

Abstract
Prostanoid receptor EP2 is emerging as a novel target for development of anti-inflammatory drugs for the treatment of chronic neurodegenerative and peripheral diseases; however, the availability of EP2 antagonist probes for exploration of peripheral disease models is very limited. We now report identification and characterization of a novel chemical class of compounds that show nanomolar potency and competitive antagonism of the EP2 receptor. A compound in this class, TG6-129, showed prolonged plasma half-life and did not cross the blood brain barrier. This compound also suppressed the induction of inflammatory mRNA markers in a macrophage cell line upon activation of EP2. Thus, this compound could be useful as a probe for a variety of peripheral chronic inflammatory diseases such as rheumatoid arthritis and chronic obstructive pulmonary disease, in which EP2 appears to play a pathogenic role.

Keywords: Inflammation, EP2 receptor, EP2 antagonist, cytokines, acrylamides, macrophage
Inflammation is an early event during the development of many chronic peripheral diseases including rheumatoid arthritis, chronic obstructive pulmonary disease, and inflammatory bowel disease.1–4 Inflammation is also a key driver of many central nervous system disorders such as Alzheimer’s and Parkinson’s disease, amyotrophic lateral sclerosis, and epilepsy.5–7 Elevation of cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), inflammatory cytokines and chemokines occurs during the early stages of the development of these diseases.1–7 COX-2 has been extensively explored as a biological target for the development of anti-inflammatory therapy.8, 9 In the past, COX-2 inhibitors including rofecoxib (Vioxx®) and valdecoxib (Bextra®) were approved for the treatment of acute pain and inflammation in arthritis patients. The COX-2 inhibitors have also been used in an attempt to prevent the progression of neurodegenerative disease such as Alzheimer’s disease (AD).10 However, although these agents have shown efficacy in AD animal models, they have not shown any benefit to patients with AD. Clinical studies from chronic use of COX-2 inhibitors for pain and inflammatory diseases resulted in cardiovascular side effects. Consequently, Vioxx® and Bextra® were withdrawn from the United States market.11
COX-2 synthesizes five prostanoids PGD2, PGE2, PGF2, PGI2, and TxA2, which activate nine G-protein coupled receptors (EP1–EP4, DP1–DP2, FP, IP and TP) and several of these receptors play protective as well as deleterious roles.12 For example, IP activation can be cardioprotective, whereas EP2 activation can be both pro and anti-inflammatory.13 Studies now conclude that COX-2 inhibition mediated toxicity is due to reduced levels of PGI2 leading to reduced IP receptor activation.14–16 Thus future anti-inflammatory strategy could involve specific prostanoid receptor inhibition rather than generic block of the entire COX-2 cascade.
Prostanoid receptor EP2 is activated by PGE2 and is emerging as an important biological target for anti-inflammatory therapy.17–20 EP2 is positively coupled though Gαs to cyclic AMP (cAMP) production. When activated by PGE2, EP2 stimulates adenylate cyclase resulting in elevation of cAMP concentration, which initiates multiple downstream events mediated by protein kinase A (PKA) or exchange protein activated by cAMP (Epac).21, 22 We have developed a set of cell based TR-FRET assays to identify EP2 selective antagonists.23 By using these assays in conjunction with high-throughput screening of 262,300 compounds we have previously identified an acrylamide class of selective antagonists.24, 25 From the same screen, we have also found a compound 1 (PubChem, SID 17503974) as a hit with IC50 = 1.6 µM at saturating concentrations (1 µM) of PGE2 used for the activation of EP2 receptors in C6 glioma (C6G) cells. Interestingly this compound up to 40 µM has shown no inhibition of EP4 or β2-Adrenergic receptors under agonist saturated conditions (Fig 1A). Compound 1 possesses a carbamothioylacrylamide moiety flanked by phenylacrylamide on one side, and a thiadiazole-sulfonamide on the other side. Initially we asked whether this scaffold is worth pursuing for further characterization because of structural similarity to thioureas, which are known to potentially form adducts with glutathione in in vitro and in vivo systems.26 While the formation of adducts (a.k.a. bioactivation) is not a stumblingblock for compound development, this process could have widespread effects on other proteins. We have searched the PubChem data base to find whether compound 1 (SID 17503974) belongs to a promiscuous class of compounds, but interestingly, we found it was only active in 19 bioassays out of 575 tested. It showed an IC50 of 10–100 µM in most of the assays, except in an assay to identify inhibitors of S. aureus NadD (saNadD), where it has an IC50 of 4.5 µM.27 Thus, this compound may not be a promiscuous hit.
Figure 1.

Selective inhibition of EP2 receptor by a carbamothioylacrylamide. (A) Compound TG6-129 (SID 17503974) showed robust inhibition of PGE2 (1 µM)-induced cAMP accumulation in C6G-EP2 cells with an IC50 = 1.6 µM, without affecting prostaglandin EP4 and β2-adrenergic receptors. Data were normalized as percentage inhibition of maximum control response; points represent mean ± SEM (n = 3). (B) TG6-129 inhibited PGE2-induced EP2 receptor activation in a concentration-dependent manner. The PGE2 EC50s were 0.79, 1.86, 15.2 and 275 nM, respectively, in the presence of vehicle, 0.01, 0.1 or 1 µM TG6-129. Data were normalized as percent maximum control (vehicle) response; points represent mean ± SEM (n = 4). (C) TG6-129 displayed competitive antagonism of the EP2 receptor as shown by Schild regression analysis, with KB = 8.8 nM and slope = 1.2. (D) TG6-129 was tested for inhibition of all 4 human Gαs-coupled prostanoid receptors. KB values: 14,600, 8.8, 3900, and 199 nM for DP1, EP2, EP4, and IP receptors, respectively.
We have resynthesized compound 1. As shown in Scheme 1. Commercially available p-nitrobenzene-sulfonyl chloride (A) and thiadiazole-2-amine (B) were coupled to form intermediate C, which on metal catalyzed reduction using tin chloride provided p-aminobenzene-sulfonamide D. The final step was carried out analogous to the reported procedure in a one-pot reaction,28 where ammonium thiocyanate was treated with 4-fluorocinnamyl chloride (E) in refluxing acetone for 15 min followed by D for 1 hr to synthesize the product (henceforth referred as TG6-129). Both synthesized and commercially available materials have shown greater than 95% purity by 1HNMR and LCMS analysis (see supporting information for characterization data), and both show bioactivity in the EP2 screen (see below).
Scheme 1.

Synthesis of (E)-N-((4-(N-(5-ethyl-1,3,4-thiadiazol-2-yl)sulfamoyl)phenyl)carbamothioyl)-3-(4-fluorophenyl)acrylamide (TG6-129). i. Pyridine, 95%. ii. Tin-chloride dihydrate in ethyl acetate at reflux, 70%. iii. Ammoniumthiocyanate in acetone at reflux, 80%.
We then demonstrated that TG6-129 inhibits PGE2 induced EP2 receptor activation in a concentration-dependent manner (Fig 1B). In this assay it displayed a competitive mechanism of antagonism of EP2 receptors as shown by Schild regression analysis with KB 8.8 nM and slope of 1.2 (Fig 1C). There are four Gαs coupled prostanoid receptors: DP1, EP2, EP4 and IP. Among these DP1 has the closest sequence homology as EP2, followed by IP, then EP4, although both EP2 and EP4 share a common endogenous ligand PGE2 for their activation.13 To evaluate the selectivity of TG6-129 for EP2 over other Gαscoupled prostanoid receptors, we created C6G-cell lines overexpressing DP1, EP4 and IP receptors and used them for counter screening. Compound TG6-129 showed 1660-fold selectivity against DP1, 443-fold for EP4, and 22-fold selectivity against IP receptors (Fig 1D), indicating it is an EP2 selective antagonist and could be a useful probe for in vitro and in vivo biochemical studies.
To develop structure activity relationships for compound TG6-129, a limited set of 8 commercially available analogs has been purchased and tested in EP2, EP4 and β2-AR assays. The EP2 Schild KB value, which represents the concentration required to cause a 2-fold increase of the EC50 of the agonist PGE2, is used for SAR comparisons below. As shown in Table 1, removal of fluorine (compound 2) resulted in 2-fold less potency on EP2 in comparison to parent TG6-129. Incorporation of a methoxy group as in 3 in place of fluorine caused 4-fold less potency. Replacement of the whole cinnamoyl group with thiocinnamoyl (4 vs 2) reduced the potency 2-fold, but 4-fold less to parent compound TG6-129. Replacement of a 4-methoxycinnamicamide with a 4-methoxyamide (5 vs 3) improved the activity by 1.5-fold, but in comparison to the parent it is still 2.6-fold less. This result indicates the acrylamide moiety is not absolutely essential and may be replaced with amides for further SAR study.
Table 1.
EP2 potency, selectivity and cytotoxicity of carbamothioylacrylamide derivativesa
| Entry | PubChem SID or vendor ID |
Structure | KB (nM) EP2 |
KB (nM) IP |
KB (µM) DP1 |
KB (µM) EP4 |
CC50 (µM) |
|---|---|---|---|---|---|---|---|
| 1 (TG6-129) | 17,503,974 | 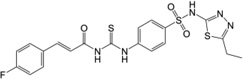 |
8.8 | 199 | 14.6 | 3.9 | 326 |
| 2 | 17,505,080 | 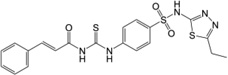 |
16.7 | 57 | 9.8 | 5.8 | 370 |
| 3 | 17,505,097 | 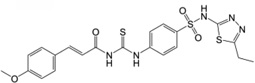 |
34.5 | 360 | 46.6 | 13.0 | 365 |
| 4 | 24,782,326 | 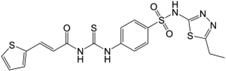 |
33.1 | 2,260 | 10.3 | 2.6 | 330 |
| 5 | 17,508,450 | 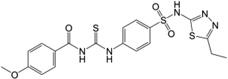 |
23.1 | 1,730 | 16.1 | 7.8 | 186 |
| 6 | 26,671,393 | 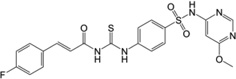 |
18.0 | 3,250 | Inactive | 18.2 | 378 |
| 7 | 17,504,270 | 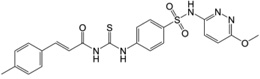 |
13.3 | 360 | Inactive | 20.5 | 150 |
| 8 | 17,504,776 | 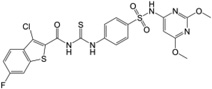 |
108 | 21,000 | 8.7 | 0.7 | 96 |
| 9 | 17,508,802 | 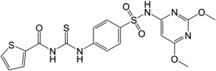 |
132 | 5900 | 25.2 | 3.1 | 258 |
EP2, EP4, IP and DP1 KB values are average from 2–5 independent experiments. In vitro half-maximal cytotoxic concentration (CC50) values are from one experiment run in quadruplicate measurements.
We have also included analogs in which the thiadiazole ring was replaced with methoxy-pirimidine (6) or methoxy-piridazine (7), and in which the methoxy-pirimidines were substituted with amides rather than acryl amides on the left side (8–9). Compound 6 and 7 showed only about 2, and 1.5-fold less potency in comparison to the parent respectively, indicating the thiadiazole ring may be replaced with other heterocycles for further SAR study. However, the amide derivatives 8 and 9 were 12–15 fold less potent than parent. Nonetheless, all these compounds (2–9) have shown nanomolar Schild KB values for EP2 receptors.
Analogs 2–9 were then subjected to a selectivity test using C6G-cells overexpressing the EP4 receptors. Interestingly a majority of them have shown low to high micromolar Schild KB for EP4 receptors, except compound 8 which showed submicromolar Schild KB value. As shown in Table 1, the selectivity index against EP4 is excellent for five compounds 2, 3, 5, 6 and 7 in the range 340 to 2000-fold, but marginal for three compounds 4, 8 and 9 (78, 7 and 24-fold respectively). The compounds 2–5 also showed high selectivity index (311–1300-fold) against DP1 receptor, compounds 6 and 7 showed no activity to DP1 (Table 1). However majority of these derivatives only show 10–75-fold selectivity index against IP receptor, except compound 6 which showed 180-fold selectivity, and compound 2 which was only 3.5-fold selective for EP2 over IP receptor. Unlike our other class of EP2 antagonist, which showed high selectivity to IP receptor (240–0-fold) and low selectivity to DP1 (~10-fold),24, 25 the present class of carbamothioyl-acrylamides (TG6-129 and 2–9) show the opposite – high selectivity to DP1 and low selectivity to IP (Table 1) suggesting a somewhat different nature of binding to the receptors. We also determined the in vitro cytotoxicity for these compounds by measuring the cell viability of C6G-cells after incubation for 48 hours. All test compounds showed low cytotoxicity with a CC50 ≥ 96 µM (Table 1). The high selectivity against EP4, DP1 and moderate selectivity to IP receptors and insubstantial cytotoxicity in vitro prompted us to characterize this scaffold for further development.
We first determined the aqueous solubility of the compound TG6-129 in PBS buffer at pH7 by nephelometry.29 It was soluble at 71 µg/mL (145 µM) indicating a moderate solubility. Then, we wanted to know whether this scaffold is stable in liver microsomal fractions. Based on the potency and selectivity, we have selected two compounds: compound TG6-129 represents a thiadiazole-sulfonamide and compound 6 represents a methoxypyrimidine-sulfonamide, both of which have shown high selectivity and low cytotoxicity (Table 1). These two compounds therefore were subjected to in vitro liver metabolism in microsomal fractions of pooled mouse or human livers at 1 and 10 µM. As shown in Figure 2, compound TG6-129 was slightly more stable in comparison to compound 6 in both mouse and human liver microsomes, with 40–60% of both compounds remaining at 60 minutes. Midazolam was used as a reference in these tests for comparison, which showed expected half-lives (< 15 min) in both liver fractions. These results prompted us to examine pharmacokinetics.
Figure 2.

Metabolic stability study of carbamothioylacrylamide compounds in liver microsomes. Two representative carbamothioylacrylamide compounds (TG6-129 and compound 6 were tested for metabolic stability at 1 µM or 10 µM in pooled mouse (A) or human (B) liver microsomes with midazolam as a control. The metabolic stability of compounds was expressed as half-life (t1/2) in liver microsomes. Data are shown as mean ± SEM (n = 3).
Based on its higher stability in liver fractions, we have chosen TG6-129 for pharmacokinetic study in mice. Compound TG6-129 was dosed into female C57Bl/6 mice at 10 mg/kg by oral gavage or 5mg/kg intraperitoneally in a vehicle consisting of 10% DMSO, 70% PEG400 and 20% sterile water. At five time points, starting from 0.5h to 8h after administration, blood was collected and processed for preparation of plasma samples. Brain samples were also collected at 1h and 2h time points for brain permeability analysis. As shown in Figure 3, TG6-129 was readily absorbed into plasma. About 12 µM (by oral route) and 30 µM (by ip route) concentrations are observed at 30 minutes. These concentrations are 1 to 3-orders of magnitude higher than its EP2 KB value (Fig 1). Although the calculated half-life in plasma is 2.7h, its concentration at 8 hr by both routes of administration is about 200–500-fold higher than its EP2 Schild KB value, indicating prolonged availability of therapeutic levels in plasma. Interestingly, analysis of brain samples revealed that at 5mg/kg by ip or 10 mg/kg by oral gavage no detectable amount is found. However, at 10mg/kg ip, very little compound at 1 hr was detected in the brain tissue with a brain-to-plasma ratio of 0.02. Prolonged plasma half-life and exclusion from the brain will be particularly attractive for using this compound as a probe for chronic peripheral disease models, in contrast to the other class of highly brain permeant EP2 antagonists we recently reported.24, 25
Figure 3.

Pharmacokinetics of TG6-129. The compound was administered to mice by i.p. (5 mg/kg) or p.o. (10 mg/kg) route for pharmacokinetics. Compound concentrations in plasma (ng/ml) were measured at different time points. Data are shown as mean ± SEM (n = 3 mice per time point).
Recently Pfizer has reported a novel class of selective EP2 antagonists with requisite plasma pharmacokinetics.30 While the Pfizer lead compound may well be useful for anti-inflammatory exploration in in vivo models, we asked whether the new scaffold discovered in the present study will also demonstrate anti-inflammatory properties. We have previously reported that the EP2 receptor regulates a host of pro- and anti-inflammatory mediators in human prostate cancer cell lines and rat primary microglia cultures.31, 32 Here, we treated P388D1 macrophages with butaprost (EP2 agonist) to induce mRNA expression of inflammatory markers. As shown in Figure 4A, COX-2, IL-1β, IL-6, IL-12 (p40), IL-17, IL-23, and TNF-α were significantly elevated upon treatment with butaprost, In contrast, only a slight increase in expression of the EP2 receptor itself was observed by butaprost in P388D1 macrophages (Fig 4B), reinforcing data recently reported for primary microglia.24, 32 As predicted, compound TG6-129 attenuated elevation of all inflammatory markers investigated (Fig 4A). Further studies are ongoing to determine this compound has an effect not only at mRNA levels, but also at protein levels and in regulation of inflammation in an in vivo model of arthritis.
Figure 4.

EP2 receptor activation induces macrophage activation. A) P388D1 macrophages were pretreated with TG6-129 (10 µM) or vehicle for 1 h, then followed by incubation with EP2 selective agonist butaprost (10 µM) for 2 h. The mRNA levels of the indicated inflammatory mediators were measured by qRT-PCR. B) EP2 mRNA levels in P388D1 macrophages stimulated with butaprost for 2h (n = 3–4, ***P < 0.001; **P < 0.01; *P < 0.05, one-way ANOVA with post hoc Bonferroni test). Data are shown as mean ± SEM.
In conclusion, we reported a novel class of highly potent EP2 antagonists, identified a compound in the class with good solubility and pharmacokinetic properties, and showed that it suppressed inflammation in vitro in a macrophage cell line after activation with EP2 agonist butaprost. Further optimization is needed to improve the selectivity against IP-receptor to obtain molecules for in vivo proof of concept studies in a variety of peripheral inflammatory disease model studies.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH/NINDS grants K99NS082379 (to J.J.) and U01NS058158 (to R.D.), and the Epilepsy Foundation (to J.J.).
Footnotes
ASSOCIATED CONTENT
Contains experimental methods including details of synthesis and characterization data and assay protocols. This material is available free of charge via internet at http:/pubs.acs.org.
Author contributions
T.G., J.J., R.S. and R.D. designed the research; T.G., J.J. and R.S. performed the research; T.G., J.J. and R.D. wrote the manuscript.
REFERENCES
- 1.Baumgart DC, Carding SR. Gastroenterology 1 - Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E(2) and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. 2008;13:1014–1021. doi: 10.1111/j.1440-1843.2008.01365.x. [DOI] [PubMed] [Google Scholar]
- 3.Lin CC, Lee IT, Yang YL, Lee CW, Kou YR, Yang CM. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: Role of TLR4-dependent NADPH oxidase activation. Free Rad. Biol. Med. 2010;48:240–254. doi: 10.1016/j.freeradbiomed.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 4.McInnes IB, Schett G. MECHANISMS OF DISEASE The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 5.Akiyama H, Barger S, Barnum S, Bradt B, et al. Neurobiol. Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Brit. J. Pharmacol. 2006;147:S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minghetti L. Role of inflammation in neurodegenerative diseases. Curr. Opin. Neurol. 2005;18:315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 8.Al-Hourani BJ, Sharma SK, Suresh M, Wuest F. Cyclooxygenase-2 inhibitors: a literature and patent review (2009–2010) Exp. Opin. Ther. Pat. 2011;21:1339–1432. doi: 10.1517/13543776.2011.593510. [DOI] [PubMed] [Google Scholar]
- 9.DeWitt DL. Cox-2-selective inhibitors: the new super aspirins. Mol. Pharmacol. 1999;55:625–631. [PubMed] [Google Scholar]
- 10.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, Study AsDC. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression - A randomized controlled trial. J. Amer. Med. Assoc. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 11.Grosser T, Yu Y, FitzGerald GA. Emotion Recollected in Tranquility: Lessons Learned from the COX-2 Saga. Annu. Rev. Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 12.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 13.Hirata T, Narumiya S. Prostanoid Receptors. Chem. Rev. 2011;111:6209–6230. doi: 10.1021/cr200010h. [DOI] [PubMed] [Google Scholar]
- 14.Arehart E, Stitham J, Asselbergs FW, Douville K, MacKenzie T, Fetalvero KM, Gleim S, Kasza Z, Rao Y, Martel L, Segel S, Robb J, Kaplan A, Simons M, Powell RJ, Moore JH, Rimm EB, Martin KA, Hwa J. Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation - Potential implications for cyclooxygenase-2 inhibition. Cir. Res. 2008;102:986–993. doi: 10.1161/CIRCRESAHA.107.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannon CP, Cannon PJ. COX-2 Inhibitors and Cardiovascular Risk. Science. 2012;336:1386–1387. doi: 10.1126/science.1224398. [DOI] [PubMed] [Google Scholar]
- 16.Egan KM, Lawson JA, Fries S, Koller B, Rader DJ, Smyth EM, FitzGerald GA. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–1957. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 17.Andreasson K. Emerging roles of PGE(2) receptors in models of neurological disease. Prostagland. & Other Lipid Mediat. 2010;91:104–112. doi: 10.1016/j.prostaglandins.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E-2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J. Exp. Med. 2006;203:325–335. doi: 10.1084/jem.20051310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheibanie AF, Khayrullina T, Safadi FF, Ganea D. Prostaglandin E-2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthrit. & Rheumat. 2007;56:2608–2619. doi: 10.1002/art.22794. [DOI] [PubMed] [Google Scholar]
- 20.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, Ganea D. The proinflammatory effect of prostaglandin E-2 in experimental inflammatory bowel disease is mediated through the IL-23 -> IL-17 axis. J. Immunol. 2007;178:8138–8147. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 21.Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- 22.de Rooij J, Zwartkruis FJT, Verheijen MHG, Cool RH, Nijman SMB, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 23.Jiang JX, Ganesh T, Du YH, Thepchatri P, Rojas A, Lewis I, Kurtkaya S, Li L, Qui M, Serrano G, Shaw R, Sun AM, Dingledine R. Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc. Natl. Acad. Sci. U S A. 2010;107:2307–2312. doi: 10.1073/pnas.0909310107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang J, Ganesh T, Du Y, Quan Y, Serrano G, Qui M, Speigel I, Rojas A, Lelutiu N, Dingledine R. Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proc. Natl. Acad. Sci. U S A. 2012;109:3149–3154. doi: 10.1073/pnas.1120195109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J, Quan Y, Ganesh T, Pouliot WA, Dudek FE, Dingledine R. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc. Natl. Acad. Sci. U S A. 2013;110:3591–3596. doi: 10.1073/pnas.1218498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalgutkar AS, Gardner I, Obach RS, Shaffer CL, Callegari E, Henne KR, Mutlib AE, Dalvie DK, Lee JS, Nakai Y, O'Donnell JP, Boer J, Harriman SP. A comprehensive listing of bioactivation pathways of organic functional groups. Curr. Drug Metabol. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- 27. http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?sid=17503974. [Google Scholar]
- 28.Miyazaki I, Simizu S, Ichimiya H, Kawatani M, Osada H. Robust and Systematic Drug Screening Method Using Chemical Arrays and the Protein Library: Identification of Novel Inhibitors of Carbonic Anhydrase II. Biosci. Biotechnol. & Biochem. 2008;72:2739–2749. doi: 10.1271/bbb.80383. [DOI] [PubMed] [Google Scholar]
- 29.Bevan CD, Lloyd RS. A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal. Chem. 2000;72:1781–1787. doi: 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- 30.af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K, Pullen N. In vitro and in vivo characterization of PF-044189948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Brit. J. Pharmacol. 2011;164:1847–1856. doi: 10.1111/j.1476-5381.2011.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang J, Dingledine R. Role of Prostaglandin Receptor EP2 in the Regulations of Cancer Cell Proliferation, Invasion, and Inflammation. J. Pharmacol. Exp. Ther. 2013;344:360–367. doi: 10.1124/jpet.112.200444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J. Biol. Chem. 2013;288:9293–9302. doi: 10.1074/jbc.M113.455816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.