

Abstract
NFκB-dependent signaling is an important modulator of inflammation in several diseases including sepsis. G-protein coupled receptor kinase-5 (GRK5) is an evolutionarily conserved regulator of NFκB pathway. We hypothesized that GRK5 via NFκB regulation plays an important role in the pathogenesis of sepsis. To test this we utilized a clinically relevant polymicrobial sepsis model in mice that were deficient in GRK5. We subjected wild type (WT) and GRK5 knockout (KO) mice to cecal-ligation and puncture (CLP)-induced polymicrobial sepsis and assessed the various events in sepsis pathogenesis. CLP induced a significant inflammatory response in the WT and this was markedly attenuated in the KO mice. To determine the signaling mechanisms and the role of NFκB activation in sepsis-induced inflammation, we assessed the levels of IκBα phosphorylation and expression of NFκB-dependent genes in the liver in the two genotypes. Both IκBα phosphorylation and gene expression were significantly inhibited in the GRK5 KO compared to the WT mice. Interestingly however, GRK5 did not modulate either immune cell infiltration (to the primary site of infection) or local/systemic bacterial load subsequent to sepsis induction. In contrast GRK5 deficiency significantly inhibited sepsis-induced plasma corticosterone levels and the consequent thymocyte apoptosis in vivo. Associated with these outcomes, CLP-induced mortality was significantly prevented in the GRK5 KO mice in the presence of antibiotics. Together, our studies demonstrate GRK5 as an important regulator of inflammation and thymic apoptosis in polymicrobial sepsis and implicate GRK5 as a potential molecular target in sepsis.
Introduction
G-protein coupled receptor kinases (GRKs) are serine/threonine kinases well known for their role in phosphorylation of G-protein coupled receptors (GPCRs) [1]. Functionally GRKs have been linked to a number of cell signaling processes, not only related to their role in GPCR phosphorylation, but also in their ability to phosphorylate or scaffold a number of intracellular signaling proteins [2]. GRKs are functionally grouped into three classes: GRK1-like (GRK1 and GRK7, otherwise known as rhodopsin kinases), GRK2-like [GRK2 and GRK3, otherwise known as βARK1 and 2 (β-adrenergic receptor kinases 1 and 2)] and GRK4-like (GRK4, GRK5 and GRK6). Even though there is some specificity in terms of their tissue distribution, GRKs especially GRK2 and GRK5 are ubiquitously expressed in many cell types including immune cells [2].
Of the seven members of the GRK family, GRK5 was first identified as a kinase that phosphorylates β2-adrenergic receptor (β2AR), m2 muscarinic cholinergic receptor, and rhodopsin [3]. Using GRK5 deficient mice, Gainetdinov et al [4] further identified a critical role for GRK5 in muscarinic receptor signaling in vivo. Subsequently, however, several studies have demonstrated a broad role for this kinase in cell signaling. For example, GRK5 has been shown to phosphorylate and/or interact with non-receptor substrates including arrestin-2 [5], F-actin [6], HDAC5 [7], Hip [8], IκBα [9], p105 [10], Lrp6 [11], nucleophosmin [12], and p53 [13]. Based on its role in cell signaling, GRK5 has been proposed to be a critical kinase in the pathogenesis of several diseases including endotoxemia [14], cancer [15], Alzheimer’s [16] and atherosclerosis [17]. In addition, GRK5 levels are modulated in a number of diseases including sepsis, heart failure, obesity, cystic fibrosis, cancer and mental disorders [2]. We recently demonstrated that the GRK5 knockout mice have attenuated ability to produce cytokines in vivo, in response to lipopolysaccharide (a Toll-like receptor-4, TLR4 ligand) [14]. We further showed GRK5 to be an important regulator of signaling from multiple Toll-like receptor ligands including TLR2 and TLR3 in vivo [18]. Interestingly, a recent study also demonstrated that GRK5 is a critical mediator of inflammation in Drosophila and Zebra fish models [19]. However, role of GRK5 in the pathogenesis of a clinically relevant model of polymicrobial sepsis is not known.
Sepsis is the leading cause of death among intensive care patients [20]. Dysregulated inflammatory response is a prominent modulator of sepsis progression, causing coagulation derangements, apoptosis of lymphoid and non-lymphoid tissues and organ dysfunction [21]. Despite the improvements in resuscitation and antibiotic supportive care, the high incidence and fatality in sepsis underscores the need for better understanding of the pathophysiology of sepsis and to identify new molecular therapeutic targets. Using a clinically relevant polymicrobial sepsis model [22], we demonstrate here that GRK5 is an important modulator of sepsis progression, inflammation, thymocyte apoptosis and mortality. We further demonstrate that GRK5 is an important regulator of sepsis-induced NFκB activation in the liver. Together, our studies implicate GRK5 as an important molecular target in the pathogenesis of polymicrobial sepsis.
Materials and Methods
Materials
Protease inhibitor cocktail tablets were from Roche Applied Science (Indianapolis, IN); pIκBα, pERK1/2, pP38 and pJNK and tubulin antibodies were from Cell Signaling Technology, Inc. (Danvers, MA) and Sigma (St. Louis, MO) respectively. Ultra pure Escherichia coli (0111:B4) LPS was from Invivogen (San Diego, CA) and Dexamethasone was from Sigma (St. Louis, MO).
Mice
GRK5 knockout mice were obtained from Jackson labs and have been previously described [14]. Animals used for experiments were 8-12 week old males. Animals were housed 4-5 mice per cage at 22-24°C with 50% humidity and a 12 hour light-dark cycle. All animal procedures were approved by Michigan State University Animal care and Use committee.
Sepsis model
Polymicrobial intra-abdominal sepsis was induced by cecal ligation puncture technique [23]. Briefly, mice were anaesthetized by administering ketamine (80mg/kg) and xylazine (5mg/kg) intraperitoneally. Cecum was exteriorized, ligated and punctured twice with 20 G needle. Sham surgeries were carried out to serve as control wherein the exteriorized cecum was neither ligated nor punctured. All animals were administered with 1ml of warm saline post surgery subcutaneously. In one set of survival experiments, antibiotics (Ceftriaxone 25mg/kg and Metronidazole 15mg/kg) were administered (intraperitoneal) 1-hour post-sepsis and every 24 hours for 5 days.
Peritoneal Lavage fluid and blood collection
Peritoneal exudate fluid was collected at different time points post sepsis as described before [24]. Briefly, peritoneal cavity was lavaged with 7 ml of RPMI media with 10% FBS and peritoneal fluid collected, centrifuged to separate the cells and the supernatants stored at -80°C until further analysis. Blood was collected by cardiac puncture and plasma was separated (by centrifugation) and stored at -80°C until further analysis.
Cytokine/chemokine measurements
Cytokines and chemokines were measured from peritoneal exudate and plasma using ELISA kits from eBiosciences, Inc. as described before [25].
Bacterial counts
Bacterial load was determined in blood, and peritoneal fluid at different time points as described before [24]. Briefly, blood, and peritoneal fluid were serially diluted and plated on Trypticase™ Soy Agar with 5% Sheep Blood (BD Biosciences) and incubated at 37°C for 48 hours. Colony forming units were counted to determine bacterial load and expressed as CFU/ml.
Determination of thymic cell numbers and apoptosis
Thymi were collected from septic and sham operated mice 20- and 36-hours post-sepsis and single cell suspension prepared as described before [26]. For determining cell number changes, cells were counted using hemocytometer. Cells were also labeled with Annexin V and propidium iodide (following manufacturer’s instructions (eBiosciences, Inc)) to determine the apoptotic cells by flow cytometry (LSRII, BD biosciences) as described before [27]. In addition, cells were labeled with anti-CD4-PE-Cy7 and anti-CD8-PE for determining CD4+ and CD8+ cells in the thymus and data were acquired using LSRII (BD Biosciences) and analyzed using Flowjo software (Tree StarInc., Ashland, Oregon).
Caspase activity assays
Thymocytes (obtained as described above) were lysed in buffer (50 mM HEPES, 0.1% CHAPS, 1 mM DTT, 0.1 mM EDTA, 0.4% Triton-X100, pH 7.4) at 4°C for 15 min. The cell lysate was collected and the protein content determined (BIO-RAD). 10 μg of the cell lysate was incubated with the fluorescent substrates (Ac-DEVD-AFC, Z-IETD-AFC and Ac-LEHD-AFC) to determine caspase-3, -8 and -9 activities, at 100 μM in the assay buffer (50 mM HEPES, 1% Sucrose, 0.1% CHAPS, 10 mM DTT, pH 7.4) as described [28]. The fluorescence of the cleaved substrates was determined spectro-fluorometrically (excitation of 400 nm and emission of 505 nm) in Tecan Spectra FluorPlus fluorescence plate reader. Data are presented as picogram of cleaved AFC per mg protein per min calculated from a standard curve plot with free AFC.
In-vitro stimulation of septic peritoneal cells
Peritoneal cells collected from wild type (WT) and GRK5 knockout (KO) mice 36 hours-post sepsis were washed with PBS and plated in 12-well plate at one million cells/well. Cells were then stimulated (or not) with LPS (0.5 μg/ml) for 12 hours and supernatants collected and assayed for the indicated cytokines by ELISA.
Restraint stress
Eight to 10 week old GRK5 WT and KO mice were subjected to stress with physical restraint as previously described [29]. Briefly, mice were placed in a 50 ml centrifuge tube with multiple openings for ventilation and held horizontally for 30 minutes. After the stipulated time, blood was collected for corticosterone measurement.
Corticosterone measurements
Plasma corticosterone levels were measured using a corticosterone EIA kit from Cayman Chemical (Ann Arbor, MI) as per the manufacturers instructions.
RNA extraction and Real-time Q-PCR
Liver and lung samples from septic and sham operated mice were collected 12 hours post-CLP, and total RNA was extracted using Qiagen’s RNeasy Mini kit. Reverse transcription was carried out with 1 μg of RNA with promega cDNA synthesis kit. Real-time Q-PCR was performed as described before for the expression of IκBα, IL-6, IL-1β, and HPRT [9]. Primers were obtained from IDT DNA Technologies. Following primers were used: IκBα-Forward: TGG CCA GTG TAG CAG TCT TG; Reverse: GAC ACG TGT GGC CAT TGT AG; IL-6- Forward: ACA AGT CGG AGG CTT AAT TAC ACA T; Reverse: TTG CCA TTG CAC AAC TCT TTT C; IL-1β-Forward: TCG CTC AGG GTC ACA AGA AA; Reverse: CAT CAG AGG CAA GGA GGA AAA C; HPRT-Forward: AAG CCT AAG ATG AGC GCA AG; Reverse: TTA CTA GGC AGA TGG CCA CA. Real-time Q–PCR was performed using ABI fast 7500 (Applied Biosystems) and all the genes were normalized to HPRT.
Western blot analysis
Cytoplasmic extracts from frozen liver tissue samples were prepared by homogenizing the tissue in lysis buffer (1M HEPES, 2M KCl, 0.5M EDTA, 0.1M EGTA along with protease and phosphatase inhibitors). The protein concentrations in the extracts were determined and equivalent amounts of protein were loaded onto the gels for western blot analysis. Immunoblotting was carried out for p-IκBα, p-ERK, p-JNK, p-p38 and tubulin as described before [14]. The bands were quantified using image-J (for chemiluminescence) or Licor’s Odyssey program (for fluorescence).
Statistical analysis
All data are presented as the mean±SEM. Two group comparisons were performed using Student’s t-test and comparisons of more than two groups were done by ANOVA with post-Bonferroni test. Survival studies were analyzed by log rank test (Mantel-Cox) [30-32] as well as by factorial analysis. All statistical analyses (except factorial analysis) were performed using GRAPHPAD PRISM Software (San Diego, CA) and p<0.05 were considered statistically significant. Factorial analysis for survival [Two genotypes (wild type and knockout) and two treatment (without and with antibiotics)] was performed in consultation with MSU’s Center for Statistical Training and Consulting and using the SPSS software program.
Results
In previous studies, we demonstrated that the GRK5 deficient mice have an attenuated inflammatory response after in vivo stimulation with TLR ligands [14,18]. Given that the progression of polymicrobial sepsis is in part dependent on stimulation of multiple TLRs, we examined the role of GRK5 in a clinically relevant model of polymicrobial sepsis. For this, we subjected wild type (WT) and GRK5 knockout (KO) mice to cecal ligation and puncture and determined the various pathogenic events including inflammatory response, immune cell infiltration, thymic apoptosis, bacterial load and mortality.
GRK5 mediates sepsis-induced cytokine production
Cytokine and chemokine levels were examined in the peritoneal fluid and plasma from the different groups of mice (sham and CLP in the WT and KO groups) at 12 hours after surgery. As shown in Fig 1A, IL-6, IL-10, TNFα and MCP-1 levels in the peritoneal fluid were significantly decreased in KO septic compared to WT septic mice. Peritoneal IL-12/23 (total p40) was also inhibited in KO mice but did not reach statistical significance. Similar to the peritoneal fluid, plasma levels of IL-6 and IL-10 were significantly decreased in KO compared to the WT septic mice (Fig 1B). Plasma levels of TNFα, IL-12/23 and MCP-1 did not significantly differ between septic groups. Note that neither of the sham groups showed any detectable levels or showed very low levels of cytokines/chemokines at the time points tested (data not shown). Together these results suggest that GRK5 mediates sepsis-induced local and systemic inflammation.
Figure 1. GRK5 mediates inflammatory response in cecal ligation and puncture model of polymicrobial sepsis.
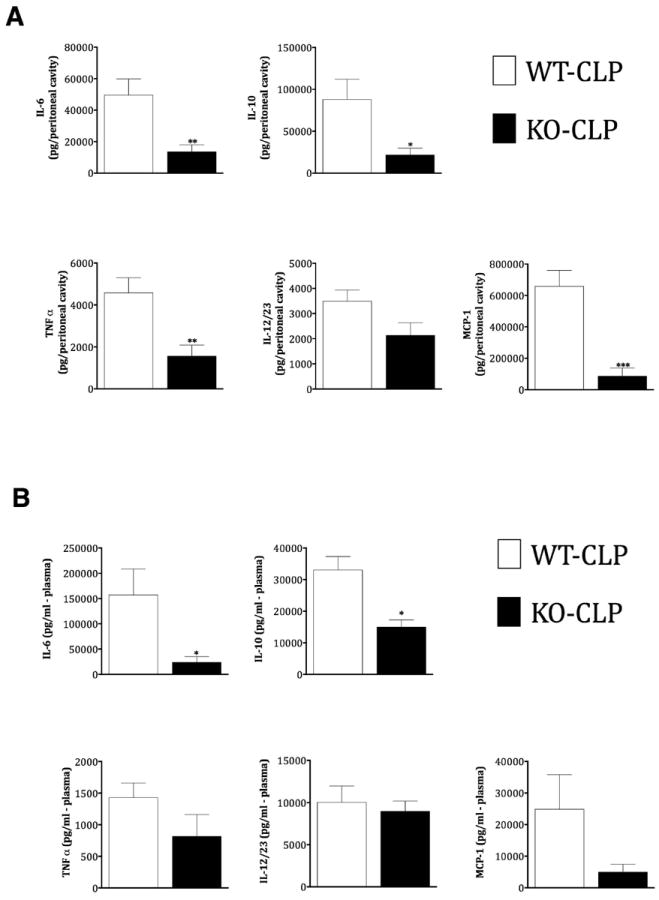
GRK5 wild type and knockout mice were subjected to CLP surgery and peritoneal (A) and plasma (B) fluids collected 12 hours after surgery as described in the methods. IL-6, IL-10, TNFα, IL12/23 (total p40), and MCP-1 were measured in these two fluid samples using ELISA kits from eBiosciences. (N=12 for wild type and N=9 for knockouts for the 12 hour timepoint).*P<0.05, **P<0.01 and ***P<0.001 compared to the corresponding septic wild type groups.
GRK5 mediates NFκB signaling in polymicrobial sepsis
Previous studies have shown that GRK5 is an important regulator of NFκB signaling and inflammatory gene expression in mouse, zebra fish and Drosophila models [14,19]. Our results in the CLP model of polymicrobial sepsis suggest that GRK5 deficiency attenuates inflammatory cytokine production both in the peritoneal cavity and in the plasma. To examine if GRK5 deficiency leads to attenuated NFκB signaling in this sepsis model, we determined phospho-IκBα levels and mRNA expression of NFκB-dependent genes in the liver from the two genotypes of mice subjected to sham or CLP surgery. As shown in Fig 2A, CLP-induced significant phosphorylation of IκBα in the liver of WT mice and this was significantly inhibited in the GRK5 knockout. To determine the expression of NFκB-dependent genes, we examined the mRNA levels of IκBα, IL-6 and IL-1β in the liver 12 hours after CLP. Consistent with the role of GRK5 in IκBα phosphorylation, mRNA expression of iκbα, il1β and il6 was significantly inhibited in the GRK5 KO mice (Fig 2B). Interestingly, this phenomenon was also observed in the lungs (Fig 2B lower panel). To rule out other signaling pathways, we also examined pERK, pJNK and pP38 and found no effect of GRK5 deficiency on these pathways (Fig 2C). Together, these results demonstrate a crucial role for GRK5 in NFκB activation in vivo and the consequent inflammatory response in this polymicrobial sepsis model.
Figure 2. GRK5 mediates IκBα phosphorylation and expression of NF-κB dependent genes in liver and lung.
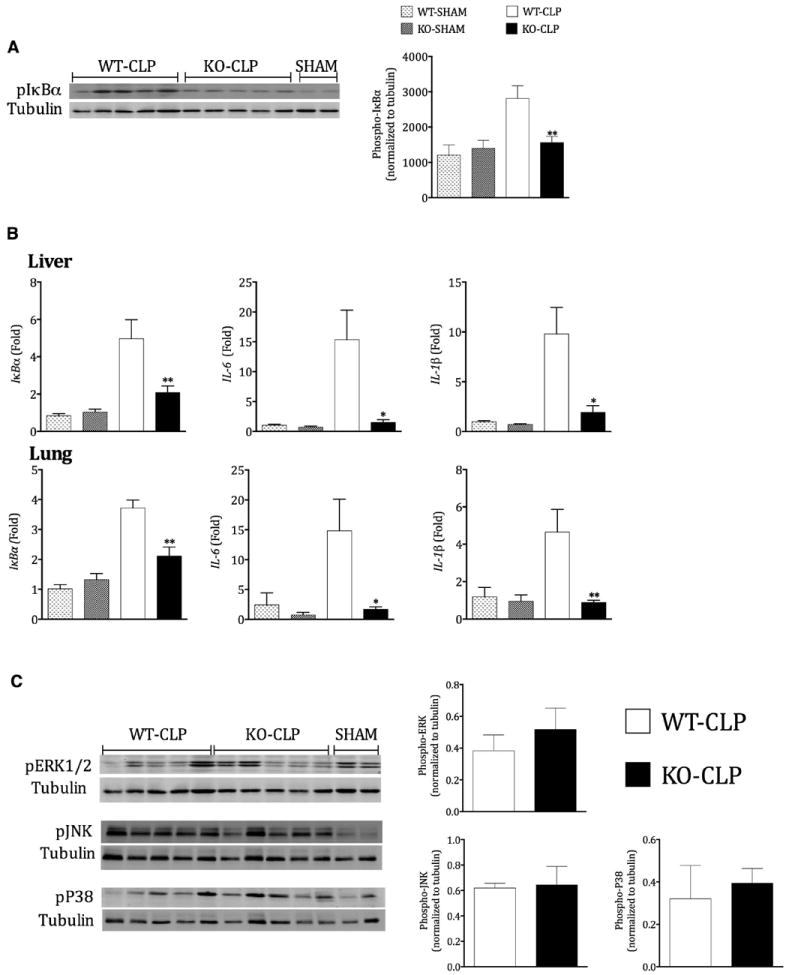
(A) Cytosolic extracts of the liver tissue from GRK5 wild type and knockout mice 12 hours post-sepsis were subjected to Western blotting for phospho-IκBα and tubulin (for normalization) as described before [14]. Representative blot is shown in the left and quantitation on the right. N=11-12 each for wild type and KO mice CLP and N=3 for shams; *P<0.05. (B) Liver and lung tissue samples from GRK5 wild type and knockout mice subjected to sham or CLP surgery were collected 12 hours post-sepsis and analyzed for the expression of NF-κB-dependent genes as described in methods. (N= 8 for CLP and 4 for sham per genotype *P<0.05, **P<0.01 compared to the corresponding septic wild type group. (C) Liver tissue extracts described in (A) above were subjected to immunoblotting for pERK, pJNK and pP38 as described in the methods. Western blot is shown on the left and quantitation on the right. N=5 each for wild type and KO mice CLP.
GRK5 does not regulate chemotaxis to the local site of injury/infection
Studies have shown that following CLP, peritoneal cell infiltration plays a critical role in the progression of sepsis [33] and that modulation of immune cell infiltration can have therapeutic consequences [34]. GRKs have been shown to be important regulators of chemokine receptor signaling and chemotaxis [35]. Therefore, to determine if GRK5 regulates immune cell infiltration into the site of injury/infection (peritoneum), we examined the number of cells in the peritoneal cavity following CLP at different time points in the two genotypes of mice. As shown in Fig 3, GRK5 deficiency did not affect immune cell infiltration into the peritoneal cavity at any of the time points tested. In addition there was no difference in infiltration of specific immune cell populations (Fig 3). Together these results suggest that GRK5 is an unlikely regulator of chemotaxis in this model of sepsis.
Figure 3. Role of GRK5 in peritoneal cell infiltration following cecal ligation and puncture.
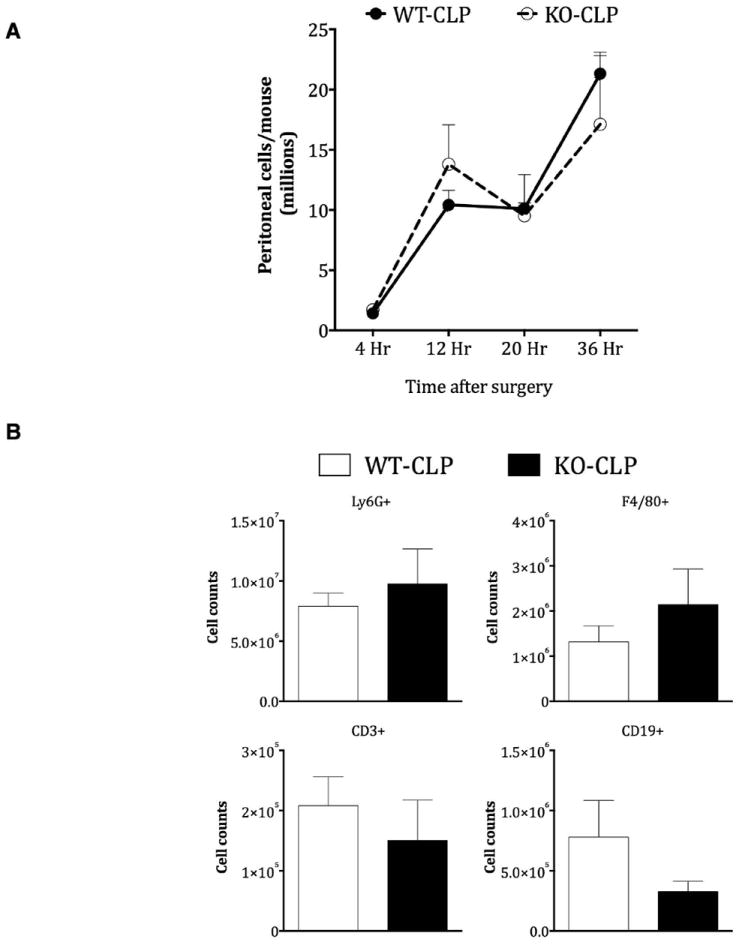
GRK5 wild type and knockout mice were subjected to CLP surgery and peritoneal cells collected by lavaging peritoneal cavity and cells counted (A) as described in the methods. The different immune populations (B) were assessed by flow cytometry as described in the methods (N=9 for 4 and 12 hour time points and N=5-6 for 20 and 36 hour time points, per genotype).
Deficiency of GRK5 does not affect bacterial load following sepsis
In order to examine if GRK5 is able to modulate bacterial load after CLP, we plated peritoneal lavage fluid, and blood samples from sham and septic mice onto 5% sheep blood agar plates and determined the colony forming units (CFU). Interestingly, bacterial load was not any different between the wild type and the KO mice at any of the time points tested (Fig 4A and 4B). Taken together, our data so far suggests that even though GRK5 mediates NFκB signaling and inflammation in sepsis, neither chemotaxis nor bacterial load are significantly affected.
Figure 4. Role of GRK5 on bacterial load following cecal ligation and puncture.
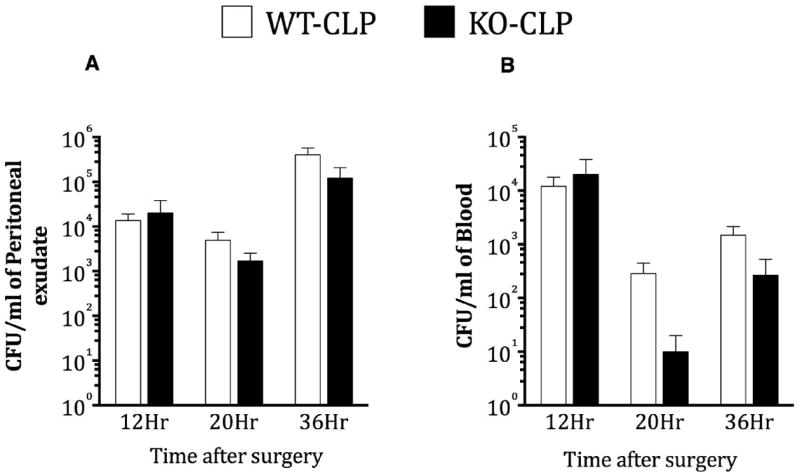
Blood and peritoneal samples from GRK5 wild type and knockout mice subjected to sham or CLP surgery were assessed for bacterial load as described in the methods. CFU counts from peritoneal (A), and blood (B) for different time points are shown from mice subjected to CLP. (N= 11-14 per genotype at 12 hour time point; N= 5 per genotype at 20 hour time point; N= 9-12 for 36 hour time point). Note that sham mice did not show any bacterial colonies (data not shown).
Regulation of thymocyte numbers by GRK5 in sepsis
It is now well established that sepsis-induced thymocyte apoptosis contributes to the pathogenic events and the consequent mortality in septic animals [36] and human patients [37]. Importantly, inhibiting thymic apoptosis has been shown to be beneficial in preventing sepsis-induced mortality in experimental models [38]. Previous studies have suggested a role for GRK5 in irradiation-induced thymocyte apoptosis via a p53-dependent pathway [13]. Furthermore, NFκB signaling in thymocytes has been shown to be an important regulator of thymocyte apoptosis [39,40]. Together, based on these rationale, we hypothesized that GRK5 could be an important regulator of sepsis-induced thymocyte apoptosis. To test this, we first assessed the number of thymocytes from WT and KO mice subjected to sham or CLP surgery. As predicted from previous studies, in the WT septic mice thymocyte numbers were significantly decreased compared to sham operated mice (Fig 5A). Interestingly however, septic KO mice had significantly higher numbers of thymocytes compared to the corresponding WT mice at both time points. In addition to the total cells, the frequency of double positive CD4+CD8+ lymphocytes (loss of these lymphocytes has been linked to poor survival [41]) was also markedly decreased in the wild type septic mice compared to the sham (Fig 5B). Importantly, decrease in these double positive CD4+CD8+ cells was significantly attenuated in the GRK5 deficient mice (Fig 5B).
Figure 5. GRK5 mediates thymocyte apoptosis in polymicrobial sepsis.
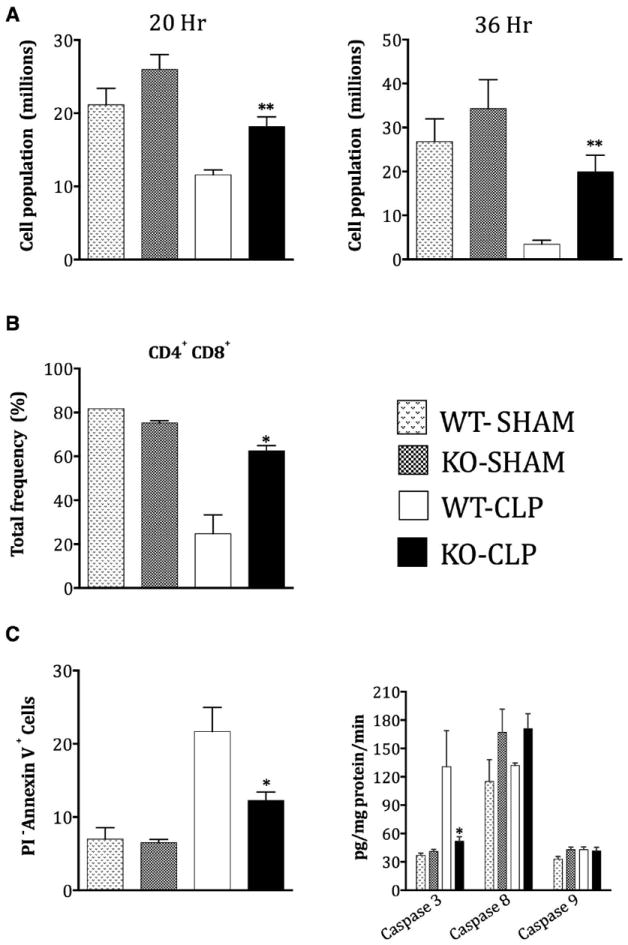
Thymi from GRK5 wild type and knockout mice subjected to sham or CLP surgery were collected 20 and 36 hours after surgery as described in the methods. Thymocytes were analyzed for total number (A), CD4+, CD8+ (B), Annexin V+ and PI- staining and for caspase activity (C) as described in the methods. (N=5-6 per genotype for assessing total thymus cell number and Annexin V+ and PI- staining; N=3-4 per genotype for the flow cytometry experiments). *P<0.05, **P<0.01 and ***P<0.001 compared to the corresponding septic wild type groups.
Regulation of thymocyte apoptosis by GRK5 in vivo
To further confirm the difference in thymocyte numbers is as a result of altered apoptosis between wild type and GRK5 KO mice, we performed two separate assays: Flow cytometry analysis of thymocytes stained with Annexin V and PI; and Caspase activity assays. Consistent with the thymocyte numbers, increased frequency of Annexin V+PI- cells (early apoptotic cells) was observed in wild type septic mice and this was significantly inhibited in the GRK5 KO mice (Fig 5C). Furthermore, CLP significantly induced caspase-3 activity in the wild type thymocytes and this was again markedly reduced in the GRK5 KO mice (Fig 5C). Unlike caspase-3, activities of caspase-8 and -9 were not significantly induced in sepsis and did not differ between the groups (Fig 5C). Together, these results implicate GRK5 in the regulation of caspase-3-mediated thymocyte apoptosis following sepsis.
Mechanism of GRK5-mediated thymic apoptosis
Previous studies have shown that sepsis-induced corticosteroids induce thymic apoptosis in the CLP model [42]. To determine whether GRK5 directly modulates corticosteroid-induced thymocyte apoptosis, we stimulated thymocytes from wild type and GRK5 KO mice ex vivo with dexamethasone (100 nM) and assessed apoptosis using flow cytometry (AnnexinV/PI). Dexamethasone treatment induced significant apoptosis in both the genotypes and surprisingly, apoptosis was equivalent between the genotypes (data not shown). Because these results ruled out any direct effect of GRK5 in thymic apoptosis (at least as induced by corticosteroids), we next examined whether the levels of corticosterone are different between the two genotypes during sepsis progression. Interestingly, plasma corticosterone levels were significantly higher in the wild type septic mice (compared to sham) and the levels were markedly attenuated in the GRK5 KO septic mice (Fig 6). This was more evident at the later time point (20 h). To determine whether the difference in the corticosterone levels between the two genotypes is specific to sepsis, we induced stress with physical restraint in both genotypes and measured plasma corticosterone levels. We found that 30 minutes of physical restraint induced significant increase in plasma corticosterone, but the levels were similar between the two genotypes (Fig 6). Together these results suggest that sepsis-induced corticosterone levels are attenuated in GRK5 deficient mice and this might lead to enhanced thymocyte survival during later stages of sepsis.
Figure 6. Sepsis-induced plasma corticosterone levels are diminished in GRK5 knockout mice.
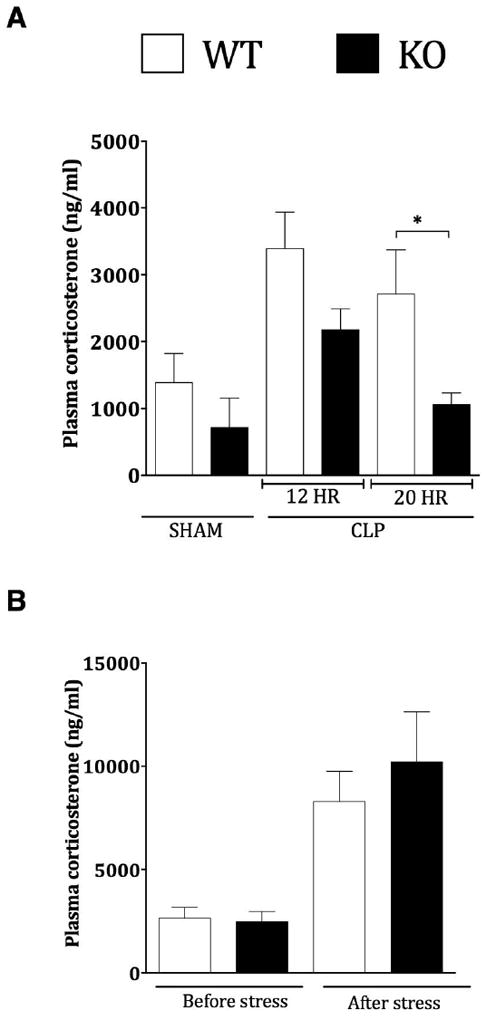
(A) Plasma samples collected 12 and 20 hours post surgery (sham and CLP) from GRK5 wild type and knockout mice, were assessed for corticosterone levels using Cayman Chemical EIA kit. (N=8 per genotype for both 12 and 20 hour time point for CLP; N=2-3 for sham at 12 hour time point). (B) Corticosterone levels in plasma samples obtained from GRK5 wild type and knockout mice before and after restraint stress (N=7 per genotype). *P<0.05 compared to the corresponding wild type groups.
GRK5 inhibits immunoresponsiveness of peritoneal cells in sepsis
Decrease in lymphocytes due to apoptosis is thought to be an important pathogenic event in the development of immunosuppression observed during sepsis progression. Because, thymic apoptosis is reduced in the GRK5 deficient mice, we hypothesized that the consequent development of immune suppression may be attenuated in GRK5 deficient mice. To test this in vitro, we obtained peritoneal cells from the two genotypes of mice subjected to sepsis (36 hours post-CLP) and assessed their immunocompetency in response to in vitro lipopolysaccharide (LPS) stimulation. Supernatants from these experiments were assayed for IL-6, IL-10, TNFα, and IL-12/23 using ELISA. As expected, unstimulated GRK5 KO cells (from septic mice) produced lower cytokine levels compared to the wild type septic mice (Fig 7). However, upon stimulation with LPS, GRK5 KO cells produced significantly enhanced pro-inflammatory cytokines (IL-6, TNFα and IL-12/23) compared to the wild type cells. This effect was restricted only to the typical pro-inflammatory group and not to IL-10. Basal IL10 level was much higher in the wild type cells from septic mice and LPS stimulation did not further enhance IL10. Together, these results show that even though the initial inflammatory response in the KO cells is attenuated, cells from these septic mice respond better than the wild type cells to in vitro LPS stimulation. This suggests that deficiency of GRK5 possibly renders the mice more immune-responsive at later stages of sepsis.
Figure 7. GRK5 suppresses immuno-responsiveness of peritoneal cells in sepsis.
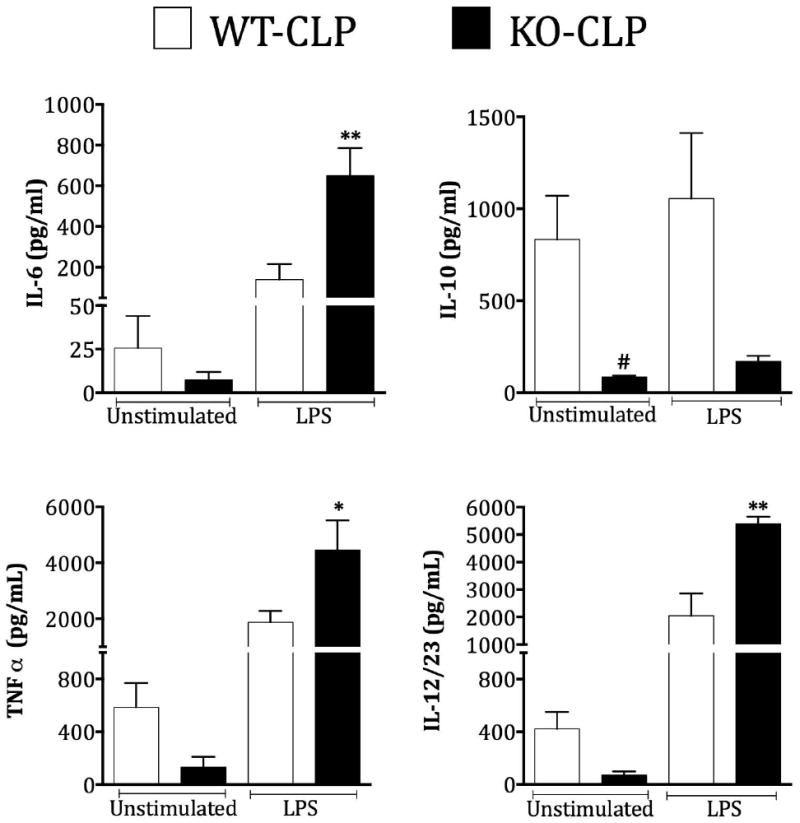
Peritoneal cells from GRK5 wild type and knockout mice subjected to sham or CLP surgery were collected 36 hours post-sepsis, stimulated with LPS and assessed for cytokine production as described in methods. (N= 7-8 per genotype). *P<0.05, **P<0.01 compared to the corresponding septic wild type peritoneal cells stimulated with LPS. #P<0.05 compared to the corresponding septic wild type peritoneal cells without LPS stimulation.
Role of GRK5 in sepsis-induced mortality
Dysregulated inflammatory response, poor bacterial clearance and excessive loss of lymphocytes have all been linked to poor survival in sepsis [21]. Our results using GRK5 deficient mice indicate that GRK5 mediates inflammatory response and thymocyte apoptosis in sepsis. Therefore, we predicted that septic GRK5 deficient mice might exhibit altered survival profile compared to the corresponding wild type mice. Contrary to our expectations, mortality following sepsis was similar between the two genotypes (Fig 8A). We then reasoned that because bacterial load was similar between the two genotypes, GRK5 deficient mice might exhibit better survival in the presence of antibiotics. To test this, we subjected wild type and GRK5 deficient mice to CLP and administered antibiotics (Ceftriaxone: 25μg/g and Metronidazole: 15μg/g body weight) intra-peritoneally 1 hour post- CLP and every 24 hours thereafter for 5 days [43]. Compared to wild type mice that did not receive any antibiotics (~80% mortality), mortality in wild type mice that received antibiotics decreased to ~45%. Interestingly however, GRK5 deficient mice receiving antibiotics had only ~10% mortality (compared to ~90% mortality in GRK5 deficient mice not receiving antibiotics) (Fig 8B). Together, these results demonstrate that in the presence of antibiotics, GRK5 deficiency protects mice from sepsis-induced mortality.
Figure 8. Role of GRK5 on survival following cecal ligation and puncture.
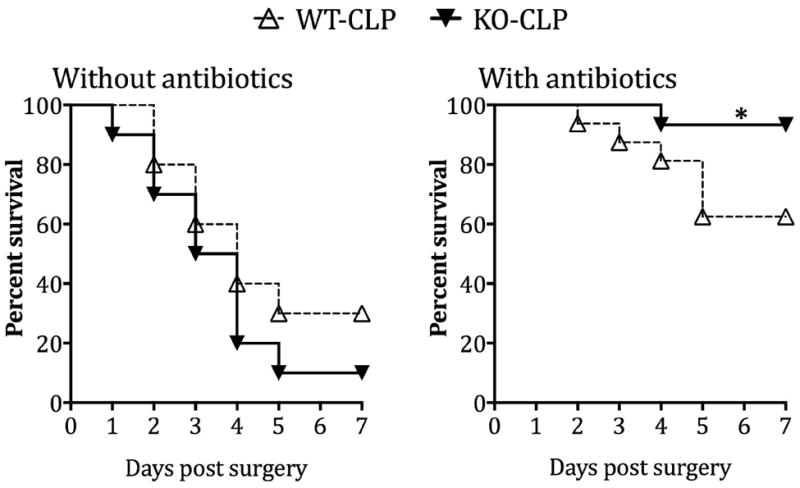
GRK5 wild type and knockout mice were subjected to sham or CLP surgery and survival was assessed for 7 days in the absence (A) or presence (B) of antibiotics as described in methods. (N=10 per genotype in A; N=15-16 per genotype in B). *P<0.05 compared to the corresponding septic wild type in the antibiotic treated group by log-rank test. Factorial design analysis of the data showed the following: Between genotypes: p=0.204; Between antibiotic and non-antibiotic groups: p=0.024; Interaction between genotypes and with antibiotics: p=0.038.
Discussion
Given the high rate of mortality in sepsis, understanding the pathophysiologic events and molecular mechanisms that mediate mortality in sepsis can help in developing new therapies and better treatment strategies. In the CLP model of polymicrobial sepsis GRK5 deficiency inhibited several but not all aspects of sepsis progression. Importantly, GRK5 deficiency significantly enhanced survival only in the presence of antibiotics. Even though GRK5 deficiency attenuated inflammatory response both at the systemic and organ levels, as well as significantly inhibited thymocyte apoptosis, these changes were not sufficient to enhance survival. It is possible that both the severity of sepsis and bacterial dissemination were too high, in spite of the favorable effects on inflammation and thymocyte apoptosis in GRK5 deficiency. This is in part supported by the observation that bacterial load per se, was not different between the two genotypes of mice at early or later time points of sepsis. Thus it is possible that at this level of severity, both antibiotics (to clear bacterial infection) and GRK5 deficiency yield better outcome. A similar phenotype was reported in MyD88 knockout mice [44] wherein deficiency of MyD88 led to a diminished inflammatory response and attenuated lymphocyte apoptosis without any effect on bacterial load following polymicrobial sepsis. Importantly, similar to our studies, these effects were not sufficient to prevent mortality following CLP. MyD88 is a critical adaptor molecule for many TLRs including TLR4. Consistent with the phenotype of MyD88 knockout mice, blocking TLR4 alone in a CLP model of sepsis did not prevent mortality because of persistent bacterial load [32]. Similar to our model, TLR4 antagonist significantly improved survival only in the presence of antibiotics [32]. Based on our previous studies [14,18] showing that GRK5 is an important regulator of TLR4 signaling, our results are consistent with these other studies in terms of the outcome of sepsis.
Studies have consistently related cytokine response as playing a major role in resolution of sepsis by activation of immune cells and subsequent clearance of microbes. However, excessive production of cytokines with aberrant activation of immune cells can have ill effects on the host. As shown in the endotoxemia model [14], we demonstrate here that GRK5 deficiency attenuates inflammatory cytokines following intra-abdominal polymicrobial sepsis. Consistent with the systemic and peritoneal cytokines, mRNA expression of NFκB-dependent genes in the liver and lungs were also attenuated at 12 hours post-sepsis. In line with our observations in LPS model as well as the demonstrated effects on bacterial infection in Drosophila and Zebra fish models, polymicrobial sepsis-induced IκBα phosphorylation in the liver was significantly inhibited in the GRK5 knockout mice. Together, these studies are consistent with previous studies showing that GRK5 regulates NFκB signaling and therefore is able to modulate NFκB-dependent gene expression.
Contrary to our results in mice and that of Valanne et al’s [19] results in Drosophila, Zebra fish and human cells, other studies have also shown that GRK5 is a negative regulator of NFκB signaling in endothelial cells [17] and vascular smooth muscle cells [17]. This effect of GRK5 has been linked to its role in stabilizing nuclear IκBα levels whereas we demonstrated previously that GRK5 is able to phosphorylate IκBα at the same sites as that of IKKβ and mediate degradation [9]. Thus it is possible that GRK5 may have multiple roles in terms of NFκB regulation but the dominance of regulation might depend on the cell type and disease being examined.
Lymphocyte apoptosis has increasingly been recognized as an important step in the pathogenesis of sepsis, by inducing a state of ‘immune paralysis’ that renders the host vulnerable to invading pathogens [41]. NFκB signaling plays a vital role in lymphocyte development, function and apoptosis. In addition, NFκB signaling can either promote survival [39] or apoptosis [40] of lymphocytes depending on the cell type involved. Even though we found GRK5 to be an important regulator of NFκB signaling in the liver, this phenomenon appeared to be tissue specific since we did not observe any difference in NFκB activity in the thymus from the two genotypes subjected to CLP (data not shown). Since previous studies have shown that sepsis-induced thymocyte apoptosis is mediated by corticosterone [42], we hypothesized that either corticosteroid signaling or its plasma levels may be differentially regulated in the two genotypes. Even though our in vitro results rule out any direct effect of GRK5 on corticosteroid-induced thymocyte apoptosis, our in vivo results demonstrate that GRK5 is an important regulator of sepsis-mediated increase in corticosterone levels. Since sepsis-induced thymocyte apoptosis has been shown to be mediated by corticosterone, lower levels of plasma corticosterone may in part explain why GRK5 KO thymocytes are protected from apoptosis in vivo. In future studies, we will determine whether the effect of GRK5 on thymocyte apoptosis in vivo is due to decreased corticosterone levels or due to differential regulation of other signals that may be involved in thymocyte apoptosis in sepsis.
Consistent with the better survival of thymocytes in the GRK5 deficient mice, septic peritoneal cells from the GRK5 knockout were more immunocompetent than the cells from wild type mice. While our studies don’t demonstrate a cause-effect relationship between thymocyte apoptosis and peritoneal cell responsiveness in the septic mice, previous studies have suggested that enhanced lymphocyte apoptosis leads to enhanced immunosuppression in animal models [45]. In addition it is possible that enhanced plasma corticosterone levels in the wild type mice might suppress LPS-induced cytokine production in the wild type cells compared to less suppression in the knockout cells [46]. These mechanisms will be dissected out in future studies. Regardless of the mechanism, our results suggest that under conditions where the innate cells are already exposed to polymicrobial injury, GRK5 deficiency is able to modulate the cells to be more immune-responsive compared to the wild type. The better immuno-responsiveness of innate cells during the stage of sepsis when cells are likely “paralysed” may lead to a beneficial outcome in sepsis [47]. This has been shown before where innate immune paralysis observed later in sepsis has been attributed to poor outcome [48]. Together, these results suggest that GRK5 deficiency could modulate sepsis at several levels or stages.
In conclusion, we demonstrate here that GRK5 is an important regulator of sepsis pathogenesis and that blocking GRK5 could result in a protective mechanism via modulating several aspects of sepsis. Even though bacterial load is not affected by GRK5, administration of antibiotics in the presence of GRK5 inhibition might lead to a better outcome in sepsis-induced mortality.
Acknowledgments
We gratefully acknowledge the support from NIH (grants HL095637, AR055726 and AR056680 to N.P.). We thank the University lab animal resources for taking excellent care of our animals, the Histopathology laboratory for their excellent service and Mr. Vernon LaLone for excellent technical assistance. We also thank the MSU’s Center for Statistical Training and Consulting for their consultation with regard to the survival analysis.
Footnotes
Authorship: N. Packiriswamy performed most of the experiments. T. Lee, P.B. Raghavendra and H. Durairaj performed some experiments. H. Wang designed the study for examining behavioral stress-induced corticosterone levels. N. Parameswaran conceptualized the study. N. Parameswaran and N. Packiriswamy designed experiments and wrote the manuscript.
References
- 1.Premont RT, Gainetdinov RR. Physiological roles of g protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 2.Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: More than just kinases and not only for gpcrs. Pharmacol Ther. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kunapuli P, Onorato JJ, Hosey MM, Benovic JL. Expression, purification, and characterization of the g protein-coupled receptor kinase grk5. J Biol Chem. 1994;269:1099–1105. [PubMed] [Google Scholar]
- 4.Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT. Muscarinic supersensitivity and impaired receptor desensitization in g protein-coupled receptor kinase 5-deficient mice. Neuron. 1999;24:1029–1036. doi: 10.1016/s0896-6273(00)81048-x. [DOI] [PubMed] [Google Scholar]
- 5.Barthet G, Carrat G, Cassier E, Barker B, Gaven F, Pillot M, Framery B, Pellissier LP, Augier J, Kang DS, Claeysen S, Reiter E, Baneres JL, Benovic JL, Marin P, Bockaert J, Dumuis A. Beta-arrestin1 phosphorylation by grk5 regulates g protein-independent 5-ht4 receptor signalling. The EMBO journal. 2009;28:2706–2718. doi: 10.1038/emboj.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Wang F, Long H, Wu Z, Ma L. Grk5 promotes f-actin bundling and targets bundles to membrane structures to control neuronal morphogenesis. The Journal of cell biology. 2011;194:905–920. doi: 10.1083/jcb.201104114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martini JS, Raake P, Vinge LE, DeGeorge BR, Jr, Chuprun JK, Harris DM, Gao E, Eckhart AD, Pitcher JA, Koch WJ. Uncovering g protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc Natl Acad Sci U S A. 2008;105:12457–12462. doi: 10.1073/pnas.0803153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker BL, Benovic JL. G protein-coupled receptor kinase 5 phosphorylation of hip regulates internalization of the chemokine receptor cxcr4. Biochemistry. 2011;50:6933–6941. doi: 10.1021/bi2005202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N. G-protein-coupled-receptor kinases mediate tnfalpha-induced nfkappab signalling via direct interaction with and phosphorylation of ikappabalpha. The Biochemical journal. 2010;425:169–178. doi: 10.1042/BJ20090908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parameswaran N, Pao CS, Leonhard KS, Kang DS, Kratz M, Ley SC, Benovic JL. Arrestin-2 and g protein-coupled receptor kinase 5 interact with nfkappab1 p105 and negatively regulate lipopolysaccharide-stimulated erk1/2 activation in macrophages. J Biol Chem. 2006;281:34159–34170. doi: 10.1074/jbc.M605376200. [DOI] [PubMed] [Google Scholar]
- 11.Niehrs C, Shen J. Regulation of lrp6 phosphorylation. Cell Mol Life Sci. 2010;67:2551–2562. doi: 10.1007/s00018-010-0329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.So CH, Michal AM, Mashayekhi R, Benovic JL. G protein-coupled receptor kinase 5 phosphorylates nucleophosmin and regulates cell sensitivity to polo-like kinase 1 inhibition. J Biol Chem. 2012;287:17088–17099. doi: 10.1074/jbc.M112.353854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Zhu H, Yuan M, Fu J, Zhou Y, Ma L. G-protein-coupled receptor kinase 5 phosphorylates p53 and inhibits DNA damage-induced apoptosis. J Biol Chem. 2010;285:12823–12830. doi: 10.1074/jbc.M109.094243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patial S, Shahi S, Saini Y, Lee T, Packiriswamy N, Appledorn DM, Lapres JJ, Amalfitano A, Parameswaran N. G-protein coupled receptor kinase 5 mediates lipopolysaccharide-induced nfkappab activation in primary macrophages and modulates inflammation in vivo in mice. J Cell Physiol. 2011;226:1323–1333. doi: 10.1002/jcp.22460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JI, Chakraborty P, Wang Z, Daaka Y. G-protein coupled receptor kinase 5 regulates prostate tumor growth. J Urol. 2012;187:322–329. doi: 10.1016/j.juro.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 16.Cheng S, Li L, He S, Liu J, Sun Y, He M, Grasing K, Premont RT, Suo WZ. Grk5 deficiency accelerates {beta}-amyloid accumulation in tg2576 mice via impaired cholinergic activity. J Biol Chem. 2010;285:41541–41548. doi: 10.1074/jbc.M110.170894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu JH, Zhang L, Fanaroff AC, Cai X, Sharma KC, Brian L, Exum ST, Shenoy SK, Peppel K, Freedman NJ. G protein-coupled receptor kinase-5 attenuates atherosclerosis by regulating receptor tyrosine kinases and 7-transmembrane receptors. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:308–316. doi: 10.1161/ATVBAHA.111.239608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Packiriswamy N, Parvataneni S, Parameswaran N. Overlapping and distinct roles of grk5 in tlr2-, and tlr3-induced inflammatory response in vivo. Cell Immunol. 2012;272:107–111. doi: 10.1016/j.cellimm.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valanne S, Myllymaki H, Kallio J, Schmid MR, Kleino A, Murumagi A, Airaksinen L, Kotipelto T, Kaustio M, Ulvila J, Esfahani SS, Engstrom Y, Silvennoinen O, Hultmark D, Parikka M, Ramet M. Genome-wide rna interference in drosophila cells identifies g protein-coupled receptor kinase 2 as a conserved regulator of nf-kappab signaling. J Immunol. 2010;184:6188–6198. doi: 10.4049/jimmunol.1000261. [DOI] [PubMed] [Google Scholar]
- 20.Hall MJ, Williams SN, DeFrances CJ, Golosinskiy A. Inpatient care for septicemia or sepsis: A challenge for patients and hospitals. NCHS Data Brief. 2011:1–8. [PubMed] [Google Scholar]
- 21.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. doi: 10.1146/annurev-pathol-011110-130327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24(Suppl 1):7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- 23.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 24.Parvataneni S, Gonipeta B, Packiriswamy N, Lee T, Durairaj H, Parameswaran N. Role of myeloid-specific g-protein coupled receptor kinase-2 in sepsis. Int J Clin Exp Med. 2011;4:320–330. [PMC free article] [PubMed] [Google Scholar]
- 25.Porter KJ, Gonipeta B, Parvataneni S, Appledorn DM, Patial S, Sharma D, Gangur V, Amalfitano A, Parameswaran N. Regulation of lipopolysaccharide-induced inflammatory response and endotoxemia by beta-arrestins. J Cell Physiol. 2010;225:406–416. doi: 10.1002/jcp.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groesdonk HV, Wagner F, Hoffarth B, Georgieff M, Senftleben U. Enhancement of nf-kappab activation in lymphocytes prevents t cell apoptosis and improves survival in murine sepsis. J Immunol. 2007;179:8083–8089. doi: 10.4049/jimmunol.179.12.8083. [DOI] [PubMed] [Google Scholar]
- 27.Shounan Y, Feng X, O’Connell PJ. Apoptosis detection by annexin v binding: A novel method for the quantitation of cell-mediated cytotoxicity. Journal of immunological methods. 1998;217:61–70. doi: 10.1016/s0022-1759(98)00090-8. [DOI] [PubMed] [Google Scholar]
- 28.Kusner LL, Sarthy VP, Mohr S. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase: A role in high glucose-induced apoptosis in retinal muller cells. Invest Ophthalmol Vis Sci. 2004;45:1553–1561. [PubMed] [Google Scholar]
- 29.Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through nmda receptor activation. Journal of neuroimmunology. 2006;171:72–85. doi: 10.1016/j.jneuroim.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 30.Leelahavanichkul A, Bocharov AV, Kurlander R, Baranova IN, Vishnyakova TG, Souza AC, Hu X, Doi K, Vaisman B, Amar M, Sviridov D, Chen Z, Remaley AT, Csako G, Patterson AP, Yuen PS, Star RA, Eggerman TL. Class b scavenger receptor types i and ii and cd36 targeting improves sepsis survival and acute outcomes in mice. J Immunol. 2012;188:2749–2758. doi: 10.4049/jimmunol.1003445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romero CR, Herzig DS, Etogo A, Nunez J, Mahmoudizad R, Fang G, Murphey ED, Toliver-Kinsky T, Sherwood ER. The role of interferon-gamma in the pathogenesis of acute intra-abdominal sepsis. J Leukoc Biol. 2010;88:725–735. doi: 10.1189/jlb.0509307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sha T, Iizawa Y, Ii M. Combination of imipenem and tak-242, a toll-like receptor 4 signal transduction inhibitor, improves survival in a murine model of polymicrobial sepsis. Shock. 2011;35:205–209. doi: 10.1097/SHK.0b013e3181f48942. [DOI] [PubMed] [Google Scholar]
- 33.Craciun FL, Schuller ER, Remick DG. Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. J Immunol. 2010;185:6930–6938. doi: 10.4049/jimmunol.1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He M, Moochhala SM, Adhikari S, Bhatia M. Administration of exogenous fractalkine, a cx3c chemokine, is capable of modulating inflammatory response in cecal ligation and puncture-induced sepsis. Shock. 2009;31:33–39. doi: 10.1097/SHK.0b013e31817789da. [DOI] [PubMed] [Google Scholar]
- 35.Arraes SM, Freitas MS, da Silva SV, de Paula Neto HA, Alves-Filho JC, Auxiliadora Martins M, Basile-Filho A, Tavares-Murta BM, Barja-Fidalgo C, Cunha FQ. Impaired neutrophil chemotaxis in sepsis associates with grk expression and inhibition of actin assembly and tyrosine phosphorylation. Blood. 2006;108:2906–2913. doi: 10.1182/blood-2006-05-024638. [DOI] [PubMed] [Google Scholar]
- 36.Wang SD, Huang KJ, Lin YS, Lei HY. Sepsis-induced apoptosis of the thymocytes in mice. J Immunol. 1994;152:5014–5021. [PubMed] [Google Scholar]
- 37.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, Karl IE. Sepsis-induced apoptosis causes progressive profound depletion of b and cd4+ t lymphocytes in humans. J Immunol. 2001;166:6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 38.Weaver JG, Rouse MS, Steckelberg JM, Badley AD. Improved survival in experimental sepsis with an orally administered inhibitor of apoptosis. FASEB J. 2004;18:1185–1191. doi: 10.1096/fj.03-1230com. [DOI] [PubMed] [Google Scholar]
- 39.Beg AA, Baltimore D. An essential role for nf-kappab in preventing tnf-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 40.Bessho R, Matsubara K, Kubota M, Kuwakado K, Hirota H, Wakazono Y, Lin YW, Okuda A, Kawai M, Nishikomori R, et al. Pyrrolidine dithiocarbamate, a potent inhibitor of nuclear factor kappa b (nf-kappa b) activation, prevents apoptosis in human promyelocytic leukemia hl-60 cells and thymocytes. Biochem Pharmacol. 1994;48:1883–1889. doi: 10.1016/0006-2952(94)90586-x. [DOI] [PubMed] [Google Scholar]
- 41.Hotchkiss RS, Tinsley KW, Swanson PE, Chang KC, Cobb JP, Buchman TG, Korsmeyer SJ, Karl IE. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci U S A. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayala A, Herdon CD, Lehman DL, DeMaso CM, Ayala CA, Chaudry IH. The induction of accelerated thymic programmed cell death during polymicrobial sepsis: Control by corticosteroids but not tumor necrosis factor. Shock. 1995;3:259–267. doi: 10.1097/00024382-199504000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Turnbull IR, Wlzorek JJ, Osborne D, Hotchkiss RS, Coopersmith CM, Buchman TG. Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock. 2003;19:310–313. doi: 10.1097/00024382-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Peck-Palmer OM, Unsinger J, Chang KC, Davis CG, McDunn JE, Hotchkiss RS. Deletion of myd88 markedly attenuates sepsis-induced t and b lymphocyte apoptosis but worsens survival. J Leukoc Biol. 2008;83:1009–1018. doi: 10.1189/jlb.0807528. [DOI] [PubMed] [Google Scholar]
- 45.Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K, Osborne DF, Clark AT, Coopersmith CM, McDunn JE, Hotchkiss RS. Il-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol. 2010;184:1401–1409. doi: 10.4049/jimmunol.0902307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Nelin LD, Kuhlman JR, Meng X, Welty SE, Liu Y. The role of map kinase phosphatase-1 in the protective mechanism of dexamethasone against endotoxemia. Life sciences. 2008;83:671–680. doi: 10.1016/j.lfs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellaban E, Bolgos G, Remick D. Selective macrophage suppression during sepsis. Cell Immunol. 2004;231:103–111. doi: 10.1016/j.cellimm.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 48.Guo RF, Riedemann NC, Ward PA. Role of c5a-c5ar interaction in sepsis. Shock. 2004;21:1–7. doi: 10.1097/01.shk.0000105502.75189.5e. [DOI] [PubMed] [Google Scholar]