ca2+-binding sequences were found to be unable to bind Ca2+ (Dai et al., 2004). This initially surprising result was explained by the atomic structure of the Syt4 C2B-domain, which showed that the canonical Ca2+-binding residues in the top loops of the domain are too far apart to be able ligate Ca2+ (Dai et al., 2004). Thus, the 16 mammalian Syts can be broadly classified into Ca2+-binding and Ca2+-independent groups (Fig. 2A). The first group can be further subclassified based on whether or not their N-terminal non-cytoplasmic sequences contain disulfide-bonded cysteine residues (Fukuda et al., 1999). Similarly, the second group can be subclassified based on evolutionary conservation and sequence homologies, which reveal two sets of evolutionarily conserved synaptotagmins formed by Syt4 and Syt11 and by Syt14–16, and into a heterogeneous set of non-conserved synaptotagmins (Syt8, Syt12, and Syt13).
Initial studies on Syt1 revealed that both of its C2-domains bind to phospholipids in a Ca2+-dependent manner (Daveltov and Südhof, 1992; Fernandez et al., 2002), and that they bind to SNARE proteins (Bennett et al., 1992; Li et al., 1995; Chapman et al., 1995). Syt1 binding to phospholipids requires Ca2+ and negatively charged phospholipids, with phosphatidyl-inositolphosphates being most effective (Pang et al., 2006). Interestingly, the intrinsic Ca2+-affinity of the Syt1 C2-domains is relatively low (millimolar), but the presence of negatively charged phospholipids increases their apparent Ca2+-affinity by 2–3 orders of magnitude. This probably occurs because the Ca2+-coordination spheres of the Syt1 C2-domains are incomplete, but are completed by the negatively charged phospholipid headgroups during Ca2+-dependent phospholipid binding (Fernandez-Chacon et al., 2001; Fernandez et al., 2002). In contrast to the Ca2+-dependent interaction of Syt1 with phospholipids, its binding to SNARE proteins and SNARE complexes is less well understood. There seems to be a direct interaction of the Syt1 C2-domains with syntaxin-1 that is greatly enhanced by Ca2+ (Li et al., 1995); moreover, Syt1 also binds to assembled SNARE complexes in a Ca2+-dependent manner (Pang et al., 2006). However, the interaction of Syt1 with SNARE complexes is highly sensitive to ionic strength in that it becomes Ca2+-independent at lower ionic strength, and at least some Ca2+-independent SNARE-complex binding by Syt1 may occur physiologically (Pang et al., 2006).
All Syt isoforms that bind Ca2+ appear to bind to phospholipids and to SNARE proteins in a Ca2+-dependent manner (Li et al., 1995; Sutton et al., 1999; Sugita et al., 2002; Hui et al., 2005). Interestingly, at least during Ca2+-dependent phospholipid binding different Syts exhibit quite different apparent Ca2+-affinities, suggesting that they normally differentially respond to Ca2+-signals (Sugita et al., 2002). The biochemical properties of most Ca2+-independent Syts remain unknown, apart from Syt12 which has been shown to bind to Syt1 on the synaptic vesicle membrane (Maximov et al., 2007).
Function of synaptotagmins in neurotransmitter release
From the very beginning, Syt1 was hypothesized to function as a Ca2+-sensor for neurotransmitter release (Perin et al., 1990; Brose et al., 1992; Davletov and Südhof, 1993). Knockout (KO) of Syt1 confirmed this hypothesis, demonstrating that the Syt1 deletion abolished fast synchronous neurotransmitter release (Fig. 3A; Geppert et al., 1994). Strikingly, not all Ca2+-stimulated release was abolished in Syt1 KO neurons, which retained a delayed asynchronous form of release that is increased upon repetitive stimulation (Maximov and Südhof, 2005). Moreover, release induced by hypertonic sucrose which is thought to cause the Ca2+-independent exocytosis of all vesicles in the readily-releasable pool of a synapse was unchanged, indicating that the Syt1 KO did not interfere with vesicle fusion as such, but only with the Ca2+-triggering of fusion (Geppert et al.., 1994).
Figure 3. Syt1, Syt2, and Syt9 function as synaptic Ca2+-sensors for neurotransmitter release.
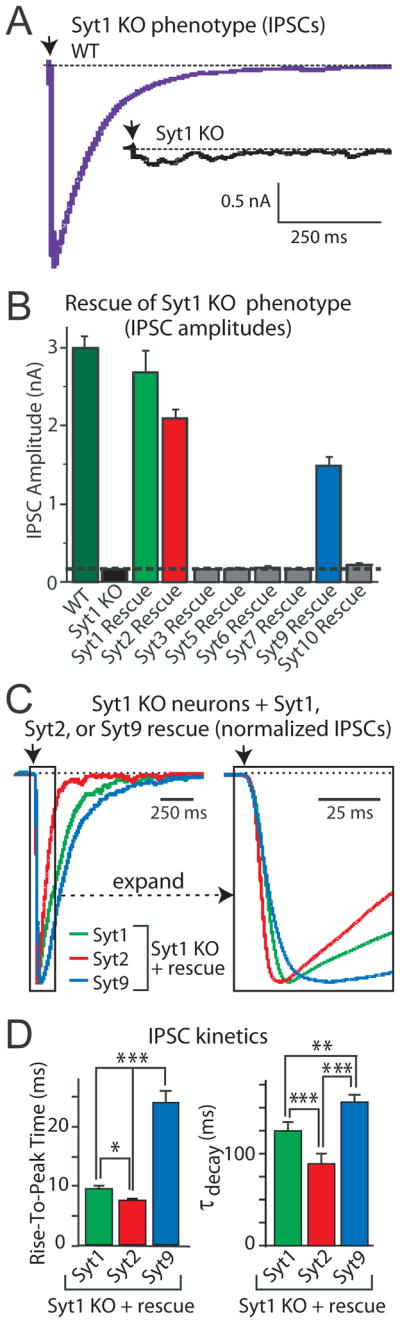
A. Deletion of Syt1 in cortical neurons blocks fast synchronous neurotransmitter release. Panels depict representative inhibitory postsynaptic currents (IPSCs) monitored in cortical neurons cultured from littermate wild-type (WT) and Syt1 knockout (Syt1 KO) neurons (arrow = action potential).These data and those shown in panels B-D were modified from Xu et al. (2008).
B. Screen of all Ca2+-binding synaptotagmin isoforms for their ability to rescue the loss of fast neurotransmitter release in Syt1 KO neurons. Data shown are means IPSC amplitudes (±SEMs); note that only Syt1, 2, and 9 are capable of rescue.
C. Syt1, Syt2, and Syt9 mediate Ca2+-triggering of release with distinct kinetics. Data shown are representative IPSCs monitored in Syt1 KO neurons expressing Syt1, 2, or 9. IPSCs were normalized for the maximal amplitude. The traces on the right display an expanded part of the overall IPSCs depicted on the left.
D. Quantitation of the kinetics of the increase in the IPSC (left, as mean rise time) and the decay of the IPSC (right, as mean time constant of the IPSC decay) triggered by Ca2+-binding to Syt1, 2, or 9. Note that IPSCs mediated by Syt2 exhibit the fastest rise and decay kinetics, whereas IPSCs mediated by Syt9 are 2-fold slower. All error bars indicate SEMs; asterisks indicate statistically significant differences as assessed by Student’s t-test (*, p<0.05; **, p<0.01; ***, p<0.001).
Given that eight Ca2+-binding Syts are expressed in neurons, which of these function in neurotransmitter release? A systematic screen of which Ca2+-binding Syt can rescue the Syt1 KO phenotype revealed that only Syt1, Syt2, and Syt9 were capable of rescue (Fig. 3B; Xu et al., 2007). This result is surprising given the close similarity of Ca2+-binding Syts with each other, but is consistent with the differential expression patterns of Syt1, Syt2, and Syt9, and also explains why Syt1 KO neurons lose all release despite expressing high levels of Syt7, Syt3, and Syt5. Strikingly, Syt1, Syt2, and Syt9 were found to trigger release with quite distinct kinetics, with Syt2 exhibiting the fastest onset and the fastest decline in release, and Syt9 the slowest onset and the slowest decline (Figs. 3C and 3D; Xu et al., 2007). The distinct kinetic properties of Syt1, Syt2, and Syt9 fit well with the expression of Syt9 primarily in the limbic system, whereas Syt2 is primarily expressed in synapses requiring very fast synaptic transmission, such as the auditory system or the neuromuscular junction.
The demonstration that Syt1 is essential for fast Ca2+-triggered neurotransmitter release was consistent with the notion that Syt1 functions as a Ca2+-sensor for release, but did not represent proof that Ca2+-binding to Syt1 triggers release, nor did it tell us which of the principal two biochemical activities of Syt1 – Ca2+-dependent phospholipid- and SNARE-binding – was involved in its function. Definitive evidence that Syt1, and by analogy other Syts, is indeed a Ca2+-sensor for release came from knockin experiments in which point mutations that selectively alter the Ca2+-affinity and specificity of Ca2+-dependent interactions were introduced into the endogenous mouse Syt1 (Fernandez-Chacon et al., 2001; Pang et al., 2006). Biochemical studies revealed that the D232N mutation in the C2A-domain of Syt1 increases the strength of the Ca2+-dependent binding of the double C2-domain fragment to SNARE complexes without altering phospholipid binding, whereas both the R233Q mutation and to a lesser degree, the D238N mutation, decrease the apparent Ca2+-affinity of the double C2-domain fragment of Syt1 during phospholipid binding without major effects on SNARE-complex binding (Fernandez-Chacon et al., 2001; Pang et al., 2006). Strikingly, the D232N mutation increased the apparent Ca2+-affinity of evoked neurotransmitter release (i.e., decreased the Kd for Ca2+), whereas the R233Q and the D232N decreased the apparent Ca2+-affinity of evoked release without changing the apparent Ca2+-cooperativity of release (Fig. 4A). Thus, changes in the apparent Ca2+-affinity of Syt1 lead to corresponding changes in the apparent Ca2+-affinity of release, proving that Syt1 is a true Ca2+-sensor. Morever, these experiments show that both SNARE- and phospholipid-binding by Syt1 are involved in release, suggesting that Ca2+-binding to Syt1 triggers release by inducing a fast simultaneous interaction of Syt1 with both SNARE complexes and with phospholipids.
Figure 4. Syt1 Ca2+-binding site point mutations illustrate Syt1 function as a Ca2+-sensor for evoked and spontaneous neurotransmitter release.

A. Evoked IPSCs measured in neurons cultured from knockin mice carrying single amino-acid substitutions in the Ca2+-binding site of the Syt1 C2A-domain. Three knockin mice, each with an individual littermate control (gray) were analyzed: mice with the D232N substitution that increases the amount of Ca2+-stimulated SNARE-complex binding by Syt1 (green; Pang et al., 2006); the R233Q substitution that greatly decreases the apparent Ca2+-affinity of Syt1 during phospholipid binding (blue; Fernandez-Chacon et al., 2001); and the D238N substitution that modestly decreases the apparent Ca2+-affinity of Syt1 (red; Pang et al., 2006). Action-potential evoked IPSCs were measured at the indicated concentrations of extracellular Ca2+; the plot on the left depicts the absolute IPSC amplitude as a function of the extracellular Ca2+-concentration, while the summary graphs on the right display the parameters for the apparent Ca2+-affinity and Ca2+-cooperativity of release obtained by fitting individual Ca2+-titration experiments to a Hill function.
B. Same as A., except that the frequency of spontaneous release events was measured. For both A and B, data are means ± SEMs (adapted from Xu et al., 2007).
Ca2+-binding to both C2-domains of Syt1 contributes to triggering release, although the two C2-domains are not equivalent. Mutations in the C2B-domain Ca2+-binding sites block release similar to the Syt1 KO, whereas mutations in the C2A-domain Ca2+-binding sites have a much less severe effect (Mackler and Reist, 2001; Stevens and Sullivan, 2003; Shin et al., 2009). Initially it was hypothesized that the C2A-domain Ca2+-binding sites are of little importance, but careful Ca2+-titrations revealed that blocking Ca2+-binding to the C2A-domain not only decreases the magnitude of release, but also greatly decreases the apparent Ca2+-cooperativity of release (Shin et al., 2009). Among others, these experiments confirmed that the two C2-domains of Syt1 functions cooperatively, and that Ca2+-binding to both C2-domains trigger release to achieve the high apparent cooperativity of Ca2+-triggering of release (Meinrenken et al., 2003).
Synaptotagmins as Ca2+-sensors for spontaneous release
In a typical central neuron, spontaneous release events add up over time to a sizable signal, and may have an important physiological role (McKinney et al., 1999; Carter and Regehr, 2002; Sutton et al., 2006). Most spontaneous release is Ca2+-dependent, suggesting it is regulated (Xu et al., 2009). Although spontaneous release was traditionally considered to be determined primarily if not exclusively by the number of synapses and the release probability of these synapses, increasing evidence indicates that spontaneous release may be derived from a vesicle pool that is different from that used for evoked release (Sara et al., 2005), and that it does not always, and possibly not even in most cases, correlate with the number of synapses and their release probability.
The Ca2+-dependence of spontaneous release suggested that a Ca2+-sensor should trigger it. The logical choice for such a Ca2+-sensor would by Syt1 and its homologs. Indeed, analysis of the Ca2+-dependence of spontaneous release in knockin mutant mice in which the affinity and binding-specificity of the Syt1 Ca2+-binding sites were altered revealed that increases in the Ca2+-dependent interaction of Syt1 with SNARE complexes induced by the D232N mutation caused an increase in the apparent Ca2+-affinity of spontaneous release (Fig. 4B; Xu et al., 2009). Conversely, decreases in the apparent Ca2+-affinity of Syt1 during phospholipid binding induced by the R233Q or the D238N mutation caused a decrease in the apparent Ca2+-affinity of spontaneous release (Fig. 4B). These results indicated that Syt1 functions as a Ca2+-sensor for spontaneous release, consistent with the previous finding that other elements of the canonical release machinery (e.g., SNARE proteins, Munc18-1, Munc13, and RIMs) are similarly involved.
However, deletion of Syt1 produces a large increase in spontaneous release frequency instead of blocking spontaneous release (Littleton et al., 1994; Maximov and Südhof, 2005), suggesting that Syt1 is a clamp for spontaneous release instead of a Ca2+-sensor. This apparently contradictory observation was explained when it was found that the increased spontaneous release in Syt1 KO neurons was also completely Ca2+-dependent, but exhibited a higher apparent Ca2+-affinity and a lower apparent Ca2+-cooperativity than spontaneous release triggered by Ca2+-binding to Syt1 (Xu et al., 2009). Thus, deletion of Syt1 unmasked a secondary Ca2+-sensor with different properties that was normally clamped by Syt1. The nature of this Ca2+-sensor remains unclear, but it may be the same as the Ca2+-sensor that triggers asynchronous release and that has similar properties (Sun et al., 2007).
In addition to Syts, Doc2 proteins were implicated in regulating spontaneous release. KO or shRNA-mediated knockdown of Doc2 proteins, which are Ca2+-binding proteins related to Syts that also bind to SNAREs and to phospholipids, decreases spontaneous release significantly (Groffen et al., 2010; Pang et al., 2011). Interestingly, the Doc2 KO and knockdown had no effect on evoked release, confirming the differential nature of spontaneous and evoked release. Although the Ca2+-binding properties of Doc2 proteins suggested that they may function as Ca2+-sensors for spontaneous exocytosis (Groffen et al., 2010), the decrease in mini frequency induced by Doc2 knockdown was fully rescued by expression of Doc2 proteins lacking Ca2+-binding sites, indicating that Doc2 proteins do not function as Ca2+-sensors for spontaneous exocytosis (Pang et al., 2011). Moreover, the Doc2 knockdown had no effect on the increased spontaneous release observed in Syt1 KO neurons, and also did not affect asynchronous release, suggesting that Doc2 proteins do not function in these types of release.
Complexins as synaptotagmin sidekicks
Complexins were identified as SNARE-complex binding proteins, giving rise to their name (McMahon et al., 1995; Ishizuka et al., 1995). Complexins are small soluble proteins of approximately 120 residues that are evolutionarily conserved in all animals similar to synaptotagmins. The crystal structure of complexin bound to the assembled SNARE complex revealed that complexin contains a central short ~20-residue α-helix that nestles into the groove formed by the synaptobrevin and syntaxin SNARE motifs in the SNARE complex (Chen et al., 2002). In addition, complexin contains a similarly short accessory N-terminal α-helix that does not contact the SNARE complex in the crystal structure, with N- and C-terminal unstructured sequences flanking the two helices. Interestingly, complexin binds to the SNARE complex in an antiparallel orientation at a membrane-proximal site of the complex, suggesting that complexin can only bind to the SNARE complex after it has assembled to more than 50%.
Mammals express four complexin isoforms, two of which are widely distributed in the body and abundant in brain (complexin-1 and -2; McMahon et al., 1995), and two additional isoforms that are enriched in retina (complexin-3 and -4; Reim et al., 2005). Complexin-3 and -4 include a C-terminal isoprenylation sequence that is absent from complexin-1 and -2 and likely attaches them to the plasma membrane. Deletion of complexin-1 and -2 in mice led to a striking phenotype that represented a milder phenocopy of the Syt1 knockout phenotype, suggesting that complexins act in the same pathway as synaptotagmins (Reim et al., 2001). Specifically, these mice exhibited a selective but only partial loss of synchronous neurotransmitter release, without a major decrease in asynchronous release. Subsequent knockdown experiments using shRNAs confirmed this phenotype, but extended it to reveal an increase in spontaneous release analogous to that observed in Syt1 knockout neurons, with the same shift to a higher apparent Ca2+-affinity and lower apparent Ca2+-cooperativity (Maximov et al., 2009; Yang et al., 2010). Thus, complexins act both as activators and as clamps of neurotransmitter release. Moreover, different from the Syt1 KO which had no effect on the size of the readily-releasable pool, the complexin knockdown produced a ~40% decrease in the readily-releasable pool (Yang et al., 2010). Rescue experiments revealed that SNARE-complex binding by complexin is essential for its function (Maximov et al., 2009). Moreover, such experiments demonstrated that the complexin N-terminus is crucial for its activating role (Yue et al., 2008), whereas its accessory α-helix mediates its clamping function (Maximov et al., 2009; Yang et al., 2010). Considerable controversy developed at one point about whether or not complexins have a clamping function, in that such function was not obvious in autapses (Reim et al., 2001), but was the only function observed in in vitro fusion assays (Giraudo et al., 2009). Although the activation function of complexin is likely more important overall than its clamping function, the fact, however, that both can be localized to distinct autonomously acting sequences in complexin (Maximov et al., 2009; Yang et al., 2010) and that both phenocopy a function observed for Syts strongly supports their physiological importance.
How do complexin and Syts act in promoting Ca2+-triggering of synaptic vesicle fusion? Biochemical data indicate that the central α-helix of complexin and Syt1 bind to SNARE complexes at overlapping sites, and that Ca2+-binding to Syt1 triggers the displacement of the central α-helix of complexin from its binding site on SNARE complexes (Tang et al., 2007). Based on the electrophysiological and biochemical studies, a plausible current model is that complexin binding to primed synaptic vesicles containing partially assembled SNARE complexes ‘superprimes’ these vesicles into an activated state, and simultaneously clamps them (Fig. 5). Ca2+-binding to synaptotagmin then triggers synaptotagmin-binding to the SNARE complex and the phospholipid bilayer, dislodging the complexin clamp and pulling on the SNARE complex, thereby opening the fusion pore. This model accounts for most of the existing data, although it offers no explanation for the clamping function of synaptotagmin, and thus is unlikely to be definitive.
Figure 5. Schematic diagram of the mechanism of action of synaptotagmin and its complexin cofactor in Ca2+-triggered exocytosis.

The top diagram displays the sequential priming of synaptic vesicles by partial SNARE/SM protein complex assembly, superpriming by binding of complexin to partially assembled SNARE complexes, and Ca2+-triggering of fusion-pore opening by Ca2+-binding to synaptotagmin. The objects in the diagram approximate real size relationships. The diagram on the bottom left depicts a cartoon of a partially assembled SNARE/SM protein complex with complexin bound. The functional domain structure of complexin is shown on the right, with a space-filling model of complexin bound to a SNARE complex at the bottom (pink, the two α-helices of complexin; yellow, syntaxin-1; red, synaptobrevin; green and blue, SNAP-25). The model and the functional assignment of complexin sequences are modified from Maximov et al. (2009); the complexin/SNARE complex crystal structure is from Chen et al. (2002).
Synaptotagmins as Ca2+-sensors for other forms of exocytosis
Many forms of exocytosis are induced by Ca2+, including hormone exocytosis in endocrine cells such as chromaffin cells, neuropeptide secretion in neurons, mast cell degranulation, and the acrosome reaction in sperm. Strikingly, for most of these forms of exocytosis the same Syts that mediate synaptic vesicle fusion (Syt1, Syt2, and Syt9) appear to be involved; in addition, Syt7 was also shown to play a central role. Specifically, for chromaffin granule exocytosis in chromaffin cells, nearly all Ca2+-triggering is mediated in equal parts by Syt1 and Syt7, such that only the double Syt1/7 KO has a major phenotype in exocytosis (Schonn et al., 2008). In contrast, in pancreatic insulin- and glucagon-secreting cells, Syt7 appears to be the dominant Ca2+-sensor (Gustavsson et al., 2008 and 2009), whereas in mast cell degranulation, Syt2 is the most important Ca2+-sensor (Melicoff et al., 2009). In addition, Syt7 has been linked to exocytosis in non-neuronal cells, although it is not quite clear whether this is a physiological event (Martinez et al., 2000; Jaiswal et al., 2003; Wang and Hilgemann, 2008). At least in chromaffin cells, complexins are obligatory cofactors for Syt-triggered exocytosis (Cai et al., 2008). Thus, despite the different kinetics and localizations, the same Syt paradigm appears to widely operate for all of these forms of exocytosis.
Up until recently, no definitive function was identified for the second class of Ca2+-binding Syts that contain N-terminal disulfide bonds which lead to constitutive dimerization (Fig. 2A). These Syts exhibit a higher apparent Ca2+-affinity than Syt1, and also interact with phospholipids and SNAREs in a Ca2+-dependent manner (Li et al., 1995; Sugita et al., 2002), suggesting that they may also function as Ca2+-sensors in exocytosis. This hypothesis was recently confirmed for Syt10, which was identified as the Ca2+-sensor for exocytosis of IGF-1 containing secretory vesicles in mitral neurons from the olfactory bulb (Cao et al., 2011). Interestingly, in the same neurons Syt1 functions as the Ca2+-sensor for synaptic vesicle exocytosis, and the two Syts act completely independently of each other (Fig. 6). Thus, different synaptotagmin isoforms can operate in the same cell to mediate the Ca2+-regulated fusion of distinct types of secretory vesicles.
Figure 6. Illustration of distinct but similar functions of different synaptotagmins: Syt10 acts as a Ca2+-sensor for IGF-1 containing vesicles in mitral neurons from olfactory bulb.

A. IGF-1 secretion from cultured olfactory bulb neurons is stimulated by depolarization with 15 mM KCl, but impaired by deletion of Syt10 (this and the following panels were modified from Cao et al., 2011).
B. Syt10 co-localizes with IGF-1 containing vesicles in the somatodendritic regions of cultured mitral neurons. Images show double immunofluorescence labeling of tagged Syt10 and either IGF-1, synapsin, or MAP2 as indicated.
C. Illustration of the distinct functions of Syt1 and Syt10 as Ca2+-sensors exclusively for synaptic and IGF-1 containing vesicles, respectively, in mitral neurons.
Concluding Remarks
Over the last two decades, a universal mechanism by which Ca2+-binding to Syts triggers fusion has emerged. This process involves a pas-de-deux of Syts and complexin which act simultaneously on SNARE complexes and phospholipid membranes. The mechanism ensures the millisecond control of neurotransmitter release from presynaptic terminals upon arrival of an action potential, but goes far beyond the synapse. The mechanism similarly applies to many other Ca2+-induced secretory events, as exemplified by the role of Syts and complexins in neuroendocrine secretion including IGF-1 exocytosis, and might further extend to all Ca2+-regulated membrane trafficking events in general.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute and by grants from the NIMH.
References
- Andrews NW, Chakrabarti S. There's more to life than neurotransmission: the regulation of exocytosis by synaptotagmin VII. Trends Cell Biol. 2005;15:626–31. doi: 10.1016/j.tcb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J Physiol. 1985;367:163–81. doi: 10.1113/jphysiol.1985.sp015819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber CF, Jorquera RA, Melom JE, Littleton JT. Postsynaptic regulation of synaptic plasticity by synaptotagmin 4 requires both C2 domains. J Cell Biol. 2009;187:295–310. doi: 10.1083/jcb.200903098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin: A Ca2+ sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Butz S, Fernandez-Chacon R, Schmitz F, Jahn R, Südhof TC. The subcellular localizations of atypical synaptotagmins: synaptotagmin III is enriched in synapses and synaptic plasma membranes but not in synaptic vesicles. J Biol Chem. 1999;274:18290–18296. doi: 10.1074/jbc.274.26.18290. [DOI] [PubMed] [Google Scholar]
- Cai H, Reim K, Varoqueaux F, Tapechum S, Hill K, Sørensen JB, Brose N, Chow RH. Complexin II plays a positive role in Ca2+-triggered exocytosis by facilitating vesicle priming. Proc Natl Acad Sci U S A. 2008;105:19538–43. doi: 10.1073/pnas.0810232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Maximov A, Südhof TC. Activity-Dependent IGF-1 Exocytosis is Controlled by the Ca2+-Sensor Synaptotagmin-10. Cell. 2011;145:300–311. doi: 10.1016/j.cell.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, Südhof TC, Rizo J. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science. 1986;233:859–866. doi: 10.1126/science.3755548. [DOI] [PubMed] [Google Scholar]
- Dai H, Shin OK, Machius M, Tomchick DR, Südhof TC, Rizo J. Structural basis for the evolutionary inactivation of Ca2+-binding to synaptotagmin 4. Nature Struct Mol Biol. 2004;11:844–849. doi: 10.1038/nsmb817. [DOI] [PubMed] [Google Scholar]
- Davletov BA, Südhof TC. A single C2-domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid-binding. J Biol Chem. 1993;268:26386–26390. [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci. 2009;12:767–76. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FATT P, KATZ B. Spontaneous subthreshold activity at motor nerve endings. J Physiol. 1952;117:109–28. [PMC free article] [PubMed] [Google Scholar]
- Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RGW, Südhof TC, Rizo J. Three-dimensional structure of the synaptotagmin 1 C2B-domain: Synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorffer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a Ca2+-regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Mikoshiba K. Conserved N-terminal cysteine motif is essential for homo- and heterodimer formation of synaptotagmins III, V, VI, and X. J Biol Chem. 1999;274:31421–31427. doi: 10.1074/jbc.274.44.31421. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: A major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Giraudo CG, Garcia-Diaz A, Eng WS, Chen Y, Hendrickson WA, Melia TJ, Rothman JE. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science. 2009;323:512–516. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Diez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ, Li C, Radda GK, Südhof TC, Han W. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci U S A. 2008;105:3992–3997. doi: 10.1073/pnas.0711700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson N, Wei SH, Hoang DN, Lao Y, Zhang Q, Radda GK, Rorsman P, Südhof TC, Han W. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+-induced glucagon exocytosis in pancreas. J Physiol. 2009;587:1169–1178. doi: 10.1113/jphysiol.2008.168005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Rhee JS, Maximov A, Lao Y, Mashimo T, Rosenmund C, Südhof TC. N-glycosylation is essential for vesicular targeting of synaptotagmin 1. Neuron. 2004;41:85–99. doi: 10.1016/s0896-6273(03)00820-1. [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci U S A. 2005;102:5210–5214. doi: 10.1073/pnas.0500941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka T, Saisu H, Odani S, Abe T. Synaphin: a protein associated with the docking/fusion complex in presynaptic terminals. Biochem Biophys Res Commun. 1995;213:1107–1114. doi: 10.1006/bbrc.1995.2241. [DOI] [PubMed] [Google Scholar]
- Jaiswal JK, Chakrabarti S, Andrews NW, Simon SM. Synaptotagmin VII restricts fusion pore expansion during lysosomal exocytosis. PLoS Biol. 2004;2:E233. doi: 10.1371/journal.pbio.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. Ionic requirements of synaptic transmitter release. Nature. 1967;215:651. doi: 10.1038/215651a0. [DOI] [PubMed] [Google Scholar]
- Li C, Ullrich B, Zhang ZZ, Anderson RGW, Brose N, Südhof TC. Ca2+-dependent and Ca2+-independent activities of neural and nonneural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Stern M, Perin M, Bellen HJ. Calcium dependence of neurotransmitter release and rate of spontaneous vesicle fusions are altered in Drosophila synaptotagmin mutants. Proc Natl Acad Sci USA. 1994;91:10888–10892. doi: 10.1073/pnas.91.23.10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- Mackler JM, Reist NE. Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J Comp Neurol. 2001;436:4–16. [PubMed] [Google Scholar]
- Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW. Synaptotagmin VII regulates Ca2+-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. 2000;148:1141–1149. doi: 10.1083/jcb.148.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous Function of Synaptotagmin 1 in Triggering Synchronous Release Independent of Asynchronous Release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Südhof TC. Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol. 2007;176:113–124. doi: 10.1083/jcb.200607021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang Z, Südhof TC. Complexin Controls the Force Transfer from SNARE complexes to membranes in Fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Missler M, Li C, Südhof TC. Complexins: cytosolic proteins that regulate SNAP-receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- Meinrenken CJ, Borst JG, Sakmann B. Local routes revisited: the space and time dependence of the Ca2+ signal for phasic transmitter release at the rat calyx of Held. J Physiol. 2003;547:665–689. doi: 10.1113/jphysiol.2002.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melicoff E, Sansores-Garcia L, Gomez A, Moreira DC, Datta P, Thakur P, Petrova Y, Siddiqi T, Murthy JN, Dickey BF, Heidelberger R, Adachi R. Synaptotagmin-2 controls regulated exocytosis but not other secretory responses of mast cells. J Biol Chem. 2009;284:19445–19451. doi: 10.1074/jbc.M109.002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Südhof TC. Cell Biology of Ca2+-Triggered Exocytosis. Curr Opinion Cell Biol. 2005;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC. Doc2 supports spontaneous synaptic transmission by a calcium-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate the Ca2+ sensitivity of the synaptic neurotransmitter release machinery. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Reim K, Wegmeyer H, Brandstätter JH, Xue M, Rosenmund C, Dresbach T, Hofmann K, Brose N. Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol. 2005;169:669–80. doi: 10.1083/jcb.200502115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET. An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron. 2005;45:563–573. doi: 10.1016/j.neuron.2004.12.056. [DOI] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sorensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Davletov BA, Sutton RB, Südhof TC, Rizo J. Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science. 1996;273:248–251. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- Shin OH, Xu J, Rizo J, Südhof TC. Differential but convergent functions of Ca2+-binding to synaptotagmin-1 C2-domains mediate neurotransmitter release. Proc Natl Acad Sci USA. 2009;106:16469–16474. doi: 10.1073/pnas.0908798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A Dual Ca2+-Sensor Model for Neuro-transmitter Release in a Central Synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. Synaptotagmins: Why so many? J Biol Chem. 2002;277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin O-H, Han W, Lao Y, Südhof TC. Synaptotagmins form hierarchy of exocytotic Ca2+-sensors with distinct Ca2+-affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton AB, Davletov BA, Berghuis AM, Südhof TC, Sprang SR. Structure of the first C2-domain of synaptotagmin I: A novel Ca2+/phospholipid binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Ernst JA, Brunger AT. Crystal structure of the cytosolic C2A-C2B domains of synaptotagmin III. Implications for Ca2+-independent snare complex interaction. J Cell Biol. 1999;147:589–98. doi: 10.1083/jcb.147.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC. A Complexin/ Synaptotagmin-1 Switch Controls Fast Synaptic Vesicle Exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Ubach J, Zhang X, Shao X, Südhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 1998;17:3921–3930. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Poser C, Ichtchenko K, Shao X, Rizo J, Südhof TC. The evolutionary pressure to inactivate: A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem. 1997;272:14314–14319. doi: 10.1074/jbc.272.22.14314. [DOI] [PubMed] [Google Scholar]
- Wang TM, Hilgemann DW. Ca-dependent nonsecretory vesicle fusion in a secretory cell. J Gen Physiol. 2008;132:51–65. doi: 10.1085/jgp.200709950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Lu JC, Bai J, Chang PY, Martin TF, Chapman ER, Jackson MB. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature. 2003;424:943–7. doi: 10.1038/nature01857. [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca2+-sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nature Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]