

Abstract
Prader-Willi syndrome (PWS) is a genetic disorder caused by deficiency of imprinted gene expression from the paternal chromosome 15q11-15q13 and clinically characterized by neonatal hypotonia, short stature, cognitive impairment, hypogonadism, hyperphagia, morbid obesity and diabetes. Previous clinical studies suggest that a defect in energy metabolism may be involved in the pathogenesis of PWS. We focused our attention on the genes associated with energy metabolism and found that there were 95 and 66 mitochondrial genes differentially expressed in PWS muscle and brain, respectively. Assessment of enzyme activities of mitochondrial oxidative phosphorylation (OXPHOS) complexes in the brain, heart, liver and muscle were assessed. We found the enzyme activities of the cardiac mitochondrial complexes II+III were upregulated in the imprinting center deletion (PWS-IC) mice compared to the wild type littermates. These studies suggest that differential gene expression, especially of the mitochondrial genes may contribute to the pathophysiology of PWS.
Introduction
Prader-Willi syndrome (PWS) is a complex neurodevelopmental disorder resulting from deficiency of imprinted gene expression from paternal chromosome 15q11-15q131, 2. PWS is a characterized by multiple clinical manifestations during development and adulthood. In utero features include reduced fetal movement and polyhydramnios and in infancy, feeding difficulties, hypogonadism, hypotonia and failure to thrive. During childhood, delayed motor speech development, behavioral abnormalities that include hyperphagia, and sleep disorder predominates. During adolescence, obesity, short stature and delayed puberty are observed, and in adulthood, infertility, hypogonadism, cognitive impairment and morbid obesity are significant problems. Dysmorphic features include bi-temporal narrowing, upslanting palpebral fissures, and small hands and feet3.
The chromosome 15q11-q13 region contains imprinted sequences that are differentially expressed depending on the parent of origin. Due to imprinting, the maternally inherited copies of these genes are virtually silent, with only the paternal copies of the genes being expressed. In 70% of individuals, PWS results from the absence of paternally expressed imprinted genes located on chromosome 15q11-q13 including SNRPN, NECDIN, MAGEL2, and MKRN3 genes along with clusters of snoRNAs: SNORD64, SNORD107, SNORD108, two copies of SNORD109, 29 copies of SNORD116 (HBII-85) and 48 copies of SNORD115 (HBII-52)4, 5. Maternal uniparental disomy in which there are two maternal and no paternal copies of this region accounts for 30% of PWS cases. Deletion of the same region on the maternal chromosome causes Angelman syndrome (AS). An imprinting center (IC) in cis regulates the establishment of parental specific allelic differences in DNA methylation, chromatin structure and expression of genes in the PWS critical region. Although the 15q11-q13 defect underlies the PWS phenotype, the genes within the region do not appear to be directly responsible for the complex phenotype. Rather, it seems likely that the PWS phenotype results from dysregulation of multiple interconnected neurological and metabolic pathways6. The PWS critical region extends over nearly half of 15q11-q13 and contains multiple paternally expressed genes. The protein coding genes, small nuclear ribonucleoprotein N (SNRPN) and its bicistronic partner SNRPN upstream reading frame (SNURF) have been mapped to the PWS critical region7, 8. Families with micro-deletions causing PWS localize the PWS-IC to 4.3 kb overlapping with SNRPN exon 19. SNURF and SNRPN are paternally expressed and encode two different proteins within a single transcript6. SNRPN/SNURF-SNRPN exon1 is embedded in a CpG island with complete methylation on the maternal allele and complete absence of methylation on the paternal allele. Recent findings have shown that that deletion of the 29 copies of the C/D box snoRNA SNORD116 (HBII-85) appears to be the primary cause of PWS10.
The PWS-IC mouse model is a powerful tool that can be efficiently used to better understand the biochemical and cellular mechanisms involved in human PWS. There are now considerable insights from various deletions and other mutations in essential regulatory elements, such as the imprinting center and one or more paternally expressed structural genes in the PWS domain that these loci are important in generating the PWS phenotype. Maternal uniparental disomy (UPD)/paternal deficiency produce neonatal lethality in 100% of mice11. Similarly, a large deletion associated with a transgenic insertion caused lethality in 100% of the mice with paternal inheritance12. A 42-kb deletion of the putative imprinting center causes complete lethality and an imprinting defect when inherited from male chimeras. Deletion of this imprinting center abolishes local paternally derived gene expression and results in PWS13. Both the position of the imprinting center and its role in the coordinate expression of genes is conserved between mouse and human, therefore indicating that the mouse is a suitable model system in which to investigate the molecular function and pathophysiological implication of imprinting defects. A complete imprinting defect (UPD and the 42-kb deletion of the mouse IC causes complete or severe lethality, whereas a partial imprinting defect (the 4.8-kb deletion of the Snrpn promoter) causes partial or lesser lethality14. Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in PWS mouse model15. Another deletion introduced into the germ line using homologous recombination removed the coding exons for both Snurf and SmN and for Ube3a and removed all of the intervening 0.5-0.7 Mb of genomic DNA. This deletion from Snrpn to Ube3a on the paternal chromosome is associated with severe growth retardation, hypotonia and ~80% lethality and position effects on expression of imprinted genes. The surviving mice with the Snrpn-Ube3a deletion were fertile and were not obese16. In all cases of lethality in the PWS mouse models, the abnormal phenotype occurs only with paternal deficiency and is associated with decreased movement and poor feeding immediately after birth.
Previous work involving whole genome microarrays using RNA isolated from lymphoblastoid cells from PWS patients has revealed significant differences in expression patterns of numerous genes related to normal neurodevelopment and function including serotonin receptor genes (e.g., HTR2B); genes involved in eating behavior and obesity (ADIPOR2, MC2R, HCRT, OXTR); STAR (a key regulator of steroid synthesis); and SAG (an arrestin family member which desensitizes G-protein-coupled receptors)17. Additionally, the same group also performed whole genome microarrays on a PWS-IC deletion mouse model on RNA extracted from whole brain tissue within 24 hours after birth. Their results demonstrated two very significant differences: first, an increased expression of Pomc and Mc5r, the former of which is believed to be involved in eating behavior and the latter in thermoregulation; and secondly, decreased expression of three relatively uncharacterized transcripts that are expressed from the paternal allele under regulatory control of the IC (AK013560, BB3144814, BB182944)17.
Mitochondria, in addition to playing perhaps the most prominent role of all organelles in energy metabolism, are central to many cellular functions including the generation of ATP, intracellular Ca2+ homeostasis, reactive oxygen species (ROS) biology, and apoptosis18. Mitochondria are one of the most abundant organelles in muscle and brain cells, responsible for producing the majority of energy through oxidative phosphorylation. Many genes associated with neurodegenerative diseases are now known to regulate mitochondrial function19. Mitochondrial dysfunction has been implicated in Parkinson’s disease20, Huntington’s disease21, Amyotrophic Lateral Sclerosis (ALS)22, Muscular Dystrophy23 , type 2 Diabetes Mellitus24, and Collagen VI Myopathies25.
Despite a lack of clinical or animal studies demonstrating efficacy, a high percentage of patients with PWS are being treated with co-enzyme Q10 (Co-Q10). A vitamin-like substance, Co-Q10 is present in most eukaryotic cells, primarily in the mitochondria where it is a component of the electron transport chain functioning as an electron acceptor from Complex I and Complex II before passing on the electron to Complex III. Unpublished data from our laboratory indicates that of the majority of patients on Co-Q10 derive some benefit mainly with regard to their energy level. To date, no double blind study has been undertaken to study the effects of Co-Q10 on mitochondrial activity.
Given that previous findings have demonstrated differentially expressed genes involved in energy metabolism; that patients derive therapeutic benefit from supplementation with a mitochondrial component of the electron transport chain; the abundance of mitochondria in neuronal, liver, and skeletal muscle tissues; and that mitochondria play a central role in energy metabolism through ATP generation and pathophysiologic manifestations through ROS generation, we hypothesized that mitochondrial dysfunction is a pivotal component of PWS pathology.
We investigated the gene expression profiles of the quadriceps muscle and brain tissues in PWS-IC transgenic mouse model. Using RNA isolated from these tissues, we performed a comprehensive analysis of differential gene expression using the microarray approach with a goal to identify altered genes and link them to specific pathways that are relevant to PWS.
Methods
PWS-IC del mouse model
Prader-Willi syndrome imprinting center deletion (PWS-IC del) mice were developed as described previously26 and were kindly provided by Dr. Resnick (College of Medicine, University of Florida, Gainesville, FL). All experiments were done with the approval of the Institutional Animal Care and Use Committee (IACUC Protocol #2007-2716-1) of the University of California, Irvine (UCI), and in accordance with the guidelines established by the NIH. All animals were housed in the vivarium of the UCI and maintained under constant temperature (22°C) and humidity with a controlled 12:12-h light-dark cycle. PWS-IC del mice weighed approximately 16-20g and wild type littermates weighed approximately 30-35 g. All animals were 6 months old females and an F1 hybrid of WT FVB females and B6 males bearing the 35-kb IC del mutation strains. Mutant mice were bred after culling all but one wild type pup within a day of birth. So, the animals were presumed to have FVB mitochondria.
Electron Microscopy
Mutant and WT mice tissues were fixed in 4% paraformaldehyde plus 0.1% glutaraldehyde in 0.1M PB for 24 hrs. Sections (100 μM) were cut with a vibratom, then collected in PBS and fixed in 1% glutaraldehyde overnight. Tissue samples were fixed in 1% Osmium for 1 hr at 4°C and dehydrated in ethanol, which are embedded in Eponate 12 resin at 65°C for 24-36 hrs. Ultrathin (~60-80 nm) sections were cut with a diamond knife and stained in 1% uranyl acetate for 30 min, followed by lead citrate for 7-10 min. Electron micrographs were taken with a Gatan UltraScan US1000 digital camera and examined on a Philips CM10 transmission electron microscope.
Mitochondrial isolation and OXPHOS enzyme analysis
Mitochondria were isolated from the liver, heart, skeletal muscle (vastus lateralis) and the whole brain by homogenization and differential centrifugation as described previously27. Mitochondrial enzyme complex activities were determined by OXPHOS enzyme assays using our standard protocols27. In brief, equal amounts of mitochondrial proteins were assayed and normalized to citrate synthase (CS) activity, which remained at unchanged levels throughout the assay.
Complex I activity assay
Eighty μl of water, 100 μl of 2x buffer (500 mM sucrose, 2 mM EDTA, 100 mM Tris-HCl at pH 7.4), 2 μl of 10 mM decylubiquinone (DB) (Sigma-Aldrich, St. Louis, MO), 0.5 μl of 2 M KCN, and 10 μl of 1 mg/mL mitochondria incubated for 5 min at 30°C. The reaction was started by adding 10 μl of 1 mM NADH, after which absorbance was measured at 340 nm for 5 min to quantify the Rotenone-sensitive activity, which was determined in the presence of 1 μl of 1 mg/mL rotenone in a parallel experiment.
Complex II+III activity assay
The rate of cytochrome c (cytc) reduction was measured at 550 nm in a reaction containing 110 μl of water, 80 μl of 100 mM potassium phosphate buffer (pH 7.4), 4 μl of 1 M succinate, 1 μl of 0.1 M EDTA, 0.5 μl of 2M KCN, and 6 μl of 1 mM cytc. Five μl of 1 mg/mL mitochondria was added and the absorption was monitored at 550 nm for 2 min.
Complex IV oxidation assay
The rate of cytochrome c (cytc) oxidation was measured at 550 nm in a reaction containing 170 μl of water, 20 μl of 100 mM potassium phosphate buffer (pH 7.4), and 10 μl of 1 mM reduced cytc. Five μl of 0.1 mg/mL mitochondria was added. The absorption at 550 nm was monitored for 2 min. Reduced cytc was prepared by adding 2 μl of 1 M dithiothreitol (DTT) to 1 mL of 1 mM cytc and incubated for 15 min.
Citrate synthase (CS) activity assay
The reduction of 5,5′-dithio-bis (2-nitrobenzoic acid) (DTNB) was measured at 412 nm in a reaction containing 160 μl of water, 20 μl of 1 M Tris-HCl (pH 8.0), 10 μl of 6 mM acetyl-CoA, and 2 μl of 10 mM DTNB by adding 5 μl of 1 mg/mL mitochondria. The absorption at 412 nm was monitored for 2 minutes.
Mitochondrial Enzyme Analysis
Measurements of mitochondrial enzyme activities were repeated 3 times for each test on each animal. The unpaired student’s t test was used to evaluate differences in wild type and mutant values. Data represent mean ± SD. An analysis with a value of p<0.05 was considered to be statistically significant.
Microarray Analysis
Skeletal (quadriceps) and whole brain samples from the mutant mice (n=3) and their wild type age-matched littermates (n=4) were analyzed by microarray technology using the Mouse Genome 430 2.0 arrays (Affymetrix). Total RNA was extracted using a TRIzol-reagent (Invitrogen) and purified with RNeasy Mini kit (Qiagen). RNA concentration was determined by spectrophotometry (Bio-Rad, Hercules, CA) and the quality of RNA samples was assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Inc). Two μg of total high-quality RNA was used to start cDNA-cRNA synthesis by In Vitro Transcription using Affymetrix GeneChip® Expression 3′Amplification One-Cycle Target Labeling and Control Reagent kit. Double-stranded cDNA and Biotinylated cRNA were purified by Gene Chip Sample Cleanup Module (Affymetrix, Santa Clara, CA). Seventeen μg of biotinylated cRNAs from samples are fragmented, 15 μg of which were placed in a hybridization mixture of which 10 μg was hybridized to an Affymetrix Gene-Chips array for 16 hours. The arrays were washed and stained on the Affymetrix Fluidics Station 450 according to manufacturer’s instructions. Briefly, non-specifically bound material was removed and followed by incubation with phycoerythrin-streptavidin to detect bound cRNA. The signal intensity was amplified by second staining with biotin labeled anti-streptavidin antibody followed by phycoerythrin-streptavidin restaining. Fluorescent images were captured using the Affymetrix Gene Chip Scanner 3000 7G.
Gene Expression Profiling Data Analysis
CEL image files were analyzed with the Probe Logarithmic Intensity Error algorithm (PLIER) (Affymetrix). Unsupervised clustering of the data from all samples was performed with Hierarchical Clustering Explorer 3 (HCE). The PLIER data set was also analyzed with the data mining program GeneSpring GX (version 7.3.1) (Silicon Genetics). Before screening for probe sets with altered expression levels, the data set, or signal intensities were normalized in 2 ways: (1) to each chip using a distribution of all genes around the 50th percentile; (2) to the mean values of infected sample’s own control, so that all probe sets from control samples had a mean value of 1, and probe sets from mutant versus control samples had a value of either greater than, less than, or equal to 1, representing upregulation, downregulation, or no change, respectively. Only probe sets that were statistically significant via one-way ANOVA t-test with a p-value cut off of p<0.001 or p<0.01 were considered further. Data were clustered as gene trees where yellow represents an expression ratio around 1, or no change (controls), and hotter colors (red=maximum) indicate an increase, and cooler colors (blue=minimum) indicate a decrease from control levels. For the generation of canonical pathways and networks, we used Ingenuity Pathways Analysis (IPA) (Ingenuity Systems, Redwood City, CA). The hyperlink of the GEO submission for study number GSE41759 is: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41759.
Quantitative Real Time PCR (qRT-PCR)
Expression levels of genes that are most significantly differentially expressed (at least >1.5-fold difference) between control and mutant samples were confirmed by quantitative real-time PCR (qRT-PCR) using Roche LightCycler 480 Real-Time PCR System (Roche Applied Science, Indianapolis, IN). Two hundred ng of total RNA were treated with DNAse I (Roche Diagnostics) before cDNA synthesis that was prepared using transcription kit and random hexamers (Applied Biosystems). RT reactions were carried out in 20 μl according to manufacturer’s protocol, and 1.8 μl of cDNA was used for each qRT-PCR reaction. In order to validate our microarray data, pre-designed fluorogenic FAM-labeled primers/probes were used to measure transcription levels of the selected genes. Probes for the qRT-PCR validation were selected from Applied Biosystems (Carlbad, CA) website, and were used as sense/antisense primers. For the muscle tissue the following assays were used: Actc1 (ID: Mm00477277_g), Igfbpl1 (ID: Mm00497621_m1), Casr (ID: Mm00443375_m1), Mppe1 (ID: Mm00553989_m1), Tbc1d1 (ID: Mm00497989_m1); and for the brain tissue we used: Mrpl15 (ID: Mm00804108_m1) and Utrn (ID: Mm01168866_m1). Mouse GAPDH (Part Number 4352932E) was used as an endogenous control. All qRT-PCR results were normalized to control samples.
Results
Differentially expressed transcripts in the PWS-IC del mice and analysis of Ingenuity networks
We analyzed gene expression from the skeletal muscle and brain tissues of the PWS-IC del mice by using whole genome microarray. Gene clustering analysis of statistically significant (p<0.01) differentially expressed genes revealed dysregulated expression of 726 genes in muscle and 332 genes in brain of PWS-IC mice as compared to littermate controls (Figure 1). We then grouped the genes that had a significant change in expression using Ingenuity Pathways Analysis software (Ingenuity Systems, Redwood City, CA) into bio-functional pathways for muscle and brain (Figure 2 and Table I). Statistically significant genes with the greatest fold change found in both muscle and brain are listed in Table II. Of the differentially expressed genes, we found 95 and 66 mitochondrial genes differentially expressed in PWS-IC muscle and brain, respectively at p<0.05 (Table III).
Figure 1. Genome microarray analysis of gene expression heat map in the skeletal muscle and brain samples of PWS-IC del mice.

(A) Gene-based clustering for the genes in the skeletal muscle (726 genes) and (B) in the brain (332 genes) of the PWS-IC del mice from the Ingenuity’s top network 1 (p<0.01), analyzed together with 6 MD groups in Plier (normalization: per gene – to controls; per chip – to 50th percentile) compared to control littermates. Red color= upregulated genes; Blue color= downregulated genes.
Figure 2. Network of bio-functional pathways in PWS-IC mouse skeletal muscle and brain.
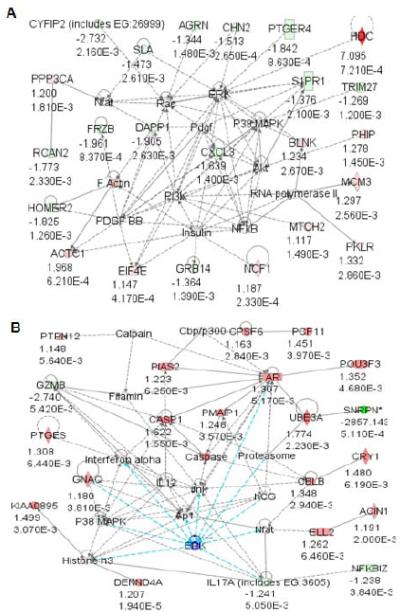
Genes with significant expression differences in (A) muscle tissue and (B) brain from PWS-IC del mice were grouped into Ingenuity networks by canonical pathways and signaling intermediates and bio-functional pathways using Ingenuity Pathways Analysis software.
Table I.
List of biological functions as identified by Ingenuity in PWS-IC mouse muscle and brain samples (p<0.05).
| TOP BIOLOGICAL FUNCTIONS (MUSCLE) | ||
|---|---|---|
| Diseases and Disorders | ||
| Name | p-Value | Number of Genes |
| Cancer | 8.02E-04 - 4.56E-02 | 29 |
| Reproductive System Disease | 8.02E-04 - 4.56E-02 | 11 |
| Developmental Disorder | 1.95E-03 - 1.64E-02 | 7 |
| Genetic Disorder | 1.95E-03 – 4.95E-02 | 28 |
| Inflammatory Disease | 3.10E-03 – 4.46E-02 | 9 |
| Molecular Cell Functions | ||
| Name | p-Value | Number of Genes |
| Amino Acid Metabolism | 3.66E-04 - 3.36E-02 | 6 |
| Small Molecule Biochemistry | 3.66E-04 - 4.19E-02 | 21 |
| Cellular Function and Maintenance | 1.95E-03 – 3.36E-02 | 3 |
| Cellular Movement | 1.95E-03 – 4.19E-02 | 18 |
| Carbohydrate Metabolism | 3.00E-03 – 3.36E-02 | 9 |
| Physiological System Development and Function | ||
| Name | p-Value | Number of Genes |
| Skeletal and Muscular System Development and Function | 1.22E-03 – 3.36E-02 | 12 |
| Tissue Morphology | 1.22E-03 – 3.36E-02 | 17 |
| Nervous System Development and Function | 1.47E-03 – 3.76E-02 | 12 |
| Tissue Development | 1.47E-03 – 3.36E-02 | 9 |
| Hematological System Development and Function | 1.95E-03 – 4.19E-02 | 5 |
| TOP BIOLOGICAL FUNCTIONS (BRAIN) | ||
| Diseases and Disorders | ||
| Name | p-Value | Number of Genes |
| Developmental Disorder | 1.511E-03-2.95E-02 | 7 |
| Genetic Disorder | 1.51E-03-3.68E-02 | 13 |
| Neurological Disease | 5.46E-03-4.40E-02 | 15 |
| Cancer | 6.20E-03-4.63E-02 | 10 |
| Reproductive system disease | 6.20E-03-4.40E-02 | 8 |
| Molecular Cell Functions | ||
| Name | p-Value | Number of Genes |
| Gene Expression | 1.5651E-04-4.40E-02 | 24 |
| Cell Morphology | 3.29E-04-4.40E-02 | 8 |
| Cellular Development | 3.29E-04-4.40E-02 | 8 |
| Cellular Growth and Proliferation | 3.29E-04-4.40E-02 | 9 |
| Lipid Metabolism | 2.53E-03-5.00E-02 | 8 |
| Physiological System Development and Function | ||
| Name | p-Value | Number of Genes |
| Cardiovascular System Development and Function | 1.741E-04-4.40E-02 | 6 |
| Organ Development | 1.74E-04-4.81E-02 | 14 |
| Reproductive System Development and Function | 3.29E-04-4.40E-02 | 4 |
| Hematological System Development and Function | 1.50E-04-4.40E-02 | 8 |
| Immune Cell Trafficking | 1.50E-03-5.00E-02 | 4 |
Table II.
Dysregulated mitochondrial genes in PWS-IC del mouse muscle and brain tissues (top 10 fold changes, p<0.05).
| Upregulated genes in PWS-IC muscle | |||
| AFFY ID | Gene Symbol | Gene Description |
Fold
Change |
| 1452771_s_at | FACL3 | fatty acid Coenzyme A ligase, long chain 3 | 1.44 |
| 1418911_s_at | FACL4 | fatty acid-Coenzyme A ligase, long chain 4 | 1.38 |
| 1417434_at | GPD2 | glycerol phosphate dehydrogenase 2, mitochondrial | 1.34 |
| 1429533_at | IMMT | inner membrane protein, mitochondrial | 1.32 |
| 1450612_a_at | PEMT | phosphatidylethanolamine N-methyltransferase | 1.31 |
| 1449137_at | PDHA1 | pyruvate dehydrogenase E1 alpha 1 | 1.268 |
| 1420044_at | NRD1 | nardilysin, N-arginine dibasic convertase, NRD convertase 1 |
1.26 |
| 1434996_at | SLC25A16 | solute carrier family 25 (mitochondrial carrier; Graves disease autoantigen), member 16 |
1.242 |
| 1453886_a_at | SLC25A26 | solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 26 |
1.24 |
| 1429971_at | TXNRD2 | thioredoxin reductase 2 | 1.24 |
| Downregulated genes in PWS-IC muscle | |||
| AFFY ID | Gene Symbol | Gene Description |
Fold
Change |
| 1452257_at | BDH | 3-hydroxybutyrate dehydrogenase (heart, mitochondrial) |
−2.55 |
| 1422905_s_at | FMO2 | flavin containing monooxygenase 2 | −2.04 |
| 1422997_s_at | MTE1 | mitochondrial acyl-CoA thioesterase 1 | −1.66 |
| 1448988_at | ACADL | acetyl-Coenzyme A dehydrogenase, long-chain | −1.56 |
| 1430969_at | MTCH2 | mitochondrial carrier homolog 2 (C. elegans) |
−1.55 |
| 1416203_at | AQP1 | aquaporin 1 | −1.52 |
| 1450387_s_at | AK4 | adenylate kinase 4 | −1.49 |
| 1422493_at | CPOX | coproporphyrinogen oxidase | −1.47 |
| 1444394_at | ACAD9 | acyl-Coenzyme A dehydrogenase family, member 9 | −1.42 |
| 1449443_at | DECR1 | 2,4-dienoyl CoA reductase 1, mitochondrial | −1.39 |
| 1431148_at | AK3L | adenylate kinase 3 alpha-like | −1.34 |
| Upregulated genes in PWS-IC brain | |||
| AFFY ID | Gene Symbol | Gene Description |
Fold
Change |
| 1451623_at | MRPL15 | mitochondrial ribosomal protein L15 | 1.98 |
| 1458404_at | NDUFB8 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 8 |
1.77 |
| 1417823_at | GCAT | glycine C-acetyltransferase (2-amino-3-ketobutyrate- coenzyme A ligase) |
1.55 |
| 1419362_at | MRPL35 | mitochondrial ribosomal protein L35 | 1.51 |
| 1421829_at | AK4 | adenylate kinase 4 | 1.46 |
| 1457790_at | ASB3 | ankyrin repeat and SOCS box-containing protein 3 | 1.45 |
| 1450948_a_at | MRPL1 | mitochondrial ribosomal protein L1 | 1.44 |
| 1421874_a_at | MRPS23 | mitochondrial ribosomal protein S23 | 1.41 |
| 1424164_at | MRPL50 | mitochondrial ribosomal protein L50 | 1.41 |
| 1422095_a_at | TYKI | thymidylate kinase family LPS-inducible member | 1.40 |
| Downregulated genes in PWS-IC brain | |||
| AFFY ID | Gene Symbol | Gene Description |
Fold
Change |
| 1422008_a_at | AQP3 | aquaporin 3 | −2100.84 |
| 1417614_at | CKM | creatine kinase, muscle | −6.85 |
| 1431739_at | MTO1 | mitochondrial translation optimization 1 homolog (S. cerevisiae) |
−1.95 |
| 1442331_at | ALAS1 | aminolevulinic acid synthase 1 | −1.82 |
| 1430969_at | MTCH2 | mitochondrial carrier homolog 2 (C. Elegans) | −1.80 |
| 1444489_at | SLC25A12 | solute carrier family 25 (mitochondrial carrier, Aralar), member 12 |
−1.72 |
| 1441030_at | RAI14 | retinoic acid induced 14 | −1.50 |
| 1453524_at | KIF5B | kinesin family member 5B | −1.40 |
| 1418987_at | PLA2G2D | phospholipase A2, group IID | −1.37 |
| 1440579_at | MIB1 | mindbomb homolog 1 (Drosophila) | −1.31 |
Table III.
Top 10 List of dysregulated genes in PWS-IC del mouse muscle and brain tissues (p<0.05) by Ingenuity.
| GENES | Fold Change |
| HDC | 7.095 |
| SYNTB1 | 4.346 |
| ONECUT2 | 2.443 |
| EGFL6 | 2.091 |
| ZNF367 | 2.028 |
| ACTC1 | 1.968 |
| IGSF3 | 1.557 |
| CILP | 1.496 |
| CASR | 1.489 |
| NUDT10 | 1.484 |
| Downregulated genes (muscle) | |
| GENES | Fold Change |
| SNRPN | −537.63 |
| NDN | −14.306 |
| ANXA10 | −13.158 |
| GZMH | −13.021 |
| NEU1 | −2.770 |
| CYFIP2 | −2.732 |
| TBC1D1 | −2.252 |
| DDAH1 | −2.247 |
| Upregulated genes (brain) | |
| GENES | Fold Change |
| MRPL15 | 1.975 |
| ALOX18 | 1.859 |
| CHRNA5 | 1.781 |
| UBE3A | 1.774 |
| AMN | 1.757 |
| XBP1 | 1.684 |
| DLEU2 | 1.675 |
| CASP1 | 1.622 |
| Downregulated genes (brain) | |
| GENES | Fold Change |
| SNRPN | −2857.1 |
| NDN | −27.100 |
| ANXA10 | −2.793 |
| SERPINA12 | −2.747 |
| GZMB | −2.740 |
| VAX2 | −2.160 |
| C90RF165 | −2.016 |
| DDXR3Y | −2.008 |
As predicted, all the known genes from the PWS critical region were downregulated and Ube3a was significantly upregulated. Previous microarray findings using a day old PWS mice 17 of which upregulation of Mc5r (melanocortin receptor) and its interaction partner Pomc were not observed. In order to validate our microarray data, we further investigated two significant genes in PWS muscle by RT-PCR. We confirmed that actin α cardiac muscle1 (Actc1) was upregulated and TBC domain family, member1 (Tbc1d1) was downregulated in the PWS-IC del mice relative to their control littermates (Figure 3).
Figure 3. Validation of microarray genes in PWS-IC mouse model by qRTPCR.
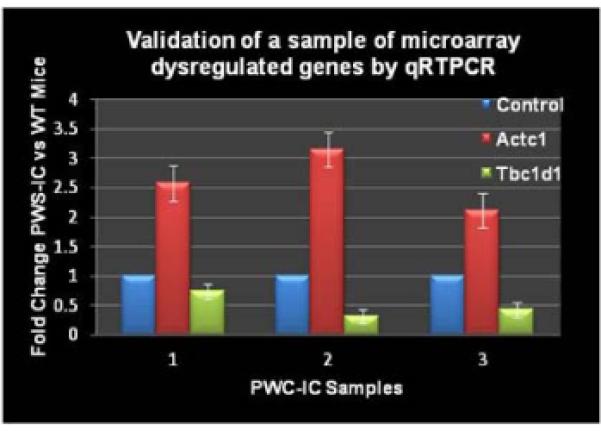
Select genes were used for qRT-PCR analysis in order to validate the microarray data of PWS-IC and WT control littermates.
Analysis of mitochondrial complexes
To assess mitochondrial bioenergetic function, we assayed the relative activity of complex I, II+III, and IV in skeletal muscle, liver, heart and brain tissues of PWS-IC mice compared to their littermate controls. The data were normalized by citrate synthase (CS) activities. The relative activities of mitochondrial complexes II+III were significantly upregulated in cardiac muscle in PWS-IC deletion mice when compared to WT mice (student t test, p<0.01). Normalized complexes I, II+III, and IV activities in the brain, liver and skeletal muscles were not significantly different between the PWS-IC deletion and WT mice (Figure 4).
Figure 4. Complexes I, II+III, and IV activities in the brain, liver, heart and skeletal muscles from PWS-IC and WT mice.
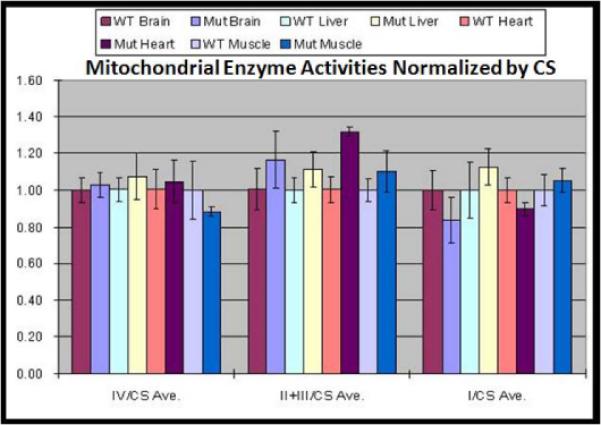
The normalized complexes I and IV activities in the brain, liver, heart and skeletal muscles and complexes II+III activities in the brain, liver and skeletal muscles were not significantly different between the PWS-IC deletion and WT mice normalized to citrate synthase activity, which remained unchanged.
Mitochondrial EM structural changes
Electron micrograph studies on cardiac and skeletal muscle tissues were reviewed in PWS-IC deletion mice compared to their WT littermate controls. We found abnormal mitochondrial proliferation, disorganized mitochondria swollen in appearance with disrupted membranes in both cardiac and skeletal muscle in PWS-IC deletion mouse as compared to littermate controls (Figure 5).
Figure 5. Electron microscopy of cardiac and skeletal muscle mitochondrial density in WT and PWS-IC del mice.

(A) Cardiac and (C) muscle tissues from WT mice and (B) cardiac and (D) muscle tissues from PWS-IC del mice. Mutant samples demonstrate ultrastructurally disorganized mitochondria, swollen in appearance with disrupted membranes (arrows) as compared to control littermates (Magnification 2950X).
Discussion
Prader-Willi syndrome results from a loss of paternal imprinted genes from chromosome 15q11-15q13, leading to a profound disturbance of neurodevelopment that manifests multiple complex phenotypes including ones involved in energy metabolism, behavioral, cognitive, and structural defects. These complex clinical manifestations are the result of not only the loss of genes found on the deleted chromosome per se, but also the result of downstream changes in gene and regulatory networks involving those genes. Previous work has demonstrated that many of the downstream affected genes are involved in various energy metabolism pathways17. Additionally, clinical evidence has shown a role for CoQ10 supplementation in patient improvement. Thus far to the best of our knowledge no one had studied the possible role of mitochondrial dysfunction in PWS, we hypothesize that a component of PWS pathology is related to mitochondrial function.
We then conducted genome microarray analysis of RNA transcripts in skeletal muscle and brain tissues of our PWS-IC deletion mice, specifically looking for altered expression of genes involved in various mitochondrial pathways. We found 95 and 66 mitochondrial genes differentially expressed in WT and PWS-IC muscle and brain. As expected, all the known genes from the PWS critical region were downregulated and Ube3a was significantly upregulated.
Focusing our attention on transcripts that demonstrated a FC of greater than 1.5 or less than −1.5, we discovered several mitochondrial genes that fall under cellular metabolism pathways by Ingenuity networks analysis. Specifically, in brain tissues Mrpl15 (mitochondrial ribosomal protein L15) was upregulated, while Alas1 (aminolevulinic acid synthase 1) was downregulated. Mrpl15 encodes a 39s subunit protein that make up a part of the mitochondrial ribosome required for mitochondrial protein synthesis28. Recent bioinformatics approaches have linked this gene to possibly playing a role in stem cell pluripotency and differentiation29. Alas1 gene product is a nuclear-encoded mitochondrial enzyme that is the rate-limiting and first enzyme in the heme biosynthetic pathway. A recent study has found downregulated levels of this enzyme in Alzheimer’s disease (AD), possibly suggesting that downregulated levels of this enzyme and dysfunctional heme metabolism could play a role in the cognitive and behavioral abnormalities seen in PWS.
In muscle tissues, we discovered the following downregulated nuclear coded mitochondrial gene expression levels; Bdh (3-hydroxybutyrate dehydrogenase) involved in fatty acid catabolism, Mte1 (mitochondrial acyl-CoA thioesterase 1), involved in lipid metabolism pointing to a possible underlying mitochondrial role in biochemical metabolism pathways in PWS pathophysiology. Acadl (acetyl-Coenzyme A dehydrogenase, long-chain) belongs to the acyl-CoA dehydrogenase family, which is a family of mitochondrial flavoenzymes involved in fatty acid and branched chain aminoacid metabolism. The resulting protein is one of the four enzymes that catalyze the initial step of mitochondrial beta-oxidation of straight-chain fatty acid. Defects in this gene are the cause of long-chain acyl-CoA dehydrogenase (LCAD) deficiency, leading to nonketotic hypoglycemia [provided by RefSeq]. Finally, variants of Mtch2 (mitochondrial carrier homolog 2) in humans have demonstrated a link with obesity30 providing yet another potential clue to the role of mitochondrial dysfunction in the pathophysiology of PWS.
We have observed in a PWS-IC mouse model changes in muscle and brain mitochondrial gene expression, mitochondrial proliferation. We found significant structural changes on EM in cardiac cells of our PWS-IC deletion mouse model in addition to an increase of complex II+III activity in cardiac tissues.
These observations are consistent with previous reports of differential expression of energy metabolism genes and provide an explanation for possible therapeutic benefit from Co-Q10 supplementation. Co-Q10 plays a pivotal role as an electron acceptor from complex II before passing on those electrons to complex III. This study provides support for our hypothesis that PWS patients have an underlying defect in mitochondrial bioenergetics that is partially compensated by the upregulation of mitochondrial biogenesis.
Conclusion
In conclusion we have demonstrated ultra-structural, enzymatic, and transcriptional changes of key mitochondrial components that point to a substantial role of mitochondrial dysfunction in the pathophysiology of PWS in the PWS-IC model. Additionally, our microarray findings have detected previously unknown candidate genes that need to be studied in more detail to fully elucidate their potential role in PWS. In conclusion, this current work supports the rationale for Co-Q10 supplementation in PWS in alleviating some of the symptoms associated with this disease. Such studies could lead to other possible novel therapeutic avenues in treating this disease.
ACKNOWLEDGMENTS
This research was supported by the Rare Diseases Clinical Research Consortia (USA), U54 RR019478, an RDCRN Post-doctoral trainee award and a Foundation of Prader-Willi Research (FPWR) grant (V.K.), California Regenerative Medicine Pre-doctoral Fellowship TI-00008 (W.F.), and the National Institute of Health (USA) grants NS21328, NS41850, AG13154, AG24373 and AG16573 (D.C.W.). We thank Fadia Haddad and Shlomit Aizik for helpful discussions.
References
- 1.Jiang Y, Tsai TF, Bressler J, Beaudet AL. Imprinting in Angelman and Prader-Willi syndromes. Current opinion in genetics & development. 1998;8(3):334–42. doi: 10.1016/s0959-437x(98)80091-9. [DOI] [PubMed] [Google Scholar]
- 2.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annual review of genomics and human genetics. 2001;2:153–75. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 3.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2011 doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 4.Cavaille J, Buiting K, Kiefmann M, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14311–6. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de los Santos T, Schweizer J, Rees CA, Francke U. Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which Is highly expressed in brain. American journal of human genetics. 2000;67(5):1067–82. doi: 10.1086/303106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray TA, Saitoh S, Nicholls RD. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(10):5616–21. doi: 10.1073/pnas.96.10.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholls RD, Saitoh S, Horsthemke B. Imprinting in Prader-Willi and Angelman syndromes. Trends in genetics : TIG. 1998;14(5):194–200. doi: 10.1016/s0168-9525(98)01432-2. [DOI] [PubMed] [Google Scholar]
- 8.Ning Y, Roschke A, Christian SL, Lesser J, Sutcliffe JS, Ledbetter DH. Identification of a novel paternally expressed transcript adjacent to snRPN in the Prader-Willi syndrome critical region. Genome research. 1996;6(8):742–6. doi: 10.1101/gr.6.8.742. [DOI] [PubMed] [Google Scholar]
- 9.Ohta T, Gray TA, Rogan PK, et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. American journal of human genetics. 1999;64(2):397–413. doi: 10.1086/302233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahoo T, del Gaudio D, German JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nature genetics. 2008;40(6):719–21. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cattanach BM, Barr JA, Evans EP, et al. A candidate mouse model for Prader-Willi syndrome which shows an absence of Snrpn expression. Nature genetics. 1992;2(4):270–4. doi: 10.1038/ng1292-270. [DOI] [PubMed] [Google Scholar]
- 12.Gabriel JM, Merchant M, Ohta T, et al. A transgene insertion creating a heritable chromosome deletion mouse model of Prader-Willi and angelman syndromes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(16):9258–63. doi: 10.1073/pnas.96.16.9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang T, Adamson TE, Resnick JL, et al. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nature genetics. 1998;19(1):25–31. doi: 10.1038/ng0598-25. [DOI] [PubMed] [Google Scholar]
- 14.Bressler J, Tsai TF, Wu MY, et al. The SNRPN promoter is not required for genomic imprinting of the Prader-Willi/Angelman domain in mice. Nature genetics. 2001;28(3):232–40. doi: 10.1038/90067. [DOI] [PubMed] [Google Scholar]
- 15.Ding F, Prints Y, Dhar MS, et al. Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models. Mamm Genome. 2005;16(6):424–31. doi: 10.1007/s00335-005-2460-2. [DOI] [PubMed] [Google Scholar]
- 16.Tsai TF, Jiang YH, Bressler J, Armstrong D, Beaudet AL. Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Human molecular genetics. 1999;8(8):1357–64. doi: 10.1093/hmg/8.8.1357. [DOI] [PubMed] [Google Scholar]
- 17.Bittel DC, Kibiryeva N, Talebizadeh Z, Butler MG. Microarray analysis of gene/transcript expression in Prader-Willi syndrome: deletion versus UPD. J Med Genet. 2003;40(8):568–74. doi: 10.1136/jmg.40.8.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual review of genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keating DJ. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. Journal of neurochemistry. 2008;104(2):298–305. doi: 10.1111/j.1471-4159.2007.04997.x. [DOI] [PubMed] [Google Scholar]
- 20.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nature neuroscience. 2000;3(12):1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 21.Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. The Journal of biological chemistry. 2005;280(35):30773–82. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- 22.Mattiazzi M, D’Aurelio M, Gajewski CD, et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. The Journal of biological chemistry. 2002;277(33):29626–33. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- 23.Angelin A, Bonaldo P, Bernardi P. Altered threshold of the mitochondrial permeability transition pore in Ullrich congenital muscular dystrophy. Biochim Biophys Acta. 2008;1777(7-8):893–6. doi: 10.1016/j.bbabio.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 24.Pagel-Langenickel I, Schwartz DR, Arena RA, et al. A discordance in rosiglitazone mediated insulin sensitization and skeletal muscle mitochondrial content/activity in Type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol. 2007;293(5):H2659–66. doi: 10.1152/ajpheart.00782.2007. [DOI] [PubMed] [Google Scholar]
- 25.Irwin WA, Bergamin N, Sabatelli P, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nature genetics. 2003;35(4):367–71. doi: 10.1038/ng1270. [DOI] [PubMed] [Google Scholar]
- 26.Chamberlain SJ, Johnstone KA, DuBose AJ, et al. Evidence for genetic modifiers of postnatal lethality in PWS-IC deletion mice. Human molecular genetics. 2004;13(23):2971–7. doi: 10.1093/hmg/ddh314. [DOI] [PubMed] [Google Scholar]
- 27.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods in enzymology. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 28.Kenmochi N, Suzuki T, Uechi T, et al. The human mitochondrial ribosomal protein genes: mapping of 54 genes to the chromosomes and implications for human disorders. Genomics. 2001;77(1-2):65–70. doi: 10.1006/geno.2001.6622. [DOI] [PubMed] [Google Scholar]
- 29.Mason MJ, Fan G, Plath K, Zhou Q, Horvath S. Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genomics. 2009;10:327. doi: 10.1186/1471-2164-10-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renstrom F, Payne F, Nordstrom A, et al. Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Human molecular genetics. 2009;18(8):1489–96. doi: 10.1093/hmg/ddp041. [DOI] [PMC free article] [PubMed] [Google Scholar]