

Abstract
Adoptive cell transfer (ACT) using ex vivo-expanded anti-tumor T cells such as tumor-infiltrated lymphocytes or genetically engineered T cells potently eradicates established tumors. However, these two approaches possess obvious limitations. Therefore, we established a novel methodology using total tumor RNA (ttRNA) to prime dendritic cells (DC) as a platform for the ex vivo generation of anti-tumor T cells. We evaluated the antigen-specific expansion and recognition of T cells generated by the ttRNA–DC–T platform, and directly modulated the differentiation status of these ex vivo-expanded T cells with a cytokine cocktail. Furthermore, we evaluated the persistence and in vivo anti-tumor efficacy of these T cells through murine xenograft and syngeneic tumor models. During ex vivo culture, IL-2 preferentially expanded CD4 subset, while IL-7 enabled homeostatic proliferation from the original precursors. T cells tended to lose CD62L during ex vivo culture using IL-2; however, IL-12 could maintain high levels of CD62L by increasing expression on effector T cells (Tem). In addition, we validated that OVA RNA–DC only selectively expanded T cells in an antigen-specific manner. A cytokine cocktail excluding the use of IL-2 greatly increased CD62Lhigh T cells which specifically recognized tumor cells, engrafted better in a xenograft model and exhibited superior anti-tumor activities in a syngeneic intracranial model. ACT using the ex vivo ttRNA–DC–T platform in conjunction with a cytokine cocktail generated potent CD62Lhigh anti-tumor T cells and imposes a novel T cell-based therapeutic with the potential to treat brain tumors and other cancers.
Keywords: Central memory T cells (Tcm), CD62L, Dendritic cells (DC), Intracranial cerebellar (IC) tumor, Adoptive cell transfer (ACT), Immunotherapy
Introduction
Dendritic cells (DCs) are potent stimulatory cells that constantly survey the antigenic environment of the host and specifically activate T and B cells responses [1, 2]. Immunotherapy using DC loaded with peptides or tumor antigen extracts has been demonstrated with potent anti-tumor activity against brain tumors [3-5], and there is accumulating evidence that RNA transfection represents a superior method for loading antigens onto DC [6]. However, vaccination with total tumor RNA (ttRNA) primed DC as a cancer therapy relies upon engendering a robust immune response to tumor targets, and it is well-documented that glioma cancer-initiating cells can suppress immunity through inhibiting T-cell proliferation and impairing effector responses [7-9].
Thus far, adoptive cell transfer (ACT) using ex vivo-expanded tumor-infiltrated lymphocytes (TILs) or peripheral blood lymphocytes (PBLs) with genetically engineered anti-tumor T-cell receptors (TCRs) or chimeric antigen receptors (CARs) have shown the most success in eradicating melanoma [10, 11] and completely removing tumor cells in leukemia [12]. However, genetically engineered receptors target only one tumor-associated antigen and often face tumor evasion from T-cell surveillance due to the loss of tumor antigen. TILs represent both potent and polyfunctional anti-tumor T cells with multiple specificities, but are limited to the treatment of melanoma due to the requirements of identifying and then successfully expanding pre-existing tumor responsive cells ex vivo. Interestingly, the infiltration of CD8 T cells in newly diagnosed glioblastoma (GBM) is associated with an increased long-term survival [13]. However, due to the size of resected tumor samples and the difficulty in identifying pre-existing tumor responsive T cells, TILs are impractical for brain tumor management. In this study, we have circumvented these technical hurdles by creating a novel ttRNA–DC–T platform for the ex vivo expansion of potent “TIL-like” anti-tumor T cells for the treatment of brain tumors.
One practical issue regarding ex vivo-generated anti-tumor T cells is the progressively terminal differentiation that enables full in vitro effector functionality but paradoxically impairs in vivo anti-tumor efficacy [14]. This differentiation status can be modulated by the following: (1) the duration of culture, (2) the addition of pharmaceuticals, and (3) the combinations of cytokines utilized in vitro [14-17]. Cytokines sharing common γc chain receptors, i.e., IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21, play an important role during immune responses in vivo and contribute to long-term T-cell memory formation [18]. Currently, many clinical ACT protocols use IL-2 for ex vivo expansion of anti-tumor T cells although IL-2 additionally expands and activates immunosuppressive CD4+CD25+FoxP3+ TRegs. In contrast, IL-7 directly inhibits TRegs function and releases the natural regulatory brakes on T cells via signaling through the high-affinity IL-7R present on both naïve and memory TRegs [19]. Except cytokines sharing γc chain receptors, IL-12 is another important cytokine, which plays a pivotal role in the regulation of T-cell differentiation and memory formation [20-22], and the priming of naïve CD8+ T cells with IL-12 selectively enhances the survival of CD8+CD62Lhigh cells and results in superior anti-tumor activity in a tolerogenic murine model [23]. Compared to IL-2, it has been demonstrated that IL-7 and IL-21 could preserve less-differentiated effector T cells and enrich for tumor-reactive CD8+ T cells [24-26].
In fact, the barriers that prevent humoral immune components from entering the brain are not an impediment to T-cell-mediated ACT as activated T cells have been clearly shown to have full access to the brain [27, 28]. Our rationale to use CD62L as a maker to indicate the differentiation status of ex vivo-cultured T cells was based upon our previous reports; in that, CD62L along with CD44 could classify the ex vivo-cultured T cells into Tem, Tcm, and Tn which mimic the in vivo antigen experienced T cells [29, 30]. In this study, we established a novel methodology to ex vivo generate anti-tumor T cells for the adoptive therapy of brain tumors. Our unique cytokine cocktail generates CD62Lhigh anti-tumor T cells which convey superior anti-tumor activities in a murine IC tumor model.
Materials and methods
Cell culture
The PBMCs were obtained from healthy donors and brain tumor patients seeking treatments at DUKE Medical Center under approved clinical protocols. Cytokines for human and murine IL-7, IL-12, IL-15, and IL-21 were purchased from PEPROTECH (Rocky Hill, NJ, USA), and IL-2 (Aldesleukin) was from PROLEUKIN (San Diego, CA, USA). Human T cells were cultured in AIM-V (Invitrogen, Grand Island, NY, USA) with 2 % human AB serum, while murine T cells were cultured in RPMI with 5 % FBS, and both medium were supplemented with 100 U/ml Penicillin/Streptomycin, 1 mM l-glutamine, 55 uM β-ME, 1 mM Sodium pyruvate, 0.1 mM NEAA, and 10 mM HEPES (Invitrogen). Murine KR158 (kindly provided by Dr. Tyler Jacks) was derived from an astrocytoma in a NF1−/−:p53−/− C57BL/6-mouse, and murine tumor lines B16F10 and B16F10OVA that were cultured in DMEM with 10 % FBS. Human brain tumor line U87 was cultured in 10 % FBS MEM zinc medium. All cells were cultured at 37 °C in a 5 % CO2 humidified incubator.
The preparation of amplified ttRNA from human brain tumor and ttRNA from human and murine tumor lines
The resected brain tumor samples from patients were fragmented and lysed according to standard protocol at Brain Tumor Immunotherapy Program (BTIP). For ttRNA amplification, the CellDirect lysate was used for 1st-strand cDNA synthesis with SMARTScribe Reverse Transcriptase (ClonTech, Mountain View, CA, USA) using CDS Primer IIIA. The 1st-strand cDNA libraries were amplified with ClonTech’s Advantage 2 Polymerase PCR kit (ClonTech) by primers CDS Primer IIIA and T7-Smart. The PCR products of pre-amplified cDNA were purified with QIA-quick PCR Purification kit (Qiaqen, Valencia, CA, USA). The ttRNA were synthesized using PCR-amplified cDNA library as templates by T7 mMessage mMachine™ Transcription Kit (Ambion, Grand Island, NY, USA). TtRNA from U87, KR158, B16F10, and B16F10OVA lines was isolated using RNeasy Maxi Kit (Qiagen) following product manual, and the quality of ttRNA was evaluated using NanaDrop at 260 nm. OVA–RNA for ex vivo expansion of murine T cells was in vitro transcribed using T7 driven pGEM4Z (Promega, Madison, WI, USA) vector harboring OVA-expressing cassette.
Preparation of human and mouse DC
The procedures to generate human DC were described previously [31]. Briefly, the autologous DC were generated from PBMCs in patients diagnosed with GBM. PBMCs from leukapheresis were further purified by Ficoll/Hypaque cushion centrifugation. After 5 extensive washes to remove platelets, 2 × 108 cells were plated into T-150 flask with 18 ml 2 % human AB serum in AIM-V for 1 h and non-adherent cells (NAs) were harvested and cryopreserved as sources for T cells. The adherent cells were cultured in 15 ml AIM-V (no serum) containing 500 U/ml human IL-4 and 800 U/ml GM-CSF (R&D Systems, Minneapolis, MN, USA) for 4 days, and then, another 15 ml medium as above was added for additional 3 days. At day 7, DC were harvested using dissociation buffer (Invitrogen) and centrifuged at 1,200 rpm, 8 min, RT. Cells were washed twice and gently re-suspended in Opti-MEM at 2.5 × 107/ml; 100 μg amplified ttRNA or 200 μg ttRNA from U87 line were added to 5 × 107 DC and thoroughly mixed. About 5 × 106 cells in 200 μl buffer were added to the 2-mm cuvette and placed into Electro Square Porator ECM 830 (BTX Harvard Apparatus), and then pulsed at 300 V for 500 μs (Mode, LV). The cells resuspended at 3 × 106 cells/ml were then immediately transferred to T150 flask in 30 ml medium containing the maturation cytokine cocktail (800 U/ml GM-CSF, 500 U/ml IL-4,10 ng/ml TNF-α, 10 ng/ml IL-1β, and 1,000 U/ml IL-6) (R&D Systems) and incubated overnight. The DC were either used as stimulators for human T cells or cryopreserved. For murine DC, the procedures were modified at BTIP based on published protocol [32]. Briefly, bone marrow cells from both rear legs including tibia, femur, and sternum were extracted following standard sterile procedures. After pooling the bone marrow cells and filtering through 70-μm cell strainer (BD Biosciences), the cells were centrifuged at 500 g for 5 min RT and re-suspended in 3 ml ammonium chloride per 5 mice harvested followed by incubation at RT for 3–4 min. The lysing for RBC was stopped by adding 30 ml cold RPMI with 10 % FBS. The cells were then filtered through 70-μm cell strainer again followed by centrifugation and re-suspended in RPMI containing 5 % FBS, GM-CSF, and IL-4 (20 ng/ml) (Peprotech), and the other supplements were as described above. The cells were plated at 3 × 106/well at 6-well plates. At day 3, media was removed and the well was gently washed three times before adding fresh media. At day 7, the cells were harvested. After centrifugation, the cells were washed and re-suspended in Opti-MEM at 2.5 × 107/ml, and 200 μl aliquots were transferred into cuvettes with 25 ug ttRNA from the KR158 line. The electroporation conditions were set up as above. The cells were collected and re-plated using 5 × 106/well at 6-well plates in RPMI medium as described above with additional 0.5 μg/ml lipopolysaccharide mitogen (LPS) (Sigma, Ronkonkoma, NY, USA) overnight. The next day the matured ttRNA–DC was collected for stimulation of murine T cells.
Co-culture of DC with T cells
Human T cells from frozen samples were recovered overnight using AIM-V medium without cytokines. After ttRNA electroporation, ttRNA–DC was co-cultured with T cells at a ratio of 1:4–10 (DC–T cells) depending on the availability of DC. The co-culture was carried out using 24-well plates with 1 × 106 T cells/well. The cytokines or their combinations were added as illustrated in each figure legend. In some experiments, T cells were labeled with 0.5 μM CFSE (Invitrogen) following manufacturer’s instructions. For murine T cell culture, the ratio of DC:T cells from spleen was kept at 1:10 due to the difficulties in generating enough numbers of ttRNA–DC. Two million T cells per well in 24-well plates were co-cultured with appropriate numbers of ttRNA–DC in RPMI-1640 medium supplemented with cytokines IL-2, IL-7, IL-12, and IL-21 (Peprotech).
Flow cytometry and analysis
Flow cytometry was performed using FACSCalibur and FACSCanto (BD Biosciences). The viability of cells was determined by a combination of forward angle light scatter and 7-AAD staining of dead cells. Cell surface expression of CD3, CD4, CD8, CD25, CD27, CD28, CD44, CD70, CD62L, and CD45RO was measured using fluorescent-conjugated antibodies (BD Biosciences). The levels for IFNγ, IL-2, and FOXP3 were determined by intracellular staining using fluorescent-conjugated IFNγ, IL-2 antibodies (BD Biosciences), and FOXP3 antibody (eBiosciences, San Diego, CA, USA). All FACS data were analyzed using FlowJo 8.1.1 software (FlowJo, Ashland, OR, USA).
Animal models
After 10 days of ex vivo expansion, 3.5 × 106 human T cells from each group were harvested and re-suspended in PBS and injected intravenously into 6–8-week-old female NSG mice. After 30 days, mice were killed and lymphocytes were extracted from lymph nodes (LN). Flow-Count Fluorospheres (Beckman Coulter, Brea, CA, USA) were added to determine the cell numbers and analyzed by flow cytometry. Female C57BL/6 mice 6–8 weeks old were used for implanting IC tumor. KR158 cells were harvested with 0.25 % trypsin containing 0.02 % EDTA and washed once in serum-containing medium and twice in PBS. The cells were mixed with an equal volume of methylcellulose in zinc option medium and loaded into a 250-μl syringe (Hamilton, Reno, NV, USA) with an attached 25-gauge needle. Thirty-thousand cells in a volume of 5 μl were implanted into the right caudate nucleus of the brain using Quitessential Stereotaxic Injector system (Stoelting Co. Wood Dale, IL, USA). Four days after IC tumor implantation, mice received 5 Gy total body irradiation using Shepherd irradiator. Next day, mice were injected with 0.5–1 × 106 ttRNA–DC intradermally (ID) and infused with 1 × 106 T cells intravenously (IV). On days 7 and 14 after ACT, mice were vaccinated twice with 0.5–1 × 106 ttRNA–DC intraperitoneally (IP). Mice were checked daily and killed as needed with humane endpoints.
Results
The ex vivo ttRNA–DC–T co-culture under each cytokine unveils the signature division
To examine ttRNA–DC as a platform for the ex vivo expansion of anti-tumor T cells, we evaluated the impact of DC on T-cell proliferation in the presence of a panel of cytokines and DC to T cell ratios (Fig. 1a, 1st, 2nd rows). We found different cytokines induced “signature division patterns.” IL-2 preferentially expanded one subset during ex vivo culture, while IL-7 enabled homeostatic division from the original precursors (Fig. 1a, 1st and 2nd columns). To determine the signature division patterns of CD4+ and CD8+ T cells (highlighted in a dotted square within Fig. 1a), we plotted the progeny of CD4+ and CD8+ T cells as histograms reflecting the percentage of each division (Fig. 1b). The summary of T cell generations cultured with IL-2 or IL-7 (Fig. 1c) clearly indicated CD4+ and CD8+ subsets undergo distinct division patterns when exposed to these cytokines (Fig. 1c). Because only IL-2 and IL-7 support the proliferation of ex vivo-cultured T cells, we sought to investigate the biological role of these cytokines in driving T-cell proliferation. The preferential expansion of one subset in the presence of IL-2 occurred from day 8 to day 10 with or without DC coculture and was identified as CD3+CD25high (data not shown). Compared to the use of IL-7, IL-2 culture yielded a significantly higher percentage of CD3+CD25high T cells and CD4+CD25highFOXP3high TRegs (data not shown).
Fig. 1.
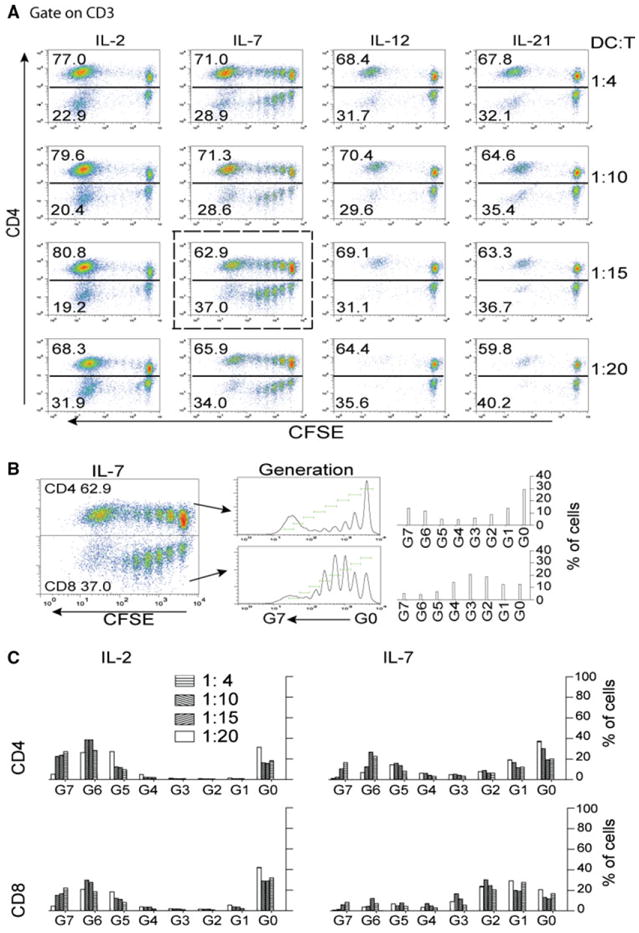
The establishment of co-culture system for ex vivo expansion of anti-tumor T cells using ttRNA–DC as a platform. a Determination of optimal ratio of DC:T cells. T cells were cultured with different cytokines as indicated on top of each column, and the ratio of DC:T cells was illustrated on right. Except the concentration of IL-2 was at 100 IU/ml, the concentration for other cytokines was 20 ng/ml. T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor the proliferation. At day 10, the cells were stained with fluorescent-conjugated antibodies and analyzed by flow cytometry. Gated on CD3, the percentage of CD4 was presented on y axis, and CFSE on x axis. b T-cell proliferation in the presence of IL-7 and DC. To caliber the generations of divided cells, a dot plot from DC to T cells at 1:15 in the presence of IL-7 (highlighted in a dotted square in a) was further analyzed using FlowJo software; during 10 days ex vivo culture, total 8 generations of divided CD4 and CD8 subsets were tracked and marked (middle panel), and the percentage reflecting each generation was illustrated (right panel). c The signature division under IL-2 and IL-7. Using the standard from b, the percentage representing each generation in the presence of IL-2 (left) and IL-7 (right) was comparatively plotted. The bars representing the different ratio of DC–T were denoted on top. Upper panel CD4 T cells, lower panel CD8 T cells
Cytokines used during co-culture determine the phenotype of ex vivo-expanded T cells
We next evaluated combinatorial cytokine cocktails (IL-2, IL-7, IL-12, and IL-21) for the optimization of ttRNA–DC driven T-cell generation. IL-12 and IL-21 play pivotal roles in memory formation and the facilitation of antigen presentation [22, 25], and IL-7 sustains homeostatic T-cell expansion after initial ttRNA–DC priming. IL-15 was not selected as its functionality is similar to IL-2, and it has shown no effect in preserving CD62L expression [30]. The purity for end products from the combinatorial cocktail (IL-7, IL-12, and IL-21) was similar to a culture with IL-2 alone as T cells were >95 % of total cells in both co-cultures (data not shown). cytokines cocktail significantly increased the percentage of CD8 T cells, versus the gold-standard of IL-2 alone; the use of IL-12 or a cytokine cocktail significantly increased the percentage of CD62L positive cells, and the use of IL-7 or IL-21 was capable of increasing the expression of CD27 (Fig. 2a). The levels of CD28 remained higher without a significant variation (Fig. 2a). Compared to dotted lines indicating the expression of each marker at day 1, we saw only the levels of CD28 remained up-regulated, while other markers stayed unchanged (CD8 and CD62L) or significantly decreased (CD27). T cells were expanded at an extent of onefold–fivefold with variations from donors to donors, and there was a trend for decrease in the IL-21 group, but no significant difference was observed among different cytokine groups (Fig. 2b).
Fig. 2.
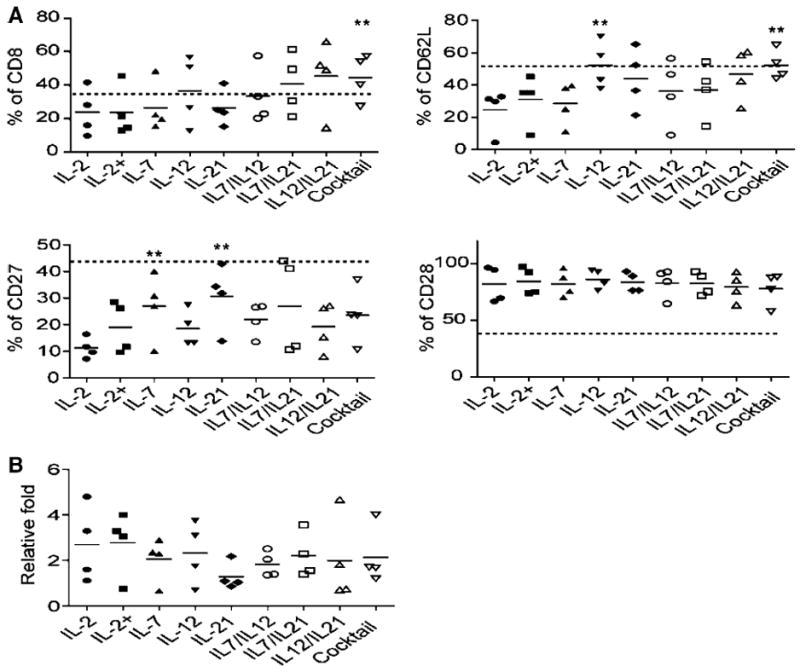
The effect of cytokines or their combinations on phenotype and proliferation of ttRNA–DC ex vivo-expanded anti-tumor T cells. a The phenotypic analysis after ex vivo expansion. The data from 4 donors were presented as average ± SD. The dashed lines represent the percentage of each marker at day 1 after overnight recovery using medium without cytokines: 37.1 % for CD8; 53.1 % for CD62L; 46.7 % for CD27; 38.9 % for CD28. The Student’s t test for mean was used for statistical analysis. b The fold expansion of ex vivo ttRNA–DC-expanded T cells for 10 days. Asterisks indicate p < 0.001 compared to IL-2 group. IL-2+ group: For the first 3 days of co-culture, IL-2 was supplemented, and then, IL-7 was added for the rest of culture
IL-12 maintains the high levels of CD62L expression in total and divided subpopulations of ex vivo-expanded T cells
A cytokine cocktail significantly enhanced the levels of CD62L expression (Fig. 2); therefore, it is pivotal if we could determine the unique role of each cytokine in orchestrating this consequence. IL-12 has been shown to possess the potent anti-tumor activity in a wide variety of murine tumor models [33-35], and the mechanisms through which IL-12 elicits its potent antitumor activity remain to be elucidated. Our previous study has identified a homing molecule CD62L that correlates with a less-differentiated phenotype of ex vivo-cultured anti-tumor T cells, which lead to an enhanced engraftment in xenograft model [29, 36]. However, the role of cytokines in ttRNA–DC–T platform has not been investigated thus far. In Fig. 3, the levels of CD62L were monitored following CFSE dye dilution on total and divided subpopulations over time. The signature division under IL-2 and IL-7 was highly reproducible (Fig. 3a). Except the addition of IL-12, the levels of CD62L kept decreased in divided subpopulations (Fig. 3a). The use of IL-2 maintained the lowest levels of CD62L, while IL-12 kept it at a higher level (Fig. 3b). In addition, the ratio of CD62L+/− (positive/negative) from the divided subpopulations in either IL-12 or cocktail group stayed higher compared to the use of IL-2, indicating the use of IL-12 was responsible for higher levels of CD62L expression (Fig. 3b, c). Furthermore, we sorted ex vivo-cultured CD8+ T cells as CD45RO+CD62L− (Tem) and CD45RO+CD62L+ (Tcm) (Fig. 3d): When we cultured the sorted cells in the presence of IL-2 or IL-12 for 7 days, the levels of CD62L on Tcm subset were remained at high levels irrelevant to the use of IL-2 or IL-12, while on Tem subset, we found the use of IL-12 led to re-expression of CD62L (Fig. 3e). Therefore, the data implied that the high levels of CD62L in the presence of IL-12 are through the re-induction of CD62L on the subset of Tem (Fig. 3).
Fig. 3.
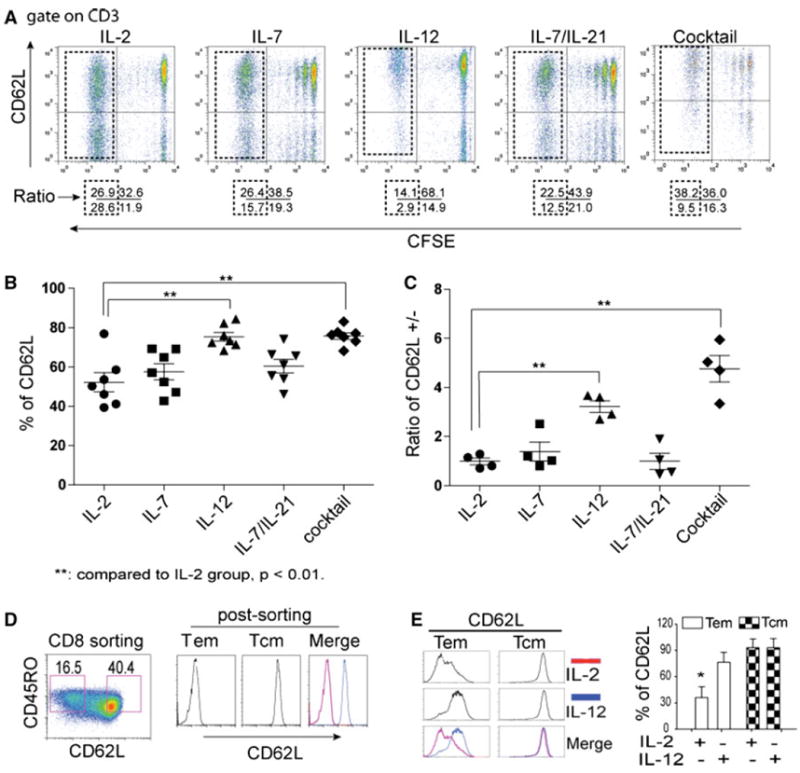
IL-12 is responsible for high levels of CD62L expression. a The effect of cytokines on dynamic expression of CD62L. T cells were stained with CFSE and co-cultured with ttRNA–DC. The proliferation in each group was monitored by CFSE dilution. The dot plots were displayed with the levels of CD62L on y axis and CFSE on x axis. The numbers below each dot plot represent the percentage of cells on each quadrant after 10 days ex vivo culture. The divided subsets were highlighted within the dotted squares. The use of cytokines or their combinations was denoted on top of each dot plot. b Statistical analysis of CD62L expression on the total population. The percentage of CD62L on the total population of T cells including divided and non-divided subsets was plotted. The data represent the average ± SD from 6 experiments. c The ratio of CD62L+ versus CD62L− subsets (CD62L+/−) from divided subsets. The ratio of CD62L+/− from divided T-cell subsets (as highlighted in the dotted squares) from 4 independent experiments was presented as average ± SD. d The sorting of CD62L+ and CD62L− subsets. The isolated CD8 T cells were stimulated with OKT3 and cultured in the presence of IL-2 for 7 days, and sorted using flow cytometry based on CD45RO+CD62L− (Tem) and CD45RO+CD62L+ (Tcm) subsets (left) and post-analyzed based on the expression of CD62L (right). e IL-12 leads to re-expression of CD62L on Tem. After sorting, Tem and Tcm were cultured in the presence of IL-2 (100 IU/ml) or IL-12 (20 ng/ml) for 5 days before the expression of CD62L was analyzed by flow cytometry. The figure on right represents the percentage of CD62L from 3 donors. Student’s t test for mean was performed, asterisks indicate p < 0.01
ttRNA–DC ex vivo-expanded human T cells commit specific recognition and lytic activity
To show the proof-of-principle that ttRNA–DC ex vivo-expanded T cells enable the specific recognition and lysis toward tumor cells, we chose two donors of HLA-A2+/HLA-A2- and electroporated with ttRNA from U87 HLA-A2+ line (Fig. 4a). After 12-day culture, we found the characteristic phenotype (as shown in Figs. 2, 3) that the cocktail culture yielded T cells with high levels of CD62L expression (Fig. 4b). Both culture conditions endowed T cells with the potential to specifically induce IFNγ when co-cultured with U87 line in a HLA-A2+ restricted manner (Fig. 4c). Moreover, U87 ttRNA–DC-expanded autologous T cells irrelevant to culture conditions committed as a specific lysis determined by HLA-A2+ restriction (Fig. 4d). A lesser extent of lysis was seen when T cells were cultured in a cocktail, and we reasoned that a substantial amount of Tn subset in this condition accounted for this phenomenon, which is consistent with our previous observation that Tn subset indeed commits a reduced lytic activity [29]. However, when co-cultured with U87, some background signals were seen in HLA-A2-T cells (Fig. 4c, d) and we did not have an explanation at this time.
Fig. 4.
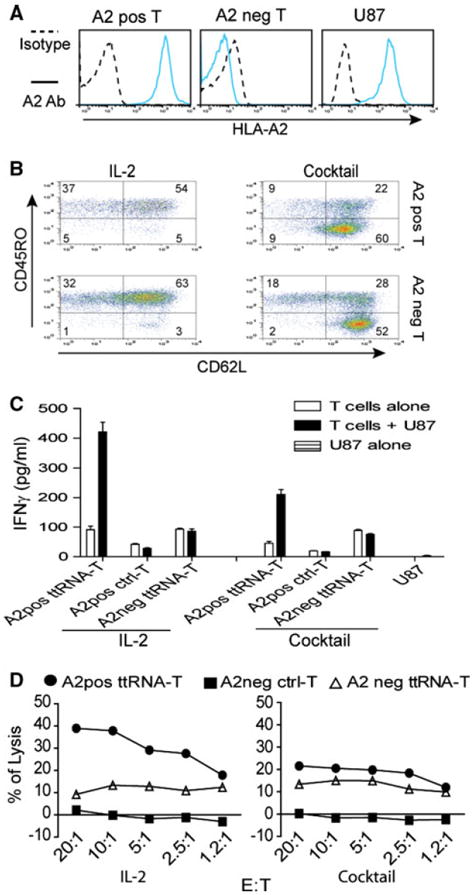
ttRNA–DC ex vivo-expanded human T cells commit specific recognition and lytic activity. a The analysis of HLA-A2 on PBMC and U87 line. HLA-A2 expression in human U87 tumor line and PBMC was measured by fluorescent-conjugated HLA-A2 antibody. For abbreviation, positive was labeled as pos and negative as neg. One representative PBMC from HLA-A2 pos/neg donors was shown. HLA-A2 expression was denoted on left of the image. b The phenotype of ex vivo ttRNA–DC-expanded T cells. Human DC were electroporated with ttRNA from U87 tumor line followed by overnight maturation as described in “Materials and methods” section; the T cells from HLA-A2+/− donors were co-cultured with ttRNA–DC in the presence of IL-2 or a cocktail for 12 days. The phenotype of T cells was analyzed using fluorescent-conjugated CD62L and CD45RO antibodies by flow cytometry. c The specific IFNγ induction. DC from both donors was electroporated with ttRNA or pseudo-electroporated, which were co-cultured with T cells from autologous donors. After 12 days expansion, T cells (2.5 × 105/well) were co-cultured with U87 at a ratio of 10:1, and the levels of IFNγ were measured by ELISA kit. d The specific lysis of U87 tumor line. T cells cultured with IL-2 (left) and a cocktail (right) were co-cultured with U87 cells at different ratio of effector to target (E:T) for 5 h before the lytic assay was performed using CytoTox 96 non-radioactive cytotoxicity assay
OVA–RNA–DC facilitate the antigen-specific T-cell expansion and recognition toward antigen-associated targets
We have shown that ttRNA–DC-expanded T cells are capable of committing specific lysis, and a cocktail culture directly modulate the less-differentiated phenotype; however, we did not have any clues that ttRNA–DC were initiating an antigen-specific expansion of T cells during ex vivo culture. The defined antigens from ttRNA remained elusive, which is hard for us to evaluate the expansion of a definite T-cell subset corresponding to a specific tumor antigen. To validate that ttRNA–DC are indeed driving an antigen-specific expansion, we chose murine OVA-specific T cells from OT-1 mice. When T cells from WT or OT-1 mice were expanded with OVA RNA–DC, we found a specific expansion only occurring at OT-1 OVA tetramer T cells (Fig. 5a). To further illustrate the antigen-specific expansion, we used OVA RNA–DC to expand the mixed T cells from WT and OT-1 mice, and showed the percentage of OVA Tetra+ T cells from one representative culture (Fig. 5b, upper panel) after co-culture with ctrl-DC for 6 days, which basically represented the ratio of initial mixture. However, we saw OVA-antigen-specific T-cell expansion when OVA RNA–DC was used, which was irrelevant to the use of IL-2 or a cocktail (Fig. 5b, lower panel). It seemed an enrichment of OVA Tetra+ T cells from WT mice as well (Fig. 5a, b); however, the extent of expansion was kept to a minimum. To test the antigen-specific recognition, we observed the specific IFNγ induction only in expanded T cells co-cultured with antigen-matched surrogate targets or tumor lines (Fig. 5c). Ctrl-DC-expanded T cells slightly recognized the surrogate targets and tumor lines at a background level, but the other expanded T cells showed an enhanced specific recognition toward antigen-associated targets (Fig. 5c).
Fig. 5.
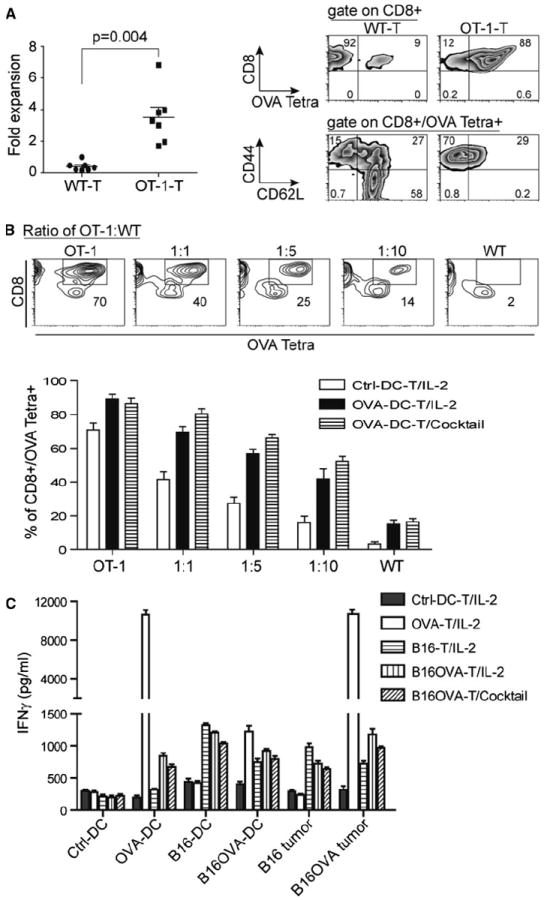
RNA–DC facilitates the antigen-specific T-cell expansion and recognition toward antigen-associated targets. a OVA RNA–DC leads to the defined expansion and enrichment of OVA-antigen-specific T cells. OVA–RNA was in vitro transcribed using pGM4Z vector harboring OVA-expressing cassette. Murine DC were electroporated with OVA RNA followed by overnight maturation; the splenic T cells from WT or OT-1 of C57bl/6 strain were co-cultured with OVA RNA–DC at a ratio of T-cell to DC, 10:1. After 6 days of ex vivo culture in the presence of IL-2 (30 IU/ml), the expansion of T cells was calculated and plotted. The percentage and phenotype of OVA Tetra+ T cells were analyzed using flow cytometry. b The specific expansion of OVA-antigen-specific T cells from a mixed population. DC was cocultured with T cells from OT-1 or WT mice or their mixture at a decreased ratio of OT-1 to WT for 6 days. Upper panel the percentage of OVA Tetra+CD8+ T cells from a representative T-cell culture with ctrl-DC was indicated in squares of each image; lower panel based upon the criteria from upper panel, the percentage of OVA Tetra+CD8+ T cells co-cultured with ctrl-DC or OVA RNA–DC in the presence of IL-2 or cocktail was shown, and the data were plotted as average ± SD from 3 independent experiments. c The specific IFNγ induction occurs only in antigen-associated targets. The different types of RNA–DC-expanded T cells were used as effector cells, and the antigen-associated targets were from B16F10 and B16F10OVA tumor lines, and from DC electroporated with RNA (OVA, B16F10, and B16F10OVA) as surrogate targets. Except the last group cultured with a cocktail, the others were cultured with IL-2. After 7 days expansion, T cells (2.5 × 105/well) were co-cultured with targets at a ratio of 10:1, and the levels of IFNγ were measured by ELISA kit
A cytokine cocktail modulated phenotype of ex vivo-expanded anti-tumor T cells promotes the engraftment in NSG mice
Using the regimen of a defined combination of cytokines as described in Fig. 2, the human T cells were ex vivo-expanded and injected intravenously into the NSG mice. Thirty days later, we killed mice and extracted lymphocytes from lung, spleen, and LN, and we could detect the enhanced engraftment of human T cells in LN from a cytokine cocktail-treated group, and the difference of persistent cells reached statistically significant when we calculated viable human T cells based on flow cytometry analysis (Fig. 6a). The phenotype of expanded T cells before infusion was consistent with previous observation; in that, the use of a cytokine cocktail increased the levels of CD62L and CD27, while the levels of CD25 and CD70 were decreased (Fig. 6a, lower panel).
Fig. 6.
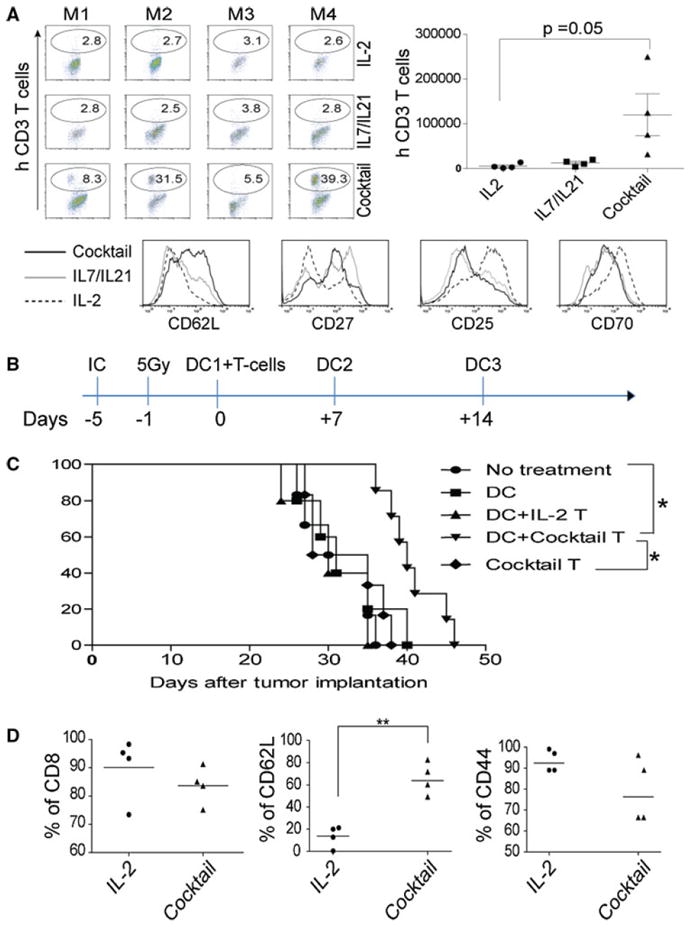
A cytokine cocktail-modulated CD62Lhigh phenotype correlates with persistence and superior anti-tumor activities in murine models. a The enhanced engraftment in LN of NSG mice. After 30 days, LN from each mouse in 3 groups was analyzed by flow cytometry using anti-human CD3 antibody on left (upper panel); the total number of engrafted human T cells were calculated using Fluorospheres, and the average ± SD from 4 mice/group was calculated and plotted on right. M1–M4 on top of each image indicates mice on each individual group. Lower panel T cells before infusion were analyzed using a panel of antibodies; the histograms representing each marker were overlaid, and the lines represent each group of cells was indicated on left. b A cytokine cocktail-modulated CD62Lhigh phenotype of anti-tumor T cells conveys the superior anti-tumor activities in a murine IC tumor model. The therapeutic procedures were schematically illustrated. C57bl/6 mice were implanted with IC tumor using KR158 cells for 5 days before ACT, and 1 day before, ACT mice received 5 Gy total body irradiation. On day 0, 1 × 106 ex vivo-cultured T cells were infused IV followed by ttRNA–DC vaccination ID. After ACT, two additional ttRNA–DC vaccinations were executed on a weekly basis by IP injection. c A cytokine cocktail-modulated T cells lead to a prolonged survival. Mice (7 mice/group) were implanted with IC tumor, and the groups indicate the use of DC alone or combined with different types of T cells were shown on right; the cocktail T group represents T cells cultured with a cocktail in the absence of DC. For survival, log-rank test was performed. Asterisks indicate p < 0.001. d A cytokine cocktail directly modulates the phenotype of ex vivo ttRNA–DC-expanded anti-tumor T cells. The phenotype of ex vivo-cultured anti-tumor T cells was analyzed before infusion using a panel of antibodies; the percentage of CD8, CD62L, and CD44 was plotted from 4 independent experiments
A cytokine cocktail-modulated CD62Lhigh phenotype of anti-tumor T cells conveys the superior anti-tumor activities in a murine IC tumor model
From a translational point of view, it was difficult for us to establish a model to evaluate the in vivo anti-tumor efficacy of these ex vivo-cultured human anti-tumor T cells. Alternatively, we sought to use a syngeneic murine IC tumor model to evaluate the cytokine cocktail-modulated anti-tumor T cells in mediating tumor regression. C57bl/6 mice were IC implanted with KR-158 murine brain tumor line for 5 days, and 1 day before T cells infusion, mice received 5 Gy irradiation and ttRNA–DC vaccination. After infusion of T cells, we executed ttRNA–DC vaccinations twice (Fig. 6b). KR-158 ttRNA–DC was used to expand T cells from murine spleen supplemented with different cytokines as we have optimized in human T cells. Compared to other groups, T cells from a cytokine cocktail culture significantly extended median survival of mice bearing pre-established IC tumor (Fig. 6c). We speculated that the cytokine cocktail-mediated CD62Lhigh phenotype and DC vaccinations were pivotal in mediating tumor regression as either T cells from IL-2 group or cocktail T group (without DC) did not extend median survival at all (Fig. 6c). Moreover, this therapeutic strategy in combining ex vivo cocktail-cultured T cells and ttRNA–DC vaccinations were independently reproducible in two additional experiments (data not shown). Consistent with the data from human T cells, the use of a cytokine cocktail significantly up-regulated the levels of CD62L (Fig. 6d), which might shed new light on how to generate a defined phenotype of anti-tumor T cells ex vivo for better tumor treatment.
Discussion
Immunotherapy using ACT has emerged as one of the most potent therapeutics toward the control of cancer. Currently, there are two major approaches for ACT including genetically re-directed anti-tumor T cells and TIL, which are limited by targeting one tumor-associated antigen and the difficulty for identifying pre-existing anti-tumor T cells. Therefore, we developed a novel methodology by recruiting ttRNA–DC–T platform to expand and enrich anti-tumor T cells using PBL from brain tumor patients and from murine IC tumor model, and optimized the culture conditions with a cytokine cocktail, which yielded T cells with a CD62Lhigh phenotype exhibiting a better engraftment in NSG and superior anti-tumor activities in a murine IC tumor model.
In response to pathogens or tumor antigens, the long-lived self-renewing memory lymphocytes are hallmark of the adaptive immune system [37, 38]. The memory compartment of T cells is heterogeneous and has been classified into two subsets based on the expression of lymph-node homing molecules CD62L and CCR7 [39]. Central memory T cells (Tcm) with high levels of CD62L and CCR7 are believed to be the stem cell-like memory subset, while the effector memory T cells (Tem) with low levels of these two molecules are proven to be terminally differentiated [40, 41]. Interestingly, the in vitro TCR engineered anti-tumor T cells exhibit distinct subsets mimicking in vivo antigen-experienced T cells, i.e., after ex vivo expansion differentiate into Tcm and Tem-like subsets [29]. In addition, ex vivo-cultured Tcm-like cells possess the feature to be long-term engrafted in NSG mice [29] and in primates [42]. In general, the high levels of CD62L expression on Tcm cells [17], naïve subset of PBL [43], and a human memory CD8+ T-cell subset [44] are better endowed phenotype of T cells possessing potent in vivo anti-tumor treatment efficacy.
Using ttRNA–DC in conjunction with a cytokine cocktail to ex vivo generate CD62Lhigh anti-tumor T cells provide a novel methodology toward the effective treatment of cancer. However, how do cytokines modulate the differentiation of ex vivo-expanded anti-tumor T cells in ttRNA–DC platform has not been systematically investigated. IL-2 is by far the best growth factor for ex vivo culture of T cells; however, it drives ex vivo-cultured T cells toward fully differentiated Tem phenotype. Tem cells possess the great potential for IFNγ secretion when encountering the associated antigens in vitro; however, Tem cells fail to engraft in xenograft model (Fig. 6a) and show a lesser potency to eradicate tumor in vivo [14, 17, 29, 30], which we believe to be the key factor to explain why IL-2-cultured T cells fail to control the tumor growth (Fig. 6). In ttRNA–DC–T system, compared to the use of IL-2, our perception toward the functions of each cytokine has been re-defined, in that IL-7 and IL-21 significantly increased the percentage of CD27+ T cells and IL-7 led to a homeostatic proliferation consisting of multiple precursors, while IL-12 constantly maintained the high levels of CD62L expression in both human and murine T cells. In this study, we focused on how cytokines modulate the CD62Lhigh phenotype of these anti-tumor T cells and the concomitant superior anti-tumor activities in a pre-clinical IC tumor model; however, the biological function of high percentage of CD4+CD25highFoxp3+ T cells from the use of IL-2 has not been thoroughly investigated. Therefore, IL-2-cultured anti-tumor T cells, although exhibited specific tumor recognition in vitro, failed to execute in vivo anti-tumor activities in this IC model, which might be partially due to the CD62Llow phenotype or the higher incidence of TRegs.
Although we proposed a novel ttRNA–DC–T platform which differs from the current genetic engineering approaches [11, 12], the in vivo anti-tumor efficacy of this system relies upon the subsequent ttRNA–DC vaccinations. We reasoned that the subsequent vaccinations provided homeostatic environment favoring the persistence of ex vivo adoptively transferred T cells, which eventually migrate to the tumor sites and kill the tumor cells. It is consistent with well-reported pmel model [45], where the in vivo anti-tumor efficacy of adoptively transferred T cells is heavily dependent upon three factors, i.e., lymphodepletion, subsequent vaccination, and the infusion of IL-2 [14].
CD62L acts as a “homing receptor” for lymphocytes to enter secondary lymphoid tissues via high-endothelial venules by ligands presented on endothelial cells. The genes-encoding CD62L for both humans and mice are arranged in tandem along chromosome 1 that reflect their common evolutionary origin [46], and the expressed proteins are highly conserved [47]. The ex vivo-cultured T cells with CD62Lhigh phenotype engrafted significantly better in LN of NSG mice than the cells grown in IL-2. Most importantly, we found a CD62Lhigh phenotype resulting from the use of a cytokine cocktail conveyed superior anti-tumor activities in an invasive, less-stringent astrocytoma murine IC model. To mimic the in vivo environment for ex vivo generating anti-tumor T cells, it is unlikely that a single cytokine can bestow onto T cells with endowed features associated with potent in vivo anti-tumor efficacy. With more and more cytokines becoming clinically available, the use of other cytokines instead of IL-2 to culture and generate anti-tumor T cells is emerging as new horizons; in that, we could directly modulate the endowed phenotype of anti-tumor T cells conveying the superior in vivo anti-tumor activities. The use of a cytokine–cocktail in this platform consistently yielded T-cell subsets exhibiting CD62Lhigh-Tcm and CD62Lhigh-Tn phenotype; at present, we are continuously investigating which subset is the “real player” in mediating tumor regression. Moreover, beyond optimizing the culture conditions with cytokines, we are further modulating the ttRNA–DC–T system by blocking negative signals between DC (PD-L1) and T cells (PD-1) [48, 49]. We believe this methodological advancement to use ttRNA–DC to ex vivo generate anti-tumor T cells would become a new epoch of medicine in cancer patient management.
Acknowledgments
We thank Dr. Kendra Congdon for proofreading of the manuscript. This study was supported by NIH grants (5P50-CA108786, 3R21CA132891-02S1, 5R01-CA135272-05, 5R21NS 067975-02, 5R21NS067980-02, 5R21NS068057-02, 3R01CA135272-02S1, R25-NS065731, 5P50-NS020023-29, 1P01-CA154291-01A1), and by Accelerate Brain Cancer Cure, National Brain Tumor Society, American Brain Tumor Association, Pediatric Brain Tumor Foundation of the United States, Goldhirsh Foundation, Brain Tumor Society, Ivy Foundation DTRI, Duke University Biomarker Factory Duke Cancer Institute, Duke Chandran.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Shicheng Yang, Division of Neurosurgery, Department of Surgery, Brain Tumor Immunotherapy Program, Duke University Medical Center, 303 Research Drive, 220 Sands Building, DUMC 3050, Durham, NC 27710, USA.
Gary E. Archer, Division of Neurosurgery, Department of Surgery, Brain Tumor Immunotherapy Program, Duke University Medical Center, 303 Research Drive, 220 Sands Building, DUMC 3050, Durham, NC 27710, USA
Catherine E. Flores, Division of Neurosurgery, Department of Surgery, Brain Tumor Immunotherapy Program, Duke University Medical Center, 303 Research Drive, 220 Sands Building, DUMC 3050, Durham, NC 27710, USA
Duane A. Mitchell, Division of Neurosurgery, Department of Surgery, Brain Tumor Immunotherapy Program, Duke University Medical Center, 303 Research Drive, 220 Sands Building, DUMC 3050, Durham, NC 27710, USA
John H. Sampson, Email: john.sampson@dm.duke.edu, Division of Neurosurgery, Department of Surgery, Brain Tumor Immunotherapy Program, Duke University Medical Center, 303 Research Drive, 220 Sands Building, DUMC 3050, Durham, NC 27710, USA.
References
- 1.Steinman RM. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. Mt Sinai J Med. 2001;68(3):160–166. [PubMed] [Google Scholar]
- 2.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 3.Iwami K, Shimato S, Ohno M, Okada H, Nakahara N, Sato Y, Yoshida J, Suzuki S, Nishikawa H, Shiku H, Natsume A, Wakabayashi T. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy. 2012;14(6):733–742. doi: 10.3109/14653249.2012.666633. [DOI] [PubMed] [Google Scholar]
- 4.Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, Yong WH, Incardona F, Thompson RC, Riedinger MS, Zhang W, Prins RM, Black KL. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61(3):842–847. [PubMed] [Google Scholar]
- 5.Liau LM, Black KL, Prins RM, Sykes SN, DiPatre PL, Cloughesy TF, Becker DP, Bronstein JM. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg. 1999;90(6):1115–1124. doi: 10.3171/jns.1999.90.6.1115. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell DA, Nair SK. RNA-transfected dendritic cells in cancer immunotherapy. J Clin Invest. 2000;106(9):1065–1069. doi: 10.1172/JCI11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, Sawaya R, Lang FF, Heimberger AB. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9(1):67–78. doi: 10.1158/1535-7163.MCT-09-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113–1125. doi: 10.1093/neuonc/noq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei J, Wu A, Kong LY, Wang Y, Fuller G, Fokt I, Melillo G, Priebe W, Heimberger AB. Hypoxia potentiates glioma-mediated immunosuppression. PLoS One. 2011;6(1):e16195. doi: 10.1371/journal.pone.0016195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26(32):5233–5239. doi: 10.1200/JCO.2008.16.5449. JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang I, Tihan T, Han SJ, Wrensch MR, Wiencke J, Sughrue ME, Parsa AT. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J Clin Neurosci Off J Neurosurg Soc Australas. 2010;17(11):1381–1385. doi: 10.1016/j.jocn.2010.03.031. j.jocn.2010.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115(6):1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gattinoni L, Klebanoff CA, Restifo NP. Pharmacologic induction of CD8+ T cell memory: better living through chemistry. Sci Transl Med. 2009 doi: 10.1126/scitranslmed.3000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, Klebanoff CA, Rosenberg SA, Leonard WJ, Restifo NP. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111(11):5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior anti-tumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. 2005;102(27):9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decaluwe H, Taillardet M, Corcuff E, Munitic I, Law HK, Rocha B, Riviere Y, Di Santo JP. Gamma(c) deficiency precludes CD8+ T cell memory despite formation of potent T cell effectors. Proc Natl Acad Sci USA. 2010;107(20):9311–9316. doi: 10.1073/pnas.0913729107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heninger AK, Theil A, Wilhelm C, Petzold C, Huebel N, Kretschmer K, Bonifacio E, Monti P. IL-7 abrogates suppressive activity of human CD4+CD25+FOXP3+ regulatory T cells and allows expansion of alloreactive and autoreactive T cells. J Immunol. 2012 doi: 10.4049/jimmunol.1201286. [DOI] [PubMed] [Google Scholar]
- 20.Lee JB, Lee KA, Chang J. Phenotypic changes induced by IL-12 priming regulate effector and memory CD8 T cell differentiation. Int Immunol. 2007;19(9):1039–1048. doi: 10.1093/intimm/dxm072. [DOI] [PubMed] [Google Scholar]
- 21.van Wely CA, Beverley PC, Brett SJ, Britten CJ, Tite JP. Expression of L-selectin on Th1 cells is regulated by IL-12. J Immunol. 1999;163(3):1214–1221. [PubMed] [Google Scholar]
- 22.Ye Z, Xu S, Moyana T, Yang J, Xiang J. Defect of CD8+ memory T cells developed in absence of IL-12 priming for secondary expansion. Cell Mol Immunol. 2008;5(2):147–152. doi: 10.1038/cmi.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz-Montero CM, El Naggar S, Al Khami A, El Naggar R, Montero AJ, Cole DJ, Salem ML. Priming of naive CD8+ T cells in the presence of IL-12 selectively enhances the survival of CD8+ CD62Lhi cells and results in superior anti-tumor activity in a tolerogenic murine model. Cancer Immunol Immunother CII. 2008;57(4):563–572. doi: 10.1007/s00262-007-0394-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115(17):3508–3519. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175(4):2261–2269. doi: 10.4049/jimmunol.175.4.2261. [DOI] [PubMed] [Google Scholar]
- 26.Albrecht J, Frey M, Teschner D, Carbol A, Theobald M, Herr W, Distler E. IL-21-treated naive CD45RA+CD8+ T cells represent a reliable source for producing leukemia-reactive cytotoxic T lymphocytes with high proliferative potential and early differentiation phenotype. Cancer Immunol Immunother CII. 2011;60(2):235–248. doi: 10.1007/s00262-010-0936-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, Sherry RM. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16(19):4892–4898. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blankenhorn EP, Stranford SA, Smith PD, Hickey WF. Genetic differences in the T cell receptor alleles of LEW rats and their encephalomyelitis-resistant derivative, LER, and their impact on the inheritance of EAE resistance. Eur J Immunol. 1991;21(9):2033–2041. doi: 10.1002/eji.1830210910. [DOI] [PubMed] [Google Scholar]
- 29.Yang S, Gattinoni L, Liu F, Ji Y, Yu Z, Restifo NP, Rosenberg SA, Morgan RA. In vitro generated anti-tumor T lymphocytes exhibit distinct subsets mimicking in vivo antigen-experienced cells. Cancer Immunol Immunother CII. 2011;60(5):739–749. doi: 10.1007/s00262-011-0977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, Rosenberg SA, Morgan RA. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother CII. 2013;62(4):727–736. doi: 10.1007/s00262-012-1378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romani N, Gruner S, Brang D, Kampgen E, Lenz A, Trockenbacher B, Konwalinka G, Fritsch PO, Steinman RM, Schuler G. Proliferating dendritic cell progenitors in human blood. J Exp Med. 1994;180(1):83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou JP, Yamamoto N, Fujii T, Takenaka H, Kobayashi M, Herrmann SH, Wolf SF, Fujiwara H, Hamaoka T. Systemic administration of rIL-12 induces complete tumor regression and protective immunity: response is correlated with a striking reversal of suppressed IFN-gamma production by anti-tumor T cells. Int Immunol. 1995;7(7):1135–1145. doi: 10.1093/intimm/7.7.1135. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, Morgan RA. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19(4):751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eisenring M, vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol. 2010;11(11):1030–1038. doi: 10.1038/ni.1947. [DOI] [PubMed] [Google Scholar]
- 36.Yang S, Dudley ME, Rosenberg SA, Morgan RA. A simplified method for the clinical-scale generation of central memory-like CD8+ T cells after transduction with lentiviral vectors encoding antitumor antigen T-cell receptors. J Immunother. 2010;33(6):648–658. doi: 10.1097/CJI.0b013e3181e311cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22(2):223–230. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 40.Fearon DT, Manders P, Wagner SD. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. 2001;293(5528):248–250. doi: 10.1126/science.1062589. [DOI] [PubMed] [Google Scholar]
- 41.Stemberger C, Neuenhahn M, Gebhardt FE, Schiemann M, Buchholz VR, Busch DH. Stem cell-like plasticity of naive and distinct memory CD8+ T cell subsets. Semin Immunol. 2009;21(2):62–68. doi: 10.1016/j.smim.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 42.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, Muranski P, Palmer DC, Scott CD, Morgan RA, Robbins PF, Rosenberg SA, Restifo NP. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117(3):808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins T, Williams A, Johnston GI, Kim J, Eddy R, Shows T, Gimbrone MA, Jr, Bevilacqua MP. Structure and chromosomal location of the gene for endothelial-leukocyte adhesion molecule 1. J Biol Chem. 1991;266(4):2466–2473. [PubMed] [Google Scholar]
- 47.Zhao L, Shey M, Farnsworth M, Dailey MO. Regulation of membrane metalloproteolytic cleavage of l-selectin (CD62l) by the epidermal growth factor domain. J Biol Chem. 2001;276(33):30631–30640. doi: 10.1074/jbc.M103748200. [DOI] [PubMed] [Google Scholar]
- 48.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, Levine BL, June CH, Medin JA, Fowler DH. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011;3(111):111–120. doi: 10.1126/scitranslmed.3003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]