

Abstract
Cocaine is the most frequently used illicit drug among individuals seeking emergency-room care, with fatal outcome most often attributable to the cardiovascular manifestations of drug abuse. While the symptomatic presentations of cocaine toxicity are increasingly understood, the molecular determinants that define outcome remain largely unknown. Here, we report that the susceptibility to cocaine-induced cardiotoxicity is genetically regulated. Targeted deletion of the KCNJ11-encoded Kir6.2 pore-forming subunit of sarcolemmal KATP channels resulted in amplified vulnerability to the toxic effects of chronic cocaine abuse. Under the hyperadrenergic stress, imposed by daily 3-week-long intraperitoneal administration of 30 mg/kg cocaine in Kir6.2-knockout mice, failure to maintain cardiac homeostasis translated into decreased exercise tolerance revealed by poor treadmill stress performance, and dilated hypokinetic left hearts with aggravated cellular hypertrophy and pathognomonic characteristics of chronic cocaine-induced cardiac toxicity. This study therefore reveals a previously unrecognized role of Kir6.2-encoded KATP channels in determining cardiovascular outcome in chronic cocaine abuse, identifying a novel molecular determinant of cocaine cardiotoxicity.
Keywords: ATP-sensitive K+ channel, disease, genetics, heart, Kir6.2, SUR2A
Introduction
Cocaine is a powerful stimulant that accounts for over 2 million of illicit-drug abusers and recurring dependents in the United States.1 The alkaloid's addictive properties are associated with near-pandemic abuse. Intoxication with cocaine is the most frequent cause of drug-related visits to the emergency department worldwide, mainly related to adverse cardiovascular manifestations of the drug. By inhibiting reuptake of catecholamines, cocaine induces an overdriven sympathomimetic effect resulting in mismatch of energy demand and supply responsible for drug-induced toxicity.2 Although introduced in medical practice as a local anesthetic, cocaine can produce irreversible structural damage to the heart, accelerate cardiovascular disease progression, and precipitate sudden cardiac death.3 Indeed, cocaine cardiopathology is the most common substrate leading to hospitalization and mortality among chronic drug abusers.4 Of note, individual variations in the severity of presentation have been documented in cases of cardiac cocaine toxicity.3 However, the underlying molecular determinants of outcome in cocaine abuse remain largely unknown.
Recently, KCNJ11 has been identified as a safeguard molecule conferring survival advantage in response to the hyperadrenergic state imposed by acute cocaine overdose.5KCNJ11 encodes the pore-forming Kir6.2 subunit of ATP-sensitive potassium (KATP) channels, recognized biosensors that adjust membrane potential-dependent processes to match metabolic demand.6–8 In the heart, Kir6.2 is integral to the make-up of sarcolemmal KATP channels.9–11 Integrated with cellular metabolic pathways,12–16 cardiac KATP channels process energetic signals of distress.6,17,18 Kir6.2 is required in cardiac adaptation to physiological and pathophysiological stress,19–22 with KATP channel malfunction implicated in heart disease.23,24 A deficit in KATP channels impairs tolerance to systemic stressors, including sympathetic surge.25,26 Genetic disruption of KATP channels compromises cardioprotection,27 while variants in channel proteins have been linked to cardiac pathology in patient populations.28–30 The increased understanding of the KATP channel involvement in regulation of heart homeostasis8,31,32 raises the hypothesis that these unique channels contribute toward protection of the myocardium from the hyperadrenergic state of cocaine cardiotoxicity.
Here, we tested the relevance of KATP channels in securing cardiac adaptation to repetitive exposures to cocaine. A chronic, sublethal cocaine administration regimen resulted in reduced exercise capacity accompanied by pathological left ventricular enlargement and impaired blood ejection velocity, as well as increased myocyte cross-sectional area in the Kir6.2-knockout compared to wild-type counterparts. Th is study identifies KATP channel functionality as a novel determinant of outcome in cocaine abuse.
Methods
Murine model of chronic cocaine abuse
Mice deficient in KATP channels (Kir6.2-KO) were generated by targeted disruption of the KCNJ11 gene encoding the pore-forming Kir6.2 subunit of sarcolemmal KATP channels, and backcrossed for five generations into a C57BL/6 background.33,34 In accordance with NIH guidelines, and following approval by the Mayo Clinic Institutional Animal Care and Use Committee, 12-week-old Kir6.2-KO and sex/age-matched C57BL/6 wild-type (WT) mice were administered a sublethal dose of cocaine (30 mg/kg i.p. every 12 hours) for 20 days (Figure 1). All mice were given standard rodent chow, were housed individually with a 12-hour day/night cycle, and were monitored daily. Body weight and food consumption were measured prior to initiation of cocaine administration (drug-free state), and following the 20 day-long cocaine administration regimen.
Figure 1. Experimental design.
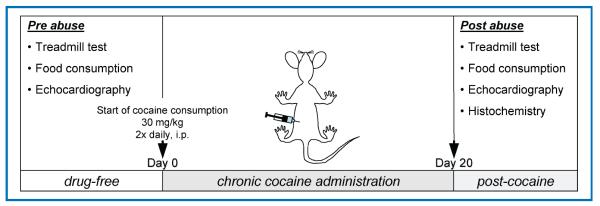
At a drug-free state, prior to initiation of cocaine administration, a treadmill stress test and baseline food consumption measurements were performed. Day 0 represents the beginning of the twice daily, 30 mg/kg intraperitoneal cocaine administration that lasted 20 days. On completion of this regimen, a second exercise test, food consumption measurements, and echocardiography were performed, followed by postmortem histopathological evaluation of heart tissue samples.
Treadmill exercise stress test
To assess the impact of chronic cocaine abuse on physical capacity in wild-type and Kir6.2-KO mice, a comparison of performance was made based on exercise stress tests at baseline and at day 21 (Figure 1). Stepwise increments of incline and velocity at 3-minute intervals on a graded treadmill (Columbus Instruments, Columbus, OH, USA) allowed the determination of total tolerated workload (W, in Joules), a composite parameter calculated as the sum of kinetic energy Ek = m · v2/2 and potential energy Ep = m · g · v · t sin(θ), where m represents animal mass, v treadmill velocity, g acceleration due to gravity, t elapsed time at a protocol level, and θ the angle of incline.20,26
Cardiac structure and function
Trans-thoracic cardiac ultrasound imaging (15L8 transducer, Sequoia c256, Acuson, Mountain View, CA, USA) was performed in lightly sedated (1.25% isoflurane) mice before and after the chronic cocaine administration protocol (Figure 1). Images were digitally acquired and stored for offline blinded analysis. Echocardiographic measurements of left ventricular dimensions were recorded at end-diastole (LVDd) and end-systole (LVDs) from three consecutive cardiac cycles, using the leading edge method. Similarly, interventricular septum (IVST) and posterior wall thickness (PWT) were measured from M-mode images. The ratio of relative wall thickness to the left ventricular end-diastolic dimension, a parameter of heart geometry, was expressed as [(IVST + PET)/LVDd]. Left ventricular fractional shortening was calculated as [(LVDd − LVSd)/LVDd]×100.20,35 Peak ejection velocity was determined from the actual pulsed-wave Doppler tracings on the parasternal long-axis view of trans-aortic flow by measuring the absolute magnitude of three consecutive tracings.36,37
Histochemistry
At 21 days, one day after the last cocaine administration, left ventricles (LV) including septum were removed, rinsed, blotted dry, and stored in 10% buffered formalin. Micrographs of 5 μm thick, paraffin-embedded, LV sections stained with hematoxylin-eosin were taken with a KS-4000 Axiocam digital camera (Carl Zeiss MicroImaging, Thornwood, NY, USA) mounted on an Axioplan 2 light microscope (Carl Zeiss MicroImaging, Thornwood, NY, USA) and analyzed using the MetaMorph software (Visitron Universal Imaging, Union City, CA, USA).38,39 All quantification was performed blinded to the sample origin.
Statistical analysis
Results are expressed as mean ± SEM. Student's t-test was used to compare variables between experimental groups and define p-values (JMP 7, SAS, Cary, NC). P-values less than 0.05 were predetermined to indicate statistical significance.
Results
Chronic cocaine administration results in exercise intolerance in KATP channel knockout
A potent sympathoadrenal stimulant, exercise amplifies the catecholamine response induced by cocaine administration increasing stress load, particularly to the heart.40 Here, under a treadmill stress-test, chronic cocaine administration rendered Kir6.2-KO mice vulnerable to physical exertion as demonstrated by reduced tolerated workload and exercise duration. These performance parameters were 25% and 11% lower, respectively (p < 0.05), than those observed at a drug-free state, an effect not seen in the wild type (WT) in which cocaine consumption enhanced tolerated workload (+27%; p < 0.05) when compared to the drug-free baseline (Figure 2A). The decreased exercise capacity of cocaine-abusing Kir6.2-KO animals was not a consequence of reduced caloric intake, as revealed by equivalent or even slightly increased food consumption compared to WT (198.7 ± 9.4 mg/day/g vs. 224.5 ± 9.0 mg/day/g before, and 204.4 ± 12.4 mg/day/g vs. 210.1 ± 6.8 mg/day/g after cocaine administration in WT vs. Kir6.2-KO, respectively; Figure 2B), nor of an accentuated body weight loss resulting from cocaine administration (30 ± 0.8 g in WT vs. 28.1 ± 0.8 g in Kir6.2-KO before, and 27.1 ± 0.9 g in WT vs. 25.5 ± 0.9 g in Kir6.2-KO after cocaine administration; Figure 2C).
Figure 2. Impaired physical capacity with cocaine challenge in the absence of KATP channels.

(A) Kir6.2 knock out compromises physical endurance and impairs cardiac performance in chronic cocaine abuse. Under exercise stress testing, Kir6.2-KO exhibited impaired endurance compared to matched cocaine-challenged WT with decreased workload tolerance (25% less than at drug-free state for Kir6.2-KO vs. 27% above baseline for WT) and exercise duration (11% decrease from baseline for Kir6.2-KO vs. no change for WT) after 20 days of cocaine administration. (B) Food consumption normalized to body weight was constant before (198.7 ± 9.4 mg/day/g vs. 224.5 ± 9.0 mg/day/g) and after (204.4 ± 12.4 mg/day/g vs. 210.1 ± 6.8 mg/day/g) cocaine administration in WT vs. Kir6.2-KO. (C) Both WT and Kir6.2-KO animals experienced a reduction in body weight. However, body weight was indistinguishable between groups before (30 ± 0.8 g in WT vs. 28.1 ± 0.8 g in Kir6.2-KO) and after (27.1 ± 0.9 g in WT vs. 25.5 ± 0.9 g in Kir6.2-KO) the cocaine administration regimen. In each group, n = 5; †p < 0.05 when comparing each group to baseline drug-free state. *p < 0.05 when comparing WT versus Kir6.2-KO.
Ventricular dilatation and impaired contractility in KATP channel knockout after cocaine abuse
Cocaine abuse can lead to clinically relevant left ventricular dysfunction.41,42 Here, after 20 days of imposed cocaine consumption, left ventricular diastolic (LVDd) and systolic (LVSd) dimensions reached 3.3 ± 0.2 mm and 2.4 ± 0.2 mm in Kir6.2-KO, respectively, significantly exceeding measurements obtained in WT (2.1 ± 0.2 mm LVDd, p < 0.01, Figure 3A; 1.3 ± 0.2 mm LVSd, p < 0.05, Figure 3B). Exaggerated left ventricular chamber dilatation was accompanied by prominent wall thinning, as the ratio between relative left ventricular wall thickness and LVDd ((IVST + PWT)/LVDd, a parameter reflecting heart geometry, was lower at 0.5 ± 0.1 in Kir6.2-KO versus 1 ± 0.1 in WT (p < 0.01; Figure 3C), as revealed by M-mode echocardiography (Figure 3D).
Figure 3. Dilated hypokinetic hearts in cocaine-challenged KATP channel deficient hearts.

Examination by ultrasound revealed dilated hearts in Kir6.2-KO compared to WT after chronic cocaine administration. (A) Left ventricular end-diastolic dimension (LVDd; 3.3 ± 0.2 mm vs. 2.1 ± 0.2 mm; n = 5) and (B) left ventricular end-systolic dimension (LVSd; 2.4 ± 0.2 mm vs. 1.3 ± 0.2 mm; n = 4) were augmented in Kir6.2-KO compared to WT after 20 days of cocaine. (C) The ratio between relative ventricular wall thickness (the sum of interventricular septum thickness, IVST, and posterior wall thickness, PWT) and LVDd was lower in Kir6.2-KO (0.5 ± 0.1; n = 5) versus WT (1 ± 0.1; n = 4) after the cocaine regimen, as shown in representative echocardiographic M-mode recordings (D). (E) Deteriorated cardiac performance following chronic cocaine injections was observed as a significantly reduced fractional shortening from the drug-free baseline in Kir6.2-KO (48.2 ± 4.2% pre- vs. 28.3 ± 4.2% post-cocaine; n = 5 in each group) in contrast to sustained function in WT (47.5 ± 4.2% pre- vs. 35.4 ± 4.7%; n = 4 in each group). (F) Peak ejection velocity appeared lower in Kir6.2-KO (0.49 ± 0.05 m/s; n = 5) versus WT (0.67 ± 0.03 m/s; n = 5), as documented by representative Doppler ultrasound (C). †denotes p < 0.05 when comparing each group to baseline drug-free state. *denotes p < 0.01 when comparing WT versus Kir6.2-KO.
Beyond structure, Kir6.2-KO also demonstrated significantly reduced cardiac performance manifested as reduction in fractional shortening after chronic cocaine administration compared to the baseline drug-free state (48.2 ± 4.2% pre- vs. 28.3 ± 4.2% post-cocaine; p < 0.05), a functional decline not developed by WT (47.5 ± 4.2% pre- vs. 35.4 ± 4.7% post-cocaine; Figure 3E). Moreover, peak ejection velocity after chronic cocaine abuse was significantly reduced in Kir6.2-KO compared to WT (0.49 ± 0.05 m/s vs. 0.67 ± 0.03 m/s, respectively; p < 0.01, Figure 3F), as demonstrated by Doppler ultrasound imaging (Figure 3G).
KATP channel deficiency precipitates cardiac myocyte enlargement after repeated cocaine administration
Activation of the sympathetic system by cocaine targets the cardiovascular system resulting in augmented cardiac workload and in turn cardiac hypertrophy.40 Here, as a result of repetitive cocaine exposure, the cross-sectional area of cardiac myocytes was enlarged in Kir6.2-KO compared to WT (460 ± 15 μm2 vs. 374 ± 23 μm2, respectively; p = 0.01) as revealed by histological evaluation of postmortem heart preparations (Figure 4A), indicating maladaptive cardiac remodeling.
Figure 4.
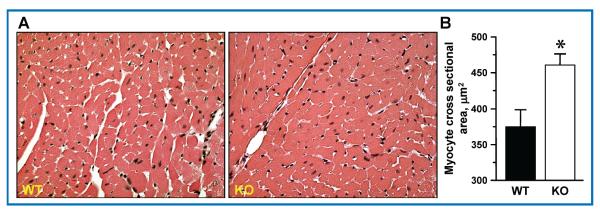
(A) Aggravated hypertrophy in cocaine-challenged KATP channel deficient cardiomyocytes. Representative images of hematoxylin-eosin-stained cardiac tissue. (B) Analysis of histological heart sections demonstrated increased myocyte cross-sectional area in Kir6.2-KO (460 ± 15 μm2, n = 4) in contrast to WT (374 ± 23 μm2, n = 4) after 20 days of cocaine i.p. injections. Bars are mean ± SEM. *denotes p ≤ 0.01.
Discussion
Cocaine is one of the most commonly used illicit drugs, and a major public health problem.2 Indeed, recent reports suggest that 3% of Americans between the ages of 15 and 64 currently abuse cocaine.43 Although the effects of cocaine intoxication are systemic, cardiovascular distress is a major substrate of adverse outcome. Cocaine exerts its cardiac effect through a sympathomimetic action by inhibiting catecholamine re-uptake and stimulating adrenergic receptors. While activation of proximal and distal components of the cardiac sympathetic axis has been associated with outcome,3,4 the present study provides evidence linking KATP channel genes as molecular determinants of cocaine cardiotoxicity.
Sarcolemmal KATP channels have the unique property to operate as high-fidelity homeostatic molecular rheostats capable of adjusting membrane excitability to match energy demand.24,30 In this study, the diminished exercise capacity of KATP channel-deficient mice was accompanied by cardiac structural and functional abnormalities on echocardiography under chronic cocaine intoxication. Kir6.2-KO developed left ventricular dilatation and relative wall thinning compared to wild type as a result of cocaine abuse, indicating a progression toward cardiomyopathy in the absence of protective KATP channel function. This maladaptation underscores the inability of KATP channel-deficient organs to withstand continuous sympathomimetic action.26 Indeed, the contractile function of Kir6.2-KO hearts deteriorates under isoproterenol challenge26 or, as shown here, with chronic administration of intraperitoneal cocaine. Fractional shortening was compromised in Kir6.2-KO following 20 days of cocaine, with a reduction in peak ejection velocity compared to WT counterparts, implicating an essential role played by these biosensors in supporting cardiac performance under chronic cocaine use. In addition, the enlarged cardiomyocyte cross-sectional area revealed by histopathological evaluation of cardiac tissue in Kir6.2-KO compared to WT, as a result of repetitive cocaine exposure, indicates a pivotal role for the KATP channel in protecting myocardial cytostructure in response to cocaine abuse.
By processing signals of cellular distress, KATP channels control cardiac action potential duration, thus limiting Ca2+ influx during cardiac contraction and protecting the heart against calcium overload and downstream calcium-dependent signaling events leading to pathological remodeling under stress.22,24,26 As one of the major mechanisms of cocaine's action is enhancement of sympathetic tone, leading to increased currents through L-type Ca2+ channels44 and aggravated myocardial oxygen demand45 with deterioration of ventricular performance,41,46 KATP channels may provide a significant safeguard. Indeed, KATP channel deficiency has been related to intracellular Ca2+-overload predisposing to malignant structural remodeling under physiological or pathophysiological stress.7 In fact, Ca2+ channel blocking drugs, successfully used in protection against the toxic effects of cocaine in the heart, have been shown to rescue the impaired cardioprotection imposed by knockout of KATP channels.19
While the pharmacodynamics of cardiac cocaine toxicity are increasingly understood,3 the notion that susceptibility to cocaine-induced mortality may be genetically controlled has been only recently documented.5 In this regard, variance in KATP channel genes could contribute in determining cardiac outcome in chronic cocaine abuse. Indeed, there is growing evidence that implicates KATP mutations or polymorphisms in the susceptibility to human cardiac disease.28–30 Studies in well-defined human cohorts will be necessary to establish the clinical implications of KATP channels in protection against cocaine cardiotoxicity.
Conclusion
In summary, this proof-of-principle study uncovers the importance of sarcolemmal KATP channels in determining the cardiovascular outcome in an experimental setting of chronic cocaine abuse. The presented data implicate the KATP channel as a candidate regulator of cardiovascular performance under the sympathomimetic effects of cocaine. This report thus identifies a previously unrecognized molecular determinant of cocaine cardiotoxicity, providing the foundation for translational studies in patient populations.
Acknowledgments
This work was supported by the National Institutes of Health (HL064822), Marriott Heart Disease Research Program, and Mayo Graduate School.
References
- 1.Research Report Series Cocaine abuse and addiction. [Accessed July 1, 2009];National Institute on Drug Abuse. 2004 Available at: http://www.drugabuse.gov/ResearchReports/Cocaine/cocaine.html.
- 2.Mendelson JH, Mello NK. Management of cocaine abuse and dependence. N Engl J Med. 1996;334(15):965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- 3.Phillips K, Luk A, Soor GS, Abraham JR, Leong S, Butany J. Cocaine cardiotoxicity: a review of the pathophysiology, pathology, and treatment options. Am J Cardiovasc Drugs. 2009;9(3):177–196. doi: 10.2165/00129784-200909030-00005. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein RA, DesLauriers C, Burda AM. Cocaine: history, social implications, and toxicity. Dis Mon. 2009;55(1):6–38. doi: 10.1016/j.disamonth.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Reyes S, Kane GC, Miki T, Seino S, Terzic A. KATP channels confer survival advantage in cocaine overdose. Mol Psychiatry. 2007;12(12):1060–1061. doi: 10.1038/sj.mp.4002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alekseev AE, Hodgson DM, Karger AB, Park S, Zingman LV, Terzic A. ATP-sensitive K+ channel channel/enzyme multimer: metabolic gating in the heart. J Mol Cell Cardiol. 2005;38(6):895–905. doi: 10.1016/j.yjmcc.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zingman LV, Alekseev AE, Hodgson-Zingman DM, Terzic A. ATP-sensitive potassium channels: metabolic sensing and cardioprotection. J Appl Physiol. 2007;103(5):1888–1893. doi: 10.1152/japplphysiol.00747.2007. [DOI] [PubMed] [Google Scholar]
- 8.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 9.Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16(5):1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 10.Lorenz E, Terzic A. Physical association between recombinant cardiac ATP-sensitive K+ channel subunits Kir6.2 and SUR2A. J Mol Cell Cardiol. 1999;31(2):425–434. doi: 10.1006/jmcc.1998.0876. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki M, Li RA, Miki T, Uemura H, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Ogura T, Seino S, Marban E, Nakaya H. Functional roles of cardiac and vascular ATP-sensitive potassium channels clarifi ed by Kir6.2-knockout mice. Circ Res. 2001;88(6):570–577. doi: 10.1161/01.res.88.6.570. [DOI] [PubMed] [Google Scholar]
- 12.Abraham MR, Selivanov VA, Hodgson DM, Pucar D, Zingman LV, Wieringa B, Dzeja PP, Alekseev AE, Terzic A. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J Biol Chem. 2002;277(27):24427–24434. doi: 10.1074/jbc.M201777200. [DOI] [PubMed] [Google Scholar]
- 13.Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, Hodgson D, Bienengraeber M, Puceat M, Janssen E, Wieringa B, Terzic A. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA. 2001;98(13):7623–7628. doi: 10.1073/pnas.121038198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jovanovic A, Alekseev AE, Terzic A. Intracellular diadenosine polyphosphates: a novel family of inhibitory ligands of the ATP-sensitive K+ channel. Biochem Pharmacol. 1997;54(2):219–225. doi: 10.1016/s0006-2952(97)00262-1. [DOI] [PubMed] [Google Scholar]
- 15.Karger AB, Park S, Reyes S, Bienengraeber M, Dyer RB, Terzic A, Alekseev AE. Role for SUR2A ED domain in allosteric coupling within the KATP channel complex. J Gen Physiol. 2008;131(3):185–196. doi: 10.1085/jgp.200709852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selivanov VA, Alekseev AE, Hodgson DM, Dzeja PP, Terzic A. Nucleotide-gated KATP channels integrated with creatine and adenylate kinases: amplifi cation, tuning and sensing of energetic signals in the compartmentalized cellular environment. Mol Cell Biochem. 2004;256–257(1–2):243–256. doi: 10.1023/b:mcbi.0000009872.35940.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park S, Lim BB, Perez-Terzic C, Mer G, Terzic A. Interaction of asymmetric ABCC9-encoded nucleotide binding domains determines KATP channel SUR2A catalytic activity. J Proteome Res. 2008;7(4):1721–1728. doi: 10.1021/pr7007847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zingman LV, Alekseev AE, Bienengraeber M, Hodgson D, Karger AB, Dzeja PP, Terzic A. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001;31(2):233–245. doi: 10.1016/s0896-6273(01)00356-7. [DOI] [PubMed] [Google Scholar]
- 19.Kane GC, Behfar A, Dyer RB, O'Cochlain DF, Liu X-K, Hodgson DM, Reyes S, Miki T, Seino S, Terzic A. KCNJ11 gene knockout of the Kir6.2 KATP channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet. 2006;15(15):2285–2297. doi: 10.1093/hmg/ddl154. [DOI] [PubMed] [Google Scholar]
- 20.Yamada S, Kane GC, Behfar A, Liu X-K, Dyer RB, Faustino RS, Miki T, Seino S, Terzic A. Protection conferred by myocardial ATP-sensitive K+ channels in pressure overload-induced congestive heart failure revealed in KCNJ11 Kir6.2-null mutant. J Physiol. 2006;577(3):1053–1065. doi: 10.1113/jphysiol.2006.119511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zingman LV, Hodgson DM, Alekseev AE, Terzic A. Stress without distress: homeostatic role for KATP channels. Mol Psychiatry. 2003;8(3):253–254. doi: 10.1038/sj.mp.4001323. [DOI] [PubMed] [Google Scholar]
- 22.Gumina RJ, O'Cochlain DF, Kurtz CE, Bast P, Pucar D, Mishra P, Miki T, Seino S, Macura S, Terzic A. KATP channel knockout worsens myocardial calcium stress load in vivo and impairs recovery in stunned heart. Am J Physiol Heart Circ Physiol. 2007;292(4):H1706–1713. doi: 10.1152/ajpheart.01305.2006. [DOI] [PubMed] [Google Scholar]
- 23.Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan C, Gumina RJ, Pucar D, O'Coclain F, Mann DL, Alekseev AE, Terzic A. Cellular remodeling in heart failure disrupts KATP channel-dependent stress tolerance. EMBO J. 2003;22(8):1732–1742. doi: 10.1093/emboj/cdg192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kane GC, Liu X-K, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005;38(6):937–943. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X-K, Yamada S, Kane GC, Alekseev AE, Hodgson DM, O'Cochlain F, Jahangir A, Miki T, Seino S, Terzic A. Genetic disruption of Kir6.2, the pore-forming subunit of ATP-sensitive K+ channel, predisposes to catecholamine-induced ventricular dysrhythmia. Diabetes. 2004;53(Suppl 3):S165–S168. doi: 10.2337/diabetes.53.suppl_3.s165. [DOI] [PubMed] [Google Scholar]
- 26.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002;99(20):13278–13283. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gumina RJ, Pucar D, Bast P, Hodgson DM, Kurtz CE, Dzeja PP, Miki T, Seino S, Terzic A. Knockout of Kir6.2 negates ischemic preconditioning-induced protection of myocardial energetics. Am J Physiol Heart Circ Physiol. 2003;284(6):H2106–H2113. doi: 10.1152/ajpheart.00057.2003. [DOI] [PubMed] [Google Scholar]
- 28.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang Y-P, Alekseev AE, Terzic A. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36(4):382–387. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, Seino S, Asirvatham SJ, Jahangir A, Terzic A. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;4(2):110–116. doi: 10.1038/ncpcardio0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes S, Terzic A, Mahoney DW, Redfield MM, Rodeheffer RJ, Olson TM. KATP channel polymorphism is associated with left ventricular size in hypertensive individuals: a large-scale community-based study. Hum Genet. 2008;123(6):665–667. doi: 10.1007/s00439-008-0519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sattiraju S, Reyes S, Kane G, Terzic A. KATP channel pharmacogenomics: from bench to bedside. Clin Pharmacol Ther. 2008;83(2):354–357. doi: 10.1038/sj.clpt.6100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashcroft FM. From molecule to malady. Nature. 2006;440(7083):440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- 33.Nelson TJ, Martinez-Fernandez A, Terzic A. KCNJ11 knockout morula re-engineered by stem cell diploid aggregation. Philos Trans R Soc Lond B Biol Sci. 2009;364(1514):269–276. doi: 10.1098/rstb.2008.0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J-i, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA. 1998;95(18):10402–10406. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson TJ, Martinez-Fernandez A, Yamada S, Perez-Terzic C, Ikeda Y, Terzic A. Repair of acute myocardial infarction by human stemness factors induced pluripotent stem cells. Circulation. 2009;120:408–416. doi: 10.1161/CIRCULATIONAHA.109.865154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kane GC, Lam CF, O'Cochlain F, Hodgson DM, Reyes S, Liu XK, Miki T, Seino S, Katusic ZS, Terzic A. Gene knockout of the KCNJ8-encoded Kir6.1 KATP channel imparts fatal susceptibility to endotoxemia. Faseb J. 2006;20(13):2271–2280. doi: 10.1096/fj.06-6349com. [DOI] [PubMed] [Google Scholar]
- 37.Yamada S, Nelson TJ, Crespo-Diaz RJ, Perez-Terzic C, Liu XK, Miki T, Seino S, Behfar A, Terzic A. Embryonic stem cell therapy of heart failure in genetic cardiomyopathy. Stem Cells. 2008;26(10):2644–2653. doi: 10.1634/stemcells.2008-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamada S, Nelson TJ, Behfar A, Crespo-Diaz RJ, Fraidenraich D, Terzic A. Stem cell transplant into preimplantation embryo yields myocardial infarction-resistant adult phenotype. Stem Cells. 2009;27(7):1697–1705. doi: 10.1002/stem.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Cochlain DF, Perez-Terzic C, Reyes S, Kane GC, Behfar A, Hodgson DM, Strommen JA, Liu XK, van den Broek W, Wansink DG, Wieringa B, Terzic A. Transgenic overexpression of human DMPK accumulates into hypertrophic cardiomyopathy, myotonic myopathy and hypotension traits of myotonic dystrophy. Hum Mol Genet. 2004;13(20):2505–2518. doi: 10.1093/hmg/ddh266. [DOI] [PubMed] [Google Scholar]
- 40.Han DH, Kelly KP, Fellingham GW, Conlee RK. Cocaine and exercise: temporal changes in plasma levels of catecholamines, lactate, glucose, and cocaine. Am J Physiol Endocrinol Metab. 1996;270(3):E438–E444. doi: 10.1152/ajpendo.1996.270.3.E438. [DOI] [PubMed] [Google Scholar]
- 41.Bertolet BD, Freund G, Martin CA, Perchalski DL, Williams CM, Pepine CJ. Unrecognized left ventricular dysfunction in an apparently healthy cocaine abuse population. Clin Cardiol. 1990;13(5):323–328. doi: 10.1002/clc.4960130505. [DOI] [PubMed] [Google Scholar]
- 42.Afonso L, Mohammad T, Thatai D. Crack whips the heart: a review of the cardiovascular toxicity of cocaine. Am J Cardiol. 2007;100(6):1040–1043. doi: 10.1016/j.amjcard.2007.04.049. [DOI] [PubMed] [Google Scholar]
- 43.United Nations office on drugs and crime . World drug report. United Nations; Vienna: 2008. [Google Scholar]
- 44.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104(4):569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 45.Hollander JE, Henry TD. Evaluation and management of the patient who has cocaine-associated chest pain. Cardiol Clin. 2006;24(1):103–114. doi: 10.1016/j.ccl.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 46.Henning RJ, Cuevas J. Cocaine activates calcium/calmodulin kinase II and causes cardiomyocyte hypertrophy. J Cardiovasc Pharmacol. 2006;48(1):802–813. doi: 10.1097/01.fjc.0000211796.45281.46. [DOI] [PubMed] [Google Scholar]