

Abstract
Background
Cetuximab is an anti-epidermal growth factor receptor (EGFR) monoclonal antibody (mAb) that prolongs survival in the treatment of head and neck cancer (HNC), but only in 10–20% of patients. An immunological mechanism of action such as natural killer (NK) cell-mediated antibody-dependent cellular cytotoxicity (ADCC) has been suggested. We investigated the effects of activating toll-like receptor (TLR)-8 to enhance activity of cetuximab-stimulated, FcγR bearing cells.
Objective
To determine the capability of TLR8-stimulation to enhance the activation and function of NK cells and dendritic cells (DC) in the presence of cetuximab-coated HNC cells.
Methods
Peripheral blood mononuclear cells (PBMC), NK, DC and CD8+ T cells were isolated and analyzed using 51Cr release ADCC, flow cytometry analysis, cytokine ELISA, and EGFR853–861 tetramer staining.
Results
TLR8 stimulation of unfractionated PBMC led to enhanced cetuximab-mediated ADCC in healthy donors (p<0.01) and HNC patients (p<0.001), which was dependent on NK cells. Secretion of Th1 cytokines TNFα(p<0.0001), IFNγ(p<0.0001), and IL-12p40(p<0.005) was increased. TLR8 stimulation of PBMC augmented cetuximab-enhanced NK cell degranulation (p<0.001). TLR8 stimulated NK cells enhanced DC maturation markers CD80, CD83, and CD86 in co-culture with cetuximab-treated HNC cells. TLR8 stimulation of NK-DC co-cultures significantly increased DC priming of EGFR-specific CD8+ T cells in the presence of cetuximab.
Discussion
VTX-2337 and cetuximab combination therapy can activate innate and adaptive anti-cancer immune responses. Further investigation in human trials will be important for determining the clinical benefit of this combination, and for determining biomarkers of response.
Keywords: Cetuximab, VTX-2337, TLR8, ADCC, Head and Neck Cancer, Immunotherapy
Introduction
Cetuximab, a chimeric mouse/human epidermal growth factor receptor (EGFR)-specific monoclonal antibody (mAb), has been used therapeutically in multiple solid tumors including head and neck cancer (HNC) [1]. Although the anti-tumor effects of EGFR signaling blockade have been demonstrated, there has been an increasing understanding and recognition of the immunological anti-tumor mechanisms of cetuximab therapy in HNC [2, 3]. In a proposed HNC model, cetuximab binds to EGFR positive HNC cells via its Fv domains while the Fc portion binds Fcγ receptors (FcγR) on natural killer (NK) cells which leads to their activation and results in antibody dependent cellular cytotoxicity (ADCC) of the HNC tumor cell [4]. The degree of affinity of the Fc portion of cetuximab to the NK cell Fc receptor, FcγRIIIa, is influenced by the FcγRIIIa-158 V/F polymorphism and explains the differential cytotoxicity effects seen among donors in vitro [5]. This initial NK cell activation may induce secondary adaptive immune responses through dendritic cell (DC) cross presentation and cytotoxic T- lymphocyte (CTL) activation for sequential and synergistic anti-tumor effects [2, 6]. Cross-presentation by licensed DC is vital for the cross-priming of anti-tumor CTL, while immature DC propagate a tolerogenic phenotype [7]. The limited efficacy of cetuximab has motivated novel combination approaches to stimulate anti-tumor immunity.
Toll-like receptors (TLRs) are primary sensors of microbial invasion and their activation results in initiation of innate immunity and secondary stimulation of adaptive immune responses via pro-stimulatory cytokine secretion [8–10]. TLR7 and TLR8 agonists have been studied in various cancer targets and have shown some promising results [11–13]. TLR8 is endosomal and its natural ligand is considered to be viral ssRNA [14, 15]. Recognition of a TLR8 agonist activates several immune cells such as myeloid DC, monocytes and macrophages [16, 17]. These activated cells are stimulated to produce Th1-polarizing cytokines such as TNFα, IFNγ, IL-12 and monocyte chemotactic protein 1 (MCP-1) and result in further recruitment of immune cells to the tumor microenvironment [8, 16]. The TLR8 selective agonist VTX-2337 has recently been observed to stimulate secretion of IL-12 and TNFα from monocytes and myeloid dendritic cells, IFNγ from NK cells, and enhance rituximab- and trastuzumab-mediated ADCC [18]. However, the effect of VTX-2337 on DC maturation and function has not been fully described. Therefore, we evaluated VTX-2337, a synthetic TLR8 selective agonist, as an immune adjuvant in cetuximab-mediated ADCC and cetuximab-mediated enhancement of NK cell-induced DC maturation and CD8+ T cell priming.
Methods
Cell lines and authentication
EGFR+ HNC cell lines (UM-22B and PCI-15B) were cultured in DMEM supplemented with 10% FBS, penicillin/streptomycin and L-glutamine at 37°C at 5% CO2 atmosphere.
Antibodies and tetramer
Cetuximab (Erbitux, BMS Imclone, Princeton NJ) was purchased from the University of Pittsburgh Hillman Cancer Center Pharmacy. A human IgG1 isotype control was purchased from Sigma Aldrich, St Louis MO. The CD16-specific mAb 3G8 was obtained from BD Biosciences (San Jose CA). The following fluorophore-conjugated antibodies/molecules were used for staining for flow cytometry: CD3-Alexa 405 was purchased from Invitrogen (Carlsbad CA); CD16-PE-Cy7, Granzyme B-FITC, EpCAM-APC, CD11c-PE-Cy7, and CD86-PE were purchased from Biolegend (San Diego CA); CD56-APC, CD8-APC, CD80-FITC, CD83-PE, CD107a-PE, HLA-A*0201-FITC, and 7-AAD were purchased from BD Pharmingen (San Diego CA).
Cellular components
Whole blood or leukapheresis products from healthy donors were purchased from the Western Pennsylvania blood bank. HNC patient blood cells were obtained from University Ear, Nose, and Throat Specialists at University of Pittsburgh Medical Center. PBMC were separated using a Ficoll-hypaque gradient (Amersham Biosciences, Uppsala, Sweden). Enriched NK and CD8+ T cells were obtained from PBMC using EasySep negative selection kits (Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer’s protocols. Purity of more than 95% was monitored using flow cytometry. DC were generated from PBMC as previously described [19]. Briefly, PBMC were adhered to tissue culture flasks for 90 minutes and adherent cells were washed with PBS and treated 6 days with 1000 IU/mL rhGM-CSF & 1000 IU/mL rhIL-4 (R&D Systems Minneapolis, MN).
FcγRIIIa expression and genotyping of effector PBMC
FcγRIIIa-158 V/F genotype from donor PBMC was determined using the quantitative PCR-based assay kit from Applied Biosystems. Genomic DNA was extracted from PBMC using the DNeasy Kit (Qiagen). Five to 50 ng of genomic DNA were used in PCR containing primers and FAM-labeled probe specific for FcγRIIIa along with 2x Taqman master mix (Applied Biosystems). The 96-well optical plates (Perkin-Elmer) were run and analyzed for allelic expression using an ABI prism 7700 sequence detection system.
Cytotoxicity assays
51Chromium release assay was used to determine cytotoxicity as described [5]. In brief, a total of 1×106 HNC cells, UM-22B or PCI-15B, were labeled with 25 μCi of Na251CrO4 for 1 h at 37°C. Cells were then washed twice with RPMI 1640 medium and incubated with cetuximab and either 250nM VTX-2337 stimulated effector cells (18 h) or untreated effector cells in RPMI at an E:T=50:1. After incubation for 4 h at 37°C, a sample of supernatant was counted on a Perkin Elmer 96-well plate gamma counter. Percentage cytotoxicity is calculated using the formula [(experimental-spontaneous)/(maximum-spontaneous)] x 100%, where spontaneous was defined as release from targets incubated with medium alone and maximum was defined as release from targets lysed by 5% Triton X-100 (Sigma Aldrich). Enhancement of cytotoxicity was considered to be >5% increase in specific lysis.
Cytokine measurements
Cytokine concentrations in the supernatants of cytotoxicity assays were determined using a multiplexed ELISA (Luminex ™). A commercially validated kit (In-vitrogen) was used to measure the cytokine concentrations and standard calibration curve was used for quantification.
Flow cytometry
Stained cells prepared as published previously [5] were analyzed with a Beckman Coulter EPICS XL flow cytometer. Data were analyzed using the Summit 4.3 software (Beckman Coulter, Fullerton, CA).
NK cell degranulation
For NK cell degranulation experiments, PBMC were incubated with VTX-2337 (250 nM), or with media for 18 h. PBMC were then co-cultured with PCI-15B HNC cells at 1:1 ratio with or without 10μg/mL cetuximab for 4 h with brefeldin A (Imgenex) and anti-CD107a-PE or PE-isotype control. PBMC were then harvested; stained for CD3, CD56 and CD16; permeabilized using a commercial kit (BD Pharmingen); and stained for intracellular granzyme B. Upon analysis, events were gated for lymphocytes, excluding doublets, that were CD3negCD56+CD16+granzyme B+ and analyzed for %CD107a+.
DC maturation
Generated DC were harvested using trypsin-EDTA and incubated in 1:1:1 co-culture with NK cells and PCI-15B HNC cells for 48 h in IMDM supplemented with 10% FBS, L-glutamine, and penicillin/streptomycin. During this time, the co-culture was incubated with media alone, VTX-2337 (800nM), cetuximab (10 μg/mL), or both. The cells were then harvested with trypsin-EDTA and stained for EpCAM-APC, CD11c-PE-Cy7 and separate sample wells were stained for either CD80-FITC & CD86-PE, or CD83-PE and analyzed by flow cytometry. Events were gated for high side scatter cells, excluding doublets, which were EpCAMnegCD11c+ and analyzed for %CD80+, %CD83+, and CD86 median FI.
CD8+ T cell in vitro stimulation
Autologous HLA-A*0201 CD8+ T cells and DC were harvested or generated, respectively. DC were matured in the same conditions as stated in the previous paragraph. CD8+ T cells were then stimulated in vitro with these differentially mature DC either once for 7 days, or twice for 7 days each (14 days total). IL-2 (10ng/mL) was included in the media. At the end of the stimulation, CD8+ T cells were harvested and stained with CD3, CD8, 7-AAD, and EGFR853–861 tetramer and analyzed by flow cytometry. Events were gated for viable (7-AADneg) lymphocytes, excluding doublets, which were CD3+CD8+ and analyzed for EGFR853–861 tetramer+/10,000 CD8+ T cells.
Statistical analysis
All statistical analyses were performed with an alpha level of 0.05. Experimental data with more than 2 comparison groups were analyzed using repeated measures ANOVA with post hoc pairwise comparisons using Bonferroni’s multiple comparison test. Two-group comparison was performed using paired t test or Wilcoxon signed rank text when the data distribution was visibly skewed.
Results
VTX-2337 enhances cetuximab-mediated cellular cytoxicity of HNC cells
To investigate the effect of TLR8 stimulation on specific cetuximab-mediated ADCC, PBMC were isolated and TLR8 stimulated with VTX-2337 (250nM) or left untreated in RPMI for 18 h, washed, and subsequently used in 51Cr release assays with or without cetuximab (10μg/mL) added to UM-22B as target HNC cells (E:T=50:1). Specific lysis was enhanced after incubating with cetuximab-coated HNC cells (p<0.05), as previously observed [5]. TLR8-stimulation of PBMC did not statistically significantly enhance lysis of control IgG1 antibody treated target HNC cells (Fig 1a). Importantly, TLR8-stimulated PBMC produced significantly greater specific lysis of cetuximab-coated UM-22B HNC cells than did untreated PBMC (p<0.01) (Fig 1a); however, purified NK cells treated with VTX-2337 did not show similar enhancement in cetuximab-mediated ADCC, even at an E:T=10:1. The cetuximab-mediated cytotoxicity of these NK cells was abolished upon treatment with anti-CD16 mAb 3G8 (Fig 1b). Moreover, cetuximab-mediated cytotoxicity of TLR8-stimulated PBMC was abolished upon treatment with anti-CD16 mAb 3G8 (data not shown).
Figure 1.

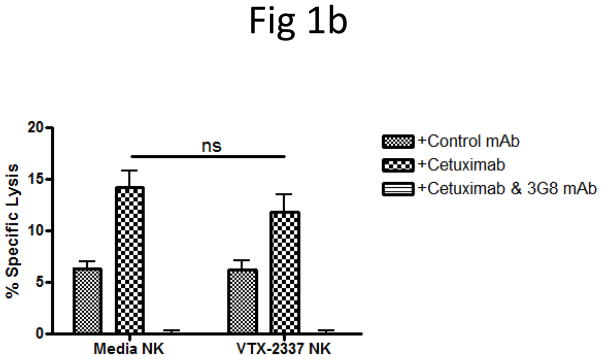
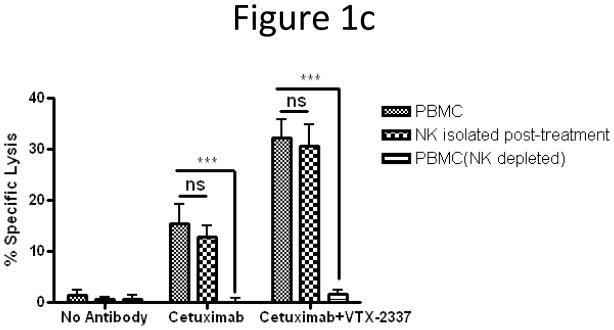
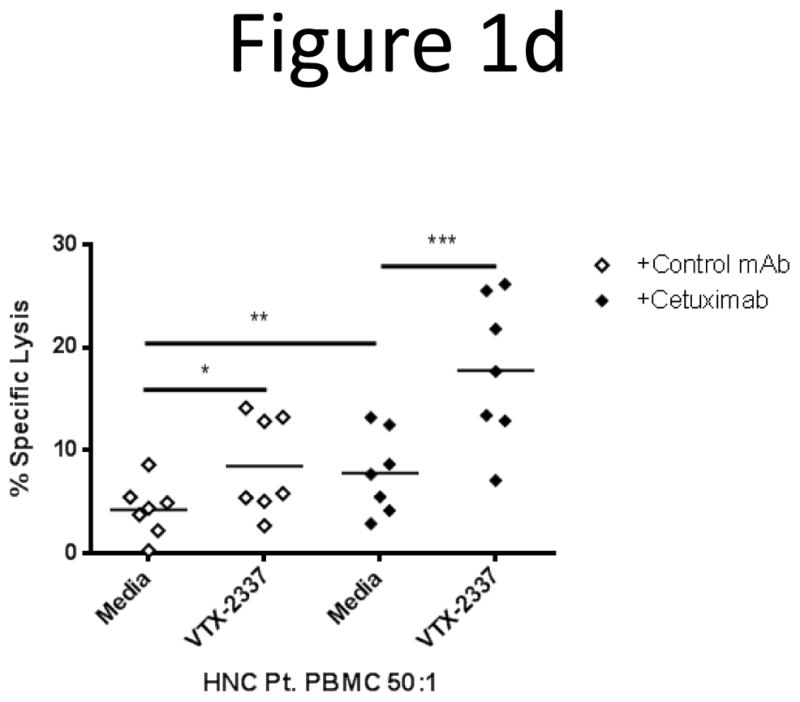
TLR8 stimulation indirectly activates NK cells to enhance cetuximab-mediated ADCC. (A) PBMC were incubated with TLR8 selective agonist VTX-2337 (250nM) or with media for 18 h, then co-cultured at E:T=50:1 for 4 h with 51Cr labeled UM-22B HNC cells coated with control IgG1 or cetuximab. Under conditions of TLR8 stimulation of PBMC followed by incubation with cetuximab-coated HNC cells, enhanced ADCC was observed compared to unstimulated PBMC against cetuximab-coated HNC cells (p<0.01). (B) Purified NK cells were treated with VTX-2337 (250nM) or media for 18 h, then co-cultured with 51Cr labeled UM-22B HNC cells for 4 h at an E:T=10:1. VTX-2337 treatment did not significantly influence NK cell cytotoxicity using control IgG1 or cetuximab coated HNC cells (p=0.13). Blockage of FcγRIIIa by 3G8 mAb abolished ADCC activity of NK cells. (C) PBMC were stimulated with VTX-2337 (250nM) or media for 18 h, then a portion of PBMC were fractionated into purified NK cells or NK-depleted PBMC and then assayed separately for ADCC activity. PBMC (E:T=50:1) and NK cells (E:T=2.5:1) demonstrated similar levels of specific lysis, while NK depletion abolished cytotoxicity of PBMC. (D) TLR8 stimulation enhanced cetuximab-mediated ADCC in PBMC from treatment naïve HNC patients with active disease. TLR8 stimulation treatment alone produced enhancement of ADCC in PBMC from HNC patients (p<0.05), whereas TLR8 stimulation plus cetuximab induced significantly greater ADCC activity of PBMC (p<0.001). Cetuximab and control IgG1 antibodies were used at 10μg/mL. *p<0.05, **p<0.005, ***p<0.001
To further determine the extent to which NK cells were responsible for the increased cetuximab-mediated ADCC by TLR8-stimulated PBMC, whole PBMC were stimulated for 18 h and a portion subsequently fractionated into purified NK cells or NK-depleted PBMC. NK cells purified after TLR8 stimulation at an E:T=2.5:1 demonstrated very similar specific lysis as whole PBMC at E:T=50:1. However, depletion of NK cells after TLR8 stimulation significantly and nearly completely abrogated cytotoxicity of cetuximab-coated HNC cells by TLR8-stimulated NK-depleted PBMC (Fig 1c, p<0.001). This was observed even when E:T was escalated to 200:1 (data not shown). VTX-2337 (250nM and 500nM) treated HNC target cells in the absence of PBMC did not manifest any measurable lysis (data not shown).
To determine if TLR8 stimulation also enhances cetuximab-mediated ADCC in PBMC from HNC patients, PBMC isolated from HNC patients were stimulated and used as effectors with the E:T=50:1. These tumor bearing HNC patients had no prior treatment, including no prior cetuximab.
Initially, the frequency of HNC patients demonstrating detectable cetuximab-mediated ADCC was 30.1% (8/26). However, when the same patients’ PBMC were TLR8 stimulated and used in cetuximab ADCC assays, 3 additional HNC patients demonstrated appreciable cetuximab-mediated ADCC. Thus, the overall cetuximab response rate improved to 42.3% (11/26) using a 5% enhancement of cytotoxicity as a positive response to TLR8 stimulation. Among the HNC patient PBMC enhanced by VTX-2337, a pattern of increased specific lysis similar to HD PBMC was observed. Importantly, TLR8 stimulated HNC patient PBMC demonstrated greater specific killing of cetuximab-treated target HNC cells than untreated PBMC (p<0.001, Fig 1d).
Cetuximab-mediated NK cell degranulation is enhanced by TLR8 stimulation
To corroborate the mechanism of increased specific lysis, PBMC were incubated with increasing concentrations of VTX-2337 (250nM and 500nM) or RPMI for 18 h and co-cultured with high EGFR expressing PCI-15B HNC cells at 1:1 ratio for 4 h with or without cetuximab. CD3negCD56+CD16+Granzyme B+ NK cells demonstrated increased degranulation marker CD107a staining in cetuximab-treated conditions (p<0.01). Indeed, TLR8 stimulation enhanced cetuximab induced %CD107a+ NK cells in a dose dependent manner (Fig 2a-b).
Figure 2.


TLR8 stimulation increases cetuximab-enhanced NK cell degranulation in a dose dependent manner. (A) PBMC were incubated with TLR8 selective agonist VTX-2337 (250nM) or with media for 18 h, then co-cultured with PCI-15B HNC cells (E:T=1:1) with or without cetuximab. Cetuximab-coated PCI-15B HNC cells increased %CD107a+ CD3negCD56+CD16+ NK cells vs uncoated PCI-15B HNC cells. TLR8 stimulation markedly enhanced %CD107a+ NK cells (p<0.005) in a dose dependent manner in the presence of cetuximab-coated PCI-15B HNC cells. (B) For a representative donor, histograms represent lymphocyte gated CD3negCD56+CD16+ cells. Cetuximab was used at 10μg/mL, and IL-2 at 50 IU/mL. *p<0.05, **p<0.01
Role of FcγRIIIa-158 polymorphism in TLR8 enhancement of cetuximab-mediated ADCC
Previously, we demonstrated a strong correlation in cetuximab-mediated cytotoxicity of HNC cells with FcγRIIIa 158 genotype (VV>VF>FF) [5]. Therefore, TLR8-stimulated PBMC from donors of each FcγRIIIa genotype were used in ADCC assays. TLR8 stimulation of PBMC enhanced cetuximab-mediated ADCC in all genotypes (FF: p<0.001, VF: p<0.001, VV: p<0.05) (Fig 3a). Following TLR8 stimulation, PBMC expressing the low affinity FcγRIIIa-158 FF genotype achieved a level of lytic activity comparable to unstimulated PBMC with FcγRIIIa-158 VF genotype with a target of cetuximab coated HNC cells (E:T=50:1). This trend was also observed in treated PBMC with FcγRIIIa-158 VF genotype versus unstimulated FcγRIIIa-158 VV PBMC genotype (Fig 3). With regards to cetuximab-mediated ADCC enhancement, the response rate of PBMC to a moderate dose of 250nM VTX-2337 was 70% (28/40) and was similar across all 3 FcγR IIIa-158 genotypes (85.7% for FF; 60% for VF and 66.7% for VV); (p=0.368).
Figure 3.
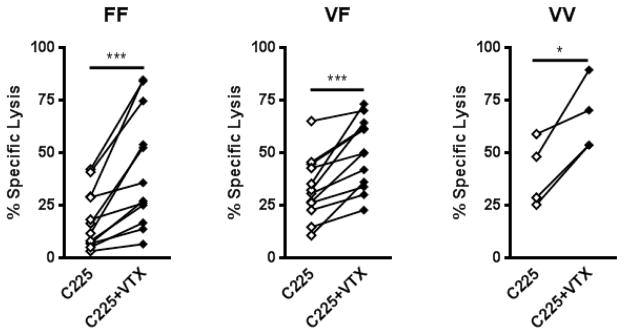
TLR8 stimulation enhances cetuximab-mediated ADCC of HNC cells in all FcγRIIIa-158 V/F genotypes. PBMC were incubated with TLR8 selective agonist VTX-2337 (250nM) or with media for 18 h, then co-cultured at E:T=50:1 for 4 h with 51Cr labeled UM-22B or PCI-15B HNC cells coated with cetuximab (10μg/mL). Genomic DNA was extracted from PBMC, and FcγRIIIa-158 V/F genotype was determined using a PCR SNP assay. TLR8 stimulation of PBMC enhanced specific lysis of cetuximab-coated UM-22B and PCI-15B HNC cell targets: FF (p<0.001), VF (p<0.001), VV (p<0.05). *p<0.05, ***p<0.001 (C225:Cetuximab VTX:VTX-2337)
TLR8 stimulation enhances cytokine secretion in supernatant from ADCC assays
Supernatants from TLR8 stimulated and untreated PBMC wells were collected and analyzed by Luminex™ for cytokines. Relevant Th1 cytokines were increased in treated conditions: TNFα (p<0.0001), IFNγ (p<0.001), and IL-12p40 (p<0.005, Fig 4). A single HD demonstrated an exaggerated enhancement of IL-12p40 level relative to the other HD. Even when this data point was removed from the data set the difference in IL-12p40 between groups remained significant (p<0.01).
Figure 4.
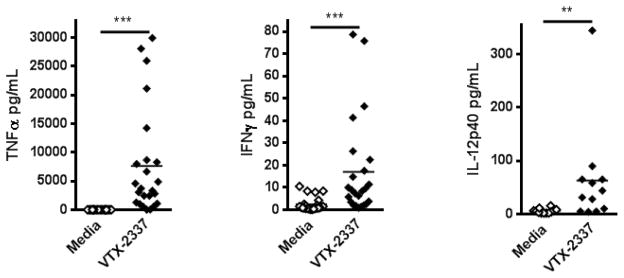
TLR8 stimulation of PBMC in presence of cetuximab coated HNC cells causes inflammatory cytokine secretion. PBMC were incubated with TLR8 selective agonist VTX-2337 (250nM) or media for 18 h, then co-cultured at E:T=50:1 for 4 h with cetuximab (10μg/mL) coated UM-22B HNC cells. Supernatants were collected from untreated control and TLR8 stimulated conditions. TNFα, IFNγ, and IL-12p40 secretion was significantly enhanced (p<0.0001, p<0.0001, and p<0.005 respectively). Of note, IL-12p40 enhancement was still significant when analyzed without the single high outlier (p<0.01). **p<0.005, ***p<0.0001
TLR8 stimulation directly matures DC and enhances cetuximab-mediated DC maturation
Monocyte derived DC are known to express TLR8 [16]. Therefore, we assessed the ability of TLR8 stimulation by VTX-2337 to directly mature DC as well as enhance cetuximab-activated, NK cell-induced DC maturation. DC were cultured with or without TLR8 stimulation (VTX-2337, 800nM, 48 h), and stained for the expression of co-stimulatory molecules. TLR8-stimulation of DC alone (in the absence of cetuximab or NK cells) enhanced %CD80+ (p=0.0001), %CD83+ (p<0.05), and CD86 median FI (p<0.005) (Fig 5a).
Figure 5.
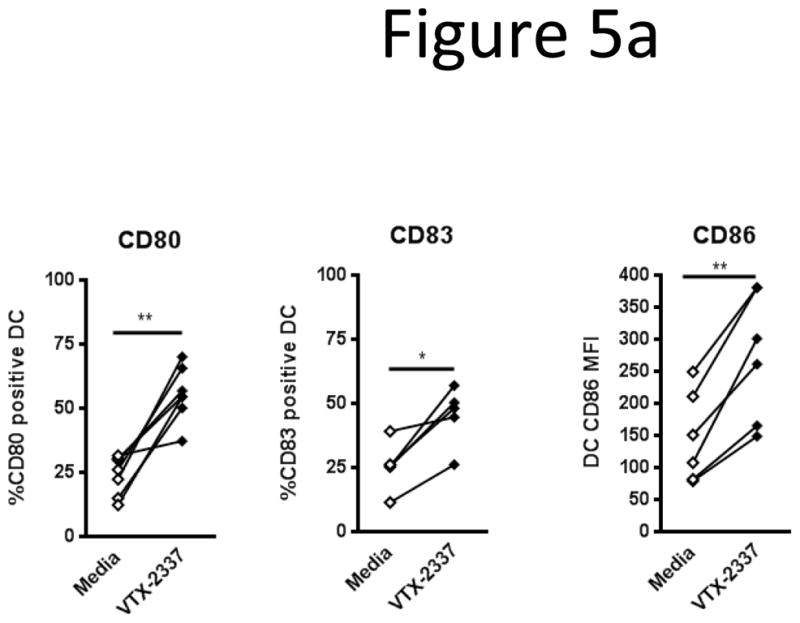

TLR8 stimulation directly matures DC and additively enhances cetuximab-mediated DC maturation. (A) DC were incubated with TLR8 selective agonist VTX-2337 (800nM) or with media for 48 h. TLR8 stimulation increased DC expression of %CD80 (p<0.001), %CD83 (p<0.05), and CD86 median FI (p<0.005). (B) DC were co-cultured with NK cells and PCI-15B HNC cells at 1:1:1 ratio. Cetuximab treatment alone enhanced DC maturation: %CD80 (p<0.01), %CD83 (p<0.001) and CD86 median FI (p<0.05), as previously observed. TLR8 stimulation alone modestly enhanced DC maturation: %CD80 (p<0.01), %CD83 (p<0.01). Addition of TLR8 stimulation to cetuximab treatment additively enhances DC maturation: %CD80 (p<0.01), %CD83 (0<0.001). Cetuximab was used at 10μg/mL. *p<0.05, **p<0.01, ***p<0.005 (C225:Cetuximab VTX:VTX-2337)
Co-cultures of DC, NK cells and PCI-15B HNC cells were incubated with media, TLR8 agonist (VTX-2337, 800nM, 48 h), cetuximab (10μg/mL), or both. Of note, PCI-15B HNC cells do not express an appreciable level of TLR8 (supplementary figure 1). The DC from the co-cultures were then stained for the expression of co-stimulatory molecules (Fig 5b). TLR8 stimulation alone significantly enhanced %CD80+ (p<0.01) and %CD83+ (p<0.01), although the absolute increase in each marker was modest. Likewise, cetuximab treatment enhanced %CD80+ (p<0.01), %CD83+ (p<0.001), and CD86 median FI (p<0.05), but cetuximab failed to increase any of these markers if either NK or PCI-15B HNC cells were absent from the co-culture (data not shown). However, the addition of TLR8 stimulation to cetuximab treatment significantly enhanced %CD80+ (p<0.01), and %CD83+ (p<0.001) to a larger extent than either condition alone.
TLR8 stimulation promotes cetuximab-mediated DC cross-priming of EGFR-specific CD8+ T cells
To determine the functional significance of enhanced DC maturation induced by TLR8 and FcγR stimulation via cetuximab coated HNC cells, we performed in vitro stimulation (IVS) of HLA-A*0201+ CD8+ T cells by incubating CD8+ T cells with autologous DC that had been matured in the presence of NK cells and PCI-15B HNC cells in the conditions mentioned above from the previous experiments. After the first 7-day IVS, neither TLR8 nor cetuximab stimulation alone significantly resulted in more EGFR-tetramer staining. However, in combination TLR8 and cetuximab stimulation resulted in an increased proportion of EGFR-specific CD3+CD8+ T cells (p<0.05, Fig 6). After the second 7-day IVS (day 14) the same pattern was observed, and the EGFR-specific CD3+CD8+ T cell frequency in the TLR8 and cetuximab stimulated condition was significantly greater than in the untreated condition (p<0.05). Although, neither TLR8 nor cetuximab stimulation alone significantly increased EGFR-specific CD3+CD8+ T cell frequency. NK cells were necessary for VTX-2337 combined with cetuximab to enhance DC cross priming of EGFR-specific CD8+ T cells (supplementary figure 2).
Figure 6.

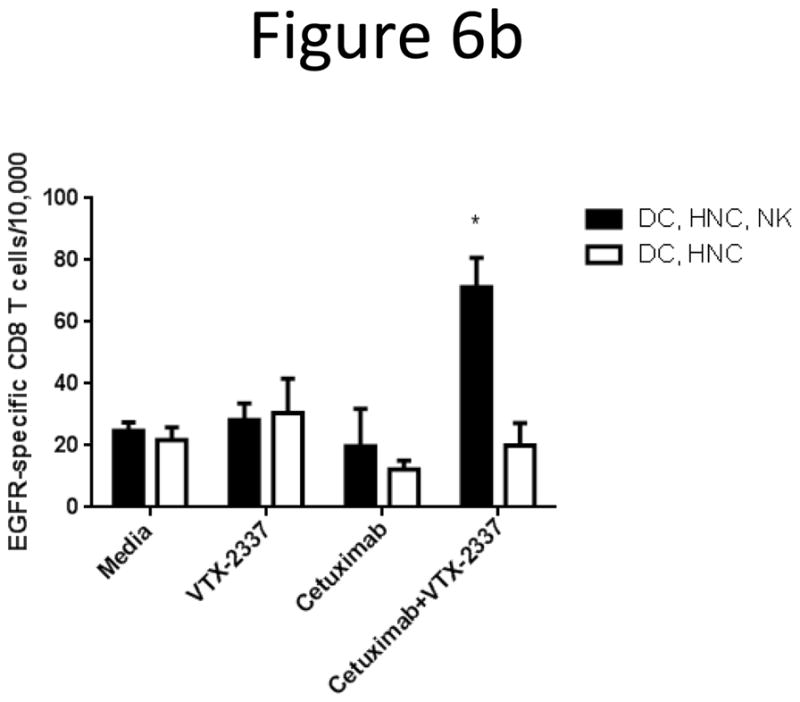
TLR8 stimulation enhances cetuximab-mediated induction of EGFR-specific CD8+ T-cells during in vitro stimulation (IVS) by VTX-2337 treated DC. (A) HLA-A*0201+ DC were incubated with NK cells and PCI-15B HNC cells at 1:1:1 ratio, then treated with media alone, VTX-2337 (800nM), cetuximab (10μg/mL), or cetuximab+VTX-2337. Thereafter, autologous CD8+ T cells were incubated with DC treated using each set of conditions for 7 days. A subset of CD8+ T cells were similarly re-stimulated for another 7 days (14 days total). DC incubation with CD8+ T cells for 7 days increased EGFR853–861-specific CD8+ T cells in all DC treatment conditions vs. unstimulated CD8+ T cells on day 0. After the first 7 day IVS, TLR8 stimulation and cetuximab treatment individually enhanced EGFR853–861 CD8+ T cell frequency, and the combination additively increased the frequency of EGFR853–861 CD8+ T cells, which enhancement was significant relative to CD8+ T cells incubated with untreated DC (p<0.05) during the first round of IVS. A second 7 day IVS with each set of DC conditions further increased specific CD8+ T cells, but the combination of TLR8 stimulation and cetuximab significantly increased EGFR853–861-specific CD8+ T cells relative to untreated DC (p<0.05), and TLR8 stimulated treated DC (p<0.05). (B) 7 day IVS using DC matured in the aforementioned treatment conditions in the presence or absence of NK cells. An increase in EGFR853–861-specific CD8+ T cells was seen when DC were matured in the presence of NK cells, but not when NK cells were absent. *p<0.05
Discussion
This is the first study to demonstrate that cetuximab-mediated ADCC can be enhanced in HNC patient PBMC upon TLR8 stimulation by VTX-2337, which suggests that it may be clinically possible to enhance the anti-tumor response rate of cetuximab in HNC patients. We also demonstrated that TLR8 stimulation is capable of enhancing an acquired immune response in vitro by inducing DC maturation directly, and enhancing cetuximab-mediated DC maturation in the presence of NK and HNC cells. Significantly, these TLR8 and cetuximab-activated NK cell matured DC were more capable of stimulating EGFR-specific CD8+ T cells in vitro. TLR7/8 agonists have recently become an area of active interest in cancer research. An early TLR7/8 agonist, imiquimod, has been FDA approved as a topical agent for the treatment of actinic keratosis and superficial basal cell carcinoma [20, 21]. Recently, a novel class of synthetic oligoribonucleotides, referred to as stabilized immune-modulatory RNA(SIMRA) compounds were described and these compounds serve as dual TLR7/8 agonists [22]. These SIMRA TLR7/8 agonists were shown to induce the production of Th1-type cytokines/chemokines, including IL-2, IL-12, IP-10, IFN-γ, and TNFα [11, 23, 24]. However, although TLR7 and TLR8 detect very similar ligands, their expression patterns and the cytokines they induced have been demonstrated to differ [18]. Therefore, there has been an increasing interest in developing and determining the utility of specific agonists.
This preclinical study sought to investigate combination immune therapy, using selective TLR8 stimulation to enhance TA-specific cellular immune responses by cetuximab activated lymphocytes. We investigated a selective, potent TLR8 agonist, VTX-2337 [18], as an immune adjuvant to demonstrate the augmentation of antibody dependent cell cytotoxicity (ADCC) against EGFR-positive cancer cells by NK cells from HNC patients in addition to HD in the presence of cetuximab. We also pursued the potential that this immune mediated cytotoxicity serves to promulgate the sequential adaptive immune responses by T cells based on NK-DC cross talk. Recruitment and activation of CTLs by DC may serve to present the lysed tumor epitopes through cross presentation. Therefore, even a modest increase in cetuximab-mediated cytotoxicity by VTX-2337 may result in a synergistic improvement in the overall immune mediated anti-tumor effects.
Our findings that TLR8 agonist VTX-2337 enhances cetuximab-mediated ADCC against EGFR positive HNC cells extend previous findings of VTX-2337 enhancing mAb cytotoxic function against cancer from other sites [18]. This effect is immune mediated since direct incubation of VTX-2337 in the absence of lymphocytes does not show any measurable cytotoxic effects. Furthermore, the enhancement of cytotoxicity was NK cell dependent since NK cell depletion abolished lysis of cancer cells and ADCC enhancement by TLR8-stimulation correlated with increased degranulation marker CD107a expression on NK cells. However, TLR8 stimulation of unfractionated PBMC was needed to initiate this effect. This suggests that NK cells were not directly activated by TLR8 stimulation, but potentially activated by TLR8-stimulated monocytes or mDC [22, 23]. This is corroborated by the observation by Lu et al. that NK cells produce IFNγ upon TLR8-stimulation of PBMC, but the presence of blocking antibody for IL-18 significantly decreases IFNγ secretion [18]. This is interesting because IL-18 is known to activate NK cell IFNγ production [25]. Secreted IL-12 also activates NK cell IFNγ production [26], and its secretion was observed to be increased by VTX-2337 TLR8-stimulation by both our study and Lu et. al. Interestingly, Lu et. al. also showed that VTX-2337 directly enhances NK cell mediated ADCC with a high dose of 500nM, but PBMC were enhanced at a lower dose of 167nM as well as 500nM. This may point to a dose dependent direct NK cell activation by VTX-2337, or possibly activation of residual non-NK cells after cell purification. At our dose of 250nM, VTX-2337 enhanced PBMC cytotoxicity and NK cell cytotoxicity when isolated from PBMC after treatment, but not in purified NK cells.
Enhancement in cetuximab-mediated ADCC by VTX-2337 is not FcγRIIIa-158 genotype specific since enhancement is seen across all 3 FcγRIIIa genotypes. This is beneficial, since donors with NK cells expressing the poor affinity FF genotype are potentially disadvantaged due to the inherent poor ability to mount a crucial ADCC innate immune response against HNC. Enhancing TAA-targeted mAb therapy in FcγRIIIa FF patients may improve clinical outcomes, as FcγRIIIa FF genotype has been suggested to be associated with decreased disease free survival in B cell lymphoma patients treated with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone [27].
We noted that HNC patient PBMC demonstrated lower levels of specific lysis than healthy donor PBMC, and we attribute this to the immunosuppressed state of the HNC patient PBMC. HNC patient PBMC can, according to our findings, be stimulated by cetuximab and VTX-2337 to perform enhanced ADCC against HNC cell targets. This may be beneficial in extending cetuximab’s benefit to those HNC patients who may not otherwise have derived a clinical benefit from cetuximab therapy. Various explanations have been brought forth to account for the lack of response to cetuximab in some cancer patients. Evidence points to the presence of immune regulatory cells such as regulatory T cells, myeloid derived suppressor cells (MDSCs) or tumor associated macrophages (TAMs) in cancer patients which are either increased in numbers or functionally more able to negate the immune effects of immunotherapy [28, 29]. These observations may explain at least a portion of the lack of response to cetuximab immunotherapy. Interestingly, TLR8 stimulation has been reported to reverse regulatory T cell function [30], which may be key to overcoming the tolerogenic nature of the tumor microenvironment. These are vital avenues to explore the mechanistic pathway of TLR8 stimulation to overcome some of these immune blockages to cetuximab immunotherapy.
TLR8 stimulation in the context of cetuximab treated HNC is capable of enhancing an acquired immune response through enhanced CD8+ T cell stimulation by DC in vitro. The presence of a strong CD8+ TA-specific cytotoxic T cell response has been associated with improved clinical outcomes [31], and this in vitro observation suggests that addition of TLR8 stimulation to mAb therapy may lead to a stronger CD8+ cytotoxic T cell response, and potentially subsequently to a more durable anti-tumor response in patients treated with cetuximab immunotherapy. It will be important to evaluate this therapeutic combination in patients, and a phase I clinical trial of VTX-2337 and cetuximab in combination to treat HNC patients is underway. We recently reported that cetuximab triggers NK:DC cross talk and TA-specific immunity in the absence of adjuvant stimulation, as we used with VTX-2337 ligation of TLR8 here. In some experiments using donors likely devoid of substantial EGFR-specific precursor CTL, cetuximab alone was insufficient to induce cross-priming of EGFR-specific CTL (Figure 6), in contrast to our observations in other systems. However, it is likely that TLR8 or other adjuvant stimulation can overcome variability in certain FcγR IIIa genotype or low/absent baseline CTL frequencies, explaining the lack of cross-presentation observed in some experiments presented here in the absence of VTX-2337 [32].
In summary, we have demonstrated that the selective TLR8 agonist VTX-2337 significantly enhances cetuximab-mediated ADCC and DC IVS of CD8+ T cells in HNC. The primary effectors for the increase in cytotoxicity are NK cells. Predominant Th1 cytokines (TNF-α, IFN-γ and IL12) are preferentially stimulated by VTX-2337 that are likely immunologically beneficial in the tumor environment. Furthermore, VTX-2337 TLR8 stimulation enhances cetuximab-mediated DC maturation and cross priming of CD8+ T cells.
Supplementary Material
Acknowledgments
Grants funding this project:
Robert L. Ferris: R01 DE19727, P50 CA097190, P30 CA047904
Ryan M. Stephenson: Doris Duke Charitable Foundation Clinical Research Fellow.
Footnotes
Disclosure of Potential Conflicts of Interest
Maura Matthews, Gregory Dietsch, and Robert Hershberg are employees of VentiRx® Pharmaceuticals.
Other authors have no conflicts to disclose.
References
- 1.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11:21–8. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 2.Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28:4390–9. doi: 10.1200/JCO.2009.27.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Overdijk MB, Verploegen S, van den Brakel JH, Lammerts van Bueren JJ, Vink T, van de Winkel JG, Parren PW, Bleeker WK. Epidermal growth factor receptor (EGFR) antibody-induced antibody-dependent cellular cytotoxicity plays a prominent role in inhibiting tumorigenesis, even of tumor cells insensitive to EGFR signaling inhibition. J Immunol. 2011;187:3383–90. doi: 10.4049/jimmunol.1003926. [DOI] [PubMed] [Google Scholar]
- 4.Lee SC, Srivastava RM, Lopez-Albaitero A, Ferrone S, Ferris RL. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res. 2011;50:248–54. doi: 10.1007/s12026-011-8231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, Ferris RL. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–64. doi: 10.1007/s00262-009-0697-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Albaitero A, Ferris RL. Immune activation by epidermal growth factor receptor specific monoclonal antibody therapy for head and neck cancer. Arch Otolaryngol Head Neck Surg. 2007;133:1277–81. doi: 10.1001/archotol.133.12.1277. [DOI] [PubMed] [Google Scholar]
- 7.McDonnell AM, Robinson BW, Currie AJ. Tumor antigen cross-presentation and the dendritic cell: where it all begins? Clin Dev Immunol. 2010;2010:539519. doi: 10.1155/2010/539519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schon MP, Schon M. TLR7 and TLR8 as targets in cancer therapy. Oncogene. 2008;27:190–9. doi: 10.1038/sj.onc.1210913. [DOI] [PubMed] [Google Scholar]
- 9.Hamm S, Rath S, Michel S, Baumgartner R. Cancer immunotherapeutic potential of novel small molecule TLR7 and TLR8 agonists. J Immunotoxicol. 2009;6:257–65. doi: 10.3109/15476910903286733. [DOI] [PubMed] [Google Scholar]
- 10.Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301–11. doi: 10.1158/1078-0432.CCR-07-1403. [DOI] [PubMed] [Google Scholar]
- 11.Dudek AZ, Yunis C, Harrison LI, Kumar S, Hawkinson R, Cooley S, Vasilakos JP, Gorski KS, Miller JS. First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clin Cancer Res. 2007;13:7119–25. doi: 10.1158/1078-0432.CCR-07-1443. [DOI] [PubMed] [Google Scholar]
- 12.Dummer R, Hauschild A, Becker JC, et al. An exploratory study of systemic administration of the toll-like receptor-7 agonist 852A in patients with refractory metastatic melanoma. Clin Cancer Res. 2008;14:856–64. doi: 10.1158/1078-0432.CCR-07-1938. [DOI] [PubMed] [Google Scholar]
- 13.Smits EL, Ponsaerts P, Berneman ZN, Van Tendeloo VF. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist. 2008;13:859–75. doi: 10.1634/theoncologist.2008-0097. [DOI] [PubMed] [Google Scholar]
- 14.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Diebold SS. Recognition of viral single-stranded RNA by Toll-like receptors. Adv Drug Deliv Rev. 2008;60:813–23. doi: 10.1016/j.addr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Lombardi V, Van Overtvelt L, Horiot S, Moingeon P. Human dendritic cells stimulated via TLR7 and/or TLR8 induce the sequential production of Il-10, IFN-gamma, and IL-17A by naive CD4+ T cells. J Immunol. 2009;182:3372–9. doi: 10.4049/jimmunol.0801969. [DOI] [PubMed] [Google Scholar]
- 17.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 18.Lu H, Dietsch GN, Matthews MA, et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res. 2012;18:499–509. doi: 10.1158/1078-0432.CCR-11-1625. [DOI] [PubMed] [Google Scholar]
- 19.Andrade Filho PA, Lopez-Albaitero A, Gooding W, Ferris RL. Novel immunogenic HLA-A*0201-restricted epidermal growth factor receptor-specific T-cell epitope in head and neck cancer patients. J Immunother. 2010;33:83–91. doi: 10.1097/CJI.0b013e3181b8f421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lebwohl M, Dinehart S, Whiting D, Lee PK, Tawfik N, Jorizzo J, Lee JH, Fox TL. Imiquimod 5% cream for the treatment of actinic keratosis: results from two phase III, randomized, double-blind, parallel group, vehicle-controlled trials. J Am Acad Dermatol. 2004;50:714–21. doi: 10.1016/j.jaad.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Oldfield V, Keating GM, Perry CM. Imiquimod: in superficial basal cell carcinoma. Am J Clin Dermatol. 2005;6:195–200. doi: 10.2165/00128071-200506030-00006. discussion 1–2. [DOI] [PubMed] [Google Scholar]
- 22.Sauder DN, Smith MH, Senta-McMillian T, Soria I, Meng TC. Randomized, single-blind, placebo-controlled study of topical application of the immune response modulator resiquimod in healthy adults. Antimicrob Agents Chemother. 2003;47:3846–52. doi: 10.1128/AAC.47.12.3846-3852.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart OM, Athie-Morales V, O’Connor GM, Gardiner CM. TLR7/8-mediated activation of human NK cells results in accessory cell-dependent IFN-gamma production. J Immunol. 2005;175:1636–42. doi: 10.4049/jimmunol.175.3.1636. [DOI] [PubMed] [Google Scholar]
- 24.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 25.Okamura H, Tsutsi H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 26.Xu M, Mizoguchi I, Morishima N, Chiba Y, Mizuguchi J, Yoshimoto T. Regulation of antitumor immune responses by the IL-12 family cytokines, IL-12, IL-23, and IL-27. Clin Dev Immunol. 2010;2010 doi: 10.1155/2010/832454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahlgrimm M, Pfreundschuh M, Kreuz M, Regitz E, Preuss KD, Bittenbring J. The impact of Fc-gamma receptor polymorphisms in elderly patients with diffuse large B-cell lymphoma treated with CHOP with or without rituximab. Blood. 2011;118:4657–62. doi: 10.1182/blood-2011-04-346411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergmann C, Strauss L, Wang Y, Szczepanski MJ, Lang S, Johnson JT, Whiteside TL. T regulatory type 1 cells in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clin Cancer Res. 2008;14:3706–15. doi: 10.1158/1078-0432.CCR-07-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whiteside TL. Immune cells in the tumor microenvironment. Mechanisms responsible for functional and signaling defects. Adv Exp Med Biol. 1998;451:167–71. [PubMed] [Google Scholar]
- 30.Peng G, Guo Z, Kiniwa Y, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–4. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 31.Hiroishi K, Eguchi J, Baba T, et al. Strong CD8(+) T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J Gastroenterol. 2010;45:451–8. doi: 10.1007/s00535-009-0155-2. [DOI] [PubMed] [Google Scholar]
- 32.Srivastava RM, Lee SC, Andrade Filho PA, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–72. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.